Submitted:
03 June 2026
Posted:
03 June 2026
You are already at the latest version
Abstract
Breast cancer (BC) is a major global public health problem. Classical therapies have limited success on the treatment of BC, therefore new therapeutic options are needed. Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules that represent a revolutionary class of new drug candidates because they induce the degradation of harmful, undruggable proteins by activating the ubiquitination machinery of cells. Their unique mechanism of action offers several advantages over conventional drugs, but also disadvantages, as most of them are large molecules with unfavorable pharmacokinetic properties, which limits their bioavailability. Vepdegestrant (VeppanuTM) is an orally administered, estrogen receptor (ER) targeting chimera that was approved by the FDA on May 1, 2026, for the treatment of adults with ESR1-mutated advanced or metastatic breast cancer. Thus, vepdegestrant became the first-ever approved PROTAC drug. In this article, we briefly summarize the structure, mechanism of action, and key available pharmacokinetic and pharmacological data of vepdegestrant.
Keywords:
vepdegestrant
; PROTAC
; breast cancer
; VeppanuTM
; ARV 471
; targeted tumor therapy
; estrogen receptor
; protein degradation
; SERD
; endocrine therapy
1. Introduction
Breast cancer (BC) is the most frequent type of cancer among women and hormone receptor positive (HR+) tumors make up the majority of breast cancer cases[1]. Estrogen receptor alpha (ERα) is a major driver of growth in hormone receptor-positive breast cancer, making ER one of the most validated targets in endocrine therapy[2]. Traditional approaches such as aromatase inhibitors (e.g. exemestane, formestane) and selective estrogen receptor modulators (SERMs, e.g. tamoxifen, toremifene) can suppress estrogen signaling, but resistance often emerges through ER mutations, pathway reactivation, or ligand-independent ER activity[3]. This has pushed the field toward approaches that degrade ER rather than merely inhibit it[4].
The first selective estrogen receptor degrader (SERD) was fulvestrant, an estradiol-derivative with a large 7α-pentafluoroalkylsulfinyl side chain which allowed it to not only antagonize the target receptor, but also destabilize its structure, leading to its degradation by the cell machinery[5] (Figure 1). While this made a much more drastic reduction in estrogen signaling reduction possible, its suboptimal pharmacokinetic properties posed problems, requiring intramuscular injections for efficacy[6]. Newer SERDs, like the recently approved imlunestrant and elacestrant, were designed for oral use, allowing for much more convenient dosing, and are also effective against ESR1-mutated, endocrine-resistant breast cancer[7].
Fulvestrant, and more generally SERDs, represent an early iteration of an emerging approach in drug design: Targeted Protein Degradation (TPD), which shifts the focus from designing inhibitors to designing molecules capable of hijacking different proteolytic mechanisms for the degradation of selected proteins[8]. Many different modalities are known by now, each with their own sets of scopes, advantages and disadvantages, but the most prominent by far is the PROTAC (Proteolysis Targeting Chimera) modality[9].
PROTACs are heterobifunctional molecules consisting of an E3-ligase ligand and a target protein ligand connected by a linker (Figure 2). E3 ligases are a large family of enzymes responsible for the ubiquitination of proteins, which can result in changes in protein localization, activity, interactions, or, most importantly, its degradation by the 26S proteasome. PROTACs hijack this system by simultaneously binding to both the target protein and a selected E3 ligase, creating a ternary complex: this forced proximity enables the polyubiquitination and subsequent degradation of the target protein[9]. The PROTAC molecule can the participate in the formation of further ternary complexes, acting catalytically unlike conventional inhibitors, which generally act stoichiometrically, with each molecule inhibiting a single protein [10].
Besides the catalytic mechanism and the complete knock-out of the target protein, PROTACs offer other advantages: they can be used against targets previously described as “undruggable” (proteins without good binding pockets), they can overcome different resistances and can offer higher target selectivity over conventional inhibitors by ternary complex optimization[11].
Dozens of PROTACs with different targets are currently in clinical trials[12], and fittingly, the first to recently gain FDA-approval was a new SERD designed to overcome resistance mechanisms that limited earlier endocrine therapies, vepdegestrant.
2. Vepdegestrant
2.1. Chemistry
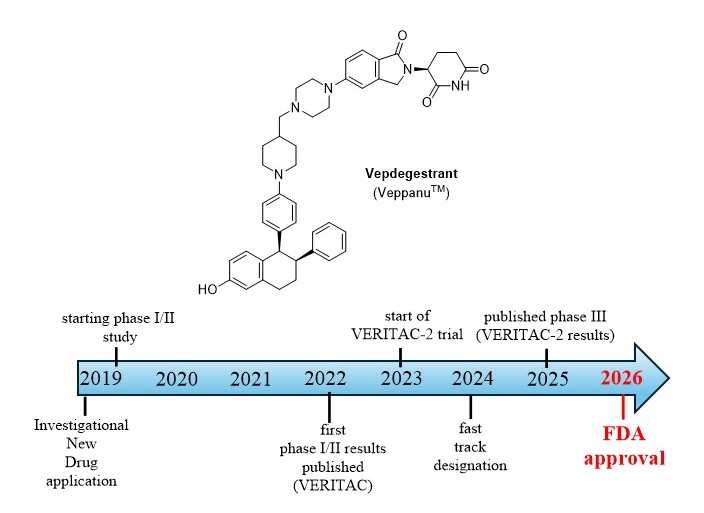
Similarly to other PROTACs, vepdegestrant consists of 3 main parts: a target protein (ER) binding part, a linker and a ligase binding part. The first component, responsible for binding to the ER is structurally similar to the 3rd generation SERM, lasofoxifene, which contains a 6-hydroxi-tetrahydronaphtalene skeleton. The linker consists of two heterocycles, a piperidine and a piperazine, making the molecules structure more rigid, which increases efficacy and improves pharmacokinetic properties. The cereblon E3 ligase (CRBN) binding part is a glutarimide-isoindolinone derivative, showing similarities with thalidomide and lenalidomide[13].
