Submitted:
29 April 2026
Posted:
02 May 2026
You are already at the latest version
Abstract
Quantitative structure–activity relationship (QSAR) modeling has traditionally relied on expert-designed molecular descriptors to encode chemical structures. DeepSnap is a descriptor-free QSAR approach that converts three-dimensional molecular structures into image representations and feeds them directly into convolutional neural networks for activity prediction. The method generates a conformer for each molecule, renders it as a color-coded molecular image, and captures omnidirectional snapshots from systematically varied viewing angles. This review traces DeepSnap from its introduction in 2018 to its current state. The method has been applied to 35 nuclear receptor endpoints from the Tox21 10K library (mean AUC 0.884), 59 molecular initiating event models spanning the full Tox21 target panel, rat hepatic clearance (ensemble AUC 0.943), and blood–brain barrier penetration (ensemble AUC 0.936). An ensemble strategy combining image-based and descriptor-based predictions has consistently outperformed either approach alone. The computational pipeline has evolved from a DIGITS/Caffe/Jmol system to a TensorFlow/Keras/PyMOL framework. Limitations include endpoint-dependent parameter sensitivity, class imbalance effects, the absence of direct comparisons with graph neural networks, and an interpretability gap addressed in part by CAM-family visualization in the AI-SHIPS platform and S-COPHY. Future directions include systematic application of explainable AI methods, automated hyperparameter optimization, and integration with graph-based approaches.
Keywords:
DeepSnap
; molecular image
; quantitative structure–activity relationship
; convolutional neural network
; transfer learning
; ensemble learning
; Tox21
; ADME prediction
; molecular informatics
; explainable artificial intelligence
1. Introduction
Quantitative structure–activity relationship (QSAR) modeling has long served as a cornerstone of computational chemistry—a topic of direct relevance to the molecular informatics community—enabling the prediction of biological activity, toxicity, and pharmacokinetic properties from molecular structure alone. In drug discovery and chemical safety assessment, QSAR provides a means to prioritize compounds for experimental testing, thereby reducing cost and animal use [1]. The Tox21 initiative, a multi-agency collaboration involving the U.S. Environmental Protection Agency, the National Institutes of Health, the National Center for Advancing Translational Sciences, and the Food and Drug Administration, accelerated this field by generating high-throughput screening data for thousands of environmental chemicals across dozens of biological targets [1,2,3]. Within the Tox21 framework, molecular initiation events (MIEs)—the initial interactions between chemicals and biological macromolecules—represent the entry points of adverse outcome pathways (AOPs), linking chemical exposure to toxicological effects at the cellular, organ, and organism levels [1,2,4]. Predicting which MIEs a given compound may trigger is therefore central to modern computational toxicology.
Traditional QSAR approaches rely on expert-designed molecular descriptors to encode structural information [5]. These descriptors range from simple physicochemical properties such as molecular weight and partition coefficient (logP) to more elaborate representations including topological indices, extended-connectivity fingerprints (ECFP) [6], and three-dimensional steric parameters such as Taft, Sterimol, and A-Value indices [2]. While these hand-crafted features have supported decades of productive modeling, they impose an intrinsic limitation: the quality and completeness of the resulting models depend on the choice of descriptors, which in turn depends on domain expertise and prior chemical knowledge. Descriptor selection introduces human bias, and it is unlikely that any finite set of precomputed features can exhaustively represent the diversity of molecular recognition events. The Tox21 Data Challenge 2014, a landmark competition in which teams from 18 countries predicted the activity of compounds across 12 MIE targets, demonstrated both the promise and the ceiling of conventional approaches: the overall winning team, DeepTox, employed deep neural networks trained on ECFP fingerprints and computed descriptors [7], yet black-box limitations and descriptor dependence remained inherent constraints.
