Submitted:
01 June 2026
Posted:
02 June 2026
You are already at the latest version
Abstract
Background: Identification of tumor-specific cell surface targets is a critical step in the development of precision oncology therapeutics, including radioligand and antibody-based approaches. However, existing strategies often rely on single-layer analyses and lack systematic integration of proteomic, genomic, and clinical metadata. Methods: We developed a multi-layered proteogenomic filtering framework integrating quantitative proteomics from colorectal cancer (CRC) cohorts with curated metadata on protein localization, normal tissue expression, and drugability. Eleven complementary filtering strategies were applied, followed by manual curation for extracellular accessibility and composite scoring based on protein rank, localization, and clinical relevance. Results: Application of the pipeline to metastatic CRC (mCRC) identified multiple high-confidence candidate targets, including GPRC5A, SLC2A1, CD47, DPEP1 and IFITM1. The average pairwise overlap between filtering strategies was low (0.11), indicating limited redundancy and complementary target identification across approaches. Importantly, candidates detected by multiple strategies were significantly enriched for established biomarkers (FAP, CEACAM5, ITGAV, ITGB4), which were exclusively found among multi-strategy candidates (10.3% vs. 0%; Fisher’s exact test, p = 0.0064), supporting overlap-based prioritization as a marker of biological and translational relevance. Composite scoring further prioritized GPRC5A as a leading candidate. Additional validation layers confirmed tumor-enriched expression, plasma membrane localization, and relevance across multiple cancer indications. Conclusion: This study presents a scalable and generalizable framework for the prioritization of cell surface therapeutic targets in solid tumors. By integrating multiple data layers and incorporating translational criteria early in the discovery process, this approach may facilitate more efficient identification of targets for downstream development, including antibody- and radioligand-based therapies.
Keywords:
proteogenomics
; target discovery
; cell surface proteins
; therapeutic targets
; colorectal cancer
; metastatic colorectal cancer
; target prioritization
1. Introduction
Target selection is a critical and rate-limiting step in the development of precision oncology therapeutics, particularly for modalities that rely on target accessibility at the cell surface, such as radioligand therapy (RLT), radioimmunotherapy (RIT), antibody–drug conjugates, and other antibody-based platforms [1,2]. The clinical success of these approaches depends on the identification of tumor-associated antigens that are abundantly expressed on the plasma membrane of cancer cells, exhibit limited expression in normal tissues, and present extracellular epitopes amenable to therapeutic binding and internalization. Failure to adequately address these criteria during early target discovery frequently leads to suboptimal efficacy or unacceptable off-target toxicity in later stages of development.
Despite major advances in genomics, transcriptomics, and proteomics, the systematic identification of clinically actionable cell surface targets in solid tumors remains challenging. Many discovery efforts rely predominantly on differential gene expression analyses or literature-driven prioritization, which do not reliably capture protein-level abundance, subcellular localization, or therapeutic accessibility [3,4]. In addition, transcriptomic signals often correlate poorly with surface protein expression, particularly for membrane proteins subject to post-transcriptional regulation, variable trafficking, or proteolytic processing [3,5]. As a result, candidate lists derived from single-layer analyses are frequently enriched for false positives and lack translational robustness.
From a therapeutic standpoint, the “ideal” cell surface target is rarely defined by a single parameter [1,6]. Instead, clinically viable targets reflect a balance between multiple, sometimes competing, criteria, including tumor-selective overexpression, restricted normal tissue distribution, stable membrane localization without extensive shedding, evidence of functional relevance in cancer biology, and feasibility for drug development. Importantly, translational considerations such as prior clinical validation, availability of biomarker data, and compatibility with specific therapeutic modalities are often evaluated late in the discovery process, increasing the risk of downstream attrition.
To address these challenges, there is a growing need for integrative discovery frameworks that combine quantitative proteomics with curated biological and clinical metadata, while explicitly accounting for uncertainty and dataset-specific variability [7,8]. Rather than relying on a single optimal ranking scheme, parallel filtering strategies can provide complementary perspectives on the data, enabling identification of candidates that are robust to parameter selection and cohort effects. Recurrent detection across independent strategies may therefore serve as an implicit robustness metric for target prioritization.
Recent pan-cancer proteogenomic studies have demonstrated the power of integrating tumor-level protein expression with genomic data to expand the landscape of candidate therapeutic targets. In particular, Savage et al. reported a large-scale analysis across multiple cancer types, establishing proteomics as a critical layer for target discovery beyond genomics and transcriptomics alone [7]. However, global target catalogs are not designed to address key translational constraints relevant to therapeutic modalities that require extracellular target accessibility, nor do they provide indication-specific prioritization or robustness assessment under alternative analytical assumptions. These considerations are particularly important for antibody-based therapies and radioligand therapy, where plasma membrane localization and normal tissue expression critically influence clinical feasibility.
Here, we present a multi-layered proteogenomic filtering pipeline designed to systematically identify and prioritize cell surface proteins for therapeutic targeting in solid tumors. The pipeline integrates quantitative proteomics from large-scale cancer cohorts with curated metadata on protein localization, normal tissue expression, expression in cancer cell lines, and clinical drugability. Eleven complementary filtering strategies were applied, followed by manual curation to confirm extracellular accessibility and composite scoring based on expression, localization, and translational relevance. Importantly, the approach emphasizes overlap and consistency across strategies rather than reliance on any single filtering criterion.
We apply this framework to metastatic colorectal cancer (mCRC) as a case study, a disease with high unmet clinical need and increasing interest in targeted radiopharmaceutical and antibody-based therapies. Using data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) [3,4], Human Protein Atlas [9], Genotype-Tissue Expression project, Cancer Cell Line Encyclopedia, and curated drug development databases, we demonstrate that this multi-layered approach identifies both established colorectal cancer biomarkers and previously underexplored candidate targets. The resulting prioritization highlights GPRC5A as a top candidate while maintaining sensitivity to diverse protein classes and biological mechanisms.
Together, this study provides a scalable and generalizable framework for cell surface target discovery that explicitly integrates proteomic evidence with translational constraints. Beyond colorectal cancer, the modular design of the pipeline enables adaptation to other solid tumor indications and therapeutic modalities, supporting early-stage decision-making in precision oncology drug development.
2. Materials and Methods
Data Sources
The pipeline integrates multiple publicly available and proprietary datasets to enable comprehensive target discovery. Quantitative protein expression data were obtained from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) COAD and COADREAD cohorts, comprising colorectal adenocarcinoma and rectal cancer samples with matched normal tissue controls [3,4]. Protein abundance in normal tissues was assessed using data from the Human Protein Atlas (HPA) and the Genotype-Tissue Expression (GTEx) project [9,10]. RNA sequencing data for colorectal cancer cell lines were retrieved from the Cancer Cell Line Encyclopedia (DepMap) [11]. Clinical metadata describing drugability and biomarker status were sourced from Clarivate Drug Discovery Intelligence (CDDI, Cortellis platform; Clarivate Analytics), while information on the number of unique drugs, associated indications, and highest clinical development stage was extracted from Swiss-Prot [12].
