Submitted:
01 May 2026
Posted:
05 May 2026
You are already at the latest version
Abstract
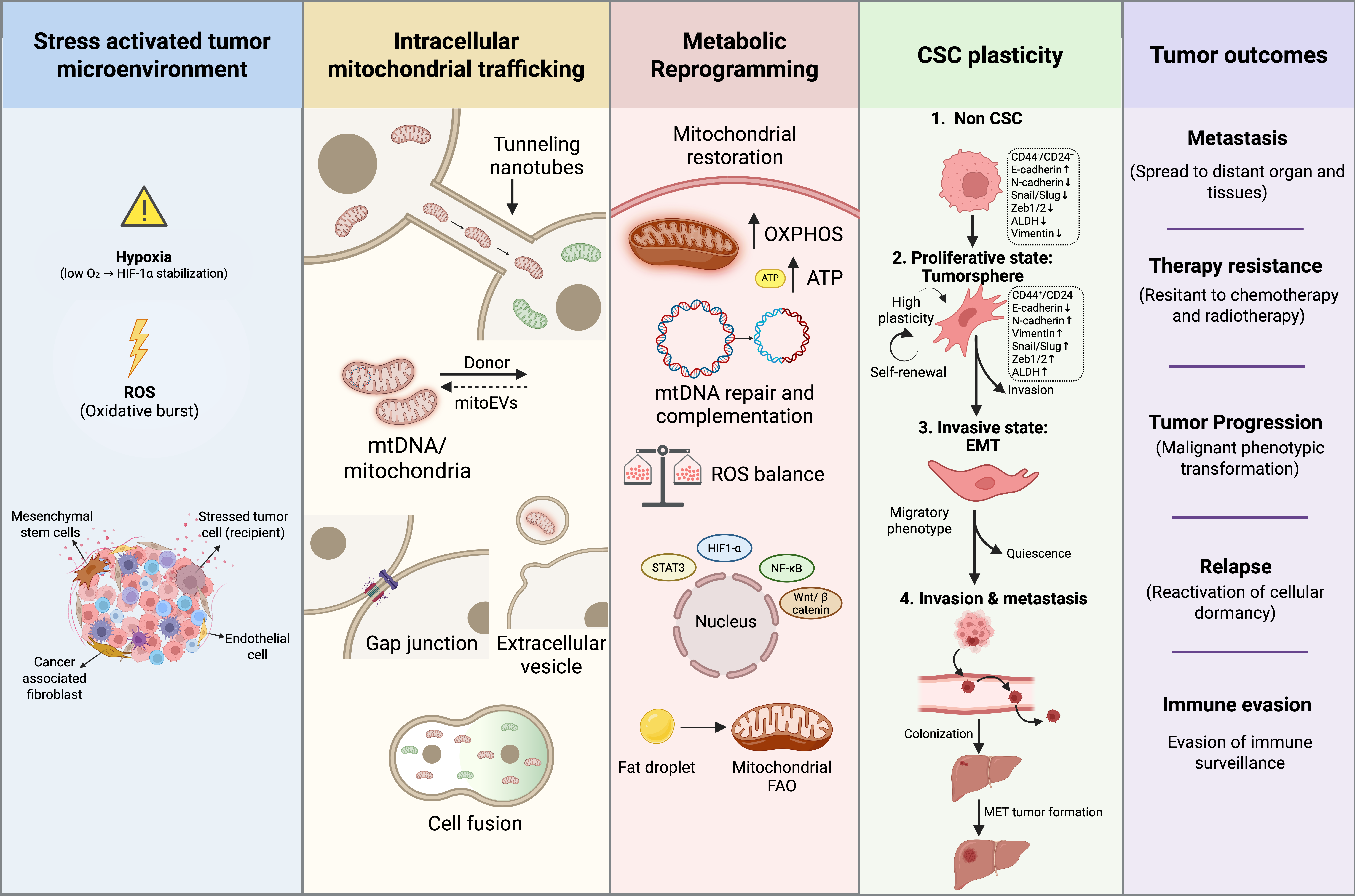
Intercellular mitochondrial trafficking has emerged as an important mechanism influencing tumor progression, metabolic adaptability, and cancer cell plasticity. Beyond their classical bioenergetic functions, mitochondria act as central regulators of redox homeostasis, signalling pathways, and epigenetic remodelling. Increasing evidences suggest that mitochondria can be transferred between tumor, stromal, and immune cells through tunnelling nanotubes (TNTs), extracellular vesicles (EVs), gap junctions, and cell fusion within the tumor microenvironment. This dynamic exchange enables metabolically compromised cancer cells to restore oxidative phosphorylation, optimize energy production, and survive under hypoxia and therapeutic stress.
Mitochondrial transfer has been increasingly associated with enhanced cellular plasticity and adaptive phenotypic transitions, including the acquisition of stem-like features that contribute to tumor heterogeneity, metastasis, and treatment resistance. In addition to bioenergetic restoration, transferred mitochondrial DNA and metabolites participate in retrograde signalling, linking metabolic state to epigenetic regulation and transcriptional reprogramming. This metabolic epigenetic interplay supports tumor cell adaptation to environmental stress and therapeutic pressure.
Although significant progress has been made, the precise mechanisms governing mitochondrial integration and their long-term impact on cellular phenotypes remain incompletely understood. A deeper understanding of these processes may reveal new therapeutic strategies to disrupt tumor adaptability and improve treatment outcomes.
Keywords:
mitochondrial transfer
; cancer stem cells
; tumor microenvironment
; metabolic reprogramming
; therapy resistance
1. Introduction
Cancer remains one of the leading causes of mortality worldwide despite significant advances in early detection and treatment [1,2]. A major challenge in achieving durable therapeutic responses is the presence of cancer stem cells (CSCs), a subpopulation of tumor cells characterized by self-renewal capacity, tumor-initiating potential, and resistance to conventional cancer treatment options e.g., chemotherapy, and radiation therapy. CSCs were first identified in Acute Myeloid Leukemia (AML) as leukemia initiating cells [3] and have since been reported in most solid tumors [4]. CSCs are now recognized as key drivers of tumor recurrence, metastasis, and therapeutic failure. CSCs exhibit distinct biological features, including metabolic adaptability, enhanced DNA repair capacity, resistance to apoptosis, and the ability to remain in a quiescent state [5]. Their slow-cycling nature allows them to evade therapies that primarily target rapidly dividing malignant cells [6].
Initially, CSCs were thought to arise within a hierarchical tumor organization [3]. However, recent evidence indicates that differentiated cancer cells can acquire stem-like characteristics under specific conditions. This phenomenon, known as cellular plasticity, represents an emerging hallmark of cancer stemness and facilitates cellular adaptation and evolution in response to the tumor microenvironment (TME) [6]. This reversible transition enables tumors to adapt to stress conditions and contributes significantly to disease progression and treatment resistance. The TME plays a central role in regulating CSC plasticity [7]. Hypoxia, metabolic stress, inflammatory signals, and therapeutic interventions can promote dedifferentiation of non-CSCs into stem-like phenotypes, activate drug efflux pathways, enhance DNA repair mechanisms, and support survival under adverse conditions [8].
Metabolic reprogramming is a key determinant of CSC behavior, highlighting the importance of mitochondrial function in maintaining stemness and adaptability [6]. Beyond their classical role as bioenergetic organelles, mitochondria function as signaling hubs that regulate redox balance, apoptosis, and cellular fate decisions [8]. Recent studies have shown that mitochondria can be transferred between cells through specialized intercellular transport mechanisms, including tunneling nanotubes, extracellular vesicles, gap junctions, and cell fusion [9]. This intercellular mitochondrial trafficking allows cancer cells to restore bioenergetic function, enhance survival, and acquire aggressive phenotypes.
Accumulating evidences suggest that mitochondrial transfer is associated with CSC plasticity, tumor progression, metastasis, and resistance to therapy [10,11]. In addition to restoring cellular energetics, transferred mitochondria and their associated genetic and metabolic components can influence intracellular signaling and epigenetic regulation, linking metabolic state to gene expression and phenotypic adaptation. However, the precise mechanisms governing mitochondrial transfer and its long-term functional consequences remain elusive.
In this review, we summarized the current understanding of intercellular mitochondrial trafficking, its role in tumor progression and cellular plasticity, and its potential as a therapeutic target.
2. Metabolic Adaptations of Cancer Stem Cells
CSCs exhibit distinct biological characteristics, including metabolic adaptability, enhanced DNA repair capacity, resistance to apoptosis, and the ability to remain in a quiescent state [12]. Their slow-cycling nature allows them to evade conventional therapies that primarily target rapidly proliferating tumor cells [6]. Among these features, metabolic reprogramming has emerged as a critical determinant of CSC function, highlighting the central role of mitochondrial activity in maintaining stemness and adaptability [6].
Beyond their classical role as bioenergetic organelles, mitochondria function as key signalling hubs that regulate redox homeostasis, apoptosis, and cellular fate decisions [13]. Oxidative phosphorylation (OXPHOS), driven by the electron transport chain (ETC), represents the primary mechanism of mitochondrial ATP generation, with oxygen acting as the final electron acceptor. In addition to ATP production, mitochondria provide essential biosynthetic intermediates required for tumor growth, including amino acids, nucleotides, and lipids, thereby supporting proliferation and survival under diverse environmental conditions [14].
CSCs exhibit pronounced metabolic flexibility, enabling them to dynamically switch between glycolysis and mitochondrial oxidative metabolism in response to environmental constraints [15]. Under nutrient-rich and oxygen-sufficient conditions, many CSCs rely on glycolysis (Warburg effect) to support rapid proliferation and biomass synthesis. However, in glucose-deprived or hypoxic environments, CSCs increasingly depend on mitochondrial oxidative metabolism to sustain energy production and survival [16]. This metabolic switching is closely associated with changes in redox balance and reactive oxygen species (ROS) levels, which act both as damaging agents and as signalling molecules regulating adaptive responses.
Hypoxia plays a pivotal role in shaping CSC metabolism through the activation of hypoxia-inducible factors (HIFs), particularly HIF-1α, which promotes stemness, survival, and therapy resistance [17]. HIF-mediated signalling regulates multiple downstream pathways, including those associated with cell surface markers such as CD44, thereby contributing to tumor aggressiveness. In parallel, metabolic alterations within CSCs can influence the tumor microenvironment. For instance, increased glycolysis and lactate secretion can suppress cytotoxic T-cell activity and promote M2-like macrophage polarization, thereby establishing an immunosuppressive niche that facilitates tumor progression [18].
Mitochondrial dysfunction, arising from hypoxia, metabolic stress, or rapid proliferative demands, further drives adaptive responses in CSCs. Activation of mitochondrial stress response pathways enables cancer cells to maintain viability and sustain tumor plasticity under adverse conditions [15]. Importantly, therapeutic interventions can also reshape CSC metabolism. For example, platinum-based chemotherapy combined with taxanes has been shown to enrich chemo-resistant ALDH⁺ CSC populations in ovarian cancer, whereas co-targeting mitochondrial oxidative metabolism using OXPHOS inhibitors can effectively prevent this enrichment and improve therapeutic response [19].
Collectively, these findings highlight metabolic plasticity as a defining feature of CSCs, with mitochondrial function serving as a central regulator of energy production, redox balance, and adaptive signalling. This metabolic adaptability not only supports tumor survival under stress but also provides a functional foundation for subsequent mitochondrial-dependent interactions within the tumor microenvironment.
3. Tumor Microenvironment as a Driver of Mitochondrial Transfer
3.1. Hypoxia
Hypoxia is a defining feature of the TME and a major driver of adaptive cellular responses that promote long-range intercellular communication and survival within the tumor microenvironment. Hypoxia can induced TNT formation via HIF signalling, facilitating direct cell-to-cell transfer of cellular components, including mitochondria [20] (Figure 1).
