Submitted:
25 May 2026
Posted:
27 May 2026
You are already at the latest version
Abstract
The concept of inflammaging — referring to the persistent, low-grade inflammatory state that accompanies biological ageing — has progressively positioned itself as a unifying framework for understanding age-related chronic disease. Unlike acute inflammation, which is adaptive, transient, and fundamentally protective, inflammaging reflects a persistent dysregulation of immune signalling. Its hallmarks include failure to resolve inflammatory signals, alongside mitochondrial decline and the gradual erosion of tissue homeostasis driven by persistent cytokine activity. Increasing evidence suggests that inflammaging is not merely a consequence of ageing, but rather an active systems-level process capable of reshaping metabolic, vascular, neurological, and musculoskeletal physiology. Persistent inflammatory activation contributes to frailty, cardiovascular disease, neurodegeneration, sarcopenia, metabolic dysfunction, and osteoarthritis through interconnected molecular and cellular pathways involving immunosenescence, cellular senescence, oxidative stress, inflammasome activation, and epigenetic remodelling [1]. Recent advances in geroscience have reframed inflammation as a multidirectional biological network integrating immune, endocrine, metabolic, microbiological, and biomechanical signalling. This perspective moves beyond reductionist cytokine models and instead conceptualises inflammation as a distributed regulatory architecture operating across tissues and organ systems. Within this framework, chronic disease may emerge not solely from isolated organ pathology, but from progressive failure of intersystem communication and adaptive resilience [2]. This invited review examines the biological foundations of inflammaging, with particular emphasis on immunosenescence, senescence-associated secretory pathways, mitochondrial dysfunction, inflammasome biology, cytokine network dynamics, and epigenetic regulation. We propose that inflammaging may be best understood as a form of multiscale biological interface failure in which persistent inflammatory signalling progressively destabilises tissue integration across physiological systems. Such a framework may facilitate the development of more precise biomarkers, systems-oriented therapeutic strategies, and translational approaches aimed at extending healthspan rather than merely prolonging survival.
Keywords:
inflammaging
; immunosenescence
; cellular senescence
; frailty
; sarcopenia
; multimorbidity
; immunometabolism
; multiscale interface failure
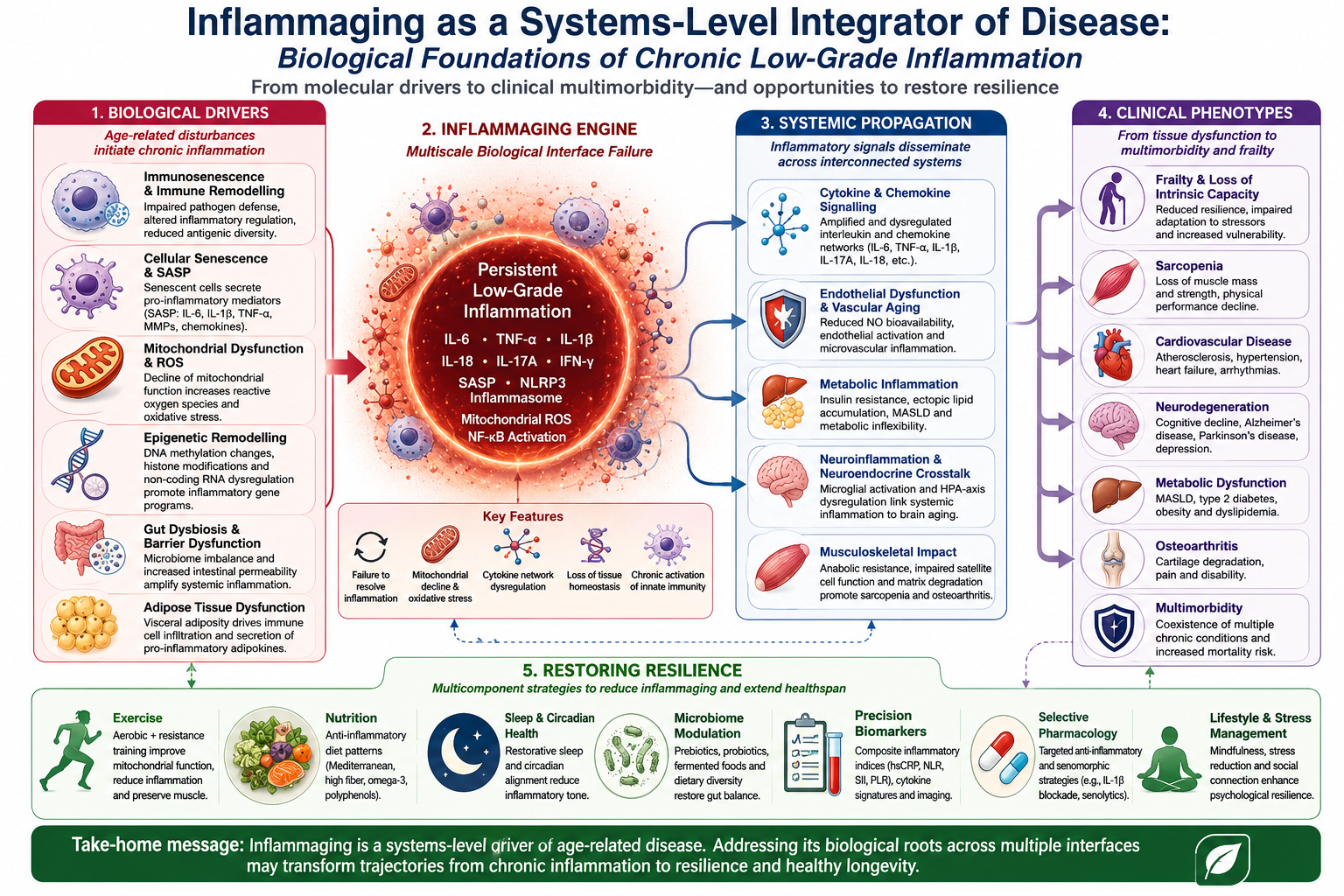
Systems-level model of inflammaging integrating biological drivers, systemic propagation, clinical
phenotypes, and resilience-oriented interventions.

Introduction
The global demographic transition toward ageing societies represents one of the defining biomedical challenges of the twenty-first century. Advances in sanitation, vaccination, antimicrobial therapy, cardiovascular medicine, and public health have dramatically increased life expectancy across much of the world. Yet increased longevity has not been uniformly accompanied by preservation of physiological resilience. Instead, modern medicine increasingly confronts a prolonged period of chronic disease accumulation characterised by multimorbidity, frailty, cognitive decline, musculoskeletal degeneration, and metabolic dysfunction.
Within this context, chronic low-grade inflammation has emerged as a unifying biological denominator linking many of the major disorders associated with ageing. The concept of inflammaging, originally proposed to describe the persistent inflammatory phenotype observed in older individuals, has progressively evolved into a broader framework integrating immune dysregulation, metabolic disturbance, tissue degeneration, and systems biology. Unlike acute inflammation, which is tightly regulated and temporally constrained, inflammaging reflects a chronic and frequently sterile activation of inflammatory pathways that persists over years or decades.
Importantly, inflammaging does not typically manifest through overt inflammatory disease. Rather than producing overt inflammatory disease, this process sustains mildly but consequentially elevated concentrations of key mediators — IL-6, TNF-α, IL-1β, and CRP — without triggering the clinical features of acute inflammation [3].
These mediators exert pleiotropic effects across vascular, neural, endocrine, adipose, and musculoskeletal tissues, progressively altering homeostatic regulation and reducing physiological reserve [4].
Ageing itself appears to generate multiple converging stimuli capable of sustaining chronic inflammatory activation. Cellular senescence, mitochondrial dysfunction, impaired autophagy, oxidative stress, microbiome dysbiosis, visceral adiposity, and accumulated tissue injury collectively contribute to a self-amplifying inflammatory milieu. Rather than functioning as isolated mechanisms, these processes interact dynamically through feedback loops that reinforce inflammatory signalling over time. In this sense, inflammaging may be viewed less as a singular pathway and more as an emergent systems-level property arising from cumulative regulatory instability.
The importance of this perspective becomes increasingly evident when examining the clinical heterogeneity of ageing-related disease. Frailty, atherosclerosis, insulin resistance, osteoarthritis, depression, sarcopenia, neurodegeneration, and chronic kidney disease appear phenotypically distinct, yet many share overlapping inflammatory signatures. This convergence suggests that chronic disease may arise from common biological architectures expressed differently according to tissue-specific vulnerabilities, genetic predisposition, environmental exposures, and biomechanical context [5].
Recent systems biology approaches have further reinforced this interpretation. Integrative clustering analyses incorporating metabolic and inflammatory biomarkers have identified distinct inflammatory-metabolic phenotypes associated with differential disease susceptibility, including digestive, cardiovascular, and neurological disorders [6]. Such findings support the concept that inflammatory signalling functions within distributed biological networks rather than isolated linear cascades.

Moreover, inflammation itself is increasingly recognised as spatially and temporally heterogeneous. Signals generated within adipose tissue, skeletal muscle, synovium, vascular endothelium, or the intestinal microbiome may propagate systemically through circulating cytokines, extracellular vesicles, metabolic intermediates, and neuroimmune communication pathways. Consequently, chronic inflammation cannot be adequately understood through organ-centred models alone. Instead, inflammaging appears to represent a distributed integrative process linking local tissue perturbations to organism-wide physiological consequences.
This review explores the biological foundations of inflammaging through a systems-oriented lens. Particular attention is given to immunosenescence, innate immune remodelling, senescence-associated secretory pathways, mitochondrial dysfunction, inflammasome activation, and cytokine network architecture. We argue that chronic low-grade inflammation may be conceptualised as a form of multiscale interface failure in which the mechanisms ordinarily coordinating communication among tissues progressively lose regulatory precision. Such a framework may provide a more coherent understanding of chronic disease pathophysiology while simultaneously identifying novel translational opportunities.