Figure 3.
Structural elements of vepdegestrant.

2.2. Pharmacodynamics and Preclinical Results
Vepdegestrant (PF-07850327 or ARV 471) simultaneously binds to ERα and CRBN, forming a ternary complex. As a result of the complex formation, CRBN ubiquitinates ERα, which leads to its degradation by the 26S proteasome. In the lack of ERα, the estrogen-dependent signaling cannot function properly, thereby blocking the growth of estrogen dependent tumors. After protein degradation, vepdegestrant is released and can bind to another target molecule. A major advantage of this mechanism of action, that it is effective against mutations such as Y537S or D538G,, which provide resistance to conventional anti-ER strategies (e.g. Y537S or D538G)[13]. The mechanism of action was examined and demonstrated in multiple levels. The formation of the ternary complex was confirmed by CRBN:probe displacement assay. The ternary complex CRBN-DDB1:ER-LBD:vepdegestrant was isolated, and the structure was studied by single-particle cryo-EM. The dominance of ER degradation over ER inhibition was investigated using A5927 (an E3 inactive ER-degrading PROTAC), which showed a tenfold lower activity (IC50 = 33.23 nM/L vs 3.057 nM/L) compared to vepdegestrant (luciferase target engagement assay). Addition of other CRBN-binding (lenalidomide), or ER-binding (lasofoxifene) molecules in 10-fold excess inhibited ER degradation induced by vepdegestrant, as did the addition of proteasome inhibitor (carfilzomib), supporting the proteasome-induced degradation mechanism[14]. In cell-free assay, vepdegestrant bound to recombinant ER with an IC50 of 0.99 nmol/L and a Ki of 0.28 nmol/L. These results are comparable to those of diethylstilbestrol and lasofoxifene[14].
In T47D-KBluc cellular luciferase assay, vepdegestrant dose-dependently decreased estradiol-bound ER/ERE-driven luciferase expression after 24 h of treatment with an IC50 value of 1.1 nmol. Vepdegestrant dose-dependently induced loss of wt ERα in MC7F cells (DC50 = 0.9 nM, Dmax = 95%), achieving above 80% reduction in ERα at 100 nM within 4 h. Western blotting revealed, that ER level was also decreased in BT474, CAMA-1, ZR-75-1, and T47D cell lines (patient derived BC cells). Vepdegestrant reduced the levels of clinically relevant mutant ERs (Y537S, D538G, Y537C, Y537N, E380Q, L536P, V422del) in T47D cells[14].
Vepdegestrant showed remarkable selectivity towards ER. Examination of MCF7 proteome after vepdegestrant treatment (7 h, 10 nmol/L) showed that the main decrease was in the level of ER, in addition PGR (progesterone receptor) and growth regulating estrogen receptor binding 1 (GREB1) levels also being decreased. The observed mild vepdegestrant-mediated degradation of PGR (IC50 of 17 nmol/L compared to 1nmol/L IC50 value against ER) may be due in part to transcriptional downregulation of the ER responder genes, as both PGR and GREB1 RNA levels were reduced[14].
The GI50 value during 5 days was 3.3 nM and 4.5 nM in MCF7 and T47D cells, respectively, with wild-type ER. T47D cells expressing Y537S and D538G were slightly less sensitive with 8 and 5.7 nmol/L GI50 values, respectively[13,14].
Vepdegestrant at 10 mg/kg (administered 3 times daily) reduced tumor ER levels by at least 90% in MCF7-bearing mice. Vepdegestrant at 30 mg/kg did not show any inherent ER agonist effect but decreased ER levels in uterine tissue by 76% (fulvestrant showed a 65% decrease in the same assay). Vepdegestrant showed robust tumor growth inhibition (TGI) in MCF7 tumor xenograft mice models (85%, 98% and 120% TGI at 3, 10 and 30 mg/kg daily dose respectively). Above 94% ER level, decrease in tumor cell lysate was observed. Vepdegestrant 10 and 30 mg/kg daily and fulvestrant 200 mg/kg (twice weekly for 2 weeks and once weekly for 2 weeks) was administered for 28 days in a Y537S mutant xenograft model. TGI was 99% and 107% for the 2 doses of vepdegestrant and 62% for fulvestrant. ER decrease was 88% for 30 mg/kg vepdegestrant and 63% for fulvestrant, respectively. Vepdegestrant was also active against palbociclib- and abemaciclib-resistant tumors and showed synergy with CDK4/6, mTOR and PI3K inhibitors[14].