Hypoxia-induced TNT formation enhances mitochondrial transfer between cells, leading to measurable increases in mitochondrial content within recipient cells. This transfer supports metabolic adaptation by restoring mitochondrial function and enabling a shift toward OXPHOS, even under oxygen-limited conditions [21]. Such metabolic reprogramming allows stressed tumor cells to maintain energy production, resist apoptosis, and sustain proliferation within the hostile microenvironment [21,22]. Notably, this adaptation reflects a dynamic balance rather than a strict dependence on either glycolysis or oxidative metabolism, underscoring the metabolic flexibility of cancer cells [23].
Beyond metabolic rescue, hypoxia-driven mitochondrial transfer contributes to immune modulation within the TME. Emerging evidence indicates that tumor cells can transfer dysfunctional mitochondria to immune cells, particularly T cells, leading to impaired mitochondrial function and the induction of a senescence-like phenotype. Concurrently, tumor cells may acquire functional mitochondria to enhance their own metabolic fitness, thereby promoting immune evasion [24]. Hypoxia acts not only as a metabolic stressor but also as a key regulator of intercellular mitochondrial trafficking, enabling tumor cells to adapt, survive, and remodel their microenvironment under conditions of limited oxygen availability.
3.2. Chemotherapy and Radiation Stress
Intercellular mitochondrial transfer has emerged as an important adaptive mechanism that enables tumor cells to withstand therapeutic stress. Cytotoxic treatments, including chemotherapy and radiation, impose significant metabolic and oxidative damage, often leading to mitochondrial dysfunction, reduced ATP production, and increased apoptosis. In response, tumor cells can acquire functional mitochondria from surrounding stromal cells, thereby restoring bioenergetic capacity and enhancing survival (Figure 1).
Evidence from hematological malignancies demonstrates that mesenchymal stem cells (MSCs) donate mitochondria to acute myeloid leukemia and T-cell acute lymphoblastic leukemia cells during treatment with chemotherapeutic agents such as cytarabine and daunorubicin [25,26]. This transfer restores OXPHOS activity and ATP levels, thereby suppressing apoptosis and promoting therapy resistance [26,27]. Similar observations have been reported in solid tumors, where stromal to tumor mitochondrial transfer contributes to resistance against agents such as doxorubicin in breast cancer models [25].
In addition to immediate metabolic rescue, mitochondrial transfer also supports long-term cellular adaptation. For instance, mitochondrial DNA (mtDNA) delivered via extracellular vesicles from cancer-associated fibroblasts (CAFs) has been shown to promote the exit of tumor cells from dormancy and enhance their proliferative capacity following therapeutic stress [28]. This suggests that mitochondrial acquisition not only aids survival during treatment but may also facilitate disease recurrence.
Radiation-induced stress similarly impacts mitochondrial function through increased ROS generation and oxidative damage. Tumor cells with restored or enhanced mitochondrial capacity exhibit improved redox buffering and reduced sensitivity to radiation-induced cytotoxicity. In this context, mitochondrial transfer may contribute to the recovery of respiratory function and stabilization of redox homeostasis, thereby increasing the threshold for apoptosis. These findings indicate that mitochondrial transfer represents a key mechanism by which tumor cells adapt to therapeutic pressure, linking metabolic recovery to both immediate survival and long-term treatment resistance (Figure 1).
3.3. Oxidative Stress
Oxidative stress, characterized by the accumulation of ROS, is a major driver of mitochondrial dysfunction and a key regulator of intercellular communication within the TME. Elevated ROS levels not only induce cellular damage but also act as signaling cues that promote the formation of tunnelling nanotubes (TNTs), thereby facilitating mitochondrial transfer between cells [22] (Figure 1).
Experimental evidence indicates that increased hydrogen peroxide levels stimulate TNT biogenesis, enabling stressed cells to acquire functional mitochondria and restore redox balance. In hematological models, mitochondrial transfer from MSCs to acute lymphoblastic leukemia (ALL) cells has been shown to reduce treatment-induced mortality by improving mitochondrial function and buffering excessive ROS [25]. Similarly, tumor cells have been reported to acquire mitochondria from platelets, leading to metabolic reprogramming toward glycolysis and improved control of the intracellular redox state, including modulation of glutathione and ROS levels [25].
The acquisition of functional mitochondria plays a critical role in mitigating oxidative damage. By improving electron transport chain efficiency, mitochondrial transfer reduces excessive electron leakage and limits ROS overproduction, thereby protecting tumor cells from oxidative stress-induced apoptosis [22]. At the same time, mitochondrial transfer does not completely eliminate ROS signalling. Instead, it maintains ROS at levels that support activation of adaptive signalling pathways, enabling tumor cells to balance survival and stress responses. Oxidative stress functions not only as a damaging stimulus but also as a key trigger for mitochondrial transfer, linking redox imbalance to metabolic adaptation and enhanced tumor cell resilience.
3.4. Metabolic Crisis in Tumor Cells
Metabolic crises in tumor cells, arising from nutrient deprivation, impaired mitochondrial function, or defects in mtDNA, represent a critical barrier to tumor growth and survival. Under such conditions, cancer cells experience severe limitations in ATP production, biosynthetic capacity, and redox balance, ultimately compromising their tumorigenic potential. Intercellular mitochondrial transfer has emerged as a key adaptive mechanism that enables tumor cells to overcome these constraints (Figure 1).
Seminal studies using mtDNAdeficient tumor cells have demonstrated that the absence of functional mitochondrial genomes severely impairs tumor formation in vivo. These cells are unable to initiate tumor growth unless they acquire intact mitochondria or mtDNA from surrounding stromal cells, thereby restoring OXPHOS and bioenergetic capacity [29,30]. For example, B16 melanoma cells lacking mtDNA regain tumorigenicity only after acquiring functional mitochondria from the TME, highlighting the essential role of mitochondrial competence in tumor initiation and progression [31].
Similarly, stromal cells within the TME actively contribute to metabolic rescue. Cancer-associated fibroblasts (CAFs), particularly those with a glycolytic phenotype, have been shown to donate mitochondria to adjacent tumor cells, enhancing their respiratory capacity and invasive potential [31]. This transfer enables energy-deficient cancer cells to meet the high metabolic demands required for proliferation, survival, and adaptation to nutrient-limited conditions [27,32].
Importantly, mitochondrial acquisition not only restores cellular energetics but also supports broader aspects of tumor cell fitness, including maintenance of stem like properties and adaptation to environmental stress. These findings underscore that mitochondrial function is not merely supportive but is a fundamental determinant of tumorigenic potential. Metabolic crises act as a strong selective pressure that drives mitochondrial acquisition, reinforcing the role of intercellular mitochondrial transfer as a critical mechanism for tumor survival and progression.
4. Modes of Mitochondrial Transfer
Mitochondrial transfer can be intracellular or extracellular and can occur through several mechanisms such as formation of tunneling nanotubes (TNTs), gap junctions (GJs), extracellular vehicles (EVs), cell fusion, and endocytosis (Figure 1). Healthy cells can transfer functional mitochondria to damaged cells, where they merge with or replace the damaged ones, helping restore normal energy functions [9]. Moreover, mitochondria can be transferred within the TME and this transfer is not only limited to tumor cells themselves but can occur between tumor cells and stromal cells, tumor cells and immune cells, tumor cells and endothelial cells, or between tumor cells and homologous normal cells [25] (Figure 2).
4.1. Tunnelling Nanotubes (TNTs)
TNTs are thin, actin-based membranous structures that facilitate direct intercellular communication through the transfer of cellular components, including proteins, lipids, and organelles such as mitochondria. First described by Rustom et al. in 2004 [33], TNTs typically range from 50–200 nm in diameter and can extend up to several hundred microns, forming transient cytoplasmic connections between distant cells [25,34]. Within the TME, TNTs frequently connect tumor cells with stromal components such as Mesenchymal Stem Cells (MSCs), Cancer Associated Fibroblasts (CAFs), endothelial cells, and immune cells enabling efficient intercellular exchange [25] (Figure 2).
The formation of TNTs is strongly induced by cellular stress signals, including ROS and metabolic stress, and involves cytoskeletal remodelling mediated by Rho GTPases such as Cdc42 and RhoA [25,35]. Mitochondrial trafficking along TNTs is facilitated by motor proteins, including kinesin and myosin, and regulated by mitochondrial transport proteins such as Miro1 (Figure 1A) [25,35]. These structures represent one of the most direct and rapid mechanisms of mitochondrial transfer, allowing the movement of intact mitochondria between connected cells without membrane fusion.
Functionally, TNT-mediated mitochondrial transfer plays a critical role in restoring metabolic competence in recipient cells under stress conditions. Using a glioblastoma model, Sriramkumar and colleagues showed that, TNTs formed under stress between tumor cells and surrounding astrocytes, facilitate the transfer of tumor-derived mitochondria into neighbouring astrocytes and this intercellular mitochondrial exchange drives metabolic reprogramming of astrocytes toward a tumor-supportive phenotype, enhancing their ability to sustain tumor growth and adapt to hypoxic tumor microenvironment. Notably, this TNT dependent crosstalk contributes to increased tumor resilience, including enhanced survival, and resistance to Temozolomide in 3D spheroid tumor models, underscoring its potential role in therapy resistance and disease progression [21,36]. Similarly, in ovarian cancer, Miro1, a mitochondrial Rho-GTPase regulated MT transfer from MSCs to tumor cells and mediated in part through TNTs restores OXPHOS and promotes chemoresistance and promotes metastatic potential [37,38,39,40]. In hematological malignancies such as multiple myeloma, tumor cells exploit TNT networks within the bone marrow microenvironment to meet high metabolic requirements, sustain growth and achieve chemoresistance [41].
Although mitochondrial transfer via TNTs is predominantly unidirectional from stromal to tumor cells, bidirectional exchange has also been reported [42]. In particular, tumor cells can transfer dysfunctional mitochondria to immune cells, impairing their function and contributing to immune evasion [17,43]. to other MT transfer mechanisms, TNTs exhibit high efficiency, with rapid mitochondrial movement occurring within minutes and connectivity observed in a substantial proportion of cells under stress conditions [21,44].
Disruption of TNT formation represents a potential therapeutic strategy. Pharmacological agents that interfere with actin polymerization, such as cytochalasin-based compounds, or inhibitors targeting mitochondrial transport regulators such as Miro1, have been shown to significantly reduce mitochondrial transfer and sensitize tumor cells to treatments [45,46]. Collectively, TNTs serve as a highly efficient and dynamic conduit for mitochondrial transfer within the TME, enabling real-time metabolic rescue and contributing to tumor progression, metastasis, and therapy resistance.
4.2. Extracellular Vesicles (EVs)
Extracellular vesicles (EVs) are membrane-bound particles that mediate intercellular communication by transporting bioactive cargo, including proteins, lipids, nucleic acids, and organelle components. Based on their biogenesis and size, EVs are broadly classified into exosomes (30–150 nm), which originate from the endosomal pathway, and ectosomes or microvesicles (100–1000 nm), which are generated through direct budding of the plasma membrane [47,48,49]. Additional subtypes include large oncosomes, apoptotic bodies, and recently described non-membranous nanoparticles such as exomeres, reflecting the heterogeneity of EV populations [47,50] (Figure 2)..
