Submitted:
01 May 2026
Posted:
06 May 2026
You are already at the latest version
Abstract
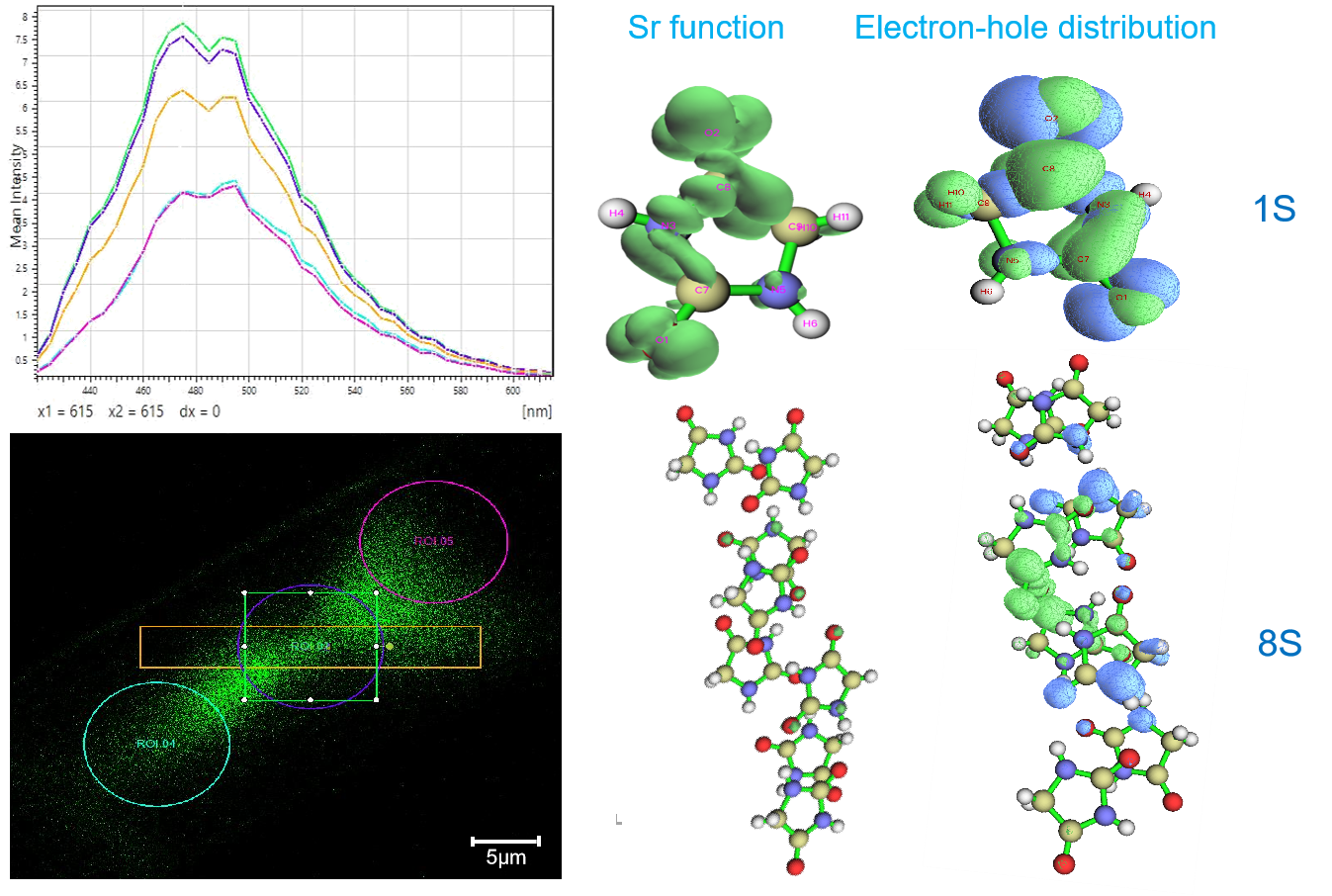
In this study, hydantoin (C₃H₄N₂O₂) was selected to investigate the photoluminescence mechanism of non-typical luminescent compounds. The emission spectra of single crystals were examined using a laser confocal microscope. Within the same crystal, the peak shape and position were consistent across different regions, while the intensity varied; this phenomenon is attributed to confinement-induced emission. For different crystal blocks, variations in molecular packing modes led to changes in both peak shape and position. Combined with theoretical calculations and analyses, the results show that: as the molecular number increases, the energy gap decreases and the excitation wavelength increases (lower excitation energy); the hole-electron attraction energy, delocalization index, and overlap degree all decrease, with the hole delocalization index decreasing faster than that of the electron; the spin-orbit coupling coefficients for high-lying triplet states are more sensitive to the molecular count; and the intersystem crossing rate increases sharply with increasing energy level. In summary, the number and mode of molecular packing in the crystal influence the excited-state electronic structure and hole-electron interactions, thereby determining the luminescence behavior of non-typical luminescent compounds.
Keywords:
laser confocal microscopy
; spectroscopy
; hole-electron
; spin-orbit coupling
; rate
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



