Submitted:
14 May 2026
Posted:
15 May 2026
You are already at the latest version
Abstract
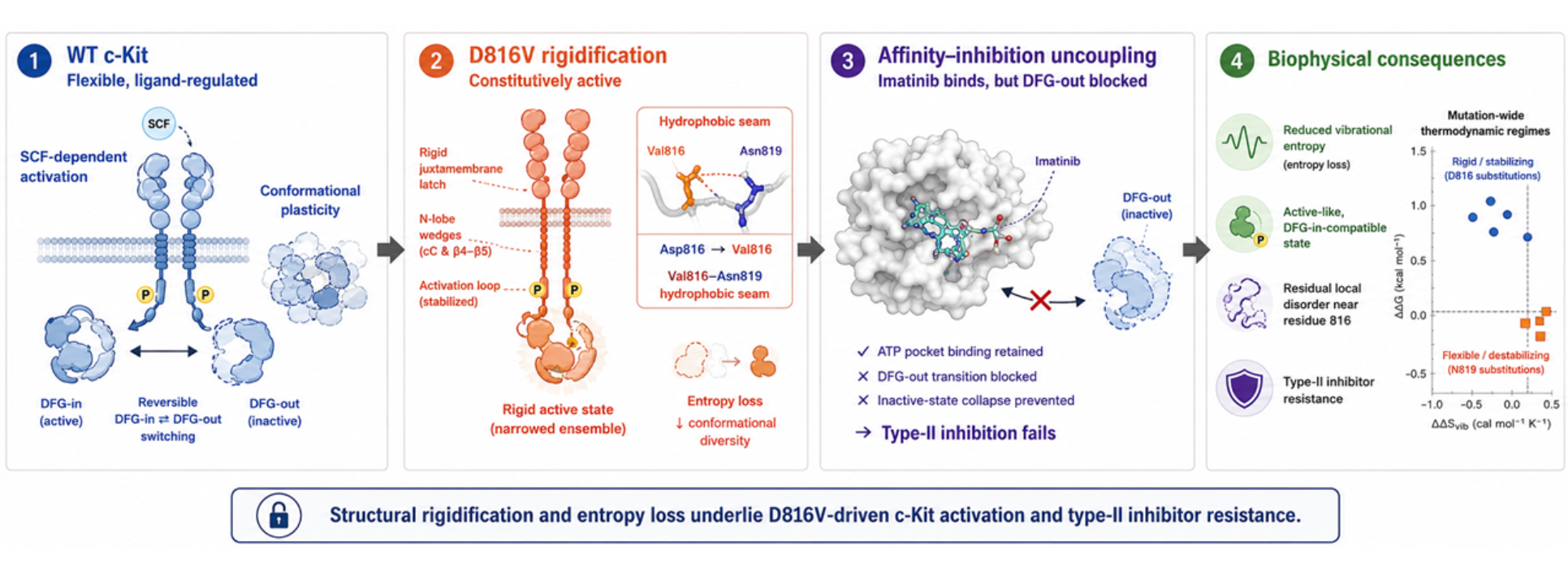
Activation-loop mutations in receptor tyrosine kinases alter catalytic-state equilibria by changing the balance between local flexibility, global allostery, and inhibitor-induced conformational selection. Here we analyze the c-Kit D816V substitution as a model for how mutation-induced entropy loss can convert a regulated kinase into a rigidified active-like enzyme that resists type II inhibition. Using vibrational-entropy mapping, normal-mode analysis, molecular dynamics simulations, docking, dimer-interface energetics, and in silico saturation mutagenesis, we identify a conformational rigidification mechanism centered on Val816. The substitution replaces the wild-type Asp816-centered hydrogen-bond network with a Val816-Asn819 hydrophobic seam that propagates rigidity through the juxtamembrane latch, N-lobe wedges, and activation loop. This architecture is predicted to pre-organize the catalytic spine and restrict access to the inactive DFG-out state. Although imatinib and dasatinib retain favorable calculated binding energies, D816V imposes a larger predicted energetic penalty on imatinib than on dasatinib, and the rigidified pocket is predicted to impair the conformational collapse required for productive type II inhibition, thereby uncoupling binding from functional shutdown. Saturation mutagenesis separates hydrophobic D816 substitutions into a rigid/stabilizing thermodynamic regime and N819 substitutions into a flexible/destabilizing regime, indicating that activation-loop variants can be classified by the conformational states they impose rather than by position alone. Residual local disorder near residue 816 and energetically permissive mutant-wild-type heterodimerization suggest additional mechanisms for signaling adaptability. These results support a testable biophysical model in which conformational entropy loss, rather than increased flexibility, drives D816-centered c-Kit activation and type II inhibitor resistance.
Keywords:
c-Kit
; KIT D816V
; receptor tyrosine kinase
; conformational rigidification
; vibrational entropy
; allostery
; DFG-out transition
; type II inhibitors
; molecular dynamics
; kinase regulation
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