EVs have emerged as stable mediators of long-distance communication within the TME, owing to their lipid bilayer structure, which protects their cargo from extracellular degradation [51,52]. Notably, EVs can also transport mitochondrial components, including mtDNA and, in some cases, carry 1–5 intact mitochondria, thereby influencing the metabolic and functional state of recipient cells (Figure 1B) [52,53]. The selective packaging of mitochondrial cargo into EVs is regulated by mechanisms involving ESCRT machinery or ceramide-dependent pathways, with proteins such as Rab27a/b and ARF6 contributing to vesicle trafficking and release [47,49].
Functionally, EV-mediated mitochondrial transfer contributes to metabolic reprogramming and cellular adaptation. CAF-derived EVs have been shown to deliver intact mtDNA and mitochondrial components to tumor cells, resulting in enhanced OXPHOS activity and increased self-renewal capacity [54]. In colorectal cancer, EVs facilitate the transfer of metabolic regulators that promote lipid metabolism and resistance to ferroptosis following chemotherapy [55]. Similarly, EV-mediated transfer of mitochondrial-related cargo has been reported to increase mitochondrial mass, oxygen consumption, and ATP production in recipient breast cancer cells [56].
Beyond local interactions, EVs enable systemic dissemination of mitochondrial signals. Tumor-derived EVs can circulate through body fluids and modulate distant microenvironments, contributing to the establishment of pre-metastatic niches. For example, EV-mediated transfer of mitochondrial components can alter stromal or immune cell function, promoting tumor-supportive conditions and facilitating disease progression [57,58].
Compared with TNTs, EV-mediated transfer is less direct but allows broader spatial distribution of mitochondrial signals, thereby amplifying tumor heterogeneity and intercellular communication across the TME. EVs represent a versatile and systemic mechanism of mitochondrial transfer that supports metabolic reprogramming, tumor progression, and adaptation to environmental stress.
4.3. Gap Junctions (GJs)
Gap junctions (GJs) are intercellular channels formed by connexin (Cx) proteins that enable direct communication between adjacent cells [59]. These channels permit the transfer of small molecules (<1.2 kDa), including ions, metabolites, and signalling molecules such as calcium and Cyclic Adenosine Monophosphate (cAMP), thereby coordinating cellular responses within the TME [60]. Among connexins, Cx43 is the most extensively studied in cancer-related intercellular communication [59] (Figure 2)..
In addition to the exchange of small molecules, emerging evidence suggests that GJs can facilitate the transfer of mitochondrial components, including mtDNA fragments and mitochondrial proteins, thereby influencing cellular metabolism and signalling (Figure 1C) [41,61,62]. This process may also involve connexosome formation, in which portions of one cell containing organelles are internalized by neighbouring cells, providing an additional route for intercellular mitochondrial exchange [63].
Physiologically, GJ-mediated communication plays a critical role in maintaining tissue homeostasis. For example, connexin-dependent coupling between granulosa cells and oocytes supports metabolic coordination during follicular development [64,65]. Similarly, in the bone marrow microenvironment, bidirectional mitochondrial exchange between hematopoietic stem/progenitor cells and MSCs via Cx43-dependent GJs contributes to cellular recovery and stress adaptation [41,61,62].
In the context of cancer, GJ-mediated communication contributes to tumor progression by enabling metabolic coupling between tumor and stromal cells. In non-small cell lung cancer, GJs between (Cancer Associated Fibroblasts) CAFs and tumor cells promote epithelial–mesenchymal transition (EMT) and invasion through calcium-dependent signalling [66]. In breast cancer, Cx43-mediated communication supports ATP maintenance under hypoxic conditions, enhancing tumor cell survival, while inhibition of GJs reduces viability [66]. In glioma, coupling between astrocytes and tumor cells facilitates DNA repair following radiation-induced damage, contributing to therapy resistance.
Compared with TNTs and EVs, GJ-mediated transfer is restricted to adjacent cells and operates through diffusion-based mechanisms, resulting in slower but sustained intercellular exchange. This localized communication contributes to micro-niche homeostasis by enabling selective exchange of metabolites and signalling cues between neighbouring cells. Emerging evidence also suggests potential coordination between GJ-mediated communication and EV-based signalling within the TME, supporting integrated intercellular responses under stress conditions [59,67,68]. GJs represent a complementary pathway of mitochondrial and metabolic communication that contributes to tumor progression, cellular adaptation, and therapy resistance within the TME.
4.4. Cell Fusion
Cell fusion represents a distinct mechanism of intercellular interaction in which two or more cells merge to form hybrid cells containing nuclear and cytoplasmic components from both parental cells. Unlike TNTs, EVs, or GJ-mediated communication, which involve partial and regulated exchange of cellular material, cell fusion results in extensive mixing of cytoplasmic contents, including mitochondria, thereby enabling large-scale cellular reprogramming [69] (Figure 2).
The molecular basis of cell fusion in human systems is primarily mediated by syncytins, a family of endogenous retroviral envelope proteins with fusogenic activity that are aberrantly expressed in malignancies such as neuroblastoma and endometrial cancer, and may serve as potential biomarkers for identifying hybrid cells [70,71]. The initiation of fusion further requires dynamic cytoskeletal remodelling driven by actin polymerization, regulated by small GTPases such as Rac1 and Cdc42, along with the Arp2/3–WASP complex [70]. Notably, overexpression of Arp2/3 has been strongly associated with metastatic progression in breast, lung, and pancreatic cancers [70]. Microenvironmental stress conditions within the TME can further enhance fusion efficiency. Hypoxia and chemotherapeutic agents, including doxorubicin, have been reported to increase fusion rates by approximately 5–10 fold through HIF-1α-dependent signalling pathways [70,71].
Fusion events have been reported between tumor cells and various stromal components within the TME, including MSCs, macrophages, fibroblasts, and endothelial cells. These interactions generate hybrid cells with increased phenotypic diversity, often combining proliferative capacity with enhanced migratory and invasive properties. In particular, fusion between tumor cells and macrophages has been associated with metastatic progression, facilitating immune evasion and altered inflammatory signalling [27,70].
Mitochondrial mixing following cell fusion plays a central role in metabolic reprogramming and adaptive fitness. The integration of mitochondria from distinct cellular origins can influence OXPHOS activity, ROS balance, and downstream signalling pathways, thereby reshaping tumor cell behaviour. These fusion events yield hybrid cells with stem-like traits and significant survival advantages [34,70]. In breast cancer, hybrid cells formed through fusion with MSCs or epithelial cells have been shown to exhibit increased chemoresistance and resistance to anoikis, as evidenced by Cre–loxP lineage tracing in patient-derived xenograft (PDX) models [72]. In glioma, fusion between CSCs and endothelial progenitors promotes vascular mimicry, thereby enhancing neovascularization and tumor progression [73,74,75]. In acute myeloid leukemia, fusion with stromal cells, although occurring at relatively low frequencies (approximately 1–5%), results in substantial transfer of functional mitochondria, restoring OXPHOS capacity and activating Wnt/β-catenin signalling pathways that support maintenance of the leukemia stem cell niche [41,76,77].
Despite its relatively low frequency compared with other mechanisms of mitochondrial transfer, cell fusion has significant biological consequences due to its ability to induce large-scale cellular remodelling. Compared with TNTs and EVs, which mediate controlled and selective transfer, cell fusion represents a more extensive process that can generate highly heterogeneous tumor cell populations. Cell fusion constitutes a potent mechanism of mitochondrial and cytoplasmic exchange that contributes to tumor progression, cellular plasticity, and adaptive evolution within the TME.
5. Metabolic Reprogramming After Mitochondrial Transfer
The transfer of mitochondria in tumor cells represents a critical adaptive mechanism that reshapes the cellular metabolic landscape in response to environmental stress and therapeutic intervention. Beyond genetic alterations in oncogenes and tumor suppressors, the metabolic state of cancer cells plays a decisive role in determining tumor aggressiveness, persistence, and progression, with mitochondrial function acting as a central regulator [78]. The acquisition of functional mitochondria and mtDNA by metabolically compromised tumor cells restores ETC activity, mitochondrial membrane potential, and ATP production, thereby promoting survival, invasion, and therapy resistance [10,11]. These effects arise from coordinated changes in oxidative phosphorylation, redox homeostasis, and metabolic flexibility (Figure 3).
5.1. Restoration of Oxidative Phosphorylation and ATP Production
Intercellular mitochondrial transfer directly restores OXPHOS in tumor cells with impaired respiratory function. Defects in mtDNA or ETC components [12] impose bioenergetic constraints that limit tumor growth and survival. Acquisition of intact mitochondria alleviates these constraints by re-establishing ETC activity, mitochondrial membrane potential, and ATP generation through oxidative metabolism. Experimental studies demonstrate that mitochondrial DNA deficient cancer cells are unable to initiate tumor growth in vivo unless they acquire functional mitochondria from host cells, establishing mitochondrial transfer as a prerequisite for bioenergetic competence rather than a secondary adaptation [10,11].
The resulting increase in ATP availability supports energy-intensive processes such as proliferation, cytoskeletal remodelling, and survival under hypoxic or nutrient-limited conditions. Moreover, aggressive and therapy-resistant tumors frequently exhibit dependence on mitochondrial respiration, further reinforcing the functional importance of OXPHOS restoration in tumor progression [79,80].
5.2. Metabolic Flexibility Through Integration of Glycolysis and OXPHOS
Tumor aggressiveness is not solely dependent on aerobic glycolysis but rather on the ability to dynamically engage both glycolytic and oxidative metabolic pathways. Functional studies in pancreatic ductal adenocarcinoma demonstrated that tumor cells surviving oncogene withdrawal remain dependent on OXPHOS, indicating that mitochondrial respiration is essential for persistence under therapeutic stress [80].
Intercellular mitochondrial transfer enhances this adaptability by restoring or augmenting respiratory capacity in recipient tumor cells. This leads to increased basal and maximal oxygen consumption while maintaining glycolytic activity, thereby establishing a hybrid metabolic state capable of responding to fluctuations in nutrient and oxygen availability [10,11]. In vivo, this metabolic plasticity enables respiration-deficient tumor cells to regain tumorigenic potential, demonstrating that mitochondrial acquisition alone is sufficient to confer adaptive metabolic flexibility [10].
Beyond glucose metabolism, restored mitochondrial function enables utilization of alternate substrates, including lactate, glutamine, and fatty acids, to sustain TCA cycle flux and ATP production. This metabolic versatility is particularly important during tumor dissemination and metastatic colonization under nutrient-limited conditions [14]. Recent in-vivo evidence further supports that mitochondrial transfer promotes simultaneous engagement of glycolysis and OXPHOS, indicating that metabolic flexibility is a direct consequence of mitochondrial acquisition rather than an intrinsic pre-existing trait [79].
5.3. mtDNA Transfer and Repair of Mitochondrial Defects
In addition to the transfer of whole organelles, intercellular mitochondrial trafficking facilitates horizontal transfer of mtDNA, providing a direct genetic mechanism for restoring respiratory competence in tumor cells. Loss or mutation of mtDNA disrupts ETC function, alters redox balance, and imposes metabolic constraints that limit tumor growth and progression. Restoration of mtDNA integrity alone has been shown to modulate tumor behaviour, establishing mitochondrial genetics as a functional determinant of malignancy [81].
Direct experimental evidence demonstrates that mtDNA-deficient tumor cells regain respiratory capacity and tumorigenic potential following acquisition of mitochondria containing intact mtDNA [11]. Importantly, mitochondrial transfer contributes to intratumoral mtDNA heterogeneity, generating metabolic diversity within tumor populations. This heterogeneity allows selection of subclones with enhanced mitochondrial function under stress conditions, thereby promoting tumor evolution and adaptation [82,83].