2.3. Pharmacokinetics, Stability and Interactions
Due to their dual targeting structure, PROTACs are inherently large, complex molecules, with high molecular weight, many rotatable bonds and potential cleavage sites during metabolism. PROTACs generally do not comply with Lipinski’s rule of five and Veber’s rule, therefore they are expected to have poor oral bioavailability[15]. Due to the aforementioned issues, pharmacokinetics (PK) is a crucial aspect of PROTAC based drug candidates, therefore PK properties of vepdegestrant have been extensively studied [15].
A sensitive LC-MS/MS method was developed and validated by Niessen et al. for the quantification of vepdegestrant in rat plasma for stability and PK studies. Vepdegestrant was shown to be stable in rat plasma after 20 min of storage at room temperature in the dark (slight instability was detected, when exposed directly to light), and after 7 weeks of storage at -20 oC. A methanolic solution of vepdegestrant was also stable at -20 oC for 8 weeks. After longer storage at room temperature, cleavage of the glutarimide ring is likely to occur. After a single i.v. dose of 1 mg/kg vepdegestrant, the AUC0-6h was 585.4 ± 43.82 h•ng/mL, while an intraduodenal bolus administration, only 73.3 ± 3.8 h•ng/mL AUC0-6h was achieved, indicating low bioavailability (F = 12.5 ± 0.6%)[16].
In another publication, they demonstrated that vepdegestrant undergoes enzymatic degradation in diluted rat intestinal fluid in vitro, in which serine protease probably plays an important role, as adding serine protease inhibitor doubled the half-life (from 5.9±3.8 h to 13.9±4.5 h). In rat serum, vepdegestrant t1/2 was measured to be 3.5±1.5 h. Effective intestinal permeability (Peff) of vepdegestrant was measured in the presence of serine protease inhibitor. Without ketoconazole, Peff was 0.23 ±0.08 x 10 -4 cm/s, addition of ketoconazole increased it more than twofold (to 0.53 ±0.15 x 10 -4 cm/s). Since ketoconazole is a P-gp inhibitor, the results suggest a major role of P-gp mediated efflux in the low intestinal permeability of vepdegestrant. After a single dose (1 mg/kg) intraduodenal (id) administration, the Cmax of vepdegestrant was 18.0±8.1 ng/mL, the tmax was 1h and the AUC0-6h was 74.4 ±12.2 h•ng/mL. Administration of the P-gp inhibitor ketoconazole (vepdegestrant Cmax= 25.9 ± 13.1 ng/mL, tmax = 1.5 h, AUC0-6h =124.4 ± 30.9 h•ng/mL) or encequidar (Cmax= 23.4 ± 8.0 ng/mL, tmax = 2.0 h, AUC0-6h = 109.0 ± 15.3 h•ng/mL) increased these values, however the increase in relative bioavailability was nonsignificant. Ketoconazole did not change the plasma AUC of vepdegestrant after i.v. administration. The concentration of vepdegestrant was higher in bile than in plasma, indicating that it is either eliminated with bile, or undergoes enterohepatic circulation. The main elimination pathway is degradation by plasma hydrolases, while CYP3A plays a minor role in elimination[15].A Korean group developed an HPLC-MS/MS method for the quantification of vepdegestrant in biological samples. The stability of vepdegestrant showed pH-dependence, as in acidic (pH = 2 puffer) solution, it remained at 82.6 % for 2 h at 37 oC. However, at pH 4-10, the amount of vepdegestrant decreased to 2.25-12.8% within 2 hours. Considering that the adsorption of vepdegestrant to the container surface is also pH/dependent, the decreased amount of vepdegestrant between pH 2-8 can be explained at least in part by adsorption. In mouse and rat plasma, vepdegestrant was stable for 4 h at 4-37 oC. This higher stability possibly can be explained by plasma protein binding, which also prevents adsorption to the container surface. Vepdegestrant showed high stability in dog, mouse and human microsomes, but low-moderate stability in rat microsomes (32.2% remained after 60 mint incubation). After i.v. administration of 2 mg/kg, clearance (CL) was 313.3 ± 44.2 mL/h/kg, the AUCinf was 6.507 ± 1.057 µg/h/mL in mice. In rats, the CL was 1053 ±49mL/h/kg and AUCinf was 1.902 ± 0.090 µg/h/mL, and the t1/2 was 3.970 ± 0.284 h. After 5 mg/kg oral dose, the AUCinf was 2.913 ± 0.707 µg/h/mL, the t1/2 was 3.637 ± 1.399 h in mice, and these values were 1.147 ± 0.446 µg/h/mL and 4.068 ± 0.418 h in rats respectively. The F value (bioavailability %) was slightly higher in rats, than in mice (24.12 ± 9.39% vs 17.91 ± 4.35%). The CL was lower in mice, than in rats, which is expected, based on the metabolic stability tests mentioned above. The possibility of an intestinal first pass effect was also raised[17].
