Submitted:
28 May 2026
Posted:
01 June 2026
You are already at the latest version
Abstract
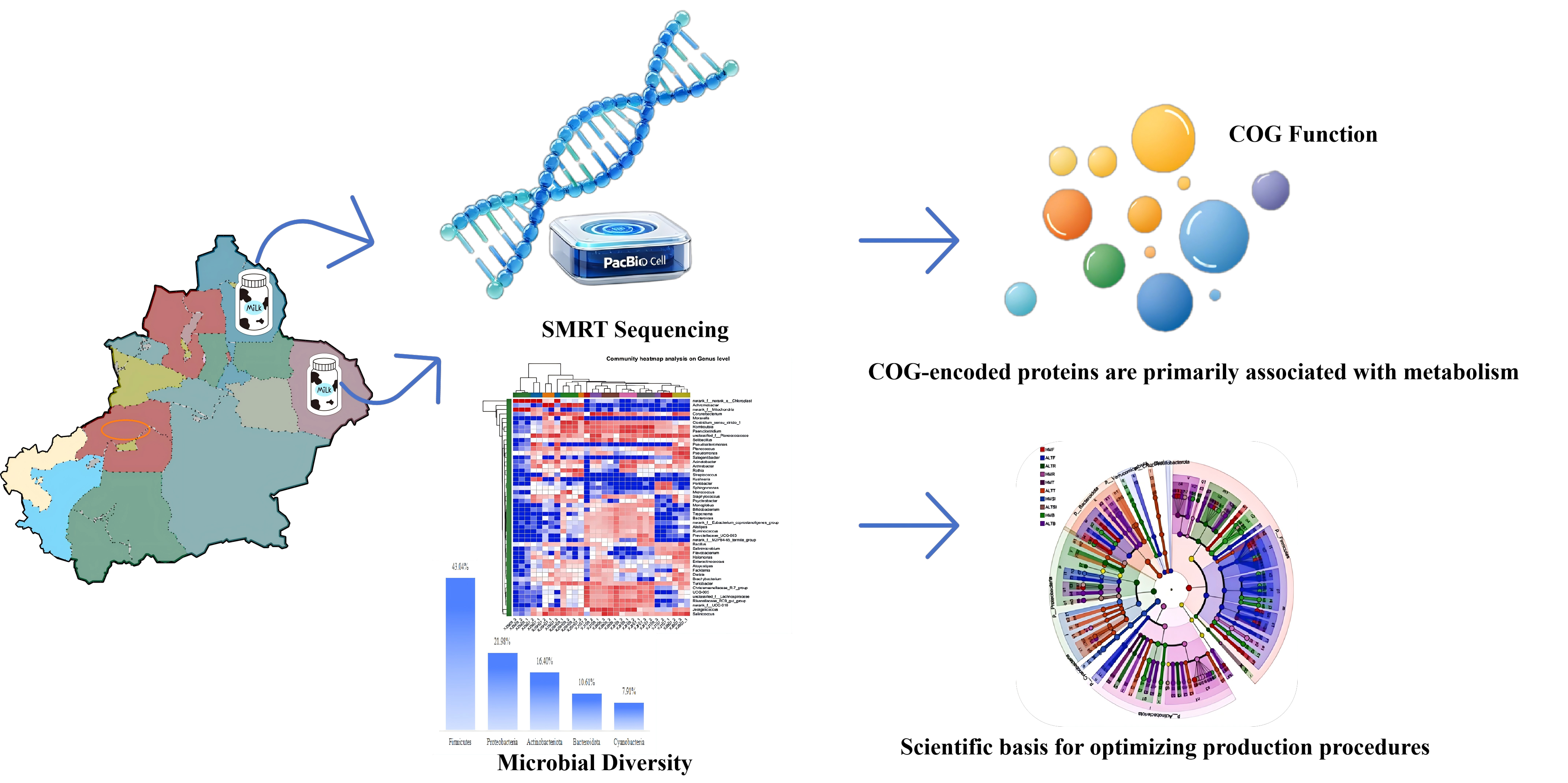
Raw milk quality serves as a pivotal determinant throughout the dairy production and processing chain, given that bacterial abundance and diversity exert direct effects on product safety and shelf life. This study employed single-molecule real-time (SMRT) sequencing to profile how production environments shape the microbial community structure of raw camel milk in the Altay and Hami regions of Xinjiang. The dominant phyla across all samples were Firmicutes (43.04%), Proteobacteria (21.98%), Actinobacteriota (16.40%), Bacteroidota (10.61%), and Cyanobacteria (7.91%). Functional profiling revealed that Clusters of Orthologous Groups (COG)-encoded proteins were predominantly enriched in metabolic pathways, particularly amino acid transport and energy conversion. These findings establish a robust scientific foundation for refining raw milk production protocols. Notably, environmental sourcing analysis identified feces and soil as primary reservoirs for key taxa, providing actionable evidence for upgrading quality surveillance systems and optimizing production practices in the camel milk industry.