5.4. Redox Recalibration and ROS Signalling
Intercellular mitochondrial transfer plays a key role in regulating intracellular redox balance by reducing excessive ROS accumulation while preserving ROS-mediated signalling required for adaptive responses. Mitochondrial dysfunction leads to increased electron leakage from the ETC, resulting in elevated ROS levels that can induce oxidative damage and apoptosis [81]. The acquisition of functional mitochondria improves ETC efficiency, thereby reducing ROS overproduction and stabilizing redox homeostasis [10,11].
Importantly, mitochondrial transfer does not eliminate ROS signalling. Instead, it maintains ROS at levels that support activation of adaptive transcriptional pathways, including NF-κB, HIF-1α, and STAT3, which promote survival, metabolic flexibility, and stress tolerance [84]. Tumor cells receiving functional mitochondria also exhibit enhanced antioxidant capacity, particularly through glutathione-dependent buffering systems, while retaining redox-sensitive signalling pathways [79].
In certain contexts, mitochondrial or mtDNA transfer can transiently increase ROS levels, thereby activating inflammatory and pro-tumorigenic signalling pathways. For example, extracellular vesicle mediated mtDNA transfer has been shown to enhance mitochondrial respiration and ROS production in recipient epithelial cells, leading to activation of TGFβ-dependent signalling and the establishment of a tumor supportive microenvironment [85].
5.5. Fatty Acid Oxidation Couples Mitochondrial Transfer to Sarcoma (Src) Tyrosine Kinase Driven Oncogenic Signalling
Mitochondrial transfer enhances the ability of tumor cells to utilize fatty acids as an energy source through fatty acid oxidation (FAO), particularly under glucose limited conditions. FAO provides a sustained supply of reducing equivalents and supports TCA cycle flux and OXPHOS activity. Importantly, FAO is not merely a compensatory pathway but actively contributes to tumor progression and invasion [86].
FAO derived energy metabolism is functionally linked to Src tyrosine kinase activation, establishing a direct connection between mitochondrial metabolism and oncogenic signalling. FAO promotes Src tyrosine kinase autophosphorylation and activation, while activated Src localizes to mitochondria and phosphorylates ETC components, thereby enhancing respiratory efficiency and reinforcing oxidative metabolism [86]. This establishes a positive feedback loop between FAO and Src kinase signalling that sustains tumor progression.
Disruption of FAO through pharmacological or genetic approaches reduces Src tyrosine kinase activation and suppresses tumor cell invasion, indicating that FAO functions upstream of Src tyrosine kinase dependent oncogenic signalling rather than serving solely as an energy source [86]. Mitochondrial transfer amplifies this FAO–Src tyrosine kinase axis by introducing mitochondria with enhanced oxidative and lipid metabolic capacity into recipient tumor cells, thereby promoting invasive behaviour.
Additionally, cybrid and mitochondrial manipulation studies demonstrated that mitochondrial genotype alone can determine FAO capacity and Src tyrosine kinase signalling strength, independent of nuclear genetic context. This highlights mitochondria as an active regulators of oncogenic signaling networks rather than passive bioenergetic organelles [87].
6. Mitochondria Derived Metabolites as Regulators of Nuclear Signaling and Epigenetic Reprogramming
Mitochondria serve as central metabolic hubs that extend their influence beyond bioenergetic functions to actively shape nuclear gene expression through a process termed metabolic epigenetics [88,89]. This mitochondria to nucleus communication, referred to as retrograde signalling, operates through the direct generation and functional transfer of mitochondria derived metabolites that act as essential co-factors or substrates for epigenetic modulating enzymes, thereby integrating cellular metabolic status with chromatin architecture and transcriptional programs [88,89,90]. This mechanism is particularly critical in CSCs, where mitochondrial metabolites directly influence epigenetic states to maintain pluripotency, plasticity, and therapy resistance [91,92]. By modulating the availability of substrates for chromatin modifying enzymes, mitochondrial metabolic flux provides a dynamic mechanism through which CSCs adapt to fluctuating stresses within the TME [88,91,93] (Figure 4).
One of the most profound examples of mitochondrial–nuclear crosstalk in oncology is the accumulation of oncometabolites resulting from mutations in TCA cycle enzymes. Mutations in isocitrate dehydrogenase (IDH1/2) result in the neomorphic production of R-2-hydroxyglutarate (R-2-HG), which acts as a competitive inhibitor of α-ketoglutarate-dependent dioxygenases, including Ten-Eleven Translocation (TET) family DNA demethylases and Jumonji-C (JmjC) histone demethylases [89,93]. In hematological malignancies and gliomas, this leads to global DNA and histone hypermethylation, trapping cells in a stem-like, undifferentiated state by silencing genes required for differentiation [89,91]. Similarly, loss of succinate dehydrogenase or fumarate hydratase results in the accumulation of succinate and fumarate, which mimic the inhibitory effects of R-2-HG. These changes drive a pseudohypoxic state by stabilizing HIF-1α and maintaining CSC niches even under normoxic conditions [61,89,91].
A fundamental mechanism linking mitochondrial metabolism to epigenetic regulation involves acetyl-CoA, a key TCA cycle-derived metabolite that serves as the direct acetyl donor for histone acetyltransferases. Because acetyl-CoA cannot directly cross the mitochondrial membrane, it is exported as citrate and reconverted into acetyl-CoA in the nucleus by ATP-citrate lyase, thereby driving histone acetylation at active promoter and enhancer regions [34,72]. Importantly, metabolic enzymes such as the pyruvate dehydrogenase complex and acetyl-CoA synthetase 2 can translocate to the nucleus, where they locally synthesize acetyl-CoA to support rapid transcriptional responses under stress conditions, including glucose deprivation [91,92,94,95,96]. Intracellular acetyl-CoA levels correlate directly with global histone acetylation, establishing mitochondrial metabolism as a rate-limiting determinant of chromatin accessibility and gene activation [89,95] (Figure 4).
This metabolic epigenetic axis promotes transcriptional activation of genes associated with EMT and CSC maintenance, thereby enhancing phenotypic plasticity and therapeutic resistance [61,91,93]. Through this mechanism, mitochondrial metabolism enables dynamic transitions between proliferative and invasive cellular states in response to microenvironmental cues.
S-adenosylmethionine (SAM), generated through mitochondrial-linked one-carbon metabolism, functions as the universal methyl donor for DNA and histone methyltransferases. Mitochondrial serine metabolism provides one-carbon units that fuel the methionine cycle, thereby regulating SAM-dependent methylation reactions [46,94,97] (Figure 4). Disruption of SAM availability or transport through mitochondrial carriers such as SLC25A26 alters the SAM/S-adenosylhomocysteine ratio, leading to epigenetic instability and aberrant gene expression in cancer [94,98].
α-Ketoglutarate (α-KG) and its associated oncometabolites represent a central regulatory axis in mitochondrial–epigenetic crosstalk. As a cofactor for TET enzymes and histone demethylases, α-KG supports active DNA and histone demethylation. However, its conversion into 2-HG or competition by accumulated succinate and fumarate inhibits these enzymes, resulting in widespread epigenetic reprogramming that contributes to EMT, maintenance of stemness, and metastatic progression through HIF-1α stabilization [91,92,99].
In acute myeloid leukemia, mitochondrial transfer from MSCs through connexin 43 (CX43) dependent mechanisms drives metabolic remodelling that sustains leukemia stem cell function [61,62]. This influx of functional mitochondria restores OXPHOS and activates Wnt/β-catenin signalling, a central regulator of self-renewal [61,77]. These transferred mitochondria act as biosynthetic sources of key metabolites, including Nicotinamide adenine dinucleotide (NAD⁺), α-KG, and acetyl-CoA, thereby reprogramming nuclear transcription toward a stem-like state [61,88,100].
NAD⁺ provides an additional link between mitochondrial metabolism and epigenetic regulation through its role as a cofactor for sirtuin deacetylases. Nuclear sirtuins (SIRT1, SIRT6, and SIRT7) regulate chromatin structure, while mitochondrial sirtuins (SIRT3–5) control metabolic enzyme activity [96,101]. Increased NAD⁺ levels enhance sirtuin mediated histone deacetylation, promoting transcriptional programs associated with stress resistance and metabolic adaptation. Conversely, nutrient-rich conditions elevate acetyl-CoA levels, favoring histone acetylation and gene activation, establishing a reciprocal regulatory balance between mitochondrial metabolism and epigenetic state [89,101,102] (Figure 4).
Collectively, mitochondria derived metabolites form a dynamic regulatory circuitry that links cellular metabolism to epigenetic remodelling and transcriptional control. This metabolic epigenetic integration enables tumor cells to rapidly adapt to environmental stress, sustain CSC plasticity, and promote tumor progression. Disruption of this axis highlights mitochondria as indispensable regulators of cellular fate determination and represents a promising therapeutic avenue (Figure 4).
7. Mitochondrial Transfer in Csc Plasticity and Tumor Progression
CSCs are characterized by their ability to self-renew, sustain continuous proliferation, and drive tumor initiation, metastasis, and intratumoral heterogeneity [2]. Although CSCs were initially identified as a subpopulation resembling normal stem cells, they are now recognized as highly dynamic entities capable of transitioning between stem-like and differentiated states, a process referred to as cellular plasticity [3]. CSCs are not uniformly distributed within tumors but are often enriched in specialized microenvironments characterized by hypoxia, acidic pH, and nutrient limitation, conditions that promote their maintenance and expansion [103].
CSC populations are defined by distinct surface markers that vary across tumor types. In breast cancer, CSCs are commonly identified by CD44⁺/CD24⁻ and ALDH1 expression, along with additional markers such as ABCG2, CD133, CD49f, LGR5, SSEA-3, CD70, and PROCR [104]. Pancreatic CSCs, although comprising less than 1% of tumor cells, express markers including CD44, EpCAM, CD133, CXCR4, CD24, ALDH1, c-MET, Dclk1, and Lgr5 [105]. In colorectal cancer, CSCs are characterized by CD44, CD133, CD24, EpCAM, LGR5, and ALDH expression [106], while in lung cancer, markers such as ALDH1, CD133, CD44, and CD90 are associated with stemness, therapeutic resistance, and intratumoral heterogeneity [107]. Notably, variants such as CD44v6 have been linked to tumor aggressiveness and relapse, particularly in oral squamous cell carcinoma, highlighting their prognostic significance [108].
CSC fate decisions are predominantly regulated by epigenetic mechanisms that control self-renewal and differentiation programs. Transcription factors such as OCT4, SOX2, NANOG, and POU5F1, along with regulators like C/EBPα, coordinate stemness and lineage commitment [109,110]. These processes are tightly linked to mitochondrial metabolism, where metabolic epigenetic coupling sustains long-term self-renewal while suppressing differentiation. Importantly, epigenetic reprogramming contributes to therapy resistance by enabling CSCs to survive cytotoxic stress and reinitiate tumor growth [103,111].
Mitochondrial transfer has emerged as a key regulator of CSC plasticity by linking metabolic adaptation to phenotypic reprogramming. Acquisition of functional mitochondria restores OXPHOS activity, redox balance, and mitochondrial signalling, thereby enabling tumor cells to overcome metabolic constraints and transition toward stem-like states. Experimental evidence demonstrates that tumor cells receiving mitochondria exhibit enhanced sphere formation, increased expression of stemness markers such as OCT4, SOX2, and NANOG, and improved tumor initiating capacity.
Beyond stemness, mitochondrial transfer actively drives tumor progression and metastasis. By restoring respiratory function, modulating redox signalling, and enhancing lipid metabolism, mitochondrial acquisition enables tumor cells to regain migratory capacity, survive dissemination, and colonize distant tissues [10,81]. These findings establish mitochondrial function as a central determinant of tumor evolution.