Vepdegestrant shows high protein binding in human and rat plasma, hepatocytes and liver microsomes. After incubation with hepatocytes, hepatic extraction ratios were 69.22% and 41.47% in humans and rats, respectively, while they were 27.62% and 48.63% after incubation with liver microsomes. These data indicate low-moderate hepatic clearance, which is consistent with other results, suggesting that the major metabolic pathway is protease-mediated degradation. After oral administration of 20 mg/kg 14C-labeled vepdegestrant, the tmax of total radioactivity (TRA) was 2.67 h, the Cmax was 2,197 ng eq./mL, the AUC0-∞ was 25,036 ng eq./ (mL*h), and the t1/2 was 12.2 h. These data are not comparable with previous results due to the different conditions (e.g. dose). Also, there were differences between the PK of the parent drug and the radiolabeled derivative. For example, vepdegestrant showed higher AUC0-∞ and t1/2 in female rats than in males, while no difference of these values was observed in TRA. More than 90% of radioactive substances excreted through feces, with <2% urine excretion. The radioactive substances were distributed widely among tissues. After 48 h, the highest radioactivity was measured in the eye, indicating potential safety issues. Faster hepatic metabolism was observed in female rats. The absorption of vepdegestrant was slower in fed rats than in fasted ones, but the absorbed amount was higher. Eleven metabolites were identified, the main metabolites being oxidized, hydrolized and glucuronidated derivatives. Cleavage of the linker was also observed[18].
Electron-activated dissociation HRMS was used to identify vepdegestrant metabolits. After incubation with dog microsomes, 12 phase I metabolites and glucuronides were detected. The main identified biotransformations were: piperazine-N-dealkylation, hydrolysis of the glutarimide ring, phenol-O-glucuronidation, piperidine oxidation and piperidine lactam formation[19].
In the oxidative metabolism of vepdegestrant, CYP3A4 plays a maior role. Therefore, in the NCT06005688 phase I study, the effect of CYP3A4 inducer (carbamazepine) on vepdegestrant PK was investigated in 12 healthy adult male participants. Administration of multiple doses of carbamazepin after a single dose of vepdegestrant decreased the AUCinf of vepdegestrant to 64.1% and the Cmax to 80.2% compared to vepdegestrant alone. The Tmax was 6.0 h in both cases, and the t1/2 was decreased from 50.1 h to 45.4 h with carbamazepine. These results indicate that CYP3A4 plays a role, however not a major role, in vepdegestrant elimination[20].
In the NCT05538312 phase I study, the effect of CYP3A4 inhibitor itraconazole on the PK of vepdegestrant was investigated in 12 participants. AUCinf was increased by 68.9%, Cmax was increased by 52.2%. t1/2 was increased from 42.6 h to 61.6 h, but this can be a consequence of unequal sampling. These changes are significant, however much smaller than those observed for drugs primarily eliminated by CYP3A4, supporting previous statements that although vepdegestrant is a substrate of CYP3A4, it is not the primary route of elimination. These changes in PK are unlikely to affect the safety of vepdegestrant[21]. It is possible that these changes are due to the P-gp inhibitory effect of itraconazole, as Niessen et al. achieved similar PK changes, using P-gp inhibitors[15,22].
Based on in vitro results, it was assumed that vepdegestrant is an inhibitor of P-gp and breast cancer resistance protein, BCRP. The effect of vepdegestrant on the PK of dabigatran, a P-gp substrate, was studied in a phase I study in 24 healthy participants (NCT05673889). Vepdegestrant increased the AUCinf of dabigatran etexilate by 97.8% and the Cmax by 92.2%. The effect of vepdegestrant on the PK of rosuvastatin, a BCRP substrate, was also studied in on a total of 12 participants (NCT05652660). The AUCinf and Cmax of rosuvastatin was also increased moderately (16.9% and 120.5% respectively[23].
The interaction between vepdegestrant and esomeprazole (NCT06275841) and midazolam (NCT06256510) were also studied[13].
In Japanese patients, median Tmax was 4.74 and 4.69 h after a single dose and after multiple QD (once daily) doses, respectively (200 mg daily). Cmax was 630.9 ng/mL after a single dose and 1056 ng/mL after multiple doses. AUC24 was 10.40 ng∙hr/mL after single dose and 18.31 ng∙hr/mL after multiple doses. The accumulation ratio Rac was 1.76 and he effective elimination half-life (t1/2eff) was 20.2 h[24].
2.4. Efficacy in Clinical Trials
In a phase I dose escalation study (NCT04072952), 83 previously treated patients with ER+HER2- advanced breast cancer received 30-700 mg of vepdegestrant daily. A reduction in mutant ESR1 circulating tumor DNA was observed. The clinical benefit rate was 37% after 24 weeks[25]. In the phase II cohort extension of the study (called VERITAC) 2 doses (200 mg and 500 mg QD) were tested. The clinical benefit rate (CBR) was 37.1% (n = 35), and the objective response rate was 8.3% (n = 24). Median progression-free survival was 3.5 months in all evaluable patients. Because of comparable efficacy, the 200 mg daily dose was chosen for further evaluation[26,27,28]. In the phase Ib cohort of the study, the combination of vepdegestrant-palbociclib (a CDK4/6 inhibitor) was investigated in 46 patients. 180-500 mg of vepdegestrant and 125 mg of palbociclib were administered QD for 21 days followed by 7 days without treatment, setting up a 28day cycle. The CBR was 63%, indicating a clinical benefit of this combination as expected based on preclinical results. The objective response rate (n=31) was 41.9%[29].