The emergence of deep learning in the early 2010s opened new avenues for molecular property prediction by enabling models to learn representations directly from raw input data, potentially bypassing the feature-engineering bottleneck. Three principal strategies for encoding molecular information in deep learning frameworks have gained prominence. First, descriptor-based deep learning extends classical QSAR by feeding precomputed fingerprints or descriptor vectors into deep neural networks, as exemplified by DeepTox [7]. Second, graph neural networks (GNNs) represent molecules as graphs in which atoms are nodes and bonds are edges, allowing message-passing operations to learn atom-level features from molecular topology. The message passing neural network (MPNN) framework [8] and its directed variant, D-MPNN (Chemprop) [9], established graph-based methods as a leading paradigm, while attention-based architectures such as AttentiveFP introduced interpretable atom-level attention maps [10]. However, most GNN approaches operate on two-dimensional molecular graphs and do not explicitly encode three-dimensional conformational information. Third, image-based approaches feed visual representations of molecules into convolutional neural networks (CNNs) originally developed for image classification. Chemception applied an Inception-ResNet CNN to two-dimensional structural drawings of molecules, demonstrating that a deep network could extract chemically relevant features from images with minimal prior chemical knowledge [11]. More recently, ImageMol employed self-supervised pretraining on 10 million molecular images using a ResNet-18 backbone, achieving competitive performance across multiple drug discovery benchmarks [12]. These 2D image-based methods, however, discard all three-dimensional spatial information because they operate on flat structural drawings.
Against this backdrop, the DeepSnap method was introduced in 2018 as a descriptor-free QSAR approach that converts three-dimensional molecular structures into image-based representations [13]. Rather than computing molecular descriptors or constructing a molecular graph, DeepSnap generates a conformer of each compound, renders it as a colored molecular image, and captures multiple two-dimensional snapshot images from systematically varied viewing angles around the x-, y-, and z-axes. These images are then fed directly into a CNN—utilizing transfer learning from an ImageNet-pretrained model [2]—for property prediction. Because the images are generated from three-dimensional conformers and are captured from multiple orientations, DeepSnap encodes spatial information that is absent from two-dimensional image and graph representations; the original study noted that the method yields distinct predictions for optical isomers, though this property has not been systematically validated [13]. The original study applied DeepSnap with an AlexNet architecture to predict mitochondrial membrane potential disruption, one of the Tox21 endpoints, and achieved an area under the receiver operating characteristic curve (AUC) of 0.921 on external validation [13]. This result was obtained without any molecular descriptor calculation, relying solely on pixel-level features learned by the CNN from three-dimensional molecular images.
Since its introduction, DeepSnap has undergone substantial technical evolution and has been applied to a broad range of biological targets. The method has been extended from a single Tox21 endpoint to 35 nuclear receptor agonist and antagonist models [14] and 59 MIE models spanning the full Tox21 target panel [15]. Applications have expanded beyond toxicological endpoints to include pharmacokinetic parameters such as hepatic clearance [16] and blood–brain barrier permeability [17]. The computational pipeline has migrated from a DIGITS/Caffe/Jmol system to a TensorFlow/Keras/PyMOL framework [15], and an ensemble strategy combining image-based and descriptor-based predictions has demonstrated consistent improvements over either approach alone [16,17]. A subsequent study within the broader DeepSnap research line adapted the method for cosmetics safety assessment and reported gradient-weighted class activation mapping (Grad-CAM) visualizations of molecular features in the S-COPHY model [18].
This review provides a comprehensive account of the DeepSnap method from its conceptual origin to its current state. Section 2 describes the rationale and the first publication. Section 3 details the core rendering and learning pipeline, including the systematic optimization of imaging parameters and the transition between computational platforms. Section 4 and Section 5 survey applications to toxicological targets and ADME parameters, respectively. Section 6 traces the technical evolution of the method and its variants, including the single-image S-COPHY adaptation and ensemble strategies. Section 7 examines limitations and unresolved issues, including hyperparameter sensitivity, class imbalance, reproducibility concerns, and the interpretability gap. Section 8 positions DeepSnap relative to descriptor-based, graph-based, and other image-based approaches. Section 9 discusses future directions, including the integration of explainable AI methods and the expansion to additional pharmacological and toxicological endpoints. Throughout, we distinguish between empirical evidence from primary studies and interpretive claims from review articles, and we flag areas where the evidence base remains incomplete.