Keywords:
single-molecule real-time sequencing
; raw milk
; production environment
; microbial diversity
1. Introduction
Raw milk harbors a complex and dynamic microbiome, thriving in a nutrient-rich aqueous environment that supports rapid microbial proliferation. These microbial consortia encompass spoilage agents, pathogens, beneficial probiotics, and technologically relevant starter cultures (Wang et al., 2019; Dufour et al., 2019; Moineau-Jean et al., 2019). Following milking and during refrigerated storage, microbial dynamics persist, ultimately influencing the microbiological quality of milk upon arrival at processing facilities (Castilho et al., 2018; Isele et al., 2019; Srisengfa et al., 2016). The raw milk microbiota is typically characterized by Gram-positive taxa, including Bacillus, Micrococcus, and Staphylococcus, alongside various lactic acid bacteria (LAB), such as Lactococcus and Streptococcus. Conversely, Gram-negative psychrotrophs, particularly Pseudomonas, Aeromonas, and Enterobacteriaceae members (e.g., Enterobacter, Hafnia, Klebsiella), pose significant spoilage risks due to their resilience. Yeasts, including Candida and Kazachstania, also contribute to the fungal diversity of raw milk (Krewinkel et al., 2015).
Psychrotrophic bacteria, such as Pseudomonas spp., Aeromonas spp., and Bacillus spp., dominate the cold-chain ecology (Qin et al., 2014). Of particular concern are the thermostable enzymes, specifically lipases and proteases, secreted by these microorganisms. These enzymes can withstand pasteurization and ultra-high temperature (UHT) treatments, representing the primary agents of spoilage that compromise the sensory and functional quality of dairy products. Specifically, lipases catalyze triglyceride hydrolysis, resulting in lipolytic rancidity and butyric acid off-flavors. Concurrently, proteases hydrolyze casein fractions, generating bitter peptides and inducing age-related gelation, thereby reducing shelf life (Xiang et al., 2024).
The taxonomic composition of raw milk is primarily dictated by direct-contact microbial reservoirs, notably the teat apex, udder skin, and milking apparatus. Secondary influences stem from indirect-contact environments, including bedding materials, feces, feed, water sources, and the broader barn and parlor environments (Stoeckel et al., 2016). Subsequent contamination can occur during milking, transportation, and storage, posing substantial risks to both the safety and processing quality of the final product. Consequently, characterizing the raw milk microbiome is critical for ensuring product safety and optimizing downstream processing protocols.
Traditional culture-independent methods, such as Single-Molecule Real-Time (SMRT) sequencing, bypass the limitations of microbial cultivation by analyzing total DNA extracted directly from samples. This technology offers superior resolution and throughput compared to conventional culture-dependent microbiological techniques (Fenelon et al., 2020). By applying SMRT sequencing to metagenomic DNA, researchers can achieve precise taxonomic classification down to the species level, significantly accelerating the diagnostic timeline (Hou et al., 2016). Furthermore, third-generation sequencing facilitates a holistic interrogation of microbial genomic architectures within environmental niches, thereby maximizing the exploitable potential of bioresources (Zhou et al., 2024). Specifically, targeting the full-length 16S rRNA gene allows for high-fidelity resolution of bacterial taxa, enabling localization to the species level (Liang et al., 2022).
Herein, we conducted a comprehensive investigation into the microbial ecology of raw camel milk and its associated production environments (i.e., feces, bedding, feed, and teat skin) to elucidate the overall microbial diversity. The primary objectives were to guide post-harvest processing strategies, optimize disease prevention protocols, and identify potential beneficial microbial resources for industrial exploitation. This work establishes a theoretical framework for understanding the dynamics of microbial contamination within camel milk production systems in Xinjiang, China. By mitigating potential contamination risks and deepening insights into raw milk microbiology, this study furnishes detailed contextual data to support dairy processing, enhance safety assurance, and facilitate novel strain discovery. Ultimately, these findings lay a robust scientific foundation for the systematic monitoring of raw milk quality and safety.
2. Materials and Methods
2.1. Sample Collection
Sampling was conducted across camel breeding farms in the Altay and Hami regions of Xinjiang, China. Environmental matrices were systematically collected, including bedding and feed from pens, feces and soil from the vicinity of the farms, and teat skin swabs from lactating camels. For each farm and sample type, three independent biological replicates were collected. Following quality control, a total of 30 samples were retained for downstream analysis. These comprised five categories (cushion, feces, teat skin, feed, and soil) from both regions, with three replicates per category. The coding scheme denoted samples from Altay as "07" and Hami as "09" , with suffixes -1 to -3 indicating replicates (See Supplementary Table S1 for detailed sample IDs).