7.1. Epithelial to Mesenchymal Transition (EMT) Driven by Mitochondrial Transfer
EMT is a key process enabling tumor cells to acquire invasive and metastatic capabilities [112]. While traditionally regulated by transcriptional programs involving TGFβ, Wnt, and Notch pathways [113], emerging evidence highlights metabolic capacity as a critical enabling factor [114]. Mitochondrial transfer supports EMT by fulfilling the energetic and signalling demands required for cytoskeletal remodelling, cell matrix interaction changes, and activation of EMT associated transcriptional programs.
Cells with impaired mitochondrial function exhibit limited invasive capacity, whereas restoration of mitochondrial competence enhances migration and metastasis [11,81]. Tumor cells acquiring mitochondria, particularly from stromal sources such as MSCs, display increased invasiveness, directly linking mitochondrial exchange to EMT-associated plasticity [10,115]. Importantly, mitochondrial transfer supports EMT-MET plasticity rather than a fixed mesenchymal state, allowing tumor cells to dynamically switch between epithelial and mesenchymal phenotypes during metastasis [11].
7.2. Migration, Invasion, and Cytoskeletal Dynamics
Tumor cell migration and invasion require coordinated energy production, signalling, and cytoskeletal remodelling [116]. Mitochondrial transfer restores these capabilities by re-establishing metabolic competence. Enhanced FAO activates Src-family kinases, linking mitochondrial metabolism to focal adhesion turnover, actin remodelling, and invasive behavior [86]. Inhibition of FAO reduces Src activation and suppresses migration, establishing mitochondrial metabolism as an upstream regulator of motility.
Additionally, acquired mitochondria reposition toward the leading edge of migrating cells, supporting localized ATP production required for lamellipodia formation and directional movement [116].
7.3. Metastatic Colonization and Niche Formation
Metastatic dissemination imposes significant stress on tumor cells due to differences in oxygen, nutrient availability, and microenvironmental conditions. Many disseminated cells fail to survive; however, mitochondrial transfer enhances survival and facilitates metastatic colonization [10]. Restoration of mitochondrial function is therefore a key determinant of metastatic competence.
Successful metastasis also requires the formation of a supportive microenvironment. Mitochondrial transfer contributes to metastatic niche formation by reprogramming stromal and immune cells. Transfer of mitochondria or mtDNA to surrounding cells induces metabolic and inflammatory changes that promote tumor-supportive conditions, thereby facilitating colonization and growth at distant sites [85,115,117,118].
7.4. Intratumoral Heterogeneity and Clonal Selection
Intratumoral heterogeneity arises from genetic, epigenetic, and metabolic variation among tumor cells [119,120]. Mitochondrial transfer introduces an additional layer of heterogeneity by generating diversity in mtDNA content, respiratory capacity, and redox states. This results in mosaic patterns of mitochondrial heteroplasmy within tumor populations [121,122].
Under selective pressures such as hypoxia, immune surveillance, and nutrient limitation, subclones with enhanced mitochondrial function gain a survival advantage, leading to clonal expansion and tumor evolution [80,122]. Thus, mitochondrial transfer not only supports tumor growth but also drives selection of aggressive phenotypes during progression and metastasis.
8. Therapeutic Implications and Targeting of Mitochondrial Transfer
The recognition of intercellular mitochondrial transfer as a key driver of tumor progression, metabolic adaptation, and CSC plasticity has opened new avenues for therapeutic intervention. Targeting mitochondrial transfer and its downstream effects represents a promising strategy to disrupt tumor survival mechanisms, overcome therapy resistance, and limit disease progression [10,81] (Figure 5).
8.1. Targeting Mitochondrial Transfer Mechanisms
One of the most direct therapeutic approaches involves inhibition of the physical mechanisms that mediate mitochondrial transfer. Disruption of TNT formation through agents that interfere with actin polymerization, such as cytochalasin-based compounds, has been shown to reduce mitochondrial trafficking and sensitize tumor cells to chemotherapy [25,45]. Similarly, targeting key regulators of mitochondrial movement, including Miro1, can impair mitochondrial transport between stromal and tumor cells, thereby limiting metabolic rescue [35].
Inhibition of EV-mediated transfer represents another potential strategy. Pharmacological agents targeting EV biogenesis pathways, such as neutral sphingomyelinase inhibitors or Rab GTPase regulators, can reduce the release and uptake of mitochondria-containing vesicles [48,49]. Additionally, blockade of cx43 dependent gap junction communication has been shown to impair mitochondrial transfer in hematological malignancies, reducing OXPHOS restoration and sensitizing tumor cells to treatment [41,61,62].
8.2. Targeting Mitochondrial Metabolism and Bioenergetics
An alternative strategy focuses on disrupting the metabolic advantages conferred by mitochondrial transfer. Inhibition of OXPHOS using agents such as mitochondrial complex I inhibitors can selectively target tumor cells that rely on mitochondrial respiration for survival [79,80]. Similarly, targeting FAO has emerged as an effective approach to suppress mitochondrial-driven signalling pathways, including the FAO–Src axis, thereby reducing tumor invasion and metastasis [86].
8.3. Targeting Mitochondrial Epigenetic Crosstalk
Given the central role of mitochondrial-derived metabolites in regulating epigenetic programs, targeting metabolic epigenetic coupling offers a powerful therapeutic strategy. Inhibitors of mutant IDH enzymes, which reduce 2-HG production, have demonstrated clinical efficacy in restoring normal epigenetic states and promoting differentiation in certain cancers [89,93]. Similarly, targeting enzymes involved in acetyl-CoA production or one-carbon metabolism may disrupt epigenetic programs that sustain CSC plasticity [94,97,123].
Modulation of NAD⁺ metabolism and sirtuin activity also represents a promising approach to influence chromatin states and tumor cell adaptation [96,101]. By altering the balance between histone acetylation and deacetylation, these strategies can potentially reverse therapy-resistant phenotypes [89,102].
8.4. Targeting the Tumor Microenvironment
The TME plays a critical role in facilitating mitochondrial transfer, making it an important therapeutic target. Strategies aimed at disrupting stromal tumor interactions, including inhibition of MSC or CAF support, may reduce the availability of donor mitochondria [10,115]. Targeting hypoxia-driven signalling pathways, particularly HIF-1α, can further limit stress-induced mitochondrial transfer and associated adaptive responses [20,124].
In addition, modulation of immune cell function within the TME may help counteract the immunosuppressive effects associated with mitochondrial transfer, thereby enhancing anti-tumor immunity [24].
8.5. Challenges and Future Perspectives
Despite the therapeutic potential, several challenges remain in targeting mitochondrial transfer. The molecular mechanisms governing selective mitochondrial packaging, transfer, and integration are not fully understood, limiting the development of highly specific inhibitors [47,49]. Furthermore, mitochondrial transfer also plays essential roles in normal physiological processes, raising concerns regarding potential toxicity and off-target effects [61].
Tumor heterogeneity and variability in mitochondrial dependence across cancer types further complicate therapeutic targeting [80,101]. Therefore, future strategies will require precise identification of patient subsets that are most likely to benefit from interventions targeting mitochondrial transfer.
9. Discussion
Intercellular mitochondrial trafficking has emerged as a fundamental mechanism driving tumor progression, metabolic reprogramming, and CSC plasticity. The dynamic horizontal transfer of mitochondria and mtDNA occurs through multiple routes, including TNTs, EVs, gap junctions, and cell fusion. This process is strongly influenced by microenvironmental stressors such as hypoxia, oxidative stress, nutrient deprivation, and cytotoxic therapies.
Upon acquisition of functional mitochondria, metabolically compromised cancer cells restore OXPHOS and ATP production, thereby re-establishing bioenergetic competence. This metabolic rescue enables tumor cells to regulate intracellular ROS levels, evade apoptosis, and survive under both therapeutic and physiological stress conditions. Beyond energy restoration, mitochondrial transfer also plays a critical role in epigenetic reprogramming through mitochondria-to-nucleus communication.
The influx of functional mitochondria alters the availability of key metabolites, including acetyl-CoA, α-ketoglutarate, S-adenosylmethionine (SAM), and NAD⁺. These metabolites serve as essential cofactors for chromatin-modifying enzymes, thereby linking cellular metabolism to gene expression programs that sustain stem-like phenotypes and CSC niches. Consequently, this metabolic-epigenetic crosstalk facilitates EMT, enhancing the migratory, invasive, and metastatic capacities of tumor cells.
Moreover, the uneven distribution of transferred mitochondria leads to mtDNA heteroplasmy, introducing an additional layer of intratumoral heterogeneity beyond nuclear genetic variation. This mitochondrial mosaicism promotes clonal selection of highly adaptable, chemoresistant, and radioresistant tumor subpopulations under selective pressures.
From a therapeutic perspective, targeting intercellular mitochondrial communication represents a promising strategy. Disruption of these transfer mechanisms using actin polymerization inhibitors to block TNTs, GW4869 to inhibit EV biogenesis, or carbenoxolone to suppress gap junction communication has shown potential in limiting mitochondrial transfer. Importantly, combining such approaches with agents targeting recipient cell dependencies, including OXPHOS or FAO, may enhance therapeutic efficacy, overcome resistance, and reduce the likelihood of disease recurrence.
10. Conclusions
Intercellular mitochondrial transfer represents a complex, non-genetic mechanism that enables cancer cells to adapt to metabolic stress and evade therapeutic pressure. Tumors with defective mtDNA or severe metabolic impairment are not inherently terminal; instead, they can regain tumorigenic and metastatic potential through acquisition of functional mitochondria from surrounding stromal cells.
Microenvironmental stressors, including hypoxia and elevated ROS levels, act as key drivers of mitochondrial transfer by promoting the formation of tunneling nanotubes and other intercellular communication pathways, effectively transforming the TME into a highly interconnected network for organelle exchange. In this context, stromal-derived mitochondrial transfer provides a critical bioenergetic and metabolic support system that contributes to multidrug resistance and tumor persistence under extreme physiological and therapeutic conditions.
More broadly, the ability of cancer cells to rely on stromal mitochondrial support highlights a fundamental shift in our understanding of tumor biology. Rather than functioning as isolated populations of genetically altered cells, tumors should be viewed as dynamic, multicellular ecosystems in which metabolic cooperation between cancer and stromal components drives progression, plasticity, and therapeutic resistance.
11. Future Directions
Since TNTs are minimally expressed under physiological conditions but markedly induced under stress, represent a highly specific and attractive therapeutic target for pharmacological intervention [21]. Future studies should further explore inhibitors of the Wnt/Ca²⁺ signalling pathway or CD38-targeted strategies, both of which have shown promise in reducing mitochondrial transfer and improving survival outcomes in preclinical models [22].
In parallel, the development of robust biomarkers is essential for clinical translation. Circulating cell-free mtDNA and mitochondrial-derived vesicles in patient plasma hold significant potential as non-invasive indicators of metabolic reprogramming and therapy resistance, enabling real-time monitoring of tumor adaptation [28].
To better understand the dynamics of mitochondrial transfer in vivo, there is a critical need for advanced imaging approaches. High-resolution techniques combined with density gradient-based analysis and computational platforms such as MERCI could allow precise quantification of the directionality, frequency, and extent of mitochondrial exchange in human tumors, moving beyond the limitations of conventional 2D and 3D co-culture systems [24].
Finally, while current research has largely focused on tumor cells acquiring functional mitochondria, future investigations should also address bidirectional mitochondrial exchange. In particular, the transfer of dysfunctional mitochondria from cancer cells to immune cells, such as T cells, may contribute to immune suppression and cellular senescence. Understanding this process could reveal new therapeutic opportunities, especially in combination with immunotherapy strategies [24,25].