Pipeline Architecture
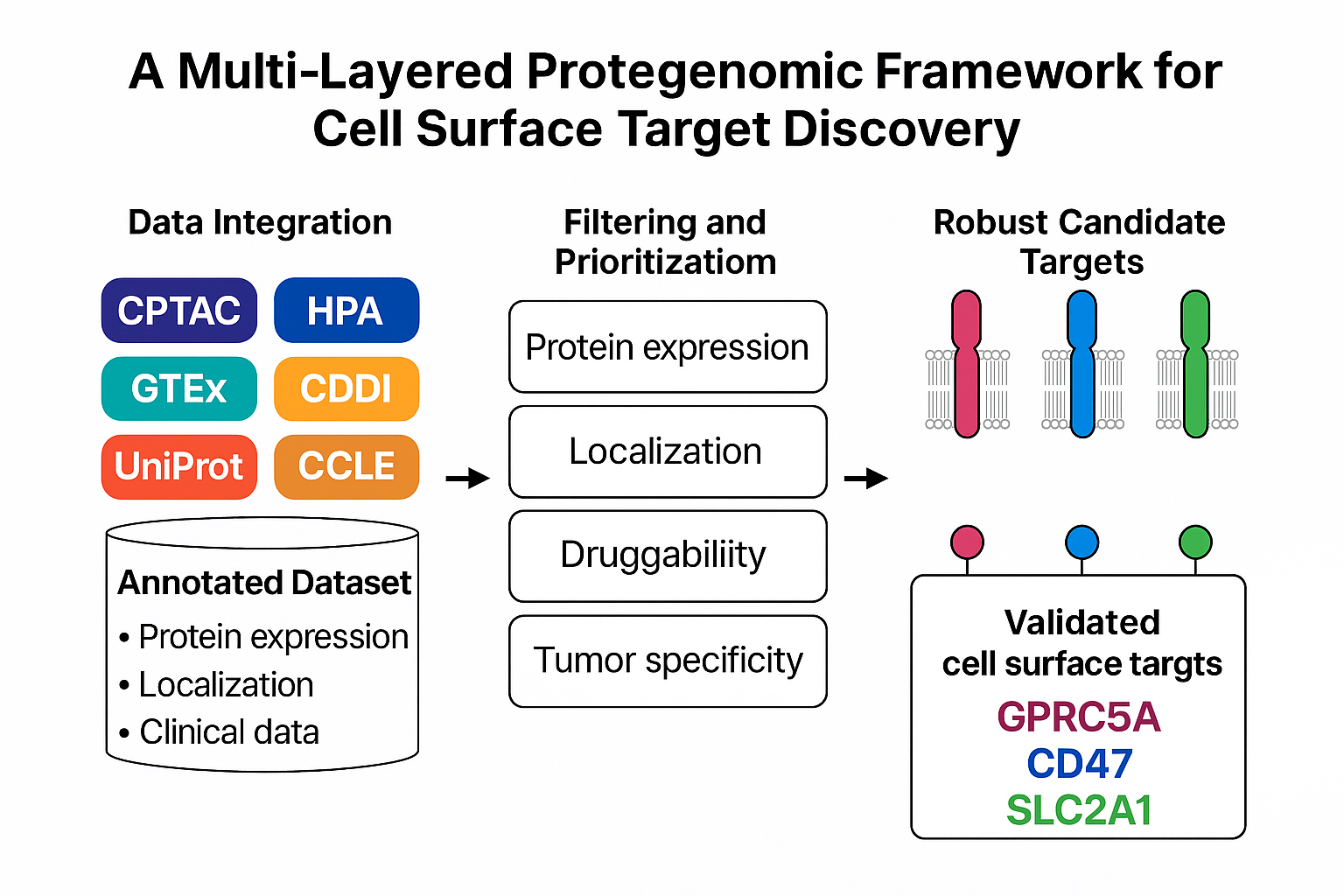
The target discovery pipeline was designed as a modular, multi-stage framework integrating quantitative proteomics with curated biological and clinical metadata. The architecture was optimized to enable systematic prioritization of cell surface proteins while accommodating dataset-specific variability and uncertainty inherent to large-scale proteomic analyses. The pipeline consists of four core stages: (i) construction of an annotated union dataset, (ii) parallel multi-parameter filtering, (iii) manual curation for extracellular accessibility, and (iv) composite scoring and prioritization (Figure 1).
Annotated Union Dataset Construction
All datasets were merged into a unified, protein-level table using standardized protein identifiers. For each protein, the annotated union table included quantitative protein expression ranks and differential expression metrics derived from CPTAC COAD and COADREAD cohorts, subcellular localization annotations obtained from UniProt, Human Protein Atlas (HPA), and the Cell Surface Protein Atlas (CSPA), normal tissue expression levels from HPA and GTEx, expression in colorectal cancer cell lines from the Cancer Cell Line Encyclopedia (DepMap), and clinical development metadata from Clarivate Drug Discovery Intelligence (CDDI) and SwissProt.
To systematically assess therapeutic accessibility, a custom localization score was generated for each protein based on curated annotations indicating plasma membrane localization, transmembrane topology, and presence of extracellular domains. Secreted proteins and proteins exclusively annotated to intracellular compartments were flagged for exclusion in later steps.
Parallel Filtering Strategies
Rather than applying a single optimal filtering scheme, the pipeline implements multiple complementary filtering strategies in parallel (Table 1, Table 2 and Table 3). Each strategy represents a distinct combination of selection criteria targeting overexpression in metastatic disease, plasma membrane localization, minimal normal tissue expression, and expression in cancer cell lines. This design reduces dependence on any single parameter threshold and allows identification of candidates that are robust to methodological variability.
Filtering strategies were grouped conceptually into three categories: (i) rank-based strategies combining protein abundance, localization scores, and normal tissue penalties; (ii) differential expression-based strategies focused on upregulated plasma membrane proteins; and (iii) cohort-driven strategies applying stricter differential expression cut-offs within individual CPTAC cohorts. All strategies shared core architectural principles but differed in stringency and parameter weighting, enabling broad exploration of the target space while preserving biological relevance.
Each filtering strategy generated an independent shortlist of candidate proteins. Proteins detected across multiple strategies were considered more robust, reflecting consistency across analytical perspectives.
Manual Curation for Extracellular Accessibility
Candidates emerging from the automated filtering steps were subjected to manual curation to confirm extracellular accessibility and therapeutic relevance. This step focused on reviewing protein topology, domain structure, and annotation consistency to exclude proteins lacking extracellular epitopes despite partial membrane association. Manual curation was essential to eliminate false positives arising from incomplete or ambiguous database annotations and to ensure compatibility with antibody-based and radioligand-based therapeutic modalities [1,6].