In the NCT05463952 phase I trial, 6 female Japanese patients with ER+ HER2- advanced breast cancer received 200 mg once daily dose of vepdegestrant for 6-28 weeks. Two patients had stable disease at week 24 and four patients had disease progression. Limitations of the study are the small sample size, lack of control, short follow-up period, and the heavily pretreated patients[24].
VERITAC-2 (NCT05654623) is a global, multicenter, randomized (randomization was stratified according to ESR1 mutation status and presence or absence of visceral disease), unblinded, open label phase III trial, comparing the efficacy and safety of vepdegestrant and a well-known SERD, fulvestrant, in ER+, HER2- advanced BC, after CDK4/6 inhibitor + endocrin therapy. Median progression-free survival as a primary endpoint was assesed by blinded independent central review for patients with ESR1 mutations and total patient groups. 200 mg vepdegestrant was per os applied QD in each 28-day cycle to 313 patients (136 with ESR1 mutations). Fulvestrant 500 mg was administered intramuscularly on the 1st and 15th days of cycle 1 and on the 1st day of each following cycles to 311 patients (134 with ESR1 mutations). Median progression-free survival was significantly longer in the vepdegestrant group than in the fulvestrant group (5.0 months vs 2.1 months) among patients with ESR1 mutations, but not in the full patient population (3.8 vs 3.6 months). The objective response rate was 18.6% vs 4.0% among ESR1 mutant patients. A limitation of the study is the relatively short follow-up period[30]. Trovato G. And Tortora G. noted the lack of results in the ER wt subgroup and performed an analysis based on the results, which showed that the median progression-free survival was 2.7 months vs 4.8 months in the vepdegestrant vs fulvestrant groups in patients with wt ER, suggesting that fulvestrant was superior in this subgroup and highlighting the differences between wt and ER mutant tumors. The results of this analysis are, however, should be treated with cautiousness. Shimoi T. and Yonemori K. also highlighted the importance of the type of ESR1 mutation, which can have serious consequences for the efficacy of the tested medicines. Additional analysis of the mutations in the trial would be useful[31].
2.5. Adverse Effects
In a dose-escalation study (NCT04072952), treatment-related adverse effects (TRAE) were observed in 70 of 83 patients. The most common TRAEs (observed in more than 1 patient) were nausea, fatigue, headache, arthralgia, constipation and hot flush. Most adverse effect was grade 1 or 2, with no grade 4 or higher adverse effects observed. Overall, vepdegestrant was well tolerated and there was no dose-limiting toxicity[25]. In the phase II expansion of the study, the most common TRAEs were fatigue, hot flush, nausea, arthralgia, increased aspartate aminotransferase and increased blood alkaline phosphatase (grade 1 or 2). No dose reduction was needed, however, 2 patients (5.7%) had to discontinue the treatment due to grade 3 QT elongation and grade 3 anemia[26,27,28]. In the phase Ib cohort, the effect of vepdegestrant+palbiciclib was studied. No dose limiting toxicity was observed. Among the 46 patients, TEAEs (Treatment-emergent adverse events) lead to dose reduction of vepdegestrant in 5 and to discontinuation of vepdegestrant in 4 cases. TEAEs leading to dose reduction or discontinuation of palbociclib occurred in 34 and 8 patients, respectively. Most common (>10%) grade 3/4 TRAEs were neutropenia (89.1%), decreased WBC (white blood cell) count (15.2%), decreased platelet count (10.9%). No grade 5 TRAE was observed, and no patient had febrile neutropenia. Altogether, the safety profile was similar to the previous data, except for grade 3/4 neutropenia, which needed monitoring and palbociclib dose reduction[29].
In the NCT05463952 phase I trial no serious adverse effects (AE) were reported, four out of six patients experienced grade 1 or grade 2 treatment emergent adverse effects. No dose limiting toxicity was observed[24].
In the NCT0600568 phase I trial (12 healthy male participants) 50% of patients reported a total of 9 TEAEs after a single dose of vepdegestrant (200 mg), and no severe adverse effects were observed. 75% of patients reported a total of 20 TEAEs after a single dose of vepdegestrant and multiple doses of carbamazepin. In 16.7% of participants, increased hepatic enzyme levels were reported, probably due to carbamazepine[20].
In the NCT05673889 trial, only mild or moderate TEAEs (e.g. headache) were reported. In the NCT05652660 trial, mild dizziness and headache were reported. There were no clinically meaningful changes in vital signs, ECGs or laboratory measurements [23]. In the NCT05538312 trial, 1 participant suffered mild abdominal pain and diarrhoea after a single dose of vepdegestrant.[21].