Figure 1.
The systems-level architecture through which inflammaging propagates from molecular drivers to tissue-interface dysfunction and clinical multimorbidity. Inflammaging emerges from the interaction of multiple interconnected biological processes that collectively drive chronic low-grade inflammation during ageing. Fundamental mechanisms include immunosenescence, cellular senescence with acquisition of the senescence-associated secretory phenotype (SASP), mitochondrial dysfunction with reactive oxygen species generation, and age-related epigenetic remodelling. These processes converge through systemic inflammatory mediators, particularly IL-6, TNF-α, and IL-1β, which propagate inflammatory signalling across organ systems. Chronic inflammation subsequently affects multiple physiological domains, including adipose tissue, the gut–immune axis, liver metabolism, skeletal muscle, and the central nervous system, thereby promoting adiposopathy, dysbiosis, metabolic dysfunction, sarcopenia, and neuroinflammation. The cumulative consequence is progressive loss of intrinsic capacity, frailty, and multimorbidity, reflecting inflammaging as a distributed systems-level pathological network rather than an isolated immune phenomenon.
Figure 1.
The systems-level architecture through which inflammaging propagates from molecular drivers to tissue-interface dysfunction and clinical multimorbidity. Inflammaging emerges from the interaction of multiple interconnected biological processes that collectively drive chronic low-grade inflammation during ageing. Fundamental mechanisms include immunosenescence, cellular senescence with acquisition of the senescence-associated secretory phenotype (SASP), mitochondrial dysfunction with reactive oxygen species generation, and age-related epigenetic remodelling. These processes converge through systemic inflammatory mediators, particularly IL-6, TNF-α, and IL-1β, which propagate inflammatory signalling across organ systems. Chronic inflammation subsequently affects multiple physiological domains, including adipose tissue, the gut–immune axis, liver metabolism, skeletal muscle, and the central nervous system, thereby promoting adiposopathy, dysbiosis, metabolic dysfunction, sarcopenia, and neuroinflammation. The cumulative consequence is progressive loss of intrinsic capacity, frailty, and multimorbidity, reflecting inflammaging as a distributed systems-level pathological network rather than an isolated immune phenomenon.

Biological Foundations of Inflammaging
Immunosenescence and Immune Remodelling
Ageing is accompanied by profound alterations in both innate and adaptive immunity. Collectively termed immunosenescence, these changes extend beyond simple immune decline and instead reflect a complex process of immune remodelling characterised by impaired pathogen defence, altered inflammatory regulation, reduced antigenic diversity, and persistent activation of innate immune pathways [7].

Progressive thymic involution curtails naïve T-cell output, narrowing the available T-cell receptor repertoire. As terminally differentiated memory T cells accumulate, immune adaptability declines further, reducing the capacity to mount effective responses against novel antigens.
B-cell function similarly declines with age, impairing antibody affinity maturation and reducing humoral adaptability.
Yet paradoxically, immunosenescence does not produce a purely immunodeficient phenotype. Instead, many ageing individuals exhibit chronic basal immune activation. Innate immune cells, particularly macrophages and neutrophils, frequently adopt pro-inflammatory phenotypes associated with exaggerated cytokine release and impaired inflammatory resolution [8]. Neutrophil dysfunction includes altered chemotaxis, defective phagocytosis, dysregulated reactive oxygen species production, and abnormal neutrophil extracellular trap formation. Such changes may contribute to persistent tissue injury and endothelial dysfunction.
Macrophage remodelling appears especially important in inflammaging biology. Age-associated macrophages demonstrate altered metabolic programming, mitochondrial dysfunction, impaired efferocytosis, and sustained activation of NF-κB signalling pathways. This phenotype promotes chronic secretion of IL-6, TNF-α, and other inflammatory mediators that perpetuate systemic inflammatory tone. Importantly, these changes are strongly influenced by local tissue microenvironments, highlighting the contextual nature of inflammatory activation.
Immunosenescence also affects regulatory mechanisms ordinarily responsible for inflammatory termination. Reduced regulatory T-cell function, impaired resolution mediator production, and altered anti-inflammatory cytokine signalling collectively diminish the capacity to restore immune equilibrium following physiological stressors. Consequently, inflammatory responses become prolonged, inefficient, and increasingly maladaptive.
Emerging evidence further suggests that immune remodelling interacts bidirectionally with metabolic dysfunction. Obesity, insulin resistance, and visceral adiposity amplify inflammatory activation through adipose tissue macrophage recruitment and altered adipokine secretion, while inflammatory cytokines themselves impair insulin signalling and mitochondrial metabolism. This reciprocal interaction forms the basis of immunometabolism, a rapidly expanding field examining the integration of metabolic and immune regulation.
Importantly, inflammaging is heterogeneous rather than uniform across individuals. Some older adults maintain relatively preserved immune function despite advanced chronological age, whereas others develop accelerated inflammatory phenotypes associated with frailty and multimorbidity. Such variability highlights the importance of biological ageing rather than chronological ageing alone.
Cellular Senescence and the Senescence-Associated Secretory Phenotype
Cellular senescence constitutes another major pillar of inflammaging biology. Initially described as an irreversible form of cell-cycle arrest, senescence is now recognised as a complex stress response triggered by telomere attrition, DNA damage, oxidative stress, oncogenic signalling, mitochondrial dysfunction, and mechanical injury.
Although transient senescence may serve adaptive functions in wound healing and tumour suppression, accumulation of senescent cells over time contributes substantially to chronic inflammation.
Although senescent cells no longer divide, they sustain a vigorous secretory programme — the SASP — through which they release inflammatory mediators, matrix-degrading proteases, chemokines, and mitogenic signals into the surrounding tissue microenvironment [9].
The SASP exerts profound paracrine effects on surrounding tissues. Pro-inflammatory mediators released by senescent cells induce neighbouring cellular dysfunction, propagate senescence into adjacent microenvironments, and amplify inflammatory signalling across tissues. IL-6 and IL-1β appear particularly important in sustaining SASP-related inflammatory loops.
Importantly, senescence is not restricted to a single tissue type. Senescent endothelial cells impair vascular homeostasis; senescent adipocytes promote metabolic inflammation; senescent chondrocytes contribute to osteoarticular degeneration; and senescent glial cells may participate in neuroinflammatory processes. Consequently, cellular senescence acts as a distributed inflammatory amplifier operating across organ systems.
The relationship between senescence and inflammation is also bidirectional. Chronic inflammatory exposure accelerates cellular senescence through oxidative damage and genomic instability, while senescent cells themselves perpetuate inflammation through SASP signalling. This self-reinforcing cycle may represent one of the fundamental engines driving biological ageing.
Recent translational interest has focused on senolytic therapies capable of selectively eliminating senescent cells. Although still investigational, such approaches highlight the growing recognition that senescence may constitute a therapeutically modifiable contributor to inflammaging rather than an inevitable consequence of ageing alone.
Mitochondrial Dysfunction and Oxidative Stress
Mitochondrial dysfunction occupies a central position within the biological architecture of inflammaging. Mitochondria regulate energy production, redox signalling, apoptosis, calcium homeostasis, and innate immune activation. Age-associated mitochondrial decline therefore exerts wide-ranging effects across multiple physiological systems.

Defective oxidative phosphorylation increases ROS production, contributing to oxidative injury affecting proteins, lipids, and nucleic acids. Elevated ROS levels additionally activate pro-inflammatory cascades via NF-κB signalling and inflammasome activation. Mitochondrial dysfunction therefore serves not merely as a marker of cellular damage but as an active mediator of inflammatory propagation. Particularly important is the relationship between mitochondrial injury and the NLRP3 inflammasome. Damaged mitochondria release mitochondrial DNA and ROS capable of activating inflammasome complexes within innate immune cells. Inflammasome activation subsequently promotes maturation of IL-1β and IL-18, thereby intensifying inflammatory signalling [10].
Mitochondrial quality control mechanisms also deteriorate with age. Impaired mitophagy permits accumulation of dysfunctional mitochondria, further amplifying oxidative stress and inflammatory activation. Simultaneously, reduced mitochondrial biogenesis limits cellular adaptive capacity under conditions of metabolic stress.
These processes appear especially relevant within metabolically active tissues such as skeletal muscle, myocardium, and neural tissue. Chronic mitochondrial dysfunction contributes to sarcopenia, insulin resistance, endothelial dysfunction, and neurodegenerative vulnerability, thereby linking cellular bioenergetics directly to systemic ageing phenotypes.
Importantly, mitochondrial dysfunction interacts closely with lifestyle factors including physical inactivity, obesity, poor sleep, and dietary quality. Such interactions reinforce the concept that inflammaging arises through convergence between intrinsic ageing mechanisms and environmental exposures rather than through genetically predetermined decline alone.
Epigenetic Regulation of Inflammatory Ageing
Epigenetic remodelling represents an additional mechanistic layer contributing to inflammaging. DNA methylation, histone modification, chromatin remodelling, and non-coding RNA regulation collectively influence inflammatory gene expression patterns across tissues.
Figure 2.
Bidirectional Senescence–SASP–Inflammation Cycle. Cellular stressors including telomere attrition, DNA damage, oxidative stress, and mitochondrial dysfunction induce irreversible cell-cycle arrest and cellular senescence. Senescent cells subsequently develop the senescence-associated secretory phenotype (SASP), characterised by secretion of pro-inflammatory cytokines, chemokines, and matrix-remodelling enzymes, including IL-6, IL-1β, and TNF-α. Persistent SASP signalling propagates paracrine senescence to neighbouring cells and activates inflammatory pathways such as NF-κB and the NLRP3 inflammasome. Chronic low-grade inflammation further amplifies oxidative injury, genomic instability, and mitochondrial dysfunction, thereby reinforcing senescence and perpetuating a self-sustaining inflammaging cycle. Downstream consequences include frailty, sarcopenia, neurodegeneration, cardiovascular dysfunction, and impaired tissue resilience.