Composite Scoring and Prioritization
Final prioritization of candidate targets was performed using a composite scoring framework integrating multiple quantitative and qualitative parameters. The composite score incorporated stage IV protein expression rank, localization score, penalties for normal tissue expression (HPA and GTEx), expression in colorectal cancer cell lines, and clinical development metrics reflecting drugability and prior biomarker use. Candidates identified by multiple filtering strategies were weighted more strongly, reflecting increased robustness. Weighting of individual parameters was intentionally kept conservative and balanced to avoid overfitting to any single dataset or feature, prioritizing robustness over optimization.
The resulting ranked list represents a balanced prioritization of candidate targets based on expression, accessibility, safety, and translational maturity, rather than optimization of any single criterion.
Validation Layers
To contextualize and illustrate the performance of the multi-layered target discovery pipeline, selected high-ranking candidate proteins were evaluated using several independent validation layers. These analyses were not intended as definitive experimental validation, but rather as orthogonal assessments addressing key translational risks, including expression robustness, localization accuracy, relevance beyond the discovery dataset, and prior clinical or biomarker evidence.
Protein Expression Visualization
Quantitative protein expression patterns were visualized across tumor stages using swarm plots of normalized protein abundance from the CPTAC COAD and COADREAD cohorts. These plots enabled assessment of stage-associated expression trends, inter-patient variability, and consistency between cohorts, with particular emphasis on overexpression in advanced and metastatic disease. Normal tissue reference samples were included where available to contextualize tumor-specific expression.
Transcriptomic Analysis
To assess concordance between protein-level signals and transcriptomic data, mRNA expression patterns for candidate targets were examined in colorectal cancer cell lines from the Cancer Cell Line Encyclopedia (DepMap) and patient tumor samples from The Cancer Genome Atlas (TCGA). Heatmaps were generated to visualize relative expression across samples and to evaluate whether proteomic signals were supported by transcriptional data, recognizing that discordance between RNA and protein expression may occur for membrane-associated proteins.
Immunohistochemistry and Subcellular Localization
Protein localization and tissue-level expression patterns were examined using immunohistochemistry data from the Human Protein Atlas (HPA). Emphasis was placed on confirming plasma membrane staining and differential expression between colorectal cancer tissue and normal colon, where available. This layer served as an independent qualitative assessment of extracellular accessibility and supported exclusion of candidates with ambiguous or predominantly intracellular localization.
Literature Mining and Biomarker Evidence
To evaluate prior evidence linking candidate targets to cancer biology and clinical investigation, systematic literature mining was performed using PubMed. The number of publications reporting associations with cancer, metastatic disease, and metastatic colorectal cancer was recorded. This analysis provided contextual insight into existing biological and clinical knowledge, without presupposing target validity or drugability.
Protein Expression in Other Cancer Types
To assess whether candidate targets exhibited broader oncologic relevance or tumor-type specificity, protein expression data from additional CPTAC cohorts were examined, including breast invasive carcinoma (BRCA; PDC000120), lung squamous cell carcinoma (LSCC; PDC000234), lung adenocarcinoma (LUAD; PDC000153), ovarian serous cystadenocarcinoma (OV; PDC000110), and pancreatic ductal adenocarcinoma (PDAC; PDC000270). Comparative analysis across indications helped contextualize colorectal cancer findings and informed prioritization based on potential therapeutic breadth or selectivity.
Clinical and Biomarker Annotation
Finally, candidate targets were evaluated using curated clinical metadata from Clarivate Drug Discovery Intelligence (CDDI) to determine whether they had previously been reported as biomarkers or explored in therapeutic development. Metrics included number of reported indications, cancer-specific usage, and highest development phase where applicable. This layer provided translational context and assisted in distinguishing well-characterized targets from emerging or underexplored candidates.
Collectively, these validation layers are intended to provide orthogonal context for target prioritization rather than functional validation, which will require dedicated experimental studies. Localization and accessibility assessments were particularly emphasized due to their importance for radiopharmaceutical and antibody-based targeting.
3. Results
3.1. Pipeline Performance Across Filtering Strategies
The multi-layered filtering pipeline was applied to proteomic and metadata from the CPTAC COAD and COADREAD cohorts [3,4] to identify cell surface proteins associated with metastatic colorectal cancer (mCRC) according to the workflow described in Figure 1. Eleven distinct filtering strategies were implemented, each varying in stringency for protein expression, localization, normal tissue expression and expression on cancer cell lines (Table 1, Table 2 and Table 3).
The number of candidate proteins identified per strategy ranged from 5 to 58 and the number of unique proteins identified for each strategy ranged from 0 to 29, reflecting differences in parameter thresholds and dataset-specific variability (Figure 2). Strategies 1–4, which combined protein rank and localization scores with normal tissue penalties, yielded between 10 and 58 candidates per iteration. Strategies 5–7, incorporating differential expression and manual curation for plasma membrane localization, produced 5–7 candidates. Cohort-driven strategies (8–11), which applied stricter log fold-change cut-offs to either COAD or COADREAD independently, identified 6–23 candidates after manual review.
Candidate targets are identified through multiple complementary filtering strategies applied in parallel, each combining criteria related to metastatic overexpression, plasma membrane localization, extracellular accessibility, and limited normal tissue expression. Manual curation is subsequently performed to confirm extracellular exposure and to exclude proteins with ambiguous or non therapeutically accessible localization. Final prioritization is achieved using a composite scoring framework that integrates expression, localization, safety related penalties, and translational relevance. Candidates supported by multiple filtering strategies are considered more robust and prioritized for downstream validation.
CPTAC=Clinical Proteomic Tumor Analysis Consortium; COAD=Colon Adenocarcinoma; COADREAD=Colon Adenocarcinoma and Rectal Adenocarincoma; CRC=Colorectal Cancer; BRCA=Breast Invasive Carcinoma; LSCC=Lung squamous cell carcinoma; LUAD=Lung Adenocarcinoma; OV=Ovarian Serous Cystadenocarcinoma; PDAC=Pancreatic Ductal Adenocarcinoma; CDDI=Cortellis Drug Discovery Intelligence; HPA=Human Protein Atlas; GTEX=The Genotype-Tissue Expression project; CSPA=Cell Surface Protein Atlas; CCLE=Cancer Cell Line Encyclopedia; GO=Gene Ontology; TCGA=The Cancer Genome Atlas.
3.2. Overlap and Robustness of Candidate Selection
To assess robustness, candidate lists from all strategies were compared. A union analysis revealed that only a subset of 43 proteins was identified by more than one strategy. A weighted scoring was implemented where candidates identified by the most stringent filtering strategies (4-10) were assigned a weight of 2 and candidates identified by less stringent strategies (1-3 and 11) were assigned a weight of 1. Figure 3 shows the resulting 48 proteins with a score of more than 1. GPRC5A emerged as the most robust candidate, appearing in 8 different strategies and all major strategy groups (2–4, 5–7, and 8–9). Other recurrent hits included IFITM1, SLC5A6, DPEP1 and LY75, each detected in 5 or more independent strategies (Figure 3).