In the VERITAC-2 trial, 86.9% of patients in the vepdegestrant group and 81.4% in the fulvestrant group reported adverse effects. In the vepdegestrant group, the most common AEs were fatigue (26.6%), increased aspartate and alanine aminotransferase level (both 14.4%) and nausea (13.5%). Grade 3 adverse events occurred in 19.2% of patients in the vepdegestrant group and 14% in the fulvestrant group, while grade 4 AEs occurred in 1.6% and 2.9% of cases, respectively. The most frequent grade 3/4 AEs were neutropenia (1.9%) and hypokalemia (1.9%) in the vepdegestrant group. In 9.9% of vepdegestrant patients, QT prolongation was reported, which lead to dose reduction in 1 patient. Altogether, 1.9% of cases in the vepdegestrant group, led to dose reduction and 2.9% to treatment discontiniuation[30].
3. Current Status and Future Perspectives
Vepdegestrant received FDA fast-track designation in 2024[17]. In 2025, Arvinas and Pfizer Inc. have submitted New Drug Application (NDA) for vepdegestrant to FDA, for the treatment of patients with ESR1 mutant ER+/HER2− advanced or metastatic BC whose disease progressed after endocrine therapy.[32] On May 1, 2026, the FDA approved vepdegestrant for the treatment of adults with ER+, HER2- ESR1 mutated advanced or metastatic breast cancer, as detected by an FDA-authorized test, with disease progression following at least one line of endocrine therapy[33].
Although the initial results are positive, there are several questions which may be answered with time, using results from greater studies with higher number of patients and longer follow-up periods. The latter should also clarify long term tolerability, and the potential occurrence of drug resistance. Current studies also highlight the possibility of potential drug-drug interactions (DDIs). Although the studied DDIs did not raise serious safety concerns, careful monitoring may be necessary during vepdegestrant treatment to avoid and manage potential future interactions.
Still there are a few ongoing clinical trials with vepdegestrant. In the TACTIVE-U program, several 1b/2 studies have been started to investigate the efficacy of vepdegestrant in combination with kinase inhibitors (abemaciclib, ribociclib and samuraciclib). In the NCT06206837 1b/2 trial, the combination of vepdegestrant and atirmociclib (PF-07220060) is investigated. The VERITAC-3 study compares vepdegestrant+palbiciclib vs letrozole+palbociclibe, but unlike the VERITAC-2, it investigates first-line settings (not pretreated patients). With approx. 1130 patients involved, VERITAC-3 could be the largest study of vepdegestrant[13].
Molecular analysis revealed that vepdegestrant has the potential to bind to rhotekin 2 (RTKN2), which is a protein, overexpressed in some gemcitabine-resistant cholangiocarcinoma[34]. Future studies in this area may lead to the discovery of another potential indication of vepdegestrant. This could be another possible route to evolve into a first-line treatment, if VERITAC-3 shows satisfying results.
Author Contributions
Conceptualization, writing and editing: A.B. Writing and editing: M.B. and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was also supported by the University of Debrecen Program for Scientific Publication.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AE | Adverse effect |
| AUC | Area under curve |
| BC | Breast cancer |
| BCRP | Breast cancer resistance protein |
| CBR | Clinical benefit rate |
| CDK 4/6 | Cyclin-dependent kinase 4 and 6 |
| CL | Clearance |
| Cmax | Maximal concentration |
| CRBN | Cereblon E3 ligase |
| CYP3A | Cytochrome P450 3A |
| DDI | Drug-drug interaction |
| DNA | Deoxyribonicleic acid |
| ER | Estrogen receptor |
| ER+ | Estrogen receptor positive |
| ESR1 | Estrogen receptor 1 gene |
| FDA | Food and Drug Administration |
| HER2- | Human epidermal growth factor receptor 2 negative |
| HPLC | High performance liquid chromatography |
| HR+ | Hormone receptor positive |
| HRMS | High-resolution mass spectrometry |
| i.d. | intraduodenal |
| i.v. | intravenous |
| MS | Mass spectrometry |
| mTOR | Mammalian target of rapamycin |
| NDA | New drug application |
| Peff | effective permeability |
| P-gp | P-glycoprotein |
| PGR | Progesterone receptor |
| PI3K | Phosphoinositide 3-kinase |
| PK | Pharmacokinetics |
| PROTAC | Proteolysis targeting chimera |
| QD | quaque die |
| Rac | Accumulation ratio |
| RTKN2 | Rhotekin 2 |
| SERD | Selective estrogen receptor degrader |
| SERM | Selective estrogen receptor modulator |
| t1/2 | Half-life |
| t1/2eff | Effective elimination half-life |
| TEAE | Treatment emergent adverse effect |
| TGI | Tumor growth inhibition |
| tmax | Time needed to reach maximal concentration |
| TPD | Targeted protein degradation |
| TRA | Total radioactivity |
| TRAE | Treatment related adverse effect |
| WBC | White blood cell |
| wt | wild type |
References
- Wilkinson, L.; Gathani, T. Understanding breast cancer as a global health concern. Br J Radiol 2022, 95, 20211033. [CrossRef]
- Rej, R.K.; Thomas, J.E.; Acharyya, R.K.; Rae, J.M.; Wang, S. Targeting the estrogen receptor for the treatment of breast cancer: recent advances and challenges. J. Med. Chem. 2023, 66, 8339-8381. [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine resistance in hormone receptor positive breast cancer–from mechanism to therapy. Front. Endocrinol. (Lausanne) 2019, 10, 245. [CrossRef]
- Haque, M.M.; Desai, K.V. Pathways to endocrine therapy resistance in breast cancer. Front. Endocrinol. (Lausanne) 2019, 10, 573. [CrossRef]
- Curran, M.; Wiseman, L. Fulvestrant. Drugs 2001, 61, 807-813. [CrossRef]
- Robertson, J.; Harrison, M. Fulvestrant: pharmacokinetics and pharmacology. Br. J. Cancer 2004, 90, S7-S10. [CrossRef]
- Sherman, S.; Sandusky, Z.M.; Russo, D.; Zak, D.; Nardone, A.; Friel, D.; Hermida-Prado, F.; Heraud, C.; Kuziel, G.; Verma, A. Imlunestrant a next-generation oral SERD overcomes ESR1 mutant resistance in estrogen receptor–positive breast cancer. JCI insight 2025, 10, e188051. [CrossRef]
- Wang, L.; Sharma, A. SERDs: a case study in targeted protein degradation. Chem. Soc. Rev. 2022, 51, 8149-8159. [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022, 21, 181-200. DOI: 0.1038/s41573-021-00371-6.