Author Contributions
Conceptualization and study design S.K., P.A., Literature review and data curation: P.A., S.T., P.M., Manuscript writing-original draft, P.A. Manuscript writing-sectional contributions: S.T., P.M., Figure preparation, P.A., P.J., Manuscript editing, S.K., S.K.Y., H.R., N.K., N.A., Supervisor & Correspondence, S.K., S.K.Y.,.
Acknowledgments
The authors thank Guru Ghasidas Vishwavidyalaya for providing computational facilities and research support. Figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin 2024; 74: 12-49. [CrossRef]
- Chu X, Tian W, Ning J, Xiao G, Zhou Y, Wang Z, Zhai Z, Tanzhu G, Yang J, Zhou R. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal Transduct Target Ther 2024; 9: 170. [CrossRef]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730-737. [CrossRef]
- Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol 2007; 18: 460-466. [CrossRef]
- Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017; 23: 1124-1134. [CrossRef]
- Mathan SV, Singh RP. Cancer Stem Cells Connecting to Immunotherapy: Key Insights, Challenges, and Potential Treatment Opportunities. Cancers (Basel) 2025; 17. [CrossRef]
- Xie Y, Ma S, Tong M. Metabolic Plasticity of Cancer Stem Cells in Response to Microenvironmental Cues. Cancers (Basel) 2022; 14. [CrossRef]
- Selvaraj NR, Nandan D, Nair BG, Nair VA, Venugopal P, Aradhya R. Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells 2025; 14. [CrossRef]
- Nakano T, Irie K, Matsuo K, Mishima K, Nakamura Y. Molecular and cellular mechanisms of mitochondria transfer in models of central nervous system disease. J Cereb Blood Flow Metab 2026; 46: 269-288. [CrossRef]
- Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A, Tan AS, Kluckova K, Judasova K, Zamecnikova K, Rychtarcikova Z, Gopalan V, Andera L, Sobol M, Yan B, Pattnaik B, Bhatraju N, Truksa J, Stopka P, Hozak P, Lam AK, Sedlacek R, Oliveira PJ, Kubista M, Agrawal A, Dvorakova-Hortova K, Rohlena J, Berridge MV, Neuzil J. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017; 6. [CrossRef]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015; 21: 81-94. [CrossRef]
- Paul R, Dorsey JF, Fan Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol Ther 2022; 231: 107985. [CrossRef]
- Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther 2023; 8: 333. [CrossRef]
- Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 2020; 11: 102. [CrossRef]
- O’Malley J, Kumar R, Inigo J, Yadava N, Chandra D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020; 6: 688-701.
- Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer 2016; 114: 1305-1312. [CrossRef]
- Bai F, Liu S, Liu X, Hollern DP, Scott A, Wang C, Zhang L, Fan C, Fu L, Perou CM, Zhu WG, Pei XH. PDGFRbeta is an essential therapeutic target for BRCA1-deficient mammary tumors. Breast Cancer Res 2021; 23: 10. [CrossRef]
- Jin X, Zhang N, Yan T, Wei J, Hao L, Sun C, Zhao H, Jiang S. Lactate-mediated metabolic reprogramming of tumor-associated macrophages: implications for tumor progression and therapeutic potential. Front Immunol 2025; 16: 1573039. [CrossRef]
- Sriramkumar S, Sood R, Huntington TD, Ghobashi AH, Vuong TT, Metcalfe TX, Wang W, Nephew KP, O’Hagan HM. Platinum-induced mitochondrial OXPHOS contributes to cancer stem cell enrichment in ovarian cancer. J Transl Med 2022; 20: 246. [CrossRef]
- Lou E, Zhai E, Sarkari A, Desir S, Wong P, Iizuka Y, Yang J, Subramanian S, McCarthy J, Bazzaro M, Steer CJ. Cellular and Molecular Networking Within the Ecosystem of Cancer Cell Communication via Tunneling Nanotubes. Front Cell Dev Biol 2018; 6: 95. [CrossRef]
- Valdebenito S, Malik S, Luu R, Loudig O, Mitchell M, Okafo G, Bhat K, Prideaux B, Eugenin EA. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci Rep 2021; 11: 14556. [CrossRef]
- Raghavan A, Rao P, Neuzil J, Pountney DL, Nath S. Oxidative stress and Rho GTPases in the biogenesis of tunnelling nanotubes: implications in disease and therapy. Cell Mol Life Sci 2021; 79: 36. [CrossRef]
- Piwocka O, Piotrowski I, Suchorska WM, Kulcenty K. Dynamic interactions in the tumor niche: how the cross-talk between CAFs and the tumor microenvironment impacts resistance to therapy. Front Mol Biosci 2024; 11: 1343523. [CrossRef]
- Chun S, An J, Kim MS. Mitochondrial Transfer Between Cancer and T Cells: Implications for Immune Evasion. Antioxidants (Basel) 2025; 14. [CrossRef]
- Guan F, Wu X, Zhou J, Lin Y, He Y, Fan C, Zeng Z, Xiong W. Mitochondrial transfer in tunneling nanotubes-a new target for cancer therapy. J Exp Clin Cancer Res 2024; 43: 147. [CrossRef]
- Jin P, Jiang J, Zhou L, Huang Z, Nice EC, Huang C, Fu L. Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management. J Hematol Oncol 2022; 15: 97. [CrossRef]
- Dong LF, Rohlena J, Zobalova R, Nahacka Z, Rodriguez AM, Berridge MV, Neuzil J. Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J Cell Biol 2023; 222. [CrossRef]
- Goncalves AC, Richiardone E, Jorge J, Polonia B, Xavier CPR, Salaroglio IC, Riganti C, Vasconcelos MH, Corbet C, Sarmento-Ribeiro AB. Impact of cancer metabolism on therapy resistance - Clinical implications. Drug Resist Updat 2021; 59: 100797. [CrossRef]
- Sheehan C, Muir A. The requirement for mitochondrial respiration in cancer varies with disease stage. PLoS Biol 2022; 20: e3001800. [CrossRef]
- Neuzil J, Berridge MV. Mitochondria break through cellular boundaries. Aging (Albany NY) 2019; 11: 4308-4309. [CrossRef]
- Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther 2021; 6: 65. [CrossRef]
- Zhang H, Menzies KJ, Auwerx J. The role of mitochondria in stem cell fate and aging. Development 2018; 145. [CrossRef]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science 2004; 303: 1007-1010. [CrossRef]
- Qin Y, Jiang X, Yang Q, Zhao J, Zhou Q, Zhou Y. The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Front Oncol 2021; 11: 672781. [CrossRef]
- Cangkrama M, Liu H, Wu X, Yates J, Whipman J, Gabelein CG, Matsushita M, Ferrarese L, Sander S, Castro-Giner F, Asawa S, Sznurkowska MK, Kopf M, Dengjel J, Boeva V, Aceto N, Vorholt JA, Werner S. MIRO2-mediated mitochondrial transfer from cancer cells induces cancer-associated fibroblast differentiation. Nat Cancer 2025; 6: 1714-1733. [CrossRef]
- Watson DC, Bayik D, Storevik S, Moreino SS, Sprowls SA, Han J, Augustsson MT, Lauko A, Sravya P, Rosland GV, Troike K, Tronstad KJ, Wang S, Sarnow K, Kay K, Lunavat TR, Silver DJ, Dayal S, Joseph JV, Mulkearns-Hubert E, Ystaas LAR, Deshpande G, Guyon J, Zhou Y, Magaut CR, Seder J, Neises L, Williford SE, Meiser J, Scott AJ, Sajjakulnukit P, Mears JA, Bjerkvig R, Chakraborty A, Daubon T, Cheng F, Lyssiotis CA, Wahl DR, Hjelmeland AB, Hossain JA, Miletic H, Lathia JD. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat Cancer 2023; 4: 648-664. [CrossRef]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 2014; 33: 994-1010. [CrossRef]
- Frisbie L, Pressimone C, Dyer E, Baruwal R, Garcia G, St Croix C, Watkins S, Calderone M, Gorecki G, Javed Z, Atiya HI, Hempel N, Pearson A, Coffman LG. Carcinoma-associated mesenchymal stem cells promote ovarian cancer heterogeneity and metastasis through mitochondrial transfer. Cell Rep 2024; 43: 114551. [CrossRef]
- Wang C, Xie C. Unveiling the power of mitochondrial transfer in cancer progression: a perspective in ovarian cancer. J Ovarian Res 2024; 17: 233. [CrossRef]
- Rickard BP, Overchuk M, Obaid G, Ruhi MK, Demirci U, Fenton SE, Santos JH, Kessel D, Rizvi I. Photochemical Targeting of Mitochondria to Overcome Chemoresistance in Ovarian Cancer (dagger). Photochem Photobiol 2023; 99: 448-468. [CrossRef]
- Guo X, Can C, Liu W, Wei Y, Yang X, Liu J, Jia H, Jia W, Wu H, Ma D. Mitochondrial transfer in hematological malignancies. Biomark Res 2023; 11: 89. [CrossRef]
- Lu J, Zheng X, Li F, Yu Y, Chen Z, Liu Z, Wang Z, Xu H, Yang W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017; 8: 15539-15552. [CrossRef]
- Ikeda H, Kawase K, Nishi T, Watanabe T, Takenaga K, Inozume T, Ishino T, Aki S, Lin J, Kawashima S, Nagasaki J, Ueda Y, Suzuki S, Makinoshima H, Itami M, Nakamura Y, Tatsumi Y, Suenaga Y, Morinaga T, Honobe-Tabuchi A, Ohnuma T, Kawamura T, Umeda Y, Nakamura Y, Kiniwa Y, Ichihara E, Hayashi H, Ikeda JI, Hanazawa T, Toyooka S, Mano H, Suzuki T, Osawa T, Kawazu M, Togashi Y. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 2025; 638: 225-236. [CrossRef]
- Ahmadian S, Lindsey PJ, Smeets HJM, van Tienen FHJ, van Zandvoort M. Spinning Disk Confocal Microscopy for Optimized and Quantified Live Imaging of 3D Mitochondrial Network. Int J Mol Sci 2024; 25. [CrossRef]
- Lambert C, Schmidt K, Karger M, Stadler M, Stradal TEB, Rottner K. Cytochalasans and Their Impact on Actin Filament Remodeling. Biomolecules 2023; 13. [CrossRef]
- Novak J, Nahacka Z, Oliveira GL, Brisudova P, Dubisova M, Dvorakova S, Miklovicova S, Dalecka M, Puttrich V, Grycova L, Magalhaes-Novais S, Correia CM, Levoux J, Stepanek L, Prochazka J, Svec D, Reguera DP, Lopez-Domenech G, Zobalova R, Sedlacek R, Terp MG, Gammage PA, Lansky Z, Kittler J, Oliveira PJ, Ditzel HJ, Berridge MV, Rodriguez AM, Boukalova S, Rohlena J, Neuzil J. The adaptor protein Miro1 modulates horizontal transfer of mitochondria in mouse melanoma models. Cell Rep 2025; 44: 115154. [CrossRef]
- Allelein S, Medina-Perez P, Lopes ALH, Rau S, Hause G, Kolsch A, Kuhlmeier D. Potential and challenges of specifically isolating extracellular vesicles from heterogeneous populations. Sci Rep 2021; 11: 11585. [CrossRef]
- Jaiswal R, Sedger LM. Intercellular Vesicular Transfer by Exosomes, Microparticles and Oncosomes - Implications for Cancer Biology and Treatments. Front Oncol 2019; 9: 125. [CrossRef]
- Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 2015; 40: 41-51. [CrossRef]
- Majood M, Rawat S, Mohanty S. Delineating the role of extracellular vesicles in cancer metastasis: A comprehensive review. Front Immunol 2022; 13: 966661. [CrossRef]
- Maas SLN, Breakefield XO, Weaver AM. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol 2017; 27: 172-188. [CrossRef]
- Wang Y, Yu HY, Yi ZJ, Qi LY, Yang JS, Xie HX, Zhao M, Liu NH, Chen JQ, Zhou TJ, Xing L, Cheng XW, Jiang HL. Super mitochondria-enriched extracellular vesicles enable enhanced mitochondria transfer. Nat Commun 2025; 16: 9448. [CrossRef]