The average pairwise overlap between filtering strategies was 0.11, indicating limited redundancy between individual approaches. Despite this low overlap, candidates detected across multiple strategies were strongly enriched for known colorectal cancer biomarkers. Specifically, FAP, CEACAM5, ITGAV, and ITGB4 were exclusively identified among multi-strategy candidates (4/39, 10.3%) and were absent among single-strategy hits (0/95), representing a significant enrichment (Fisher’s exact test, p = 0.0064). These findings support cross-strategy recurrence as a marker of biological and translational relevance.
The goal for this analysis was to find unique targets for radiopharmaceuticals. Strategy 5-11 did not contain criteria for exclusion of targets that were already exploited in this field. Therefore, a new round of selection was performed on the data in Figure 3 to remove the candidates where we could find that it had been used as a target for a therapeutic radiopharmaceutical agent. This analysis resulted in exclusion of ITGB4 [13], CEACAM5 [14] Bioorg Med Chem Lett, 2024], FAP [15], CD46 [16] and SLC7A5 [17]. Supplementary Figure 1 shows the resulting bar chart. Importantly, this exclusion was performed as a secondary analysis step and was not incorporated into the core pipeline, ensuring that initial target identification remained unbiased.
3.3. Composite Scoring and Prioritization
Final candidate ranking was based on a composite score integrating mean stage IV protein rank, localization score, normal tissue expression penalty, and protein expression in cancer cell lines. Table 4 shows the resulting Top-10 candidates. GPRC5A achieved the highest composite score (4.5), followed by SLC2A1 (4.0), CD47 (2.5), DPEP1 (1.9) and IFITM1 (1.9). These candidates represent diverse protein classes, including a G-protein coupled receptor, a solute carriers, an immune checkpoint regulator, an enzyme and an antiviral effector, highlighting the pipeline’s capacity to capture biologically distinct targets.
3.4. Validation Layers
To illustrate the translational potential of the pipeline, selected candidates were evaluated using multiple complementary validation layers. PubMed citation analysis showed that all top-ranked candidates have previously been reported in cancer-related contexts, with most also described in metastatic disease, supporting their relevance as established or emerging biomarkers (Figure 4). Among these, GPRC5A has been associated with adverse clinical outcomes and tumor progression across multiple cancer types, supporting its biological relevance as a therapeutic candidate [18]. In colorectal cancer specifically, GPRC5A expression has been reported to promote tumor growth and invasion through non-coding RNA–mediated regulatory mechanisms [19].
Additional top-ranked candidates were likewise supported by independent literature evidence. SLC2A1 is frequently overexpressed in colorectal cancer and has been linked to disease progression and immune modulation, while its plasma membrane localization and central role in tumor metabolism make it an attractive therapeutic target [20,21]. CD47 represents a well-established innate immune checkpoint in colorectal cancer, and its recovery among the top-ranked candidates provides an internal validation of the pipeline’s ability to capture clinically actionable cell-surface targets [22,23]. DPEP1 is a GPI-anchored surface enzyme with high specificity for colorectal cancer, where it has been shown to promote invasion and metastasis and to associate with adverse clinical outcome [24,25]. IFITM1 is overexpressed in metastatic colorectal cancer and functionally promotes invasion, and recent work has demonstrated its suitability as a surface imaging and targeting antigen in vivo [26,27].
Immunohistochemistry data from the Human Protein Atlas further verified plasma membrane localization for GPRC5A, SLC2A, CD47, DPEP1, IFITM1, DSC2 and IFITM3 in colorectal cancer tissues (Figure 5). In addition, protein expression analysis across other tumor types demonstrated that most of the top-10 ranked candidates were differentially expressed in multiple cancer indications (Figure 6). Finally, clinical annotation revealed that all top-ranked candidates have previously been used as biomarkers, including across different cancer types, further supporting their translational relevance (Figure 7).
3.5. Summary of Pipeline Robustness
The integration of proteogenomic data with curated metadata and iterative filtering strategies enabled the identification of high-confidence cell surface targets for mCRC. The recovery of known biomarkers alongside novel candidates demonstrates the validity and generalizability of the approach. The pipeline’s modular design allows adaptation to other solid tumor indications and therapeutic modalities.
4. Discussion
The identification of tumor-specific cell surface targets remains a major challenge in oncology drug development, particularly for modalities such as radioligand therapy (RLT) and radioimmunotherapy (RIT), where off-target toxicity can severely limit clinical applicability. Our study introduces a multi-layered proteogenomic filtering pipeline that addresses key limitations of existing approaches by integrating quantitative proteomics with curated metadata on protein localization, normal tissue expression, and clinical drugability.
Recent pan-cancer proteogenomic atlases have demonstrated the power of large-scale integration of proteomic and genomic data for expanding the landscape of candidate cancer drug targets, most notably the comprehensive analysis reported by Savage et al. [7]. While such efforts provide invaluable resources for global target enumeration, their objectives differ from the translational challenges addressed here. Our study focuses on indication-specific prioritization by explicitly incorporating extracellular accessibility, normal tissue expression penalties, and robustness across multiple complementary filtering strategies. This distinction is particularly relevant for antibody-based therapies and radiopharmaceuticals, where plasma membrane localization and on-target toxicity are dominant determinants of clinical feasibility [1,2].
Advantages of the Pipeline
The pipeline’s strength lies in its modular design and multi-parameter filtering strategy. By combining eleven complementary filtering strategies with manual curation, we achieved robust candidate selection while minimizing reliance on single-layer analyses that are prone to false positives. This is supported by the consistent recovery of clinically validated targets such as FAP and CEACAM5 across independent filtering strategies. In addition, candidates identified by multiple strategies were enriched for known cancer-associated surface proteins, suggesting that overlap across filters represents a practical proxy for biological robustness. Importantly, the identification of less-characterized candidates such as GPRC5A and SLC5A6 highlights the ability of the pipeline to move beyond canonical targets while retaining translational relevance.
Comparison with Existing Approaches
Traditional target discovery pipelines often prioritize candidates based on differential expression alone, which can lead to high false-positive rates and poor translational performance. This may partly explain the high attrition rate observed when targets identified through transcriptomic profiling alone are advanced toward therapeutic development. Our method incorporates additional layers of evidence, including plasma membrane localization and normal tissue expression penalties, which are critical for reducing on-target toxicity. Furthermore, the integration of clinical metadata enables prioritization of targets with established drugability, facilitating downstream development.
Limitations
Despite its strengths, the pipeline has limitations. First, the approach relies on the quality and completeness of underlying datasets. Differences in normalization and technology platforms between CPTAC cohorts introduced variability in differential expression analysis, necessitating separate cohort-driven strategies. Second, manual curation remains essential for confirming extracellular accessibility, as database annotations do not always provide sufficient resolution. This step, while improving accuracy, limits scalability and introduces subjectivity. Third, the current study focuses on target identification and prioritization and does not include functional validation of candidate targets in vitro or in vivo. Such experiments will be required to confirm target dependency, internalization properties, and therapeutic relevance.
