Submitted:
03 May 2026
Posted:
06 May 2026
You are already at the latest version
Abstract
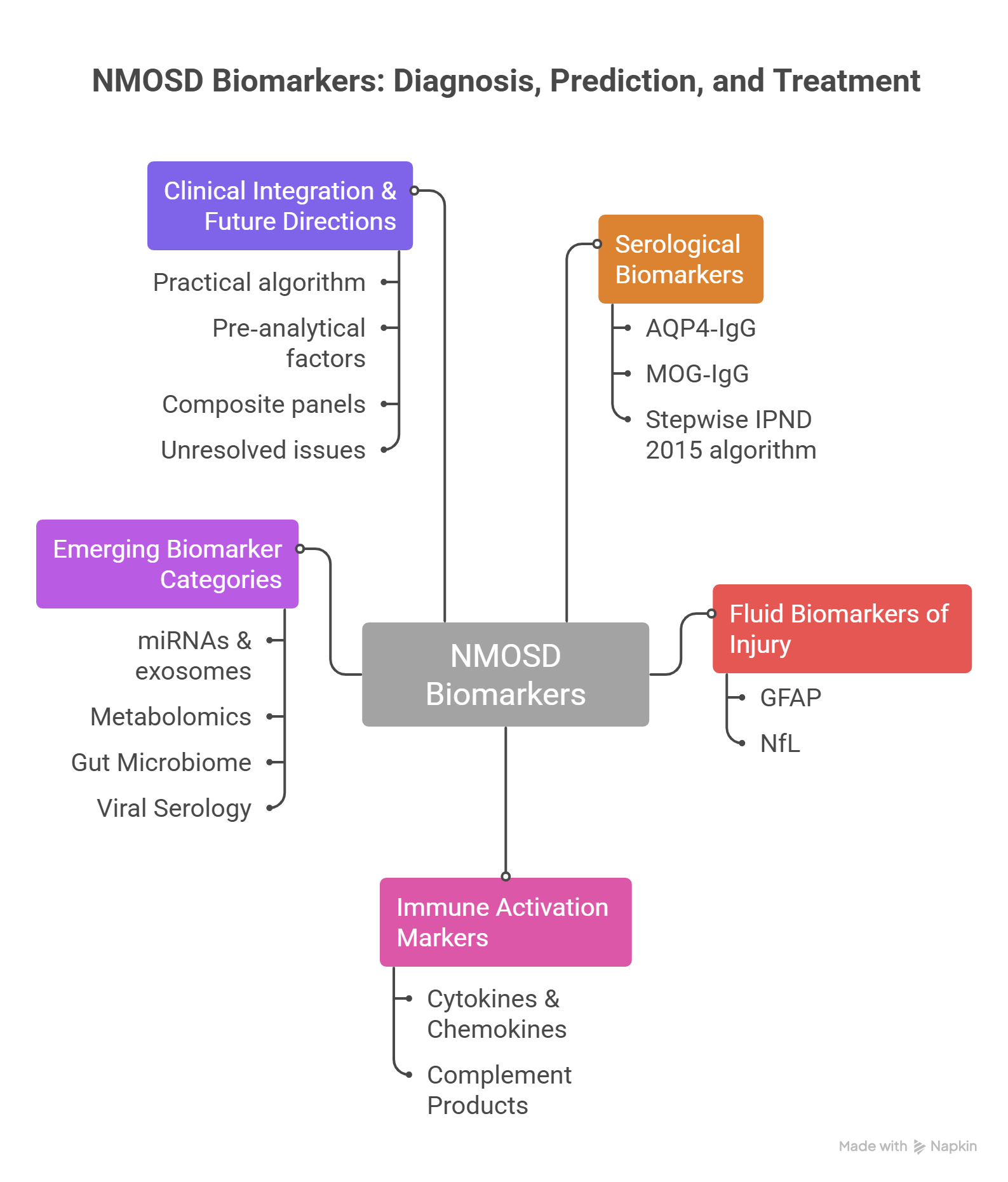
Neuromyelitis optica spectrum disorder (NMOSD) is a severe autoimmune astrocytopathy of the central nervous system, characterized by recurrent optic neuritis and longitudinally extensive transverse myelitis. Due to overlapping features with multiple sclerosis (MS), misdiagnosis remains common, yet treatment strategies differ fundamentally between these conditions. This narrative review synthesises recent advances in diagnostic and prognostic biomarkers for NMOSD, focusing on serological, cellular, molecular, and microbiological indicators.A targeted literature search was conducted in PubMed, Scopus and Web of Science (January 2015 – June 2025) using keywords related to NMOSD and biomarkers. Of over 500 initially identified articles, 72 key references were selected based on scientific relevance, methodological quality and clinical applicability, consistent with a narrative review approach.Established biomarkers – aquaporin‑4 immunoglobulin G (AQP4‑IgG) and myelin oligodendrocyte glycoprotein antibodies (MOG‑IgG) – remain central to diagnosis and phenotypic classification. Emerging markers, including glial fibrillary acidic protein (GFAP), neurofilament light chain (NfL), cytokines (interleukin‑6, CXCL13, CXCL10), complement components (C3, C4, sC5b‑9), microRNAs, metabolomics profiles, gut microbiome signatures and viral serology (Epstein–Barr virus, human herpesvirus 6), show promise in differentiating NMOSD from MS and MOGAD, predicting relapses, and guiding therapy. Integration of antibody assays with markers of astrocytic and axonal injury, immune activation and microbiological signatures could improve diagnostic accuracy and enable personalised treatment. However, clinical application remains limited by assay variability, lack of standardised cut-offs and small study populations. Future progress requires validated multimarker panels and harmonised measurement platforms across diverse populations.
Keywords:
NMOSD
; AQP4-IgG
; MOG-IgG
; GFAP
; neurofilament light
; cytokines
; complement
; microRNA
; metabolomics
; gut microbiome
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