- Xia K, Zhang S, Peng H, Chen H, Yang C, Yu J, Luo P, Lu Q, Chen H, Huang L, Xiong Y, Zhao L, Jia L, Li L, Qiu Y, Guo Y, Liu C, Fan H, Dai Z, Liu G, Ke Q, Wang T, Li W, Chen L, Deng C, Xiao H, Xiang AP. An extracellular vesicle-mediated mitochondrial transfer network critical for testosterone synthesis. Nat Cell Biol 2026. [CrossRef]
- Zhou L, Zhang W, Hu X, Wang D, Tang D. Metabolic Reprogramming of Cancer-Associated Fibroblast in the Tumor Microenvironment: From Basics to Clinic. Clin Med Insights Oncol 2024; 18: 11795549241287058. [CrossRef]
- Rahmati S, Moeinafshar A, Rezaei N. The multifaceted role of extracellular vesicles (EVs) in colorectal cancer: metastasis, immune suppression, therapy resistance, and autophagy crosstalk. J Transl Med 2024; 22: 452. [CrossRef]
- Xiong L, Wei Y, Jia Q, Chen J, Chen T, Yuan J, Pi C, Liu H, Tang J, Yin S, Zuo Y, Zhang X, Liu F, Yang H, Zhao L. The application of extracellular vesicles in colorectal cancer metastasis and drug resistance: recent advances and trends. J Nanobiotechnology 2023; 21: 143. [CrossRef]
- Chen Y, Kleeff J, Sunami Y. Pancreatic cancer cell- and cancer-associated fibroblast-derived exosomes in disease progression, metastasis, and therapy. Discov Oncol 2024; 15: 253. [CrossRef]
- Zhou P, Du X, Jia W, Feng K, Zhang Y. Engineered extracellular vesicles for targeted reprogramming of cancer-associated fibroblasts to potentiate therapy of pancreatic cancer. Signal Transduct Target Ther 2024; 9: 151. [CrossRef]
- Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer 2016; 16: 775-788. [CrossRef]
- Ruch R. Gap Junctions and Connexins in Cancer Formation, Progression, and Therapy. Cancers (Basel) 2020; 12. [CrossRef]
- Fu H, Xie X, Zhai L, Liu Y, Tang Y, He S, Li J, Xiao Q, Xu G, Yang Z, Zhang X, Liu Y. CX43-mediated mitochondrial transfer maintains stemness of KG-1a leukemia stem cells through metabolic remodeling. Stem Cell Res Ther 2024; 15: 460. [CrossRef]
- Singh AK, Prasad P, Cancelas JA. Mesenchymal stromal cells, metabolism, and mitochondrial transfer in bone marrow normal and malignant hematopoiesis. Front Cell Dev Biol 2023; 11: 1325291. [CrossRef]
- Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol 2009; 1: a002576. [CrossRef]
- Kidder GM, Vanderhyden BC. Bidirectional communication between oocytes and follicle cells: ensuring oocyte developmental competence. Can J Physiol Pharmacol 2010; 88: 399-413. [CrossRef]
- Rapani A, Nikiforaki D, Karagkouni D, Sfakianoudis K, Tsioulou P, Grigoriadis S, Maziotis E, Pantou A, Voutsina A, Pantou A, Koutsilieris M, Hatzigeorgiou A, Pantos K, Simopoulou M. Reporting on the Role of miRNAs and Affected Pathways on the Molecular Backbone of Ovarian Insufficiency: A Systematic Review and Critical Analysis Mapping of Future Research. Front Cell Dev Biol 2020; 8: 590106. [CrossRef]
- Luo M, Luo Y, Mao N, Huang G, Teng C, Wang H, Wu J, Liao X, Yang J. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell Physiol Biochem 2018; 51: 315-336. [CrossRef]
- Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys 2001; 34: 325-472. [CrossRef]
- Su J, Song Y, Zhu Z, Huang X, Fan J, Qiao J, Mao F. Cell-cell communication: new insights and clinical implications. Signal Transduct Target Ther 2024; 9: 196. [CrossRef]
- Zhou X, Merchak K, Lee W, Grande JP, Cascalho M, Platt JL. Cell Fusion Connects Oncogenesis with Tumor Evolution. Am J Pathol 2015; 185: 2049-2060. [CrossRef]
- Dittmar T, Hass R. Intrinsic signalling factors associated with cancer cell-cell fusion. Cell Commun Signal 2023; 21: 68. [CrossRef]
- Lu X, Kang Y. Cell fusion as a hidden force in tumor progression. Cancer Res 2009; 69: 8536-8539. [CrossRef]
- Augimeri G, Gonzalez ME, Paoli A, Eido A, Choi Y, Burman B, Djomehri S, Karthikeyan SK, Varambally S, Buschhaus JM, Chen YC, Mauro L, Bonofiglio D, Nesvizhskii AI, Luker GD, Ando S, Yoon E, Kleer CG. A hybrid breast cancer/mesenchymal stem cell population enhances chemoresistance and metastasis. JCI Insight 2023; 8. [CrossRef]
- Chen YS, Chen ZP. Vasculogenic mimicry: a novel target for glioma therapy. Chin J Cancer 2014; 33: 74-79. [CrossRef]
- Mao JM, Liu J, Guo G, Mao XG, Li CX. Glioblastoma vasculogenic mimicry: signaling pathways progression and potential anti-angiogenesis targets. Biomark Res 2015; 3: 8. [CrossRef]
- Sun C, Dai X, Zhao D, Wang H, Rong X, Huang Q, Lan Q. Mesenchymal stem cells promote glioma neovascularization in vivo by fusing with cancer stem cells. BMC Cancer 2019; 19: 1240. [CrossRef]
- Mohammadalipour A, Dumbali SP, Wenzel PL. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front Cell Dev Biol 2020; 8: 603292. [CrossRef]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010; 327: 1650-1653. [CrossRef]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab 2016; 23: 27-47. [CrossRef]
- Hoover G, Gilbert S, Curley O, Obellianne C, Lin MT, Hixson W, Pierce TW, Andrews JF, Alexeyev MF, Ding Y, Bu P, Behbod F, Medina D, Chang JT, Ayala G, Grelet S. Nerve-to-cancer transfer of mitochondria during cancer metastasis. Nature 2025; 644: 252-262. [CrossRef]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014; 514: 628-632. [CrossRef]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008; 320: 661-664. [CrossRef]
- Shibata T, Hoshida Y. Mitochondrial genome diversity drives heterogeneity in HCC. Hepatology 2025; 82: 12-13. [CrossRef]
- Welch DR, Foster C, Rigoutsos I. Roles of mitochondrial genetics in cancer metastasis. Trends Cancer 2022; 8: 1002-1018. [CrossRef]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015; 11: 9-15. [CrossRef]
- Guan B, Liu Y, Xie B, Zhao S, Yalikun A, Chen W, Zhou M, Gu Q, Yan D. Mitochondrial genome transfer drives metabolic reprogramming in adjacent colonic epithelial cells promoting TGFbeta1-mediated tumor progression. Nat Commun 2024; 15: 3653. [CrossRef]
- Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, Bhat VB, Bhowmik SK, Gupta V, Arora K, Wu D, Tsouko E, Zhang Y, Maity S, Donti TR, Graham BH, Frigo DE, Coarfa C, Yotnda P, Putluri N, Sreekumar A, Lewis MT, Creighton CJ, Wong LC, Kaipparettu BA. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep 2016; 14: 2154-2165. [CrossRef]
- Woytash JA, Lefebvre A, Zhang Z, Xu B, Harchenko SA, Le HT, McColloch AR, Shi X, Digman MA, Razorenova OV. CDCP1/mitochondrial Src axis increases electron transport chain function to promote metastasis in triple-negative breast cancer. Br J Cancer 2025; 133: 1265-1277. [CrossRef]
- Liu X, Si W, He L, Yang J, Peng Y, Ren J, Liu X, Jin T, Yu H, Zhang Z, Cheng X, Zhang W, Xia L, Huang Y, Wang Y, Liu S, Shan L, Zhang Y, Yang X, Li H, Liang J, Sun L, Shang Y. The existence of a nonclassical TCA cycle in the nucleus that wires the metabolic-epigenetic circuitry. Signal Transduct Target Ther 2021; 6: 375. [CrossRef]
- English J, Son JM, Cardamone MD, Lee C, Perissi V. Decoding the rosetta stone of mitonuclear communication. Pharmacol Res 2020; 161: 105161. [CrossRef]
- Jo C, Park S, Oh S, Choi J, Kim EK, Youn HD, Cho EJ. Histone acylation marks respond to metabolic perturbations and enable cellular adaptation. Exp Mol Med 2020; 52: 2005-2019. [CrossRef]
- Ambrosini G, Cordani M, Zarrabi A, Alcon-Rodriguez S, Sainz RM, Velasco G, Gonzalez-Menendez P, Dando I. Transcending frontiers in prostate cancer: the role of oncometabolites on epigenetic regulation, CSCs, and tumor microenvironment to identify new therapeutic strategies. Cell Commun Signal 2024; 22: 36. [CrossRef]
- Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022; 13: 877-919. [CrossRef]
- Haws SA, Leech CM, Denu JM. Metabolism and the Epigenome: A Dynamic Relationship. Trends Biochem Sci 2020; 45: 731-747. [CrossRef]
- Bernasocchi T, Mostoslavsky R. Subcellular one carbon metabolism in cancer, aging and epigenetics. Front Epigenet Epigenom 2024; 2. [CrossRef]
- Russo M, Pileri F, Ghisletti S. Novel insights into the role of acetyl-CoA producing enzymes in epigenetic regulation. Front Endocrinol (Lausanne) 2023; 14: 1272646. [CrossRef]
- Ottens F, Franz A, Hoppe T. Build-UPS and break-downs: metabolism impacts on proteostasis and aging. Cell Death Differ 2021; 28: 505-521. [CrossRef]
- Leung J, Gaudin V. Who Rules the Cell? An Epi-Tale of Histone, DNA, RNA, and the Metabolic Deep State. Front Plant Sci 2020; 11: 181. [CrossRef]
- Anselme M, He H, Lai C, Luo W, Zhong S. Targeting mitochondrial transporters and metabolic reprogramming for disease treatment. J Transl Med 2025; 23: 1111. [CrossRef]
- Sarkar S, Chang CI, Jean J, Wu MJ. TCA cycle-derived oncometabolites in cancer and the immune microenvironment. J Biomed Sci 2025; 32: 87. [CrossRef]
- Kim S, Ramalho TR, Haynes CM. Regulation of proteostasis and innate immunity via mitochondria-nuclear communication. J Cell Biol 2024; 223. [CrossRef]
- Xu S, Zhang X, Liu C, Liu Q, Chai H, Luo Y, Li S. Role of Mitochondria in Neurodegenerative Diseases: From an Epigenetic Perspective. Front Cell Dev Biol 2021; 9: 688789. [CrossRef]
- Kincaid JW, Berger NA. NAD metabolism in aging and cancer. Exp Biol Med (Maywood) 2020; 245: 1594-1614. [CrossRef]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501: 328-337. [CrossRef]
- Zhang X, Powell K, Li L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers (Basel) 2020; 12. [CrossRef]
- Fenu G, Grinan-Lison C, Etzi F, Gonzalez-Titos A, Pisano A, Toledo B, Farace C, Sabalic A, Carrillo E, Marchal JA, Madeddu R. Functional Characterization of miR-216a-5p and miR-125a-5p on Pancreatic Cancer Stem Cells. Int J Mol Sci 2025; 26. [CrossRef]
- Zhou Y, Xia L, Wang H, Oyang L, Su M, Liu Q, Lin J, Tan S, Tian Y, Liao Q, Cao D. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018; 9: 33403-33415. [CrossRef]
- Prabavathy D, Swarnalatha Y, Ramadoss N. Lung cancer stem cells-origin, characteristics and therapy. Stem Cell Investig 2018; 5: 6. [CrossRef]
- Patel S, Shah K, Mirza S, Shah K, Rawal R. Circulating tumor stem like cells in oral squamous cell carcinoma: An unresolved paradox. Oral Oncol 2016; 62: 139-146. [CrossRef]
- Feitelson MA, Arzumanyan A, Kulathinal RJ, Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML, Nawroth R, Sanchez-Garcia I, Sharma D, Saxena NK, Singh N, Vlachostergios PJ, Guo S, Honoki K, Fujii H, Georgakilas AG, Bilsland A, Amedei A, Niccolai E, Amin A, Ashraf SS, Boosani CS, Guha G, Ciriolo MR, Aquilano K, Chen S, Mohammed SI, Azmi AS, Bhakta D, Halicka D, Keith WN, Nowsheen S. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin Cancer Biol 2015; 35 Suppl: S25-S54. [CrossRef]
- Galassi C, Manic G, Esteller M, Galluzzi L, Vitale I. Epigenetic regulation of cancer stemness. Signal Transduct Target Ther 2025; 10: 243. [CrossRef]