Figure 2.
Bidirectional Senescence–SASP–Inflammation Cycle. Cellular stressors including telomere attrition, DNA damage, oxidative stress, and mitochondrial dysfunction induce irreversible cell-cycle arrest and cellular senescence. Senescent cells subsequently develop the senescence-associated secretory phenotype (SASP), characterised by secretion of pro-inflammatory cytokines, chemokines, and matrix-remodelling enzymes, including IL-6, IL-1β, and TNF-α. Persistent SASP signalling propagates paracrine senescence to neighbouring cells and activates inflammatory pathways such as NF-κB and the NLRP3 inflammasome. Chronic low-grade inflammation further amplifies oxidative injury, genomic instability, and mitochondrial dysfunction, thereby reinforcing senescence and perpetuating a self-sustaining inflammaging cycle. Downstream consequences include frailty, sarcopenia, neurodegeneration, cardiovascular dysfunction, and impaired tissue resilience.

Age-associated epigenetic drift alters transcriptional regulation of cytokine pathways, oxidative stress responses, and innate immune signalling. Persistent activation of NF-κB-related transcriptional programmes appears particularly important in sustaining chronic inflammatory phenotypes. Similarly, altered STAT signalling contributes to dysregulated cytokine production and impaired immune resolution.
Epigenetic mechanisms additionally mediate interactions between environmental exposures and biological ageing. Diet, smoking, pollution, psychosocial stress, physical activity, and sleep quality may all influence inflammatory gene expression through epigenetic pathways. Consequently, inflammaging reflects not only intrinsic biological ageing but also cumulative environmental imprinting across the lifespan.
Emerging evidence suggests that some epigenetic alterations may be reversible. This possibility has generated considerable interest in interventions targeting chromatin regulation, metabolic pathways, and inflammatory transcriptional programmes as potential anti-ageing strategies.
Cytokine Network Architecture
Inflammatory signalling in ageing cannot be adequately understood through isolated cytokine measurements alone. Rather, cytokines function within highly interconnected networks characterised by feedback amplification, redundancy, compensatory regulation, and context-dependent effects.
IL-6 occupies a particularly central role within inflammaging biology. Elevated IL-6 concentrations correlate strongly with frailty, sarcopenia, cardiovascular disease, disability, and mortality [11]. Importantly, IL-6 exerts both pro-inflammatory and adaptive functions depending upon signalling context, tissue origin, and receptor dynamics.
TNF-α contributes to endothelial dysfunction, insulin resistance, mitochondrial impairment, and tissue catabolism, whereas IL-1β plays a critical role in inflammasome-mediated inflammatory amplification. Persistent activation of these cytokine networks progressively alters vascular integrity, musculoskeletal homeostasis, and neuroimmune communication.
The systemic consequences of chronic cytokine exposure become particularly evident in frailty syndromes. Elevated inflammatory biomarkers correlate with reduced muscle strength, impaired mobility, diminished intrinsic capacity, and loss of physiological resilience [12]. Chronic low-grade inflammation therefore appears not merely associated with frailty, but mechanistically involved in its development.
Increasingly sophisticated analytical approaches now emphasise composite inflammatory signatures rather than isolated biomarkers. Machine-learning models integrating cytokines, metabolic markers, microbiome-derived metabolites, and physiological variables may provide more accurate representations of biological ageing trajectories and disease risk.
Ultimately, inflammaging appears to emerge from cumulative dysregulation across multiple interconnected systems rather than from singular inflammatory pathways. This systems-level perspective may prove essential for developing more effective preventive and therapeutic strategies in ageing medicine.
Tissue Interfaces: Where Chronic Inflammation Acquires Clinical Form
If inflammaging begins as a disturbance in immune regulation, it becomes clinically meaningful only when it is translated through tissues. Ageing tissues are not passive targets of inflammatory mediators; they are active participants in a distributed biological conversation. Adipose tissue, liver, skeletal muscle, vascular endothelium, kidney, brain, gut, and synovium each interpret inflammatory signals according to their own structural constraints, metabolic demands, and regenerative capacity. This is why chronic low-grade inflammation does not produce a single disease, but a spectrum of phenotypes: sarcopenia in muscle, accelerated brain ageing in neural tissue, vascular complications in diabetes, fibrosis in myocardium, dysbiosis in the gut, and functional decline in frailty syndromes.
This tissue-level perspective is essential. It prevents the reduction of inflammaging to a circulating-cytokine problem and instead frames it as a disturbance in biological communication. In this view, inflammation is not simply “present” or “absent”; it is spatially organised, temporally dynamic, and biologically interpreted. A modest rise in IL-6 or C-reactive protein may carry different implications depending on whether it arises in visceral adipose tissue, damaged muscle, metabolically stressed liver, an inflamed vascular bed, or dysbiotic intestine. The clinical phenotype depends not only on the inflammatory signal, but on the interface receiving it.
This point is illustrated by large-scale metabolic-inflammatory clustering studies. In a UK Biobank analysis of nearly 400,000 participants, integrated metabolic and inflammatory phenotyping identified distinct risk profiles for digestive diseases, with metabolomic signatures mediating part of the association between inflammatory-metabolic clusters and disease susceptibility [13]. This finding is conceptually important because it demonstrates that inflammation becomes more informative when embedded within metabolic context. The biomarker alone is not the disease; the network in which it operates is the disease substrate.
Adipose Tissue: The Immunometabolic Amplifier
Adipose tissue is a central anatomical mediator of inflammaging. In younger individuals, it operates as an adaptive energy-buffering compartment, whereas ageing and chronic nutritional surplus drive structural and immunological remodelling.
Visceral adiposity expands, adipocytes hypertrophy, local hypoxia develops, macrophages accumulate, and adipokine secretion becomes dysregulated. The tissue gradually shifts from energy storage to inflammatory signalling.
This transformation is particularly relevant in older adults, in whom excess adiposity often coexists with sarcopenia, insulin resistance, and vascular disease. Obesity in late life cannot be understood merely through body mass index. The distribution, inflammatory activity, and cellular composition of adipose tissue are more relevant than mass alone. Visceral and ectopic fat depots behave as immunometabolic organs, secreting cytokines, chemokines, leptin, resistin, and other mediators that interact with vascular and metabolic systems.
The clinical implications are considerable. In elderly populations, obesity-related inflammatory biomarkers are associated with cardiometabolic risk, and the coexistence of hypertension or diabetes appears to intensify this inflammatory phenotype [14]. This supports the concept of adipaging: the convergence of adipose dysfunction and biological ageing into a shared inflammatory state. Adipose tissue does not merely accompany ageing-related disease; it helps to organise it.
Adipose inflammation also provides a mechanistic bridge between metabolic disease and vascular injury. Insulin resistance increases lipolysis, free fatty acid flux, endothelial stress, and oxidative injury. Inflammatory cytokines further impair insulin receptor signalling and promote endothelial dysfunction. A reciprocal loop is established: metabolic dysfunction produces inflammation, and inflammation worsens metabolic dysfunction. This loop is not confined to adipose tissue but extends to liver, myocardium, skeletal muscle, and brain.
The concept of diabesity captures this convergence particularly well. In older cardiovascular patients, obesity and type 2 diabetes combine to promote myocardial inflammation, epicardial adipose expansion, fibrosis, neurohumoral activation, and cardiometabolic remodelling [15]. Epicardial fat is especially instructive because it lacks a fascial barrier separating it from myocardium. Its inflammatory secretome can act directly on adjacent coronary arteries and cardiac muscle, making it a local interface between systemic metabolism and structural heart disease.
Liver, MASLD, and Brain Ageing: Metabolic Inflammation Becomes Neurological Risk
Metabolic dysfunction-associated steatotic liver disease (MASLD) has become one of the most relevant examples of how low-grade inflammation links metabolic organs to distant systems. The liver sits at the crossroads of nutrient flux, lipid handling, insulin signalling, immune surveillance, and inflammatory mediator production. When hepatic metabolism becomes disordered, the liver becomes not only a metabolic victim but also an inflammatory broadcaster.
In population-based imaging studies, MASLD has been associated with accelerated brain ageing, even in middle-aged adults and in individuals without obvious neurological disease. Low-grade systemic inflammation partially mediated this association, suggesting that hepatic-metabolic dysfunction may influence brain structure through inflammatory pathways [16]. This finding strengthens the argument that inflammaging is not a compartmentalised process. A liver phenotype may become a brain phenotype because inflammation provides the systemic communication channel.
The mechanism is likely multifactorial. Hepatic steatosis promotes insulin resistance, dyslipidaemia, endothelial dysfunction, and release of pro-inflammatory mediators. These processes can impair cerebral microvasculature, alter blood–brain barrier integrity, and contribute to neuroinflammatory activation. Brain ageing, therefore, may partly reflect the cumulative inflammatory burden of peripheral metabolic organs.
This does not mean that inflammation is the only mediator linking MASLD to brain ageing. Vascular stiffness, glycaemic variability, lipid toxicity, and mitochondrial dysfunction almost certainly contribute. But the mediation signal is important because it identifies inflammation as a measurable and potentially modifiable pathway within a broader metabolic-neurological axis.