2. Origin and Rationale of DeepSnap
Quantitative structure–activity relationship (QSAR) modeling has traditionally relied on molecular descriptors—numerical values computed from chemical structures that encode physicochemical, topological, and electronic properties. While such descriptors have proven effective in many settings, their design requires expert knowledge and their expressiveness is bounded by the assumptions embedded in each descriptor definition. In parallel, circular fingerprints such as ECFP provide a substructure-based encoding that captures local connectivity patterns but largely discards three-dimensional (3D) spatial information. A fundamental question therefore arises: can deep learning extract predictive features directly from molecular representations without the intermediary of hand-engineered descriptors?
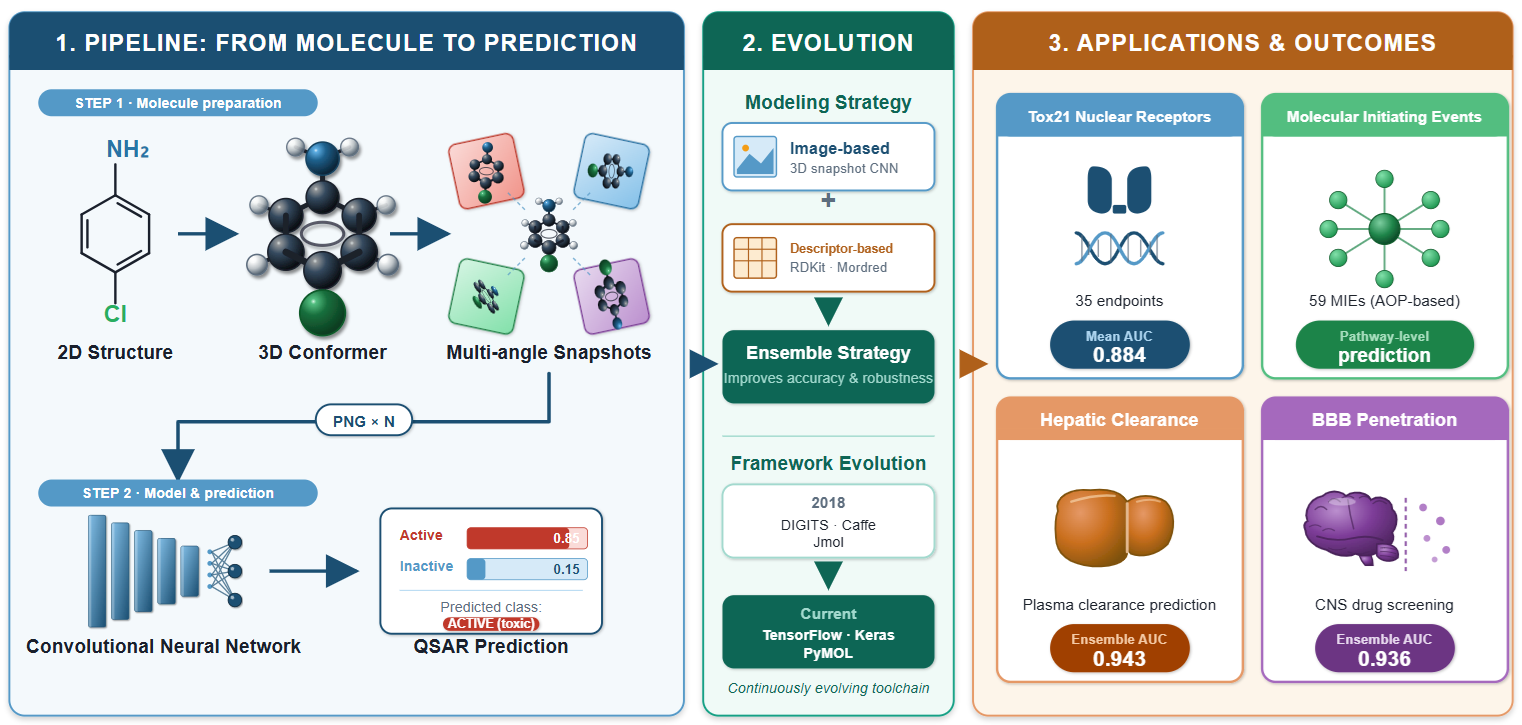
The DeepSnap method was introduced in 2018 as an answer to this question [13]. Rather than computing numerical descriptors or fingerprints, DeepSnap converts each molecule into a set of color-coded 3D molecular images that are fed directly into a convolutional neural network (CNN). Although ball-and-stick renderings were used in the initial and subsequent implementations, the DeepSnap concept is not restricted to a single visualization style. The term “Deep Snap” refers to the omnidirectional snapshot capture procedure: starting from a single 3D conformer, the molecular structure is rotated systematically around the x-, y-, and z-axes at a fixed angular increment, and an image is rendered at each orientation [13,19]. The resulting image set provides the CNN with multiple viewpoints of the same molecule, and aggregating per-image predictions across orientations approximates orientation invariance without explicit descriptor computation. Figure 1 illustrates how a single three-dimensional conformer yields distinct two-dimensional snapshots depending on viewing direction.
The Inaugural Study
The first DeepSnap study targeted mitochondrial membrane potential (MMP) disruption, one of the endpoints in the Tox21 Data Challenge 2014 [13]. Three-dimensional conformations were generated from SMILES strings using CORINA Classic (Molecular Networks GmbH), and ball-and-stick model images were rendered with Jmol, where each atom type was displayed in a distinct color [13]. Images were saved as 256 x 256 pixel PNG files [13]. With a 45-degree angular increment applied to all three rotation axes, the procedure yielded 512 images per molecule (8 orientations per axis, 8^3 = 512 combinations) [13]. A coarser setting of 90 degrees was also tested, producing 64 images per molecule [13].