Future Directions
Future iterations of the pipeline should focus on automation of curation steps through advanced protein topology prediction tools and machine learning-based annotation. Integration of additional omics layers, such as phosphoproteomics and mutational data, may further enhance target prioritization by linking expression patterns to functional relevance. Finally, expanding the pipeline to other solid tumor indications will validate its generalizability and support its application in diverse therapeutic contexts, including antibody-drug conjugates and bispecific antibodies.
Clinical and Translational Implications
The pipeline provides a systematic framework for identifying high-confidence targets for precision oncology. By structuring target prioritization around clinically relevant parameters such as tissue specificity and accessibility, this framework may support more efficient transition from discovery to preclinical validation, thereby reducing the number of non-viable targets entering early development pipelines. Its adaptability to different cancer types and therapeutic modalities positions it as a valuable tool for next-generation drug development. In particular, the prioritization of cell surface proteins with low normal tissue expression aligns closely with the requirements for radioligand therapy, where target accessibility and tumor specificity are critical determinants of therapeutic index. Although radioligand development was not performed in this study, the identified candidates provide a rational starting point for future ligand design and evaluation.
Among the prioritized candidates, GPRC5A is of particular interest given its reported overexpression in multiple epithelial malignancies and potential role in tumor progression. Similarly, SLC5A6 represents a transporter protein with limited characterization in cancer but favorable surface accessibility, making it an intriguing candidate for targeted delivery approaches. Further investigation of these targets may provide insight into both tumor biology and therapeutic vulnerability.
5. Conclusions
We developed and validated a multi-layered proteogenomic filtering pipeline for the systematic identification of cell-surface therapeutic targets in solid tumors. Using metastatic colorectal cancer (mCRC) as a case example, the pipeline integrates quantitative proteomics with curated metadata on protein localization, normal tissue expression, and clinical drugability to address key limitations of traditional target discovery approaches. Eleven complementary filtering strategies, combined with manual curation and composite scoring, enabled robust and reproducible candidate selection while explicitly incorporating translational constraints early in the discovery process.
Application of this framework consistently identified high-confidence targets, including GPRC5A, SLC2A1, CD47, DPEP1 and IFITM1, several of which are established biomarkers or emerging therapeutic candidates. Overlap analysis demonstrated that candidates detected across multiple independent strategies were enriched for known cancer-relevant surface markers, supporting the validity and robustness of the approach. In addition, the use of composite scoring provided a transparent and reproducible mechanism for prioritization, facilitating comparison of candidates across heterogeneous analytical dimensions and reducing reliance on single-parameter ranking.
By integrating multiple data layers and emphasizing robustness and therapeutic accessibility, this pipeline reduces false-positive target nomination and helps bridge the gap between discovery-stage proteogenomics and translational decision-making. Its modular design allows straightforward adaptation to other cancer indications and therapeutic platforms, including radioligand therapy, antibody–drug conjugates, and bispecific antibodies. Future development will focus on automating currently manual curation steps, incorporating additional omics layers such as phosphoproteomics and mutational data, and validating performance across diverse tumor types. Together, these enhancements are expected to improve scalability and further accelerate the translation of novel cell-surface targets into clinical applications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Candidate proteins (N=43) not used for therapeutic radiopharmaceuticals with a weighted score greater than 1, indicating support from either multiple filtering strategies or at least one high-stringency filtering strategy within the multi-layered prioritization pipeline. The weighted score is calculated as the sum of strategy-specific weights, where candidates identified by stringent filtering strategies (strategies 4–10) are assigned a weight of 2 and candidates identified by less stringent strategies (strategies 1–3 and 11) are assigned a weight of 1. Bars represent the total weighted score for each protein, reflecting the combined strength and breadth of evidence across filtering strategies. By excluding candidates with a weighted score of 1, the visualization focuses on proteins with enhanced robustness, either through cross-strategy recurrence or selection under higher stringency criteria. These candidates are prioritized for downstream analysis and experimental validation.; Table S1: Drugability and clinical efforts for top 10 candidates.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, J.D. and S.P.; methodology, J.D. and S.P.; software, J.D. and S.P.; validation, J.D. and S.P.; formal analysis, J.D. and S.P.; investigation, J.D. and S.P.; resources, J.D. and S.P.; data curation, J.D. and S.P.; writing—original draft preparation, J.D.; writing—review and editing, J.D.; visualization, J.D.; supervision, J.D:; project administration, J.D.; funding acquisition, J.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Acknowledgments
During the preparation of this manuscript/study, the author(s) used M365 Copilot for the purposes of improving English writing and making Figure 2 and the graphical abstract. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| COAD | Colon Adenocarcinoma |
| COADREAD | Colon Adenocarcinoma and Rectal Adenocarcinoma |
| CRC | Colorectal Cancer |
| BRCA | Breast Invasive Carcinoma |
| LSCC | Lung squamous cell carcinoma |
| LUAD | Lung Adenocarcinoma |
| OV | Ovarian Serous Cystadenocarcinoma |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| CDDI | Cortellis Drug Discovery Intelligence |
| HPA | Human Protein Atlas |
| GTEX | The Genotype-Tissue Expression project |
| CSPA | Cell Surface Protein Atlas |
| CCLE | Cancer Cell Line Encyclopedia |
| GO | Gene Ontology |
| TCGA | The Cancer Genome Atlas |
| PC | Prostate Cancer |
| FC | Fold Change |
References
- Di Meo, F.; et al. Mapping the cancer surface proteome in search of target antigens for immunotherapy. Mol. Ther. 2024, 32(9), 2892–2904. [Google Scholar] [CrossRef]
- Crunkhorn, S. Proteogenomics identifies anticancer targets. Nat. Rev. Drug Discov. 2024, 23(9), 660. [Google Scholar] [CrossRef]
- Vasaikar, S.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177(4), 1035–1049 e19. [Google Scholar] [CrossRef]
- Zhang, B.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513(7518), 382–7. [Google Scholar] [CrossRef]
- Wang, J.; et al. Pan-Cancer Proteomics Analysis to Identify Tumor-Enriched and Highly Expressed Cell Surface Antigens as Potential Targets for Cancer Therapeutics. Mol. Cell Proteom. 2023, 22(9), 100626. [Google Scholar] [CrossRef]
- Leung, K.K.; et al. Engineered Proteins and Chemical Tools to Probe the Cell Surface Proteome. Chem. Rev. 2025, 125(8), 4069–4110. [Google Scholar] [CrossRef]
- Savage, S.R.; et al. Pan-cancer proteogenomics expands the landscape of therapeutic targets. Cell 2024, 187(16), 4389–4407 e15. [Google Scholar] [CrossRef]
- Shraim, R.; et al. ImmunoTar-integrative prioritization of cell surface targets for cancer immunotherapy. Bioinformatics 2025, 41(3). [Google Scholar] [CrossRef]