The Gut–Immune Axis: Dysbiosis, Barrier Failure, and Systemic Propagation
The gut is not merely a digestive organ; it is an immune organ, a metabolic organ, and a microbial ecosystem. Its relevance to inflammaging arises from three interdependent functions: maintaining epithelial barrier integrity, regulating host–microbial immune tolerance, and producing metabolites that shape systemic physiology. Ageing disrupts all three.
Gut dysbiosis in older adults is characterised by reduced microbial diversity, loss of beneficial butyrate-producing taxa, enrichment of pathobionts, and altered microbial metabolism. These changes may increase epithelial permeability, allowing microbial products such as lipopolysaccharide to enter the circulation and activate innate immune pathways. The result is a form of metabolic endotoxaemia that can contribute to chronic low-grade inflammation.
Diet strongly shapes this axis. In older American men, inflammatory and insulinaemic dietary patterns were associated with gut microbial composition and circulating metabolic biomarkers, suggesting that diet influences inflammation partly through microbial ecology [17]. This is clinically important because it links a modifiable exposure—dietary pattern—to a biological interface that regulates systemic inflammation.

The gut–immune axis is also relevant to hypertension. In elderly Chinese patients with hypertension, gut microbiota dysbiosis was associated with systemic inflammatory features, including elevated pro-inflammatory cytokines and altered abundance of taxa linked to short-chain fatty acid production [18]. These findings suggest that vascular ageing may be partly shaped by microbial-inflammatory interactions. Hypertension, traditionally framed as a haemodynamic disorder, may also represent an immune-metabolic-microbial phenotype.
The therapeutic implication is not simply “give probiotics”. Microbiome interventions are highly context-dependent. The same intervention may produce different effects according to baseline diet, medication exposure, host genetics, comorbidity, and existing microbial structure. Future microbiome-directed strategies will probably require phenotyping rather than generic supplementation.
Skeletal Muscle: Inflammation, Sarcopenia, and Physical Reserve
Skeletal muscle is both a target and regulator of inflammation. It represents a large endocrine organ capable of producing myokines, regulating glucose disposal, and influencing systemic metabolism. With ageing, muscle mass and strength decline, but this process is not simply mechanical wasting. Chronic inflammation contributes to impaired proteostasis, mitochondrial dysfunction, anabolic resistance, and reduced regenerative capacity.
Sarcopenia is therefore not only a musculoskeletal condition; it is an inflammatory-metabolic syndrome.
Dietary inflammatory patterns, reduced physical activity, oxidative stress, and cytokine excess converge on muscle tissue to impair contractile function and structural resilience. In older Polish adults, a higher dietary inflammatory index was associated with sarcopenia development, impaired gait speed, reduced six-minute walk performance, and inflammatory profiles involving IL-1β, IL-6, TNF-α, CRP, and cell-free DNA [19].
The relevance of cell-free DNA is particularly interesting. It suggests that inflammatory assessment in sarcopenia may eventually extend beyond conventional cytokines toward damage-associated molecular signals reflecting tissue stress. Such markers may better capture the interface between cellular injury and systemic inflammation.
High-sensitivity CRP has also been associated with reduced muscle strength in large population studies. Among Korean adults, elevated hsCRP was independently linked with low handgrip strength, particularly in middle-aged women and older adults [20]. Although observational, these findings support the concept that low-grade inflammation contributes to functional decline before severe disability becomes clinically apparent.
In patients undergoing peritoneal dialysis, sustained CRP elevation was associated with loss of lean tissue over time, despite similar baseline body composition [21]. This provides a clinically powerful example of inflammation as a catabolic force. The inflammatory state did not merely correlate with poor health; it predicted structural deterioration of muscle compartments in a vulnerable population.
Frailty and Intrinsic Capacity: Inflammation as Loss of Resilience
Frailty is one of the most clinically important expressions of inflammaging because it converts molecular dysregulation into observable vulnerability. It is not defined by a single organ failure, but by reduced reserve across systems. This makes frailty an ideal clinical model for understanding inflammaging as systems failure.
Intrinsic capacity offers a complementary framework. Rather than focusing only on deficits, it captures locomotion, cognition, vitality, psychological function, and sensory capacity. In community-dwelling octogenarians, intrinsic capacity–frailty phenotypes showed a biological gradient of subclinical inflammation, with CRP, IL-6, and TNF-α increasing from robust to frail phenotypes [22]. Locomotion and vitality emerged as particularly inflammation-sensitive domains.
This finding has conceptual importance. It suggests that inflammation maps not only onto disease categories but onto functional architecture. In other words, inflammatory burden may be better understood as a measure of declining resilience than as a disease-specific marker. This shift is crucial for geriatric medicine, where the goal is not merely disease control but preservation of function.
Frailty also illustrates the limitation of single-organ medicine. A patient may have modest elevations of inflammatory markers, mild sarcopenia, early cognitive slowing, reduced gait speed, and subclinical vascular disease. None alone may appear dramatic. Together, they define a vulnerable biological state. Inflammaging provides a framework for understanding that state as integrated rather than fragmented.
Neuroinflammation and Brain Ageing
The nervous system is highly sensitive to inflammatory signalling. Although the brain has long been described as immunologically privileged, it is now clear that systemic inflammation can influence neural function through endothelial activation, blood–brain barrier permeability, microglial priming, altered neurotransmitter metabolism, and vascular dysfunction.
Brain ageing studies increasingly use imaging-derived brain age gap as a surrogate for neurobiological ageing. Poor sleep health has been associated with older brain age, and systemic inflammation mediated part of this association [23]. This reinforces the idea that behavioural exposures, inflammatory biology, and neural ageing are linked through measurable pathways.
Sleep is particularly relevant because it regulates immune activity, glymphatic clearance, neuroendocrine rhythms, and metabolic homeostasis. Chronic sleep disruption may therefore amplify inflammaging not merely by raising inflammatory markers but by impairing the nocturnal repair systems that ordinarily maintain neural resilience.
Parkinson’s disease offers another example of selective neuroimmune vulnerability. In a perioperative cytokine study, patients with Parkinson’s disease showed evidence compatible with chronic low-grade inflammation, although acute perioperative cytokine responses were not globally amplified compared with controls [24]. This nuance matters. Inflammaging is not always a state of exaggerated response; in some contexts, it may involve selective immune dysregulation, exhaustion, or impaired adaptability.
Long COVID further complicates the inflammatory model. In longitudinal biomarker studies, an early phase of low-grade inflammation in long COVID appeared to shift toward reduced pro-inflammatory biomarkers later, suggesting possible immune suppression or exhaustion rather than persistent hyperinflammation alone [25]. This finding is important because it challenges a simplistic model in which chronic disease equals continuously elevated cytokines. Dynamic dysregulation may be more informative than static elevation.
Cardiovascular Disease: Inflammation as Risk Modifier and Tissue Driver
Cardiovascular disease remains one of the strongest clinical domains for studying chronic low-grade inflammation. Atherosclerosis is fundamentally inflammatory, involving endothelial activation, leukocyte recruitment, lipid oxidation, plaque remodelling, and thrombo-inflammatory signalling. However, recent evidence suggests that inflammation may not only contribute to baseline risk, but also modify the pathogenicity of specific lipid-related factors.
In a secondary prevention cohort, IL-6 modified the association between lipoprotein(a), oxidised phospholipids, and cardiovascular risk, whereas hsCRP did not show the same interaction [26]. This distinction is important. It suggests that inflammatory biomarkers are not interchangeable and that IL-6 may capture risk biology not reflected by CRP alone.
Peripheral artery disease and chronic limb-threatening ischaemia offer another example of inflammatory risk stratification. In patients undergoing revascularisation, higher comorbidity-polypharmacy scores were associated with increasing neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio, and systemic immune-inflammation index [27]. Such markers are imperfect but clinically accessible, making them attractive candidates for pragmatic inflammatory phenotyping in high-risk vascular populations.
Subclinical myocardial inflammation also appears relevant in metabolic cardiac disease. In hypertrophic cardiomyopathy, abnormal glucose metabolism was associated with myocardial fibrosis and imaging features consistent with subclinical inflammation on cardiac magnetic resonance [28]. These findings suggest that glycaemic status can shape myocardial tissue biology through inflammatory and fibrotic pathways even before overt clinical decompensation.
Depression, Metabolic Dysfunction, and the Neurovascular Interface
Depression in older adults increasingly appears to involve inflammatory, metabolic, vascular, and neurodegenerative components [29]. Yet the relationship is not straightforward. In a large prospective cohort examining type 2 diabetes and incident depressive symptoms, microvascular dysfunction, neurodegeneration, advanced glycation end products, and arterial stiffness partly mediated the association, whereas low-grade inflammation did not independently mediate it [30].
This finding is valuable precisely because it complicates the narrative. It reminds us that inflammation is not always the dominant pathway and should not be invoked indiscriminately. In some disease relationships, vascular and neurodegenerative mechanisms may carry more explanatory weight than circulating inflammatory biomarkers.
By contrast, in cognitively healthy older adults, insulin resistance showed a sex-specific association with depressive symptoms, with vascular endothelial growth factor emerging as a potential mediator in women [31]. This suggests that inflammatory-metabolic signatures may differ by sex, vascular biology, and neuroendocrine context. Precision inflammaging will therefore require stratification by biological subgroup rather than broad disease labels.
Subclinical Inflammation and Imaging Phenotypes
One of the most important trends in current inflammaging research is the movement from circulating markers toward imaging-defined inflammatory phenotypes. Subclinical inflammation may be spatially localised and clinically silent, yet biologically consequential.
In giant cell arteritis, FDG-PET/CT detected subclinical optic nerve inflammation in patients, most of whom lacked visual symptoms [32]. Although this disease differs from low-grade inflammaging, the principle is relevant: inflammatory activity may exist below the threshold of clinical detection while still carrying prognostic implications.