2.2 DNA Extraction
Genomic DNA was extracted from all samples using the TIANamp Bacterial DNA Kit (Tiangen Biotech, Beijing, China), following the manufacturer’s protocol. The concentration and purity (A260/A280 ratio) of the extracted DNA were quantified using a NanoPhotometer® N60 (Implen GmbH, Munich, Germany). Subsequently, DNA integrity and fragment size were verified by electrophoresis on a 1% (w/v) agarose gel. Only samples with high purity (OD260/280 ratio between 1.8 and 2.0) and intact high-molecular-weight bands were selected for subsequent library construction.
2.3 PCR Amplification
Specific barcoded primers were synthesized to target the hypervariable V4 region of the 16S rRNA gene. To ensure data reproducibility and minimize PCR bias, amplification was performed using the minimum number of cycles required to obtain sufficient product, with strict consistency in cycle numbers across all samples. A preliminary trial was conducted on representative samples to determine the optimal cycle threshold for adequate yield.
PCR amplification was carried out using a ProFlex PCR System (GeneAmp PCR System 9700, Applied Biosystems, Foster City, CA, USA) with TransStart Fastpfu DNA Polymerase (TransGen Biotech, Beijing, China). Each sample was amplified in triplicate under standard cycling conditions. Replicate amplicons for each sample were pooled and visualized via 2% agarose gel electrophoresis. Target DNA fragments were excised from the gel and purified using the AxyPrep DNA Gel Recovery Kit (Axygen Biosciences, Union City, CA, USA). The purified products were eluted with Tris-HCl buffer (pH 8.8) and subsequently verified by 2% agarose gel electrophoresis.
Library quantification was performed using the QuantiFluor™ ST Blue Fluorescence System (Promega, Madison, WI, USA), ensuring a robust two-tiered quality control (QC) strategy prior to SMRT sequencing. Finally, libraries were normalized and pooled in equimolar ratios according to the sequencing platform's requirements.
2.4. Fluorescence Quantification
Library concentrations were accurately determined using the QuantiFluor™ ST Blue Fluorescence System (Promega, Madison, WI, USA). Subsequently, libraries were normalized and pooled in equimolar ratios based on the sequencing platform's requirements.
2.5. Library Construction and Sequencing
SMRTbell libraries were constructed using the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA) following the manufacturer’s instructions. Briefly, the PCR amplicons were purified using AMPure PB beads (Pacific Biosciences) to remove short fragments and primers. Size selection was performed via 2% agarose gel electrophoresis to ensure the integrity of the inserts. The purified DNA was eluted in TE buffer and quantified using a Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA). Finally, the double-stranded DNA templates were denatured and bound to the polymerase to form sequencing-ready complexes for Single-Molecule Real-Time (SMRT) sequencing.
2.6. SMRT Sequencing
Sequencing was performed on a PacBio Sequel II system (Pacific Biosciences, Menlo Park, CA, USA) following the manufacturer’s guidelines. The sequencing process relies on Single-Molecule Real-Time (SMRT) technology, wherein DNA polymerase complexes are immobilized at the bottom of zero-mode waveguides (ZMWs). Sequencing primers and the SMRTbell templates were bound to form sequencing-ready complexes. Upon loading into the SMRT Cell, the diffusion of nucleotides allowed for real-time monitoring of fluorescence pulses as phospholinked nucleotides were incorporated. This setup enabled the generation of high-fidelity reads, facilitating comprehensive profiling of the microbial communities.
2.7. Bioinformatics Analysis
Raw subreads generated from SMRT sequencing were processed using the ccs (Circular Consensus Sequencing) algorithm to generate High-Fidelity (HiFi) reads. Following rigorous quality filtering and primer trimming, demultiplexed reads were denoised and clustered into Amplicon Sequence Variants (ASVs) or Operational Taxonomic Units (OTUs) using the QIIME2 pipeline (version 2023.2). Taxonomic assignment was performed by querying the sequences against the SILVA reference database (release 138).
Alpha diversity metrics (including Shannon, Simpson, and Chao1 indices) and beta diversity analyses (such as Principal Coordinates Analysis, PCoA) were calculated based on the ASV/OTU table to evaluate microbial richness and structural variations. Taxonomic composition was statistically analyzed across all hierarchical levels (from phylum to genus) to characterize the microbial landscape. Furthermore, multivariate statistical analyses (e.g., ANOSIM, PERMANOVA) and differential abundance testing (e.g., DESeq2, LEfSe) were conducted to identify significant variations in microbial composition between sample groups.
3. Results
3.1. Overall Microbiome Analysis
Taxonomic profiling revealed a total of 35 bacterial phyla and 100 genera across all samples. The relative abundance of the dominant phyla is illustrated in Figure 1A, while the genus-level distribution is presented in Figure 1B (taxa with an abundance < 1.0% or those remaining unclassified were aggregated as "Others").