- Uhlen, M.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347(6220), 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; et al. A subcellular map of the human proteome . Science 2017, 356(6340). [Google Scholar] [CrossRef]
- Barretina, J.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483(7391), 603–7. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51(D1), D523–D531. [Google Scholar]
- Aung, W.; et al. Combined treatment of pancreatic cancer xenograft with (90)Y-ITGA6B4-mediated radioimmunotherapy and PI3K/mTOR inhibitor. World J. Gastroenterol. 2017, 23(42), 7551–7562. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; et al. In vivo-stable bis-iminobiotin for targeted radionuclide delivery with the mutant streptavidin. Bioorg Med. Chem. Lett. 2024, 108, 129803. [Google Scholar] [CrossRef]
- Zboralski, D.; et al. Preclinical evaluation of FAP-2286 for fibroblast activation protein targeted radionuclide imaging and therapy. Eur. J. Nucl. Med. Mol. Imaging 2022, 49(11), 3651–3667. [Google Scholar] [CrossRef]
- Bidkar, A.P.; et al. Treatment of Prostate Cancer with CD46-targeted 225Ac Alpha Particle Radioimmunotherapy. Clin. Cancer Res. 2023, 29(10), 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; et al. Astatine-211-Labeled Therapy Targeting Amino Acid Transporters: Overcoming Drug Resistance in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2025, 26(21). [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Jin, X.; Liu, Z. Prognostic and clinicopathological significance of GPRC5A in various cancers: A systematic review and meta-analysis. PLoS ONE 2021, 16(3), e0249040. [Google Scholar] [CrossRef]
- He, A.; Liao, F.; Lin, X. Circ_0007351 Exerts an Oncogenic Role In Colorectal Cancer Depending on the Modulation of the miR-5195-3p/GPRC5A Cascade. Mol. Biotechnol. 2025, 67(2), 617–627. [Google Scholar] [CrossRef]
- Liu, X.S.; et al. SLC2A1 is a Diagnostic Biomarker Involved in Immune Infiltration of Colorectal Cancer and Associated With m6A Modification and ceRNA. Front Cell Dev. Biol. 2022, 10, 853596. [Google Scholar] [CrossRef]
- Ren, Z.; et al. Targeting Glucose Transporter 1 (GLUT1) in Cancer: Molecular Mechanisms and Nanomedicine Applications. Int. J. Nanomed. 2025. 20, 11859–11879. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; et al. Role of CD47 gene expression in colorectal cancer: a comprehensive molecular profiling study. J. Immunother. Cancer 2024, 12(11). [Google Scholar] [CrossRef]
- Li, Q.; et al. Expression and Clinical Significance of CD47 in Colorectal Cancer: A Review. Cancers 2025, 18(1). [Google Scholar] [CrossRef]
- Eisenach, P.A.; et al. Dipeptidase 1 (DPEP1) is a marker for the transition from low-grade to high-grade intraepithelial neoplasia and an adverse prognostic factor in colorectal cancer. Br. J. Cancer 2013, 109(3), 694–703. [Google Scholar] [CrossRef]
- Park, S.Y.; et al. Dehydropeptidase 1 promotes metastasis through regulation of E-cadherin expression in colon cancer. Oncotarget 2016, 7(8), 9501–12. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; et al. Interferon-induced transmembrane protein 1 (IFITM1) is required for the progression of colorectal cancer. Oncotarget 2016, 7(52), 86039–86050. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; et al. IFITM1-targeted NIR-II fluorescence imaging enables.
Figure 1.
Architecture and workflow of the multi layered proteogenomic filtering pipeline for cell surface target discovery. The pipeline integrates quantitative proteomic data from CPTAC colorectal cancer cohorts with curated biological and clinical metadata to enable systematic prioritization of cell surface therapeutic targets. Multi source datasets, including CPTAC COAD and COADREAD proteomics, Human Protein Atlas (HPA), Genotype Tissue Expression (GTEx), Cancer Cell Line Encyclopedia (DepMap), UniProt, Cell Surface Protein Atlas (CSPA), and Clarivate Drug Discovery Intelligence (CDDI), are merged into an annotated union table containing protein expression metrics, subcellular localization annotations, normal tissue expression levels, tumor cell expression, and drugability information.
Figure 1.
Architecture and workflow of the multi layered proteogenomic filtering pipeline for cell surface target discovery. The pipeline integrates quantitative proteomic data from CPTAC colorectal cancer cohorts with curated biological and clinical metadata to enable systematic prioritization of cell surface therapeutic targets. Multi source datasets, including CPTAC COAD and COADREAD proteomics, Human Protein Atlas (HPA), Genotype Tissue Expression (GTEx), Cancer Cell Line Encyclopedia (DepMap), UniProt, Cell Surface Protein Atlas (CSPA), and Clarivate Drug Discovery Intelligence (CDDI), are merged into an annotated union table containing protein expression metrics, subcellular localization annotations, normal tissue expression levels, tumor cell expression, and drugability information.

Figure 2.
Number of candidate hits per filtering strategy, unique candidates, and overlap between strategies. The chord diagram visualizes pairwise overlap in candidate proteins across eleven filtering strategies used in the multi-layered pipeline. Each arc represents one filtering strategy, with arc length proportional to the total number of retained candidate proteins. Chords connecting strategies indicate shared proteins, with chord thickness reflecting the extent of overlap and chord color corresponding to the originating filtering strategy. Proteins uniquely retained by a given strategy are depicted as an inner band adjacent to each arc, with band thickness proportional to the number of unique candidate proteins for that strategy. The numbers in parentheses after the filter indexes show the total number and the unique number of hits for each filter. Strategies 1–4, based on combined protein ranking, subcellular localization scores, and penalties for normal tissue expression, show limited mutual overlap with later filters. Strategies 5–7, which incorporate differential expression and manual curation for plasma membrane localization, display intermediate connectivity. Strategies 8–11, derived from cohort-driven analyses with increasingly stringent log fold-change thresholds, show increased internal overlap, reflecting shared candidate prioritization under higher stringency. Candidates detected across multiple strategies are considered more robust and are prioritized for downstream validation, while unique candidates highlight potential strategy-specific discoveries.
Figure 2.
Number of candidate hits per filtering strategy, unique candidates, and overlap between strategies. The chord diagram visualizes pairwise overlap in candidate proteins across eleven filtering strategies used in the multi-layered pipeline. Each arc represents one filtering strategy, with arc length proportional to the total number of retained candidate proteins. Chords connecting strategies indicate shared proteins, with chord thickness reflecting the extent of overlap and chord color corresponding to the originating filtering strategy. Proteins uniquely retained by a given strategy are depicted as an inner band adjacent to each arc, with band thickness proportional to the number of unique candidate proteins for that strategy. The numbers in parentheses after the filter indexes show the total number and the unique number of hits for each filter. Strategies 1–4, based on combined protein ranking, subcellular localization scores, and penalties for normal tissue expression, show limited mutual overlap with later filters. Strategies 5–7, which incorporate differential expression and manual curation for plasma membrane localization, display intermediate connectivity. Strategies 8–11, derived from cohort-driven analyses with increasingly stringent log fold-change thresholds, show increased internal overlap, reflecting shared candidate prioritization under higher stringency. Candidates detected across multiple strategies are considered more robust and are prioritized for downstream validation, while unique candidates highlight potential strategy-specific discoveries.