- Hu, Z.; Crews, C.M. Recent developments in PROTAC-mediated protein degradation: from bench to clinic. ChemBioChem 2022, 23, e202100270. [CrossRef]
- Lu, B.; Ye, J. Commentary: PROTACs make undruggable targets druggable: challenge and opportunity. Acta Pharmaceutica Sinica B 2021, 11, 3335-3336. [CrossRef]
- Cai, H.; Zhang, T.; Hu, Y. Global landscape of PROTAC: Perspectives from patents, drug pipelines, clinical trials, and licensing transactions. Eur. J. Med. Chem. 2025, 118055. [CrossRef]
- Tang, C.; Tian, B.; Zhang, B.; Zhang, Y.; Ke, C.; Chen, M.; Wei, M.; Wang, W.; Deng, X.; Zhang, Q. Insights Into Vepdegestrant (ARV-471): The First-in-Class Estrogen Receptor Proteolysis-Targeting Chimera Approaching Food and Drug Administration Approval for Breast Cancer. ChemMedChem 2026, 21, e202501111. [CrossRef]
- Gough, S.M.; Flanagan, J.J.; Teh, J.; Andreoli, M.; Rousseau, E.; Pannone, M.; Bookbinder, M.; Willard, R.; Davenport, K.; Bortolon, E. Oral estrogen receptor PROTAC vepdegestrant (ARV-471) is highly efficacious as monotherapy and in combination with CDK4/6 or PI3K/mTOR pathway inhibitors in preclinical ER+ breast cancer models. Clin Cancer Res. 2024, 30, 3549-3563. [CrossRef]
- Niessen, J.; Arendt, N.; Sjöblom, M.; Dubbelboer, I.R.; Borchardt, T.; Koziolek, M.; Hedeland, M.; Lennernäs, H.; Indulkar, A.; Dahlgren, D. A comprehensive mechanistic investigation of factors affecting intestinal absorption and bioavailability of two PROTACs in rats. Eur. J. Pharm. Biopharm. 2025, 211, 114719. [CrossRef]
- Niessen, J.; Nilsson, J.M.; Peters, K.; Indulkar, A.; Borchardt, T.; Koziolek, M.; Lennernäs, H.; Dahlgren, D.; Hedeland, M. Development and validation of LC-MS/MS methods for the pharmacokinetic assessment of the PROTACs bavdeglutamide (ARV-110) and vepdegestrant (ARV-471). J Pharm Biomed Anal. 2024, 249, 116348. [CrossRef]
- Choi, H.-I.; Choi, J.; Kim, J.W.; Lee, Y.H.; Cho, K.H.; Koo, T.-S. Stability Evaluation and Pharmacokinetic Profiling of Vepdegestrant in Rodents Using Liquid Chromatography–Tandem Mass Spectrometry. Molecules 2024, 29, 4048. [CrossRef]
- He, Y.; Zhu, C.; Lei, P.; Yang, C.; Zhang, Y.; Zheng, Y.; Diao, X. Characterization of preclinical radio ADME properties of ARV-471 for predicting human PK using PBPK modeling. J Pharm Anal. 2025, 15, 101175. [CrossRef]
- He, Y.; Hou, P.; Long, Z.; Zheng, Y.; Tang, C.; Jones, E.; Diao, X.; Zhu, M. Application of electro-activated dissociation fragmentation technique to identifying glucuronidation and oxidative metabolism sites of vepdegestrant by liquid chromatography-high resolution mass spectrometry. Drug Metab Dispos. 2024, 52, 634-643. DOI: 10.1124/dmd.124.001661.