- French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer 2021; 148: 2884-2897. [CrossRef]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871-890. [CrossRef]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell 2016; 166: 21-45. [CrossRef]
- Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 2016; 7: 13041. [CrossRef]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 2013; 11: 94. [CrossRef]
- Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 2013; 104: 2077-2088. [CrossRef]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438: 820-827. [CrossRef]
- Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, Sloan EK, Parker BS, Bowtell DD, Smyth MJ, Moller A. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res 2012; 72: 3906-3911. [CrossRef]
- Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883-892. [CrossRef]
- Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 2021; 27: 212-224. [CrossRef]
- Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, Bolli N, Behjati S, Tarpey PS, Nangalia J, Massie CE, Butler AP, Teague JW, Vassiliou GS, Green AR, Du MQ, Unnikrishnan A, Pimanda JE, Teh BT, Munshi N, Greaves M, Vyas P, El-Naggar AK, Santarius T, Collins VP, Grundy R, Taylor JA, Hayes DN, Malkin D, Group IBC, Group ICMD, Group IPC, Foster CS, Warren AY, Whitaker HC, Brewer D, Eeles R, Cooper C, Neal D, Visakorpi T, Isaacs WB, Bova GS, Flanagan AM, Futreal PA, Lynch AG, Chinnery PF, McDermott U, Stratton MR, Campbell PJ. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014; 3. [CrossRef]
- Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, Yang Y, Martincorena I, Creighton CJ, Weinstein JN, Xu Y, Han L, Kim HL, Nakagawa H, Park K, Campbell PJ, Liang H, Consortium P. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet 2020; 52: 342-352. [CrossRef]
- A FCL. Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes. Clin Epigenetics 2020; 12: 182. [CrossRef]
- Roehlecke C, Schmidt MHH. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers (Basel) 2020; 12. [CrossRef]
Figure 1.
Factors driving intracellular mitochondrial transfer. This figure illustrates the multifaceted drivers of intercellular mitochondrial transfer within the TME. Factors such as hypoxia, therapeutic interventions like chemotherapy and radiotherapy, and oxidative stress act as potent triggers, inducing structural adaptations like TNTs and the release of EVs. These mechanisms facilitate the transfer of mitochondria from donor cells to metabolically compromised recipient tumor cells. This transfer restores OXPHOS, reinforces redox homeostasis, and promotes metabolic flexibility, collectively enhancing CSC plasticity, therapy resistance, and tumor progression.
Figure 1.
Factors driving intracellular mitochondrial transfer. This figure illustrates the multifaceted drivers of intercellular mitochondrial transfer within the TME. Factors such as hypoxia, therapeutic interventions like chemotherapy and radiotherapy, and oxidative stress act as potent triggers, inducing structural adaptations like TNTs and the release of EVs. These mechanisms facilitate the transfer of mitochondria from donor cells to metabolically compromised recipient tumor cells. This transfer restores OXPHOS, reinforces redox homeostasis, and promotes metabolic flexibility, collectively enhancing CSC plasticity, therapy resistance, and tumor progression.

Figure 2.
Routes of intercellular mitochondrial transfer. (A) TNT-mediated transfer: Intercellular communication via actin-based TNTs, where motor proteins (kinesin/dynein) and Miro1 adapter proteins facilitate the active transport of mitochondria between cells. (B) EVs: Dissemination of mitochondrial fragments, mtDNA, or whole organelles through exosomes and microvesicles, which are internalized by recipient cells via endocytosis or membrane fusion. (C) GJs: Direct organelle transfer facilitated by connexin 43 (CX43) channels or membrane engulfment, triggering the restoration of oxidative phosphorylation (OXPHOS) in recipient cells. (D) Cell fusion: The merging of two distinct cells to create a hybrid cell with a mixed mitochondrial population, leading to rapid metabolic and phenotypic reprogramming.
Figure 2.
Routes of intercellular mitochondrial transfer. (A) TNT-mediated transfer: Intercellular communication via actin-based TNTs, where motor proteins (kinesin/dynein) and Miro1 adapter proteins facilitate the active transport of mitochondria between cells. (B) EVs: Dissemination of mitochondrial fragments, mtDNA, or whole organelles through exosomes and microvesicles, which are internalized by recipient cells via endocytosis or membrane fusion. (C) GJs: Direct organelle transfer facilitated by connexin 43 (CX43) channels or membrane engulfment, triggering the restoration of oxidative phosphorylation (OXPHOS) in recipient cells. (D) Cell fusion: The merging of two distinct cells to create a hybrid cell with a mixed mitochondrial population, leading to rapid metabolic and phenotypic reprogramming.

Figure 3.
Tumor microenvironment-driven mitochondrial transfer induces metabolic reprogramming and cancer stem cell plasticity. Stressed tumor cells exhibit reduced ATP, elevated ROS, and glycolysis dependence (left). Mitochondrial transfer restores OXPHOS, increases ATP production, and rebalances redox status (right). This metabolic shift activates key signaling pathways, including HIF-1α, STAT3, NF-κB, and Wnt. Enhanced fatty acid oxidation (FAO) further promotes Src signaling, linking metabolism to invasive behavior. Collectively, these changes drive cancer stem cell-associated outcomes, including self-renewal, EMT–MET plasticity, tumorsphere formation, and therapy resistance.
Figure 3.
Tumor microenvironment-driven mitochondrial transfer induces metabolic reprogramming and cancer stem cell plasticity. Stressed tumor cells exhibit reduced ATP, elevated ROS, and glycolysis dependence (left). Mitochondrial transfer restores OXPHOS, increases ATP production, and rebalances redox status (right). This metabolic shift activates key signaling pathways, including HIF-1α, STAT3, NF-κB, and Wnt. Enhanced fatty acid oxidation (FAO) further promotes Src signaling, linking metabolism to invasive behavior. Collectively, these changes drive cancer stem cell-associated outcomes, including self-renewal, EMT–MET plasticity, tumorsphere formation, and therapy resistance.

Figure 4.
Mitochondrial retrograde signaling drives epigenetic reprogramming and cancer stem cell phenotypes. Mitochondrial dysfunction and metabolic flux influence chromatin remodeling through retrograde signaling, promoting CSC traits. Acetyl-CoA derived from mitochondrial metabolism enhances histone acetylation via HATs, supporting EMT and stemness. TCA cycle disruption generates oncometabolites (e.g., 2-HG, succinate, fumarate) that inhibit α-KG–dependent demethylases, leading to DNA and histone hypermethylation. One-carbon metabolism produces SAM, driving DNMT- and HMT-mediated methylation and chromatin remodeling. NAD⁺-dependent sirtuins regulate histone deacetylation and metabolic homeostasis. Collectively, these pathways induce a stem-like, therapy-resistant, and plastic tumor state.
Figure 4.
Mitochondrial retrograde signaling drives epigenetic reprogramming and cancer stem cell phenotypes. Mitochondrial dysfunction and metabolic flux influence chromatin remodeling through retrograde signaling, promoting CSC traits. Acetyl-CoA derived from mitochondrial metabolism enhances histone acetylation via HATs, supporting EMT and stemness. TCA cycle disruption generates oncometabolites (e.g., 2-HG, succinate, fumarate) that inhibit α-KG–dependent demethylases, leading to DNA and histone hypermethylation. One-carbon metabolism produces SAM, driving DNMT- and HMT-mediated methylation and chromatin remodeling. NAD⁺-dependent sirtuins regulate histone deacetylation and metabolic homeostasis. Collectively, these pathways induce a stem-like, therapy-resistant, and plastic tumor state.

Figure 5.
This figure illustrates therapeutic interventions designed to disrupt intercellular mitochondrial trafficking, a mechanism critical for sustaining cancer cell survival and therapy resistance. The strategies are organized into four key domains: (A) Targeting transfer mechanisms, where inhibitors of actin polymerization (e.g., cytochalasin-based compounds), Miro1/TRAK-mediated organelle transport, and connexin-GJs effectively block the physical routes such as TNTs, EVs, and GJ used for mitochondrial transfer. (B) Targeting metabolism and bioenergetics, which seeks to neutralize the energy advantage conferred by transferred mitochondria by inhibiting OXPHOS, fatty acid oxidation (FAO), and exploiting redox signaling dependencies. (C) Targeting mitochondrial-epigenetic crosstalk, which disrupts the link between mitochondrial-derived metabolites such as Acetyl-CoA, NAD+, and α-KG and nuclear epigenetic programming that maintains stemness. (D) Targeting the TME, which employs HIF-1⍺ inhibition and immune modulation to break the pro-tumorigenic support provided by stromal cells, collectively preventing CSC plasticity and disease recurrence.
Figure 5.
This figure illustrates therapeutic interventions designed to disrupt intercellular mitochondrial trafficking, a mechanism critical for sustaining cancer cell survival and therapy resistance. The strategies are organized into four key domains: (A) Targeting transfer mechanisms, where inhibitors of actin polymerization (e.g., cytochalasin-based compounds), Miro1/TRAK-mediated organelle transport, and connexin-GJs effectively block the physical routes such as TNTs, EVs, and GJ used for mitochondrial transfer. (B) Targeting metabolism and bioenergetics, which seeks to neutralize the energy advantage conferred by transferred mitochondria by inhibiting OXPHOS, fatty acid oxidation (FAO), and exploiting redox signaling dependencies. (C) Targeting mitochondrial-epigenetic crosstalk, which disrupts the link between mitochondrial-derived metabolites such as Acetyl-CoA, NAD+, and α-KG and nuclear epigenetic programming that maintains stemness. (D) Targeting the TME, which employs HIF-1⍺ inhibition and immune modulation to break the pro-tumorigenic support provided by stromal cells, collectively preventing CSC plasticity and disease recurrence.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