Figure 3.
Candidate proteins (N=49) with a weighted score greater than 1, indicating support from either multiple filtering strategies or at least one high-stringency filtering strategy within the multi-layered prioritization pipeline. The weighted score is calculated as the sum of strategy-specific weights, where candidates identified by stringent filtering strategies (strategies 4–10) are assigned a weight of 2, and candidates identified by less stringent strategies (strategies 1–3 and 11) are assigned a weight of 1. Bars represent the total weighted score for each protein, reflecting the combined strength and breadth of evidence across filtering strategies. By excluding candidates with a weighted score of 1, the visualization focuses on proteins with enhanced robustness, either through cross-strategy recurrence or selection under higher stringency criteria. These candidates are prioritized for downstream analysis and experimental validation.
Figure 3.
Candidate proteins (N=49) with a weighted score greater than 1, indicating support from either multiple filtering strategies or at least one high-stringency filtering strategy within the multi-layered prioritization pipeline. The weighted score is calculated as the sum of strategy-specific weights, where candidates identified by stringent filtering strategies (strategies 4–10) are assigned a weight of 2, and candidates identified by less stringent strategies (strategies 1–3 and 11) are assigned a weight of 1. Bars represent the total weighted score for each protein, reflecting the combined strength and breadth of evidence across filtering strategies. By excluding candidates with a weighted score of 1, the visualization focuses on proteins with enhanced robustness, either through cross-strategy recurrence or selection under higher stringency criteria. These candidates are prioritized for downstream analysis and experimental validation.

Figure 4.
Validation of top 10 ranked candidates by number of pubmed articles for searches on candidates AND cancer, candidates AND cancer AND metastatic, or candiadates AND cancer AND metastatic AND colorectal.
Figure 4.
Validation of top 10 ranked candidates by number of pubmed articles for searches on candidates AND cancer, candidates AND cancer AND metastatic, or candiadates AND cancer AND metastatic AND colorectal.

Figure 5.
Validation of top ranked candidates by protein expression data of targets by immunohistochemistry confirmed plasma membrane localization and differential expression in colorectal cancer patients vs. healthy colon. Target is visualized in brown staining, the stronger the brown color, the higher the target expression. Tumor expression for CD82 and SLC27A2 could not be confirmed and IHC images were not available for RRP12. Representative images from Human Protein Atlas, proteinatlas.org [9]. Staining intensity was evaluated semi-quantitatively based on HPA annotation (low, medium, high).
Figure 5.
Validation of top ranked candidates by protein expression data of targets by immunohistochemistry confirmed plasma membrane localization and differential expression in colorectal cancer patients vs. healthy colon. Target is visualized in brown staining, the stronger the brown color, the higher the target expression. Tumor expression for CD82 and SLC27A2 could not be confirmed and IHC images were not available for RRP12. Representative images from Human Protein Atlas, proteinatlas.org [9]. Staining intensity was evaluated semi-quantitatively based on HPA annotation (low, medium, high).

Figure 6.
Validation of top ranked candidates by protein expression in other tumor types. Data from CPTAC. BRCA (Breast Invasive Carcinoma, PDC000120), LSCC (Lung Squamous Cell Carcinoma, PDC000234), LUAD (Lung Adenocarcinoma, PDC000153), OV (Ovarian Serous Cystadenocarcinoma, PDC000110) and PDAC (Pancreatic Ductal Adenocarcinoma, PDC000270). * adj. p-value < 0,001, ** adj. p-value < 0,05. Data were not available for DPEP1 and IFITM3.
Figure 6.
Validation of top ranked candidates by protein expression in other tumor types. Data from CPTAC. BRCA (Breast Invasive Carcinoma, PDC000120), LSCC (Lung Squamous Cell Carcinoma, PDC000234), LUAD (Lung Adenocarcinoma, PDC000153), OV (Ovarian Serous Cystadenocarcinoma, PDC000110) and PDAC (Pancreatic Ductal Adenocarcinoma, PDC000270). * adj. p-value < 0,001, ** adj. p-value < 0,05. Data were not available for DPEP1 and IFITM3.

Figure 7.
Validation of top ranked candidates by protein used as biomarker. A) Number of indications protein has been used as biomarker across indications, cancers, CRC and times used. B) Times reported as biomarker for different tumor types. BRCA=Breast Invasive Carcinoma; LSCC=Lung squamous cell carcinoma; LUAD=Lung Adenocarcinoma; OV=Ovarian Serous Cystadenocarcinoma; PDAC=Pancreatic Ductal Adenocarcinoma; PC=Prostate Cancer.
Figure 7.
Validation of top ranked candidates by protein used as biomarker. A) Number of indications protein has been used as biomarker across indications, cancers, CRC and times used. B) Times reported as biomarker for different tumor types. BRCA=Breast Invasive Carcinoma; LSCC=Lung squamous cell carcinoma; LUAD=Lung Adenocarcinoma; OV=Ovarian Serous Cystadenocarcinoma; PDAC=Pancreatic Ductal Adenocarcinoma; PC=Prostate Cancer.



Table 1.
Filtering strategies 1-4 using protein expression, localization and excretion data for biopsies and tumor cells as well as normal cell expression and information about use in RLT.
Table 1.
Filtering strategies 1-4 using protein expression, localization and excretion data for biopsies and tumor cells as well as normal cell expression and information about use in RLT.
| Filter | Rationale | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|---|
| Sum of scaled localization ranks (UniProt, HPA & CSPA) | Ensure localization on PM | >0,44 | >= 0,1 | > 0,4 | ||
| Mean protein rank | Overexpression of protein | >= 0,5 | ||||
| Mean stage 4 protein rank | Proteins with stage 4 protein expr | > 0 | > 0 | > 0,3 | ||
| Targeted by RIT | Protein with other RITs removed | = 0 | ||||
| UniProtKB & HPA secreted | No secreted proteins | = 0 | ||||
| GTEx mean abundance | Low normal tissue expr | < 2 | < 1,8 | |||
| HPA max abundance | Low normal tissue expr | = 0,1, 2* | ||||
| HPA mean abundance | Low normal tissue expr | < 2* | < 1,8 | |||
| Max abundance on CCLE | Expressed on tumor cells | > 0,46* | >1* | > 0,5* | > 2* | |
| Mean abundance on CCLE | Expressed on tumor cells | > 0,4* | >0,5* | > 0,4* | > 1* | |
| N detected (CRC) in CCLE | Expressed on tumor cells | > 0* | ||||
| Mean abundance CCLE (CRC) | Expressed on tumor cells | >= 1,5* | >=1,4* | >= 1,5* | ||
*Proteins with no data >(blanks) included. Black cells: Filter not used. HPA=Human Protein Atlas; GTEX=The Genotype-Tissue Expression project; CSPA=Cell Surface Protein Atlas; CCLE=Cancer Cell Line Encyclopedia;.