The majority of OTUs were assigned to three predominant phyla: Firmicutes (43.20%), Proteobacteria (20.12%), and Actinobacteriota (12.02%). In the Altay region, Firmicutes was the predominant phylum (43.04%). At the genus level, Rothia (20.63%) and sequences identified as Chloroplast (18.54%) represented the most abundant taxa. Similarly, Firmicutes dominated the Hami region (46.80%), although a high abundance of Chloroplast (18.23%) and Mitochondria (9.22%) sequences was also observed (Fig. 1A, B). It is noteworthy that the high abundance of chloroplast and mitochondrial sequences likely reflects the presence of plant-derived material and eukaryotic cells in the samples, which is a common occurrence in 16S rRNA amplicon sequencing of environmental samples.
3.2. Microbiota Diversity Based on Alpha and Beta Analysis
Alpha diversity was evaluated using the Chao1 and ACE indices (richness), alongside the Shannon and Simpson indices (diversity and evenness). As illustrated in Figure 2, the ACE index revealed a significantly higher community richness in soil samples from Altay compared to those from Hami (P < 0.05). In contrast, no significant differences were observed in Shannon or Simpson indices across most sample types between the two regions (P > 0.05).
Specifically, fecal samples exhibited significantly higher Chao1 and ACE values in the Altay region than in the Hami region (P < 0.05, Table 1). Similarly, feed samples showed significantly greater richness in Altay based on the ACE index, suggesting regional disparities in the abundance of microbial species.
To visualize structural variations, Principal Coordinates Analysis (PCoA) based on Bray-Curtis dissimilarity was employed. The PCoA plot (Fig. 2B) revealed distinct clustering patterns between the Altay and Hami groups. The ANOSIM test confirmed that the inter-group differences were significantly larger than the intra-group variations (R = 0.7503, P = 0.001), indicating distinct microbial profiles between the two regions. Furthermore, the NMDS ordination yielded a low stress value of 0.0094 (Fig. 2C), indicating an excellent fit of the data in the reduced dimensional space.
The heatmap visualization of genus-level abundance (Fig. 3) revealed distinct clustering patterns between the Altay and Hami regions, reflecting significant differences in microbial composition. Achromobacter was consistently detected across nipple skin samples from both regions. In contrast, sequences identified as mitochondria (likely representing eukaryotic contaminants or degraded host DNA) were exclusively found in feed samples from Hami.
Anaerobic taxa, including Clostridium sensu stricto, Romboutsia, and Paeniclostridium, exhibited a broad distribution across soil and fecal samples in both regions, with an extended presence in feed and bedding samples specifically within the Hami region. Conversely, specific genera, such as Bifidobacterium, Treponema, and Bacteroides, were predominantly restricted to soil, bedding, and fecal samples in the Hami region, with sporadic detection in Altay feces.
Certain taxa demonstrated regional specificity; for instance, Bacillus was exclusively identified in fecal, soil, and bedding samples from the Altay region. Similarly, Salinimicrobium, Flavobacterium, and Halomonas were unique to the soil and bedding of Altay. In Hami, Christensenellaceae R-7 group and Rikenellaceae RC9 group were prevalent in soil and bedding. The genus Jeotgalicoccus was primarily associated with nipple skin and bedding in Hami, whereas Salinicoccus showed a ubiquitous presence across bedding and skin in both regions. These distinct distribution profiles underscore the feasibility of differentiating bacterial communities between the two regions based on environmental reservoirs.
Figure 3.
Heat map of clustering of bacterial genera in all samples from both regions.

3.3. Common and Difference Analysis of Bacterial Compositions
Venn diagram analysis revealed a core microbiome comprising 21 shared genera and 23 shared species across both regions, representing 84.02% and 82.95% of the total taxonomic units identified, respectively (Figure 4A).
Fecal samples were dominated by Romboutsia (10.77%), followed by Paeniclostridium (8.10%) and Jeotgalicoccus (4.85%) (Figure 4B). Nipple skin samples exhibited a distinct profile, with Achromobacter spp. (22.23%) and Moraxella (6.29%) being predominant, alongside Clostridium sensu stricto (5.00%) (Figure 4C). Feed samples were characterized by a high abundance of chloroplast sequences (54.47%) and mitochondrial sequences (17.08%), likely reflecting plant-derived material and eukaryotic DNA, along with members of the Planococcaceae family (3.63%) (Figure 4D). Soil samples were primarily composed of Planococcaceae (7.05%), Clostridium sensu stricto (4.16%), and Romboutsia ilealis (2.95%) (Figure 4E). Finally, bedding samples were dominated by Corynebacterium maris (5.98%), Jeotgalicoccus spp. (5.61%), and Salinicoccus spp. (4.62%) (Figure 4F).