The future of inflammaging research will likely integrate circulating biomarkers, imaging, omics, and functional outcomes. A single blood test cannot fully capture inflammatory geography. Conversely, imaging without systemic biomarker context may miss the organism-wide inflammatory state. The most informative models will combine both.
Synthesis: From Tissue Injury to Distributed Disease
The evidence reviewed in this phase supports a central argument: inflammaging is not a uniform bloodstream phenomenon but a distributed tissue-interface disorder. Adipose tissue transforms metabolic excess into inflammatory signalling. The gut converts dysbiosis and barrier dysfunction into immune activation. Skeletal muscle translates inflammation into weakness and sarcopenia. The liver links metabolic dysfunction to brain ageing. The vasculature converts inflammatory tone into endothelial dysfunction and atherosclerotic risk. The brain interprets systemic inflammatory stress through microglia, vascular permeability, and neuroendocrine signalling.
This framework helps explain why the same inflammatory mediator may be associated with multiple diseases without implying that all diseases are the same. IL-6 in vascular disease, CRP in muscle weakness, inflammatory dietary index in sarcopenia, and microbiome dysbiosis in hypertension represent different expressions of a shared principle: inflammation becomes pathogenic through context.
The clinical future of inflammaging will therefore depend on mapping inflammatory context. Who is inflamed? Where is the signal generated? Which tissue is interpreting it? Is the inflammatory state adaptive, maladaptive, exhausted, or compensatory? Does it predict disease, mediate disease, or merely accompany disease? These questions move the field beyond biomarker enumeration toward pathophysiological interpretation.
Modifying Inflammaging: Interventions, Translation, and Future Directions
The clinical value of inflammaging depends not only on its ability to explain disease, but on whether it can be modified. A purely descriptive theory of chronic inflammation would be intellectually useful but therapeutically limited. The central translational question is therefore whether chronic low-grade inflammation represents an actionable biological state. The answer appears increasingly to be yes, but with qualifications. Inflammaging is modifiable, but it is not a single therapeutic target. It is a systems-level process shaped by movement, nutrition, sleep, adiposity, microbiome ecology, psychological stress, pharmacology, and tissue injury. Its treatment therefore cannot rely on a single anti-inflammatory drug or biomarker threshold.
This point is particularly important because the history of anti-inflammatory medicine has often been disease-specific. Rheumatology, dermatology, gastroenterology, and cardiovascular medicine have all developed targeted approaches for overt inflammation. Inflammaging, by contrast, is usually subclinical, diffuse, and entangled with ageing biology. Its modulation must therefore be conceived less as suppression and more as recalibration. The goal is not to eliminate inflammation, which remains indispensable for defence and repair, but to restore the temporal and spatial precision of inflammatory responses.
The therapeutic framework that follows is therefore organised around four principles: first, interventions should restore physiological resilience rather than merely reduce biomarkers; second, the same intervention may have different effects depending on baseline inflammatory phenotype; third, multimodal strategies are likely to outperform isolated interventions; and fourth, future clinical translation will require composite biomarkers capable of tracking inflammatory trajectories rather than single measurements.
Exercise as an Anti-Inflammatory Signal
Exercise is the most compelling non-pharmacological intervention for inflammaging because it engages nearly every relevant biological interface: skeletal muscle, adipose tissue, endothelium, mitochondria, neuroendocrine regulation, and immune cell function. Unlike pharmacological suppression, exercise produces controlled physiological stress followed by adaptive repair. Inflammatory signalling during and after exercise is therefore not inherently pathological; it is part of the adaptive programme through which tissues remodel, mitochondria are renewed, and immune responses are recalibrated.
Meta-analytic evidence supports this biological plausibility. In older adults, resistance training has been shown to reduce CRP and TNF-α, with a trend toward lower IL-6, across randomised trials involving individuals aged 60 years or older [33]. The clinical implication is not merely that exercise lowers inflammatory markers, but that muscle contraction can shift systemic inflammatory tone through repeated adaptive signalling.
The anti-inflammatory effects of exercise are mediated through several converging pathways. Improved muscle mass increases glucose disposal and reduces insulin resistance. Reduced visceral adiposity decreases adipose-derived cytokine production. Enhanced mitochondrial function reduces oxidative stress. Repeated mechanical loading improves muscle quality, vascular function, and endothelial nitric oxide bioavailability. Myokines released during contraction may further exert anti-inflammatory effects by promoting metabolic flexibility and immune regulation.
However, the interaction between exercise and ageing is not uniformly favourable. Older muscle may recover more slowly from exercise-induced damage, partly because of chronic low-grade inflammation, extracellular matrix stiffening, satellite-cell dysfunction, and mitochondrial impairment [34]. This observation is clinically important: the same exercise stimulus that is adaptive in younger tissue may be excessive or inefficient in older tissue unless appropriately prescribed. Inflammation therefore modifies not only disease risk but also treatment response.
Combined interventions may offer additional advantages. A recent trial in healthy older adults found that resistance training combined with minimal high-intensity interval training improved lean mass, strength, aerobic capacity, metabolic health, and attenuated selected acute exercise-induced inflammatory responses; polyphenol supplementation alone had more limited effects [35]. This pattern reinforces the primacy of structured physical activity as a systems-level intervention, while suggesting that nutritional adjuncts may be supportive rather than sufficient.
The practical message is clear. Exercise should not be framed as an optional lifestyle recommendation appended to pharmacological care. In inflammaging, it is a biological therapy acting across immune, metabolic, vascular, and musculoskeletal interfaces. The challenge is to tailor dose, mode, progression, and recovery to the inflammatory and functional phenotype of the individual.
Nutrition, Protein, and Dietary Inflammatory Load
Nutrition is the second major modifiable axis of inflammaging. Diet shapes inflammatory biology through energy balance, adipose tissue mass, micronutrient sufficiency, oxidative stress, glycaemic variability, lipid metabolism, and microbiome composition. The clinical literature is heterogeneous, but the direction of the evidence is consistent: dietary patterns that improve metabolic health and microbial ecology tend to attenuate inflammatory burden.
Systematic review evidence indicates that exercise and dietary supplementation in older adults may reduce pro-inflammatory cytokines, although the magnitude and consistency of effects vary across interventions and study designs [36]. This heterogeneity should not be viewed as failure. It reflects the fact that inflammaging is biologically heterogeneous. A supplement may have little measurable effect in a well-nourished older adult with low baseline inflammation, but may be meaningful in a frail, sarcopenic, metabolically stressed patient.
Protein supplementation is especially relevant because muscle decline is one of the main clinical expressions of inflammaging. A meta-analysis of randomised trials found that whey protein reduced circulating IL-6 and soy protein reduced TNF-α, with stronger effects in participants with sarcopenia or pre-frailty [37]. These findings support the concept that nutrition can modulate inflammation through muscle proteostasis, not only through classical immune pathways.
Dietary inflammatory load also matters. Diets rich in ultra-processed foods, refined carbohydrates, saturated fats, and low fibre may promote metabolic endotoxaemia, dysbiosis, oxidative stress, and adipose inflammation. Conversely, diets rich in vegetables, legumes, whole grains, unsaturated fats, fermented foods, and polyphenol-containing foods may reduce inflammatory tone through both metabolic and microbiome-mediated mechanisms.
That said, not all anti-inflammatory nutritional hypotheses translate into positive trials. In postmenopausal women with overweight or obesity, high-dose green tea extract taken for one year did not significantly reduce CRP, IL-6, or TNF-α, and COMT genotype did not modify the effect [38]. This negative result is useful because it reminds us that isolated bioactive compounds may not reproduce the effects of dietary patterns. The organism responds to food architecture, timing, energy balance, microbial fermentation, and tissue state—not merely to single molecules.
Traditional or botanical interventions also remain of interest, although evidence quality varies. In metabolic syndrome, Huanglian Jiedu decoction added to lifestyle guidance was associated with reductions in body weight, metabolic indices, IL-6, IL-17A, and TNF-α over three months [39]. Such studies should be interpreted cautiously because reproducibility, standardisation, and generalisability remain concerns; nonetheless, they illustrate the broader principle that metabolic inflammation can be pharmacologically or nutraceutically modulated.
Microbiome-Directed Interventions
The gut microbiome is an attractive target for inflammaging because it is modifiable, mechanistically plausible, and linked to immune regulation. However, microbiome intervention remains less mature than the enthusiasm surrounding it. The field must distinguish between three levels of claim: association, modulation, and clinical benefit. Many studies demonstrate association; fewer demonstrate durable microbial modulation; fewer still demonstrate meaningful clinical outcomes.
A randomised, double-blind, placebo-controlled trial of Lactiplantibacillus plantarum HEAL9 in older adults with chronic low-grade inflammation found modest effects, including reduced faecal calprotectin, a trend toward lower serum CRP, and possible cognitive signals that did not reach statistical significance [40]. This type of result is probably representative of the field: microbiome interventions may work, but the effects are often modest, strain-specific, and dependent on baseline inflammatory state.
Microbiome-directed therapy should therefore not be reduced to generic probiotic use. More plausible future strategies include targeted prebiotics, resistant starches, fermented foods, synbiotics, postbiotics, microbiome-informed dietary counselling, and perhaps personalised microbial restoration. The key is ecological rather than additive thinking. A probiotic strain introduced into an unfavourable dietary and microbial ecosystem may fail to engraft or function. Conversely, dietary fibre and substrate availability may allow beneficial taxa to expand without the need for exogenous organisms.
Microbial metabolites may ultimately be more clinically informative than taxonomy alone. Butyrate, propionate, bile acid derivatives, indoles, and other metabolites regulate epithelial integrity, immune tolerance, metabolic signalling, and neuroimmune communication. Future trials should therefore measure not only microbial composition but also functional metabolic output.