Table 2.
Filtering strategies 5-7 centered on differential expression of plasma membrane proteins looking at data from the COAD and COADREAD cohorts in sequence. The derived shortlists were investigated in later comparative analysis to results from strategies 1-4, where a sum score for mean stage 4 protein rank, localization rank, mean abundance normal tissue and clinical develop ment stage was calculated.
Table 2.
Filtering strategies 5-7 centered on differential expression of plasma membrane proteins looking at data from the COAD and COADREAD cohorts in sequence. The derived shortlists were investigated in later comparative analysis to results from strategies 1-4, where a sum score for mean stage 4 protein rank, localization rank, mean abundance normal tissue and clinical develop ment stage was calculated.
| Filter | Rationale | 5 | 6 | 7 | |
|---|---|---|---|---|---|
| Stage IV protein expression in COAD and COADREAD cohorts | Proteins with stage IV protein expression | At least upregulated in one cohort and up-regulated or non-differentially expressed in the other* | |||
| UniProt localization score** | Enrichment of candidate proteins with cell surface localization | >0 | >0 | ||
| Sum of scaled localization ranks (UniProt, HPA & CSPA) | Ensure localization on PM | >0,5 | >0 | ||
| Penalty for secreted proteins | Ensure localization on PM | Shedded proteins excluded | |||
| 1.6x Up-regulation observed at min in COAD cohort | Increasing selection stringency | LogFC COAD >=0.2 | |||
| 1.6x Up-regulation observed at min in one cohort | Increasing selection stringency | logFC(COAD or COADREAD)>=0.2 | |||
| Penalty for mitochondria, nucleus and peroxisome localization | Ensure localization on PM | Exclusion of intra-cellular proteins with membrane association | |||
| Mean abundance HPA | Low normal tissue expression | <=1,5 | |||
| Manual review for no extracellularly exposed proteins | Ensure relevant PM expression | Manual curation | |||
*Proteins with no data >(blanks) included. **Sum of UniProt annotation scores for 1) plasma membrane localization + topologies, 2) Transmembrane domain, 3) Intramembrane domain, 4) Extracellular domain, 5) Cytoplasmic domain.
Table 3.
Filtering strategies 8-11 are driven by either COAD or COADREAD cohorts alone. The derived shortlists were investigated in later comparative analysis to results from strategies 1-4, where a sum score for mean stage 4 protein rank, localization rank, mean abundance normal tissue and clinical development stage was calculated.
Table 3.
Filtering strategies 8-11 are driven by either COAD or COADREAD cohorts alone. The derived shortlists were investigated in later comparative analysis to results from strategies 1-4, where a sum score for mean stage 4 protein rank, localization rank, mean abundance normal tissue and clinical development stage was calculated.
| Filter | Rationale | 8 | 9 | 10 | 11 | ||
|---|---|---|---|---|---|---|---|
| Stage IV protein expression in COAD | Proteins with stage IV protein expression | Upregulated | |||||
| Stage IV protein expression in COADREAD | Proteins with stage IV protein expression | Upregulated | |||||
| UniProt localization score** | Enrichment of candidate proteins with cell surface localization | >0 | >0 | ||||
| Manual review for no extracellularly exposed proteins | Ensure relevant PM expression | Manual curation | |||||
| Sum of scaled localization ranks (UniProt, HPA & CSPA) | Ensure localization on PM | >0 | |||||
| Penalty for secreted proteins | Ensure localization on PM | Shedded proteins excluded | |||||
| Up-regulation observed in COADREAD cohort | Increasing selection stringency | logFC >=2 | logFC >=1.4 | ||||
*Proteins with no data >(blanks) included. **Sum of UniProt annotation scores for 1) plasma membrane localization + topologies, 2) Transmembrane domain, 3) Intramembrane domain, 4) Extracellular domain, 5) Cytoplasmic domain. FC = Fold Change.
Table 4.
Top-10 ranked candidates and composite scores. Sum weighted score was calculated by adding the sum of scaled localization ranks, mean stage IV protein rank, scaled sum of CDDI ranks, and half of the mean abundance score in CCLE and CRC CCLE. Normal tissue expression in HPA and GTEX were subtracted. Higher sum weighted scores indicate stronger prioritization for translational development.
Table 4.
Top-10 ranked candidates and composite scores. Sum weighted score was calculated by adding the sum of scaled localization ranks, mean stage IV protein rank, scaled sum of CDDI ranks, and half of the mean abundance score in CCLE and CRC CCLE. Normal tissue expression in HPA and GTEX were subtracted. Higher sum weighted scores indicate stronger prioritization for translational development.
| Symbol | Description | Sum of scaled localization ranks | Mean stage 4 protein rank | Scaled sum of CDDI ranks | MAS* in HPA | MAS* in GTEX | MAS* in CCLE | MAS* (CRC) in CCLE | Sum weighted score |
|---|---|---|---|---|---|---|---|---|---|
| GPRC5A | G protein-coupled receptor class C group 5 member A | 1,84 | 1,00 | 0,83 | 0,71 | 0,44 | 1,70 | 2,19 | 4,47 |
| SLC2A1 | Solute carrier family 2 member 1 | 2,72 | 0,16 | 0,92 | 0,29 | 1,1 | 1,54 | 1,57 | 3,965 |
| CD47 | CD47 molecule | 2,32 | 0,19 | 0,92 | 1,22 | 1,33 | 1,59 | 1,56 | 2,46 |
| DPEP1 | Dipeptidase 1 | 0,36 | 0,30 | 0,83 | 0,42 | 0,28 | 0,59 | 1,67 | 1,92 |
| IFITM1 | Interferon induced transmembrane protein 1 | 1,5 | 0,32 | 0,83 | 0,78 | 1,87 | |||
| CD82 | CD82 molecule | 1,24 | 0,51 | 0,50 | 0,58 | 1,31 | 1,31 | 1,48 | 1,76 |
| SLC27A2 | Solute carrier family 27 member 2 | 0,25 | 0,24 | 0,00 | 0,49 | 0,45 | 1,49 | 2,41 | 1,50 |
| DSC2 | Desmocollin 2 | 1,61 | 0,23 | 0,00 | 1,31 | 0,97 | 1,65 | 2,15 | 1,46 |
| RRP12 | Ribosomal R processing 12 homolog | 0,83 | 0,41 | 0,00 | 1,30 | 1,52 | 1,52 | 1,46 | |
| IFITM3 | Interferon induced transmembrane protein 3 | 1,00 | 0,29 | 0,00 | 1,14 | 1,33 | 1,26 | 1,45 |
*MAS: Mean abundance score.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