To identify the specific bacterial taxa driving the significant differences in community composition between the Altay and Hami regions, biomarker analysis was performed using the LEfSe method. Taxa labeled as "uncultured," "unranked," or "unclassified" were excluded from the dataset prior to analysis. Biomarker discovery was then conducted using Linear Discriminant Analysis (LDA) Effect Size (LEfSe), which effectively discriminates between biologically relevant groups by identifying features with significant effect sizes.
As illustrated in Figure 5A–C, Firmicutes was the dominant phylum in fecal samples from Altay, whereas Actinobacteriota predominated in those from Hami. A similar pattern was observed in teat skin samples (Firmicutes in Altay vs. Actinobacteriota in Hami). In soil samples, Actinobacteriota dominated in Altay, while Firmicutes was predominant in Hami. Feed samples lacked a dominant phylum in Altay but were dominated by Firmicutes in Hami. Additionally, bedding samples in Altay were characterized by Actinobacteriota, whereas those in Hami were dominated by Firmicutes.
These findings are consistent with the overall beta-diversity trends observed in previous sections. Furthermore, LEfSe identified 10 bacterial phyla with LDA scores exceeding 4.0, highlighting distinct taxonomic signatures between the regions.
3.4. Model Prediction and Predictive Functions
Functional profiling based on the Clusters of Orthologous Groups (COG) database revealed that the majority of genes were annotated to metabolic functions, including global and overview maps, as well as secondary metabolite biosynthesis (Fig. 6A). Predominant pathways specifically encompassed amino acid transport and metabolism, alongside the transport and metabolism of inorganic ions. Furthermore, secondary metabolite biosynthesis was linked to energy production and conversion, as well as translation, ribosomal structure, and biogenesis.
These results highlight the interconnected nature of the cellular metabolic network, emphasizing systemic functionality over isolated biochemical reactions (Fig. 6B). Comparative analysis indicated highly consistent COG functional profiles across both regions, suggesting conserved core metabolic potentials despite variations in taxonomic composition
Figure 6.
Predictive functional analysis of microbial communities based on the COG database. (A) Similar COG functional classification profiles across all samples; (B) Key metabolic pathways associated with COG-encoded proteins and the structure of the overall cellular metabolic network.
Figure 6.
Predictive functional analysis of microbial communities based on the COG database. (A) Similar COG functional classification profiles across all samples; (B) Key metabolic pathways associated with COG-encoded proteins and the structure of the overall cellular metabolic network.

4. Discussion
This study provides a comprehensive characterization of the microbial ecology across the raw camel milk production chain in the Altay and Hami regions of Xinjiang. The dominant phyla identified—Firmicutes (43.04%), Proteobacteria (21.98%), and Actinobacteriota (16.40%)—differ significantly from those reported in pasteurized milk studies. For instance, Ding et al. (2020) observed that Proteobacteria and Firmicutes each exceeded 50% in HTST-treated milk, with Pseudomonas dominating the genera. The lower relative abundance of these taxa in our raw samples suggests that pasteurization selectively eliminates sensitive strains, thereby enriching thermotolerant genera.
When compared to other non-bovine milks, our results align with the trends reported by Luoyizha et al. (2020) for donkey milk, particularly regarding the abundance of Bacteroidota (~10%). However, unlike Zhang et al. (2023), who found Firmicutes to constitute 96% of mare's milk, our camel milk samples exhibited a more balanced distribution among the top phyla. This variation likely stems from species-specific factors, regional climates, and dietary differences. Similarly, Ouyang et al. (2022) reported Proteobacteria dominance (43.3%–84.0%) in seawater, noting Cyanobacteria (0.3%–10.4%); our observed Cyanobacteria (7.91%) falls within this range, although in the context of ruminant feces/feed, these signals likely represent plant-derived chloroplast sequences rather than free-living cyanobacteria (Sánchez-Baracaldo, 2025).
Environmental sourcing analysis revealed distinct distribution patterns. In Altay, Firmicutes was highly prevalent in teat skin (49.58%) and soil (48.65%), suggesting these as primary reservoirs. Conversely, in Hami, fecal samples showed high abundances of Cyanobacteria (68.34%) and Firmicutes (29.07%). These findings corroborate Dong et al. (2021), who identified feces as a critical vector for Firmicutes transmission to milking equipment and storage tanks. Furthermore, Zhang et al. (2023) highlighted that Proteobacteria often originate from raw milk and water sources, which aligns with our observation of high Proteobacteria in Hami feed (21.80%) and soil (20.46%). The high prevalence of Cyanobacteria (often indicative of chloroplast sequences from plant matter) in feces and feed highlights the influence of roughage composition on the microbial landscape.