Sleep, Stress, and Neuroendocrine Modulation
Inflammaging is often discussed in immune and metabolic terms, but neuroendocrine regulation is equally central. Sleep, circadian rhythm, cortisol dynamics, sympathetic tone, and psychological stress shape inflammatory signalling. This is clinically relevant because older adults frequently experience fragmented sleep, reduced circadian amplitude, social isolation, fear of falling, pain, and multimorbidity-related stress.

Poor sleep health has already been linked to older brain age partly through systemic inflammation. The translational implication is that sleep may be a modifiable inflammatory intervention, not merely a symptom domain. Sleep interventions could plausibly reduce neuroinflammatory and cardiometabolic risk by restoring circadian immune regulation, improving glymphatic clearance, and reducing neuroendocrine stress.
The relationship between psychological stress and inflammation is complex. In the FEARFALL study, perceived control over falling was associated with cortisol dynamics, whereas inflammatory markers were more closely related to age and body mass index than to fear-related measures [41]. This nuance matters. Psychological factors may influence stress-system regulation even when they do not directly map onto CRP or IL-6. In clinical practice, this suggests that inflammatory resilience includes neuroendocrine adaptability, not only cytokine reduction.
Depression, frailty, and anxiety-like syndromes in older adults may therefore require integrated assessment. Treating depressive symptoms without assessing frailty, mobility, sleep, inflammation, and medication burden risks missing the systems-level nature of the problem.
Conversely, improving physical function and perceived control may indirectly improve inflammatory and neuroendocrine regulation.
Pharmacological Modulation: Promise and Caution
Pharmacological modulation of inflammaging is conceptually appealing but clinically challenging. Targeted anti-cytokine therapies have transformed autoimmune and autoinflammatory disease, yet chronic low-grade inflammation differs fundamentally from overt inflammatory pathology. In older adults, indiscriminate immune suppression may increase infection risk, impair tissue repair, and worsen resilience. The goal should therefore be selective modulation of maladaptive inflammatory pathways while preserving host defence.
Anti-inflammatory interventions for depression illustrate both promise and uncertainty. A systematic review and meta-analysis in older adults found that anti-inflammatory interventions reduced depressive scores compared with placebo, with subgroup signals for omega-3 fatty acids and botanical or dietary interventions [42]. However, heterogeneity was substantial, and baseline inflammatory status likely influenced response. This supports a biomarker-stratified model rather than universal anti-inflammatory prescribing.
Senolytic and senomorphic strategies represent another frontier. By targeting senescent cells or modifying the SASP, these interventions aim upstream of cytokine excess. The theoretical advantage is that removing or reprogramming senescent inflammatory sources may produce broader effects than blocking a single cytokine. The risk is that senescence also serves protective functions in wound healing and tumour suppression. Timing, dose, tissue specificity, and patient selection will therefore be critical.
Cell-based approaches also raise translational questions. In older adults with Parkinson’s disease and preserved kidney function, repeated allogeneic mesenchymal stem-cell infusions were associated with improved kidney-function measures in secondary analyses of a clinical trial [43]. Although preliminary and disease-context-specific, such findings suggest that immunomodulatory cellular therapies may eventually intersect with inflammaging research. At present, however, they should be considered investigational rather than clinically established.
Composite Biomarkers and Precision Inflammation Medicine
A recurring limitation throughout inflammaging research is the reliance on single biomarkers. CRP is useful, inexpensive, and prognostic, but it is not the whole inflammatory state. IL-6 is informative, but not always superior. TNF-α and IL-1β may be mechanistically important but difficult to interpret in isolation. Ratios such as NLR, PLR, and SII are accessible, yet non-specific. The next phase of the field requires composite, context-aware biomarkers.
Recent work on digital health and longevity has proposed inflammation-centred frameworks integrating hsCRP, IL-6, TNF-α, microbiome diversity, butyrate, insulin resistance, and lipid ratios into composite indices such as the Longevity-Inflammation Index [44]. This approach is conceptually attractive because it recognises that inflammaging is not purely immunological; it is immune-metabolic-microbial.
However, composite indices must be validated rigorously. They require construct validity, predictive validity, responsiveness to intervention, population norms, and clinical interpretability. A score that predicts mortality but cannot guide intervention may be epidemiologically useful but clinically limited. Conversely, a score that changes with lifestyle but does not predict outcomes may be motivational but not medically meaningful.
Digital platforms may eventually permit repeated inflammatory phenotyping by combining laboratory biomarkers, wearable data, diet logs, sleep metrics, physical activity, and microbiome assessments. Yet this future must be approached carefully. Digital health can empower personalised prevention, but it can also generate noise, anxiety, and over-testing. Inflammation medicine must avoid becoming another domain of unregulated biomarker enthusiasm.
The ideal model would combine periodic validated biomarkers with clinically meaningful endpoints: gait speed, grip strength, cognitive performance, sleep quality, vascular events, metabolic control, and patient-centred function. Inflammaging should not be treated as an abstract laboratory construct. It matters because it alters how people move, think, recover, and live [45].
Figure 3.
Pharmacological modulation of inflammaging as a balance between inflammatory control and preservation of biological resilience during ageing. The figure illustrates inflammaging as a dynamic systems-level process in which therapeutic interventions must reduce maladaptive chronic inflammation while preserving host defence, tissue repair and physiological adaptation. Major therapeutic domains include targeted anti-cytokine therapies, senolytic and senomorphic strategies, nutritional and anti-inflammatory interventions, and cell-based immunomodulatory therapies. The central hub emphasises the concept of selective modulation of maladaptive inflammation, balancing inflammatory suppression against maintenance of resilience and adaptive immune function. Potential benefits include reduced frailty, improved functional ageing and attenuation of multimorbidity progression, whereas excessive suppression may increase infection susceptibility, impair regeneration and promote immune exhaustion. The right-side panel highlights the emerging paradigm of precision inflammaging, integrating biomarker-guided therapy, inflammatory phenotypes and tissue-specific responses within personalised geroscience approaches.
Figure 3.
Pharmacological modulation of inflammaging as a balance between inflammatory control and preservation of biological resilience during ageing. The figure illustrates inflammaging as a dynamic systems-level process in which therapeutic interventions must reduce maladaptive chronic inflammation while preserving host defence, tissue repair and physiological adaptation. Major therapeutic domains include targeted anti-cytokine therapies, senolytic and senomorphic strategies, nutritional and anti-inflammatory interventions, and cell-based immunomodulatory therapies. The central hub emphasises the concept of selective modulation of maladaptive inflammation, balancing inflammatory suppression against maintenance of resilience and adaptive immune function. Potential benefits include reduced frailty, improved functional ageing and attenuation of multimorbidity progression, whereas excessive suppression may increase infection susceptibility, impair regeneration and promote immune exhaustion. The right-side panel highlights the emerging paradigm of precision inflammaging, integrating biomarker-guided therapy, inflammatory phenotypes and tissue-specific responses within personalised geroscience approaches.


Clinical Trial Design: Who Should Be Enrolled?
A major obstacle in inflammaging research is trial heterogeneity. Many studies enrol broad older populations without sufficient inflammatory enrichment. If participants have low baseline inflammation, an intervention may appear ineffective despite biological plausibility. Future trials should stratify participants by inflammatory phenotype, metabolic state, frailty status, adiposity distribution, microbiome features, and functional reserve.
Trial endpoints should also evolve. CRP reduction alone is insufficient. More meaningful endpoints include preservation of muscle mass, improved intrinsic capacity, reduced frailty progression, delayed cognitive decline, improved vascular function, and reduced hospitalisation. In high-risk groups, hard outcomes may be feasible. In healthier populations, intermediate functional and biological endpoints will remain necessary.
Duration is another key issue. Inflammaging develops over years, whereas many intervention trials last weeks or months. Short trials may detect cytokine changes but miss structural and functional outcomes. Longer trials are more expensive and difficult, but they are essential if the field is to move beyond proof of mechanism.
Multimodal intervention trials may be particularly relevant. The biological architecture of inflammaging suggests that exercise, diet, sleep, stress reduction, microbiome modulation, and medication optimisation may act synergistically. Single interventions may produce modest effects because they target only one node in a network. Multidomain interventions, if well designed, may better restore system-level resilience.

A Practical Clinical Framework
For clinicians, inflammaging should not become an excuse for ordering indiscriminate cytokine panels. A pragmatic approach begins with phenotype. Does the patient show frailty, sarcopenia, metabolic syndrome, recurrent infections, chronic pain, poor sleep, depression, functional decline, or accelerated vascular disease? Are there modifiable inflammatory drivers such as visceral adiposity, periodontal disease, sleep disruption, physical inactivity, uncontrolled diabetes, chronic kidney disease, or polypharmacy?
Initial assessment can reasonably include conventional markers such as hsCRP, full blood count with differential, albumin, glucose metabolism, lipid profile, renal function, and liver enzymes. More specialised markers may be appropriate in research settings or selected clinical contexts, but their routine use remains premature.
Management should prioritise interventions with broad benefit and low risk: progressive resistance and aerobic exercise, adequate protein intake, dietary quality, weight management where appropriate, sleep optimisation, treatment of periodontal and chronic infectious sources, medication review, vaccination, and management of cardiometabolic disease. Pharmacological anti-inflammatory strategies should be reserved for defined indications or research protocols.
This approach does not trivialise inflammaging. Rather, it grounds the concept in actionable medicine. The clinician does not need to “treat IL-6”; the clinician needs to identify why the patient’s biological systems are generating persistent inflammatory tone and intervene at the relevant interface.