- Wang, H.; Winton, J.A.; Matschke, K.T.; Stouffs, A.; Lee, K.C.; Zhang, Y.; Qiao, W.; Tan, W. Effect of carbamazepine on the pharmacokinetics of vepdegestrant, a PROteolysis TArgeting Chimera estrogen receptor degrader, in healthy adults. Br. J. Clin. Pharmacol. 2026. [CrossRef]
- Tran, L.; Winton, J.A.; Matschke, K.T.; Stouffs, A.; Lee, K.C.; Zhang, Y.; Tan, W. The Effect of Itraconazole on the Pharmacokinetics of Vepdegestrant, a PROteolysis TArgeting Chimera Estrogen Receptor Degrader, in Healthy Adult Participants. Clin Ther. 2026. [CrossRef]
- Niessen, J.; Lennernäs, H.; Dahlgren, D. Letter to the Editor Regarding “The Effect of Itraconazole on the Pharmacokinetics of Vepdegestrant, a PROteolysis TArgeting Chimera Estrogen Receptor Degrader, in Healthy Adult Participants” Recently Published by Tran and Colleagues. Clin. Ther. 2026. [CrossRef]
- Yang, D.Z.; Zhou, L.; Winton, J.A.; Matschke, K.T.; Kantaridis, C.; Doyle, K.; Lee, K.C.; Chen, N.; Zhang, Y.; Tan, W. Effect of vepdegestrant, a PROTAC oestrogen receptor degrader, on dabigatran and rosuvastatin pharmacokinetics in healthy participants. Br J Clinical Pharmacol. 2025, 91, 3501-3510. [CrossRef]
- Iwata, H.; Naito, Y.; Hattori, M.; Yoshimura, A.; Yonemori, K.; Aizawa, M.; Mori, Y.; Yoshimitsu, J.; Umeyama, Y.; Mukohara, T. Safety and pharmacokinetics of vepdegestrant in Japanese patients with ER+ advanced breast cancer: a phase 1 study. Int J Clin Oncol. 2025, 30, 72-82. [CrossRef]
- Hamilton, E.; Han, H.; Schott, A.; Tan, A.; Nanda, R.; Juric, D.; Hunter, N.; Munster, P.; Fang, B.; Zahrah, G. 390P Vepdegestrant, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, in ER+/human epidermal growth factor receptor 2 (HER2)-advanced breast cancer: Update of dose escalation results from a phase I/II trial. Ann Oncol. 2023, 34, S344. [CrossRef]
- Hurvitz, S.; Schott, A.; Ma, C.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.; Telli, M.; Mesias, J.A.; Jeselsohn, R. Abstract PO3-05-08: Updated results from VERITAC evaluating vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader, in ER–positive/human epidermal growth factor receptor 2 (HER2)–negative advanced breast cancer. Cancer Res. 2024, 84, PO3-05-08-PO03-05-08. [CrossRef]
- Schott, A.F.; Hurvitz, S.A.; Ma, C.; Hamilton, E.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.; Telli, M.; Mesias, J.; Jeselsohn, R.; Munster, P.; Lu, H.; Gedrich, R.; Mather, C.; Parameswaran, J.; Han, H.S. ARV-471, a PROTAC® estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: phase 2 expansion (VERITAC) of a phase 1/2 study. Cancer Res. 2022. 83 (5_suplement): GS3-03. [CrossRef]
- Hurvitz, S.; Schott, A.; Ma, C.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.; Telli, M.; Anampa, J.; Jeselsohn, R. 205P VERITAC update: Phase II study of ARV-471, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader in ER+/human epidermal growth factor receptor 2 (HER2)-advanced breast cancer. ESMO open 2023, 8. [CrossRef]
- Hamilton, E.; Jeselsohn, R.; Hurvitz, S.; Juric, D.; Han, H.; Telli, M.; Zahrah, G.; Nanda, R.; Zhang, Y.; Tan, W. Abstract PS15-03: Vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader, plus palbociclib in ER–positive/human epidermal growth factor receptor 2 (HER2)–negative advanced breast cancer: phase 1b cohort. Cancer Res. 2024, 84, PS15-03-PS15-03. [CrossRef]
- Campone, M.; De Laurentiis, M.; Jhaveri, K.; Hu, X.; Ladoire, S.; Patsouris, A.; Zamagni, C.; Cui, J.; Cazzaniga, M.; Cil, T. Vepdegestrant, a PROTAC estrogen receptor degrader, in advanced breast cancer. N Engl J Med. 2025, 393, 556-568. [CrossRef]
- Campone, M. Vepdegestrant in Advanced Breast Cancer. N Engl J Med. 2025, 393, 1862. [CrossRef]
- Ma, Z.; Zhou, J. NDA submission of vepdegestrant (ARV-471) to US FDA: the beginning of a new era of PROTAC degraders. J Med Chem. 2025, 68, 14129-14136. [CrossRef]
- Administration, U.S.F.D. FDA approves vepdegestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-vepdegestrant-er-positive-her2-negative-esr1-mutated-advanced-or-metastatic-breast (accessed on 2026.05.12.).
- Kidoikhammouan, S.; Mahalapbutr, P.; Ma-In, P.; Lert-Itthiporn, W.; Deenonpoe, R.; Suriya, U.; Wongkham, S.; Seubwai, W. Integrated transcriptomic and molecular docking analysis identifies Rhotekin 2 as a promising therapeutic target for overcoming gemcitabine resistance in cholangiocarcinoma. Biomed Rep. 2026, 24, 52. [CrossRef]
Figure 1.
Previously approved SERD medicines.

Figure 2.
Mechanism of action of PROTACs.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