The deep learning architecture employed was AlexNet [20], implemented within the Caffe framework [13]. Transfer learning with pretrained Caffe models was used in the DeepSnap-DL pipeline from the beginning of the study series, and later methodological papers described this implementation explicitly [21]. The training set comprised 7,320 molecules from the Tox21 Data Challenge, divided equally into training and validation subsets, while the external test set contained 647 compounds corresponding to the challenge’s final evaluation data [13]. For each molecule, the median of the 512 per-image predicted probabilities was taken as the representative prediction, a design choice that would persist throughout later DeepSnap studies [13].
Performance and Comparators
On the external test set, the DeepSnap model with the 45-degree increment achieved an area under the receiver operating characteristic curve (AUC) of 0.921, an accuracy of 0.836, and a sensitivity of 0.867 [13]. The 90-degree increment model yielded a lower but still competitive AUC of 0.898 [13]. These results were compared against three alternative approaches evaluated on the same dataset: a deep learning model trained on ECFP fingerprints using the H2O framework (AUC 0.888), a random forest (RF) model on ECFP fingerprints (AUC 0.901), and an RF model on 3D molecular descriptors computed by MOE (AUC 0.907) [13]. The DeepSnap model surpassed all three comparators in AUC despite operating without any explicit molecular descriptors or fingerprints.
This result was particularly notable because the Tox21 Data Challenge 2014 winning model for the same MMP disruption endpoint had achieved an AUC of 0.95 [13]. Although DeepSnap did not match this competition benchmark, the difference was 0.029 AUC units, and the AlexNet architecture had been used without hyperparameter tuning—the author noted that further optimization could close the remaining gap [13]. Importantly, the winning model relied on conventional molecular descriptors or fingerprints, whereas DeepSnap derived its input solely from rendered molecular images.