Future Directions
The next generation of inflammaging research should move beyond biomarker association toward causal architecture. Several priorities are clear. First, longitudinal studies should identify inflammatory trajectories that precede disease, rather than merely measuring inflammation after disease is established. Second, mechanistic trials should test whether changing inflammatory state changes functional outcomes. Third, tissue-specific and imaging-based approaches should be integrated with circulating biomarkers. Fourth, microbiome studies should emphasise function and metabolites rather than taxonomy alone. Fifth, clinical trials should enrich for inflammatory phenotypes and use patient-centred endpoints.
Artificial intelligence and systems modelling may help, but only if grounded in biologically meaningful data. Machine-learning models can identify patterns, but they do not automatically explain mechanisms. The best use of computational tools will be to integrate multi-omic, physiological, behavioural, imaging, and clinical data into interpretable inflammatory phenotypes.
Equity is also important. Inflammaging is shaped by social determinants: nutrition, pollution, infection burden, stress, work conditions, healthcare access, and physical environment. A purely molecular account of inflammaging risks ignoring the social biology of inflammation. Chronic adversity is biologically embedded. Public health interventions may therefore be as anti-inflammatory as drugs.
Final Integrative Model: Inflammation as Multiscale Interface Failure
Across the three phases of this review, a coherent model emerges. Inflammaging begins with immune remodelling, cellular senescence, mitochondrial dysfunction, epigenetic drift, and cytokine network dysregulation. It acquires clinical form through tissue interfaces: adipose tissue, liver, gut, skeletal muscle, vasculature, brain, kidney, and joint. It becomes modifiable through interventions that restore adaptive signalling: movement, diet, sleep, microbial ecology, metabolic control, and selective pharmacology. The key conceptual shift is to view inflammation not as a linear pathway but as an architecture of communication. Biological systems require inflammatory signalling to detect danger, repair tissue, regulate metabolism, and adapt to stress. Disease emerges when this signalling loses precision: when local signals become systemic, when acute responses fail to resolve, when repair becomes fibrosis, when defence becomes catabolism, and when adaptation becomes exhaustion.
This is what is meant by multiscale interface failure. At the molecular scale, cytokine and inflammasome signalling become dysregulated. At the cellular scale, senescent and innate immune cells amplify inflammatory tone. At the tissue scale, adipose, gut, muscle, vascular, neural, and osteoarticular compartments interpret inflammation through local vulnerabilities. At the organismal scale, the result is frailty, multimorbidity, cognitive decline, and reduced resilience. Such a framework does not replace disease-specific medicine. Rather, it provides the connective tissue linking diseases that have too often been studied separately. It explains why a patient with visceral adiposity, poor sleep, sarcopenia, depression, diabetes, and osteoarthritis should not be understood as a collection of unrelated diagnoses. The shared biology is not identical in every tissue, but the network is connected.
The translational promise of inflammaging lies in this integrative power. If measured wisely and treated contextually, chronic low-grade inflammation may become a practical organising principle for preventive geroscience, multimorbidity care, and healthspan extension. The challenge now is to ensure that the field moves from elegant association to clinically meaningful intervention.
Conclusions
Inflammaging is no longer a marginal concept in ageing biology. It is increasingly central to the understanding of chronic disease, frailty, neurodegeneration, cardiometabolic dysfunction, sarcopenia, and osteoarthritis. Yet its clinical value depends on conceptual discipline. Chronic low-grade inflammation should not be invoked as a universal explanation for every age-related phenomenon, nor should it be reduced to a single biomarker. It is a dynamic, contextual, and multiscale process. The evidence reviewed here supports a systems-level interpretation. Inflammaging arises from the interaction of immune remodelling, cellular senescence, mitochondrial dysfunction, metabolic stress, microbiome dysbiosis, neuroendocrine regulation, and tissue injury. Its clinical expression depends on the interfaces through which these signals are interpreted. Its treatment will require interventions that restore resilience rather than merely suppress inflammation.
The future of inflammaging medicine will not be defined solely by drugs or solely by lifestyle interventions.
It will combine phenotyping, prevention, functional assessment, digital monitoring, targeted therapy, and public health.
In that sense, inflammaging may become not only a mechanism of ageing but a language through which modern medicine learns to understand chronic disease as integrated biology.
Figure 4.
Inflammaging as a Multiscale Interface Failure Network. This conceptual framework illustrates inflammaging as a distributed systems-level process emerging from the interaction of biological ageing mechanisms, tissue interfaces, and network-wide inflammatory dysregulation. Principal origins and drivers include immune remodelling, cellular senescence, mitochondrial dysfunction, epigenetic drift, and cytokine network instability. These processes propagate across multiple tissue interfaces, including adipose tissue, liver–gut interactions, skeletal muscle, vasculature, brain, kidney, and joints, thereby generating interconnected patterns of chronic low-grade inflammation. Inflammaging is conceptualised as a form of multiscale interface failure operating across molecular, cellular, tissue, and organismal levels, ultimately contributing to frailty, multimorbidity, and loss of physiological resilience. The framework further highlights inflammaging as a shared biological communication network characterised by impaired signalling precision and connective inflammatory architecture between chronic diseases. Potentially modifiable interventions include physical activity, diet, sleep optimisation, microbial ecology modulation, metabolic control, and selective pharmacological approaches. Translational implications include preventive geroscience, integrated multimorbidity care, and extension of healthspan.
Figure 4.
Inflammaging as a Multiscale Interface Failure Network. This conceptual framework illustrates inflammaging as a distributed systems-level process emerging from the interaction of biological ageing mechanisms, tissue interfaces, and network-wide inflammatory dysregulation. Principal origins and drivers include immune remodelling, cellular senescence, mitochondrial dysfunction, epigenetic drift, and cytokine network instability. These processes propagate across multiple tissue interfaces, including adipose tissue, liver–gut interactions, skeletal muscle, vasculature, brain, kidney, and joints, thereby generating interconnected patterns of chronic low-grade inflammation. Inflammaging is conceptualised as a form of multiscale interface failure operating across molecular, cellular, tissue, and organismal levels, ultimately contributing to frailty, multimorbidity, and loss of physiological resilience. The framework further highlights inflammaging as a shared biological communication network characterised by impaired signalling precision and connective inflammatory architecture between chronic diseases. Potentially modifiable interventions include physical activity, diet, sleep optimisation, microbial ecology modulation, metabolic control, and selective pharmacological approaches. Translational implications include preventive geroscience, integrated multimorbidity care, and extension of healthspan.

Author Contributions
Both authors contributed equally to this work. The authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Declaration of Generative AI and AI-assisted Technologies in the Writing Process
AI-assisted language tools were used during manuscript preparation for language editing, structural organisation, and editorial refinement. The authors critically reviewed, revised, and approved the final text and accept full responsibility for the scientific content of the article.
Conflicts of Interest
AMF serves as Medical Director for a Colombian pharmaceutical manufacturer and receives professional fees for this role. No product from the institutional portfolio is mentioned or discussed in this manuscript. This relationship did not influence the conception, design, interpretation, writing, or reporting of the article. AMR declares no conflicts of interest.
Abbreviations
CRP, C-reactive protein; FDG-PET/CT, fluorodeoxyglucose positron emission tomography/computed tomography; hsCRP, high-sensitivity C-reactive protein; IL-1β, interleukin-1 beta; IL-6, interleukin-6; IL-17A, interleukin-17A; IL-18, interleukin-18; Lp(a), lipoprotein(a); MASLD, metabolic dysfunction-associated steatotic liver disease; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLR, neutrophil-to-lymphocyte ratio; NLRP3, NOD-like receptor protein 3; PLR, platelet-to-lymphocyte ratio; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SII, systemic immune-inflammation index; STAT, signal transducer and activator of transcription; TNF-α, tumour necrosis factor-alpha; VEGF, vascular endothelial growth factor.