Functional profiling based on the COG database indicated that the microbiome is heavily geared towards metabolic activities, including amino acid transport and energy conversion. The consistent functional profiles across both regions, despite taxonomic variations, suggest a degree of functional redundancy within these ecosystems. Notably, the enrichment of Firmicutes and Bacteroidota is relevant to the production of short-chain fatty acids (SCFAs) like butyrate and propionate, which can suppress pro-inflammatory cytokines and strengthen epithelial barriers (Zhi et al., 2022). However, the presence of psychrotrophic taxa implies risks associated with heat-stable lipases and proteases (Xiang et al., 2024), which can lead to lipolytic rancidity and age-related gelation even after pasteurization (Fanelli et al., 2020).
Ensuring raw milk safety is paramount for public health and industry sustainability. Microbial contamination not only increases spoilage risks—often driven by Pseudomonas spp. and other psychrotrophs—but also undermines consumer confidence (Palii et al., 2020; Martin et al., 2021). Our study identifies critical control points, particularly the management of fecal contamination and bedding hygiene, supporting the notion that centrifugation and heat treatment can reduce microbial loads prior to processing (Peruzi et al., 2019; Jia et al., 2021). By mapping the environmental sources of key taxa, this work provides actionable evidence for upgrading quality surveillance systems and optimizing production practices in Xinjiang's dairy sector (Pierezan et al., 2022). Ultimately, the integration of SMRT sequencing data with traditional microbiological parameters offers a robust scientific foundation for regulatory bodies to implement precision monitoring across the entire dairy supply chain (Yuan et al., 2022).
5. Conclusions
This study systematically mapped the microbial landscapes of raw camel milk production chains in Xinjiang's Altay and Hami regions, resolving key taxa to the species level during the same seasonal window. We elucidated the primary environmental reservoirs of these microbes and assessed their implications for raw milk safety and processing stability, proposing targeted intervention strategies.
Furthermore, by characterizing bacterial diversity across critical nodes of the production continuum, this work deepens our understanding of how environmental factors shape the raw milk microbiome. These findings underscore the dual value of this research: advancing dairy safety protocols while identifying potential probiotic candidates or starter cultures. Ultimately, this integrated analysis provides a robust scientific foundation for regulatory bodies to implement precision monitoring and optimize quality assurance systems across the entire dairy supply chain.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1.
Author Contributions
Conceptualization: Yating Wu, Shuai Wang, Data curation: Lu Meng, Xiaoxiao Lou. Formal analysis: He Chen, Cheng Wang, Yankun Zhao. Methodology: He Chen, Cheng Wang, Yankun Zhao. Writing-original draft: Yating Wu, Shuai Wang, Xianlan Ma. Writing-review & editing: Nan Zheng, Wei Shao, Lu Meng, He Chen, Cheng Wang, Yankun Zhao.
Funding
This research was funded by the Key R&D Project of the Autonomous Region (2023B02034-1), the Dairy Industry and Technology System of XJARS (XJARS-11-09), the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01B166), the Ministry of Agriculture and Rural Affairs ((xjnkywdzc2026003-10-kt1), and the National Key Research and Development Program (2022YFD1600100).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| The following abbreviations are used in this manuscript: |
| ASV | Amplicon Sequence Variant |
| COG | Clusters of Orthologous Groups |
| HTST | High-Temperature Short-Time |
| LAB | Lactic Acid Bacteria |
| LEfSe | Linear Discriminant Analysis Effect Size |
| NMDS | Non-metric Multidimensional Scaling |
| OTU | Operational Taxonomic Unit |
| PCoA | Principal Coordinates Analysis |
| SMRT | Single-Molecule Real-Time (sequencing) |
| UHT | Ultra-High Temperature |
References
- Andréanne Moineau-Jean, C.P.; Roy, D.; LaPointe, G. Effect of Greek-style yogurt manufacturing processes on starter and probiotic bacteria populations during storage. Int. Dairy J. 2019, 93, 35–44.
- Castilho, N.P.A.; Colombo, M.; Todorov, S.D.; Nero, L.A. Beneficial properties of lactic acid bacteria naturally present in dairy production. BMC Microbiol. 2018, 18, 219.
- Celano, G.; Costantino, G.; Vacca, M.; D’Auria, M.; Calabrese, F.M.; De Angelis, M.; Gobbetti, M. Effect of Seasonality on Microbiological Variability of Raw Cow Milk from Apulian Dairy Farms in Italy. Microbiol. Spectr. 2022, 10, e0051422.
- Ding, R.; Liu, Y.; Yue, X.; Wang, Y.; Zhang, Y.; Chen, Q. High-throughput sequencing provides new insights into the roles and implications of core microbiota present in pasteurized milk. Food Res. Int. 2020, 137, 109586.
- Dong, L.; Pang, X.; Tang, S.; Wang, J.; Zhang, Y. High-throughput sequencing reveals bacterial diversity in raw milk production environment and production chain in Tangshan city of China. Food Sci. Anim. Resour. 2021, 41, 452–467.