The comparison was not uniformly favorable across all metrics, however. The RF model with ECFP achieved a higher accuracy (0.915) and specificity (0.986) than DeepSnap (accuracy 0.836, specificity 0.832), while DeepSnap showed substantially higher sensitivity (0.867 vs 0.350) [13]. This pattern—high sensitivity at the cost of specificity—suggested that the image-based model may encode information complementary to that captured by fingerprints [13].
Conceptual Rationale
The underlying rationale of DeepSnap rests on two premises. First, 3D molecular images encode spatial and electronic information—atom types, bond arrangements, steric shape, and relative atomic positions—in a format directly compatible with the feature-extraction capabilities of CNNs that were originally developed for natural image recognition [2]. The color coding of atom types (e.g., oxygen in red, nitrogen in blue, carbon in gray) provides the network with chemical identity information within the pixel space [13]. Second, the omnidirectional snapshot procedure mitigates the viewpoint dependence inherent in projecting a 3D object onto a 2D image plane; by aggregating predictions from many orientations, the model approximates a viewpoint-invariant representation [13].
This image-based strategy differs fundamentally from graph neural networks (GNNs), which represent molecules as mathematical graphs with atoms as nodes and bonds as edges [2,22]. GNNs operate on topological connectivity and learned node embeddings, whereas DeepSnap operates on pixel-level visual features rendered from explicit 3D coordinates. As noted in a subsequent book chapter, “while the GCNs use structural information based on the 2D structure of a graph as input data, DeepSnap can provide more information as a feature amount. Because it can use 3D structure information as input data” [2]. Whether this theoretical advantage translates into consistent empirical superiority remains an open question, as no head-to-head comparison between DeepSnap and GNNs on the same datasets has been reported to date [22].
Early Recognition of Limitations
The inaugural publication also identified several limitations that would shape subsequent research directions. The computational cost of generating hundreds of images per molecule was acknowledged as “decidedly more costly” than descriptor-based methods [13]. The reliance on a single conformer from CORINA Classic, without molecular dynamics or force-field refinement, was noted as a potential source of error for flexible molecules [13]. Additionally, only a single toxicological endpoint had been tested, leaving the generalizability of the approach undemonstrated [13]. These limitations set the agenda for the systematic parameter optimization, architecture upgrades, and multi-endpoint applications that followed in subsequent years.
3. Core Rendering and Learning Pipeline
The prediction performance of DeepSnap depends on the interplay of rendering parameters, CNN architecture, hyperparameter tuning, and post-processing strategies. Over the course of several studies, these components were systematically optimized, first within a DIGITS/Caffe-based pipeline and later within a redesigned TensorFlow/Keras system.
Rendering Parameter Optimization
The DeepSnap image generation procedure is governed by six rendering parameters: molecules per SDF (MPS), zoom factor (ZF), atom size (AT), bond radius (BR), minimum bond distance (MBD), and bond tolerance (BT). Matsuzaka and Uesawa conducted a systematic grid search over each of these parameters for the CAR agonist endpoint, revealing that each parameter exhibited a quadratic relationship with validation loss (R-squared greater than 0.90 for all six) [21]. The optimal values identified were MPS 150, ZF 80%, AT 22%, BR 20 milliangstroms, MBD 0.4 angstroms, and BT 0.8 angstroms [21]. These results demonstrated that rendering parameters are not arbitrary but have well-defined optima that can be located through systematic tuning. The study also showed that multi-color atom representations yielded lower validation loss than monotone gray or monotone white renderings (validation loss 0.442 versus 0.468 and 0.467, respectively), though a two-color scheme (gray plus white) achieved an intermediate validation loss of 0.437 [21].