References
- Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between inflammageing and immunosenescence during ageing. Cells 2022, 11(3), 359. [Google Scholar] [CrossRef] [PubMed]
- Andonian, B.J.; Hippensteel, J.A.; Abuabara, K.; et al. Inflammation and aging-related disease: a transdisciplinary inflammaging framework. Geroscience 2025, 47, 515–542. [Google Scholar] [CrossRef]
- Tylutka, A.; Walas, Ł.; Zembron-Lacny, A. Level of IL-6, TNF, and IL-1β and age-related diseases: a systematic review and meta-analysis. Front Immunol. 2024, 15, 1330386. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell Mol. Immunol. 2022, 19(2), 177–191. [Google Scholar] [CrossRef]
- Wissler Gerdes, E.O.; Vanichkachorn, G.; Verdoorn, B.P.; et al. Role of senescence in the chronic health consequences of COVID-19. Transl. Res. 2022, 241, 96–108. [Google Scholar] [CrossRef]
- García-Vega, D.; González-Juanatey, J.R.; Eiras, S. Diabesity in elderly cardiovascular disease patients: mechanisms and therapeutic implications. Int. J. Mol. Sci. 2022, 23(14), 7886. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Liu, X.; Chen, D.; Wang, M.; Jiang, X.; Xiong, Z. The Roles of the Gut Microbiota and Chronic Low-Grade Inflammation in Older Adults With Frailty. Front Cell Infect. Microbiol. 2021, 11, 675414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14(10), 576–590. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; Miller, A.H.; Mantovani, A.; Weyand, C.M.; Barzilai, N.; Goronzy, J.J.; Rando, T.A.; Effros, R.B.; Lucia, A.; Kleinstreuer, N.; Slavich, G.M. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25(12), 1822–1832. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferrucci, L.; Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15(9), 505–522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, Z.; Chen, Q.; Zhou, L.; Ye, K.; Liu, Z.; Jiang, W.; Luo, L.; Wang, Y.; Ye, X.; Yu, C.; Shen, Z. Integrated Metabolic and Inflammatory Clustering Reveals Distinct Risk Profiles for Digestive Diseases. Adv. Sci. (Weinh) 2025, 12(44), e11000. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153(6), 1194–217. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N Y Acad. Sci. 2000, 908, 244–54. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Chen, Q.; Zhou, L.; Ye, K.; Liu, Z.; Jiang, W.; Luo, L.; Wang, Y.; Ye, X.; Yu, C.; Shen, Z. Integrated Metabolic and Inflammatory Clustering Reveals Distinct Risk Profiles for Digestive Diseases. Adv. Sci. (Weinh) 2025, 12(44), e11000. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- García-Vega, D.; González-Juanatey, J.R.; Eiras, S. Diabesity in Elderly Cardiovascular Disease Patients: Mechanisms and Regulators. Int. J. Mol. Sci. 2022, 23(14), 7886. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Yang, R.; Miao, Y.; Zhang, X.; Paillard-Borg, S.; Fang, Z.; Xu, W. Metabolic Dysfunction-Associated Steatotic Liver Disease Is Associated With Accelerated Brain Ageing: A Population-Based Study. Liver Int. 2025, 45(6), e70109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nepal, S.; Shi, N.; Hoyd, R.; Spakowicz, D.J.; Orwoll, E.; Shikany, J.M.; Napoli, N.; Tabung, F.K. Role of insulinemic and inflammatory dietary patterns on gut microbial composition and circulating biomarkers of metabolic health among older American men. Gut Microbes 2025, 17(1), 2497400. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheng, Y.; Huang, Y.; Zhu, Z.; Ding, W.; Liu, X.; Xu, X.; Chen, Y.; Wang, L.; Zhao, L.; Ling, Z.; Ye, S. Gut microbiota dysbiosis and systemic inflammation in elderly Chinese hypertensive patients: a case-control study. Front Immunol. 2025, 16, 1662578. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morawin, B.; Białorudzki, M.; Nicikowski, J.; Tylutka, A.; Wawrzyniak-Gramacka, E.; Zembron-Lacny, A. Association between dietary inflammatory index and sarcopenia development in Polish population. Sci. Rep. 2025, 15(1), 44455. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Choi, B.H.; Shin, S. Association Between Serum High Sensitivity C-Reactive Protein Levels and Low Muscle Strength Among Korean Adults. Nutrients 2025, 17(16), 2698. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davenport, A. The effect of low-grade inflammation on body composition in peritoneal dialysis patients. J. Ren. Nutr. 2025, 35(6), 787–794. [Google Scholar] [CrossRef] [PubMed]
- Cacciatore, S.; Calvani, R.; Prokopidis, K.; Schlögl, M.; Russo, A.; Tosato, M.; Anton, S.D.; Leeuwenburgh, C.; Batsis, J.A.; Marzetti, E.; Landi, F. Intrinsic capacity-frailty phenotypes and subclinical inflammation in community-dwelling octogenarians: A cross-sectional analysis from the ilSIRENTE study. Exp. Gerontol. 2026, 214, 113026. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Wang, J.; Li, X.; Guo, J.; Ekblom, M.M.; Sindi, S.; Zhang, Q.; Dove, A. Poor sleep health is associated with older brain age: the role of systemic inflammation. EBioMedicine 2025, 120, 105941. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, J.W.; Yoo, S.A.; Choi, Y.; Jeong, G.H.; Lee, J.; Joo, J. Perioperative Inflammatory Cytokines in Parkinson's Disease. Biomolecules 2026, 16(2), 261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kallaste, A.; Kisand, K.; Aart, A.; Salumets, A.; Kisand, K.; Peterson, P.; Lember, M. Long COVID and Biomarker Dysregulation-A Shift Toward Immune Exhaustion? Medicina 2025, 61(6), 996. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mohammadnia, N.; van Broekhoven, A.; Bax, W.A.; Eikelboom, J.W.; Mosterd, A.; Fiolet, A.T.L.; Tijssen, J.G.P.; Thompson, P.L.; de Kleijn, D.P.V.; Tsimikas, S.; Cornel, J.H.; Yeang, C.; El Messaoudi, S. Interleukin-6 modifies Lipoprotein(a) and oxidized phospholipids associated cardiovascular disease risk in a secondary prevention cohort. Atherosclerosis 2025, 405, 119211. [Google Scholar] [CrossRef] [PubMed]
- Steens, I.L.M.; Schram, M.T.; Houben, A.J.H.M.; Berendschot, T.T.J.M.; Jansen, J.F.A.; Backes, W.H.; Koster, A.; Bosma, H.; Eussen, S.J.P.M.; de Galan, B.E.; van Sloten, T.T. Type 2 diabetes and depression via microvascular dysfunction, neurodegeneration, inflammation, advanced glycation end products (AGEs), and arterial stiffness. Diabetes Obes. Metab. 2025, 27(9), 4847–4858. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pasqui, E.; Casilli, G.; Anichini, T.; Cerbini, E.; Pasquetti, L.; Galzerano, G.; de Donato, G. Linking Comorbidity and Inflammation: The Role of Comorbidity Polypharmacy Score in Chronic Limb-Threatening Ischemia. Ann. Vasc. Surg. 2026, 122, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Mancinetti, F.; Labarile, F.; Bastiani, P.; Scamosci, M.; Alunno, M.; Cecchetti, R.; Mecocci, P.; Boccardi, V. A novel sex-specific association between insulin resistance and depressive symptoms in older adults: The potential mediating role of Vascular Endothelial Growth Factor. J. Affect Disord. 2025, 382, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Gernert, M.; Schmalzing, M.; Fröhlich, M.; Zhi, Y.; Strunz, P.P.; Labinsky, H.; Hartrampf, P.E.; Buck, A.K.; Bley, T.A.; Guggenberger, K.V.; Werner, R.A.; Serfling, S.E. [18F]-FDG-PET/CT detects subclinical optic nerve inflammation in giant cell arteritis. Front Immunol. 2026, 17, 1802935. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, S.D.; Yeun, Y.R. Effects of Resistance Training on C-Reactive Protein and Inflammatory Cytokines in Elderly Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Env. Res. Public Health 2022, 19(6), 3434. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kunz, H.E.; Lanza, I.R. Age-associated inflammation and implications for skeletal muscle responses to exercise. Exp. Gerontol. 2023, 177, 112177. [Google Scholar] [CrossRef] [PubMed]
- Flensted-Jensen, M.; Weinreich, C.M.; Kleis-Olsen, A.S.; Hansen, F.; Skyggelund, N.S.; Pii, J.R.; Whitlock, R.; Abrahamsen, M.B.; Petersen, T.I.; Karlsen, A.; Reihmane, D.; Dela, F. Effects of resistance-based training and polyphenol supplementation on physical function, metabolism, and inflammation in aging individuals. Geroscience 2026, 48(2), 2945–2968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hernández-Lepe, M.A.; Ortiz-Ortiz, M.; Hernández-Ontiveros, D.A.; Mejía-Rangel, M.J. Inflammatory Profile of Older Adults in Response to Physical Activity and Diet Supplementation: A Systematic Review. Int. J. Env. Res. Public Health 2023, 20(5), 4111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prokopidis, K.; Mazidi, M.; Sankaranarayanan, R.; Tajik, B.; McArdle, A.; Isanejad, M. Effects of whey and soy protein supplementation on inflammatory cytokines in older adults: a systematic review and meta-analysis. Br. J. Nutr. 2023, 129(5), 759–770. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cunningham, A.; Gomes, A.; Meng, L.; Shapses, S.; Byham-Gray, L.; Samavat, H. Effects of Green Tea Extract Supplementation on Inflammatory Cytokines Among Postmenopausal Women with Overweight or Obesity-A Secondary Analysis of a Randomized Controlled Trial. Nutrients 2026, 18(1), 143. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pingyuan, X.U.; Heng, Z.; Ruonan, Z.; Yaping, W.; Ziwei, Z.; Fangyuan, X.U.; Yingying, X.; Yue, C.; Lixuan, S.; Ziwei, W.; Yingying, X.; Xizhong, Y.U.; Penghua, F.; Wenbin, S. Beneficial effects of Huanglian Jiedu decoction ( ) on metabolic syndrome: a prospective randomized controlled clinical trial. J. Tradit. Chin. Med. 2026, 46(2), 411–417. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lazou-Ahrén, I.; Björklund, M.; Molin, G.; Xu, J.; Önning, G.; Elmståhl, S.; Jeppsson, B. Probiotic-Reduced Inflammaging in Older Adults: A Randomized, Double-Blind, Placebo-Controlled Trial. Probiotics Antimicrob. Proteins 2025, 17(5), 3429–3439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Müller, A.; Kob, R.; Sieber, C.C.; Freiberger, E.; Rohleder, N.; Britting, S. Relationship between stress systems and inflammation in older adults concerned about falling - Findings from the FEARFALL study. Exp. Gerontol. 2025, 211, 112899. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Su, W.J.; Deng, S.L.; Luo, J.; Du, Z.L.; Luo, Y.; Lv, K.Y.; Zhu, D.M.; Fan, X.T. Anti-inflammatory interventions for the treatment and prevention of depression among older adults: a systematic review and meta-analysis. Transl. Psychiatry 2025, 15(1), 114. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martinez-Lemus, J.D.; Molony, D.A.; Suescun, J.; et al. Allogeneic bone marrow-derived mesenchymal stem cells in the aging kidney: secondary results of a Parkinson's disease clinical trial. Stem Cell Res. Ther. 2025, 16(1), 493. [Google Scholar] [CrossRef]
- Adibi, S. A conceptual digital health framework for longevity optimization. Nutrients 2026, 18(2), 231. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Kroemer, G. Hallmarks of health. Cell. 2021, 184(1), 33–63. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