- Dufour, S.; Bernier Gosselin, V.; Adkins, P.R.F.; Middleton, J.R.; Barkema, H.W. Persistence of coagulase-negative staphylococcal intramammary infections in dairy goats. J. Dairy Res. 2019, 86, 211–216.
- Fanelli, F.; Cho, G.S.; Böhnlein, C.; Kabisch, J.; Franz, C.M.A.P. Microbial quality and safety of milk and milk products in the 21st century. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2013–2049.
- Fenelon, M.A.; Gleeson, D.; Hill, C.; Cotter, P.D.; Guinane, C.M. Tracking the Dairy Microbiota from Farm Bulk Tank to Skimmed Milk Powder. mSystems. 2020, 5, e00226-20.
- Ferrer, M.; Lanza, F.; Bargiela, R.; Almansa, C.; Puchades-Carrasco, L.; Moya, A. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226.
- Hou, Q.; Bian, Y.; Zhang, W.; Liu, X.; Huang, L.; Deng, Z. Using PacBio Long-Read High-Throughput Microbial Gene Amplicon Sequencing To Evaluate Infant Formula Safety. J. Agric. Food Chem. 2016, 64, 6993–7001.
- Isele, D.; Hahne, J.; Berning, J.; Baur, C.; Krewinkel, M.; von Neubeck, M.; Wenning, M.; Scherer, S. The contribution of fast growing, psychrotrophic microorganisms on biodiversity of refrigerated raw cow's milk with high bacterial counts and their food spoilage potential. Food Microbiol. 2019, 79, 11–19.
- Jia, Z.; Huang, L.; Wei, Z.; Wang, X. Dynamic kinetic analysis of growth of Listeria monocytogenes in pasteurized cow milk. J. Dairy Sci. 2021, 104, 2654–2667.
- Krewinkel, M.; Kranz, B.; von Neubeck, M.; Wenning, M.; Scherer, S. Biodiversity of refrigerated raw milk microbiota and their enzymatic spoilage potential. Int. J. Food Microbiol. 2015, 211, 57–65.
- Liang, L.; He, L.; Qu, T.; Wang, J. Single-molecule real-time sequencing reveals differences in bacterial diversity in raw milk in different regions and seasons in China. J. Dairy Sci. 2022, 105, 5669–5684.
- Luoyizha, W.; Wu, X.; Zhang, M.; Wang, J. Compared analysis of microbial diversity in donkey milk from Xinjiang and Shandong of China through high-throughput sequencing. Food Res. Int. 2020, 137, 109684.
- Martin, N.H.; Wiedmann, M.; Torres-Frenzel, P. Invited review: Controlling dairy product spoilage to reduce food loss and waste. J. Dairy Sci. 2021, 104, 1251–1261.
- Ouyang, L.; Chen, X.; Zhang, W.; Wang, J. High throughput sequencing reveals distinct bacterial communities and functional diversity in two typical coastal bays. J. Mar. Sci. Eng. 2022, 10, 1878.
- Palii, A.P.; Paliy, A.P.; Rodionova, K.O. Microbial contamination of cow’s milk and operator hygiene. Ukr. J. Ecol. 2020, 10, 392–397.
- Peruzi, G.A.S.; Bruzaroski, S.R.; Conti, A.C.M.; Nero, L.A. Short communication: Effect of bactofugation of raw milk on counts and microbial diversity of psychrotrophs. J. Dairy Sci. 2019, 102, 7794–7799.
- Pierezan, M.D.; Maran, B.M.; Maran, E.M.; Nero, L.A. Relevant safety aspects of raw milk for dairy foods processing. Adv. Food Nutr. Res. 2022, 100, 211–264.
- Qin, X.; Cheng, J.; Qiu, Y.; Wang, J. Characterization of psychrotrophic and thermoduric bacteria in raw milk using a multi-omics approach. Microb. Genom. 2014, 10, 1311.
- Sánchez-Baracaldo, P. Blue-green algae (cyanobacteria). Nat. Ecol. Evol. 2025, 9, 2174.
- Srisengfa, Y.; Laird, M.; McLeod, J.; Griffiths, M.W. The Core and Seasonal Microbiota of Raw Bovine Milk in Tanker Trucks and the Impact of Transfer to a Milk Processing Facility. mBio. 2016, 7, e00836-16.
- Stoeckel, M.; Lidolt, M.; Achberger, V.; Hinrichs, J. Growth of Pseudomonas weihenstephanensis, Pseudomonas proteolytica and Pseudomonas sp. in raw milk: Impact of residual heat-stable enzyme activity on stability of UHT milk during shelf-life. Int. Dairy J. 2016, 59, 20–28.
- Wang, N.I.; Yuan, L.; Sadiq, F.A.; He, G. Insights into Psychrotrophic Bacteria in Raw Milk: A Review. J. Food Prot. 2019, 82, 1148–1159.
- Xiang, Q.; Xia, Y.; Fang, S.; Wang, J. Enzymatic debittering of cheese flavoring and bitterness characterization of peptide mixture using sensory and peptidomics approach. Food Chem. 2024, 440, 138229.
- Yuan, H.; Han, S.; Zhang, Y.; Wang, J. Microbial Properties of Raw Milk throughout the Year and Their Relationships to Quality Parameters. Foods. 2022, 11, 3077.
- Zhang, X.; Chang, L.; Zhang, J.; Wang, J. 16S rDNA high throughput sequencing bacterial diversity analysis of cow mastitis. Indian J. Anim. Res. 2023, 57, 1578–1583.
- Zhi, W.; Chen, R.; Tang, K.; Wang, J. Research on the gut microbiota of Hainan black goat. Animals. 2022, 12, 3129.
- Zhou, X.; Chen, L.; Song, Z.; Wang, J. Pathogenic bacteria and fungi in bioaerosols from specialized hospitals in Shandong province, East China. Environ. Pollut. 2024, 341, 122922.
Figure 1.
Relative abundance of phylum (A) and genus in samples (B).

Figure 2.
Principal component analysis based on the Euclidean distance algorithm (PCA, A), principal coordinate analysis (PCoA, B), and non-metric multidimensional scaling analysis (NMDS, C).
Figure 2.
Principal component analysis based on the Euclidean distance algorithm (PCA, A), principal coordinate analysis (PCoA, B), and non-metric multidimensional scaling analysis (NMDS, C).


Figure 4.
Venn diagrams of bacteria at the genus and species level in samples from two regions of Xinjiang (A); Venn diagrams of endemic bacterial genera in fecal samples from two regions of Xinjiang (B); Venn diagrams of endemic bacterial genera in nipple skin samples from two regions of Xinjiang (C); Venn diagrams of endemic bacterial genera in feed samples from two regions of Xinjiang (D); Venn diagrams of endemic bacterial genera in soil samples from two regions of Xinjiang (E); Venn diagrams of endemic bacterial genera in cushion samples from two regions of Xinjiang (F).
Figure 4.
Venn diagrams of bacteria at the genus and species level in samples from two regions of Xinjiang (A); Venn diagrams of endemic bacterial genera in fecal samples from two regions of Xinjiang (B); Venn diagrams of endemic bacterial genera in nipple skin samples from two regions of Xinjiang (C); Venn diagrams of endemic bacterial genera in feed samples from two regions of Xinjiang (D); Venn diagrams of endemic bacterial genera in soil samples from two regions of Xinjiang (E); Venn diagrams of endemic bacterial genera in cushion samples from two regions of Xinjiang (F).


Figure 5.
Linear discriminant analysis (LDA) effect size (LEfSe) method (A); Developmental dendrogram of samples from two regions representing significant differences with an LDA threshold of 3.9. LDA discriminant histograms of genera of different abundance between the two regions (B and C).
Figure 5.
Linear discriminant analysis (LDA) effect size (LEfSe) method (A); Developmental dendrogram of samples from two regions representing significant differences with an LDA threshold of 3.9. LDA discriminant histograms of genera of different abundance between the two regions (B and C).



Table 1.
α-analytic indices of bacteria in environmental samples from two different regions.
| Sample | Chao 1 index | Ace index | Shannon index | Simpson index |
|---|---|---|---|---|
| XJB07 | 335.4882±45.27831 | 342.8485±49.18487 | 3.9087±0.11812 | 0.0380±.00298 |
| XJB09 | 350.5417±43.95742 | 348.2407±44.89349 | 3.7657±0.24476 | 0.0530±0.02124 |
| XJF07 | 327.4690±31.61595a | 326.2175±25.03794a | 3.9393±0.10067 | 0.0400±0.00960 |
| XJF09 | 235.5032±5.88348a | 239.0675±6.60897a | 3.6523±0.17569 | 0.0431±0.00744 |
| XJSH07 | 333.6703±76.73708 | 0.1464±0.06650 | 323.3333±74.76853 | 3.1366±0.41805 |
| XJSH09 | 309.4772±21.66326 | 0.0672±.02330 | 294.0000±17.08801 | 3.4327±0.19210 |
| XISI07 | 335.7717±64.94269 | 0.2589±0.19308a | 249.3333±10.69268 | 2.4649±0.75893 |
| XJSI09 | 160.5022±36.37846 | 0.5116±.00464a | 110.3333±29.56913 | 0.8969±.09049 |
| XJT07 | 335.7717±64.94269 | 0.2589±0.19308 | 249.3333±10.69268 | 2.4649±.75893 |
| XJT09 | 160.5022±36.37846 | 0.5116±.00464 | 110.3333±29.56913 | 0.8969±0.09049 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