First-Generation Pipeline
The original DeepSnap pipeline consisted of four discrete steps. Molecular structures were first cleaned using MOE (protonation state assignment, salt/fragment removal), then converted to three-dimensional conformations by CORINA Classic, and finally rendered as 256 by 256 pixel ball-and-stick PNG images using Jmol [21,23]. Deep learning was performed using AlexNet within the DIGITS (version 4.0.0) interface running on the Caffe framework, with transfer learning from ImageNet pretrained weights [21]. Training typically ran for 30 epochs with SGD at a base learning rate of 0.01, and the epoch with the lowest validation loss was selected for test prediction [21]. The overall workflow of the first-generation DeepSnap pipeline, from SMILES input and three-dimensional conformer generation to multi-angle molecular image rendering and CNN-based prediction, is summarized in Figure 2.
Transition from AlexNet to GoogLeNet
A direct comparison of AlexNet and GoogLeNet on the CAR agonist endpoint showed that GoogLeNet achieved an AUC of 0.886 versus 0.857 for AlexNet under the same pre-CORINA-optimization conditions [23]. The adoption of GoogLeNet [24], a 22-layer Inception-based CNN pretrained on the ILSVRC 2012 dataset (1.2 million images, 1000 classes), became standard from this point forward [23,25]. All subsequent DeepSnap studies employed GoogLeNet with transfer learning from ImageNet pretrained weights, run through DIGITS/Caffe in the first-generation system [25,26].
CORINA Wash Conditions
The quality of three-dimensional input structures proved to be a dominant factor in prediction performance. Ten wash combinations (three protonation states crossed with three coordinate systems, plus one mixed condition) were evaluated for the CAR endpoint; the neut_CORINA condition (neutralization followed by CORINA Classic conformation generation) achieved the highest mean AUC of 0.995 across nine learning rates [23]. However, when the same wash comparison was performed for the PR antagonist endpoint, the domi_3D condition yielded a best AUC of 0.9971, while none_2D performed worst [25]. This finding that the optimal conformational preparation varies across endpoints was explicitly noted by the authors and represents an important caveat against adopting a single universal wash protocol [25].
Angle Increment as Key Hyperparameter
The viewing angle increment, which determines both the number of snapshot images generated per molecule and the diversity of perspectives captured, emerged as one of the most influential hyperparameters. For the CAR endpoint, 92 different angle configurations were scanned, with the best single-angle configuration (176 degrees) yielding an AUC of 0.910 before wash optimization [23]. The CAR model demonstrated robustness across data split ratios under the neut_CORINA wash condition: at the 176-degree angle, both 1:1:1 and 4:4:1 splits achieved comparable AUC values (0.998 ± 0.002 and 0.999 ± 0.001, respectively), with corresponding MCC values of 0.954 ± 0.026 and 0.966 ± 0.023 [23]. For the PR endpoint, angles from 120 to 300 degrees all achieved AUC values of 0.996 or higher, while the 360-degree condition (a single image) dropped to an AUC of 0.855 at 1:1:1, demonstrating that multi-angle snapshots are essential for high performance [25]. Even with as few as four pictures at 280 degrees, the CAR model retained an AUC of 0.999 and MCC of 0.967 [23].
Second-Generation Pipeline
In 2021, the DeepSnap system underwent a comprehensive migration. The DL framework was changed from DIGITS/Caffe to TensorFlow with Keras, the rendering software from Jmol to PyMOL, and the three-dimensional structure generation from the MOE/CORINA Classic pipeline to the SMILES_TO_SDF program [15]. The MMFF (Merck Molecular Force Field) was used in the PyMOL-based pipeline for conformational optimization [27]. These changes were integrated into a one-step sequential pipeline that improved throughput by automating all stages from SMILES input to statistical evaluation [15]. However, the initial 59-MIE model set constructed with the new system achieved a mean ROC AUC of 0.818 on validation, compared with 0.884 for a previous 35 nuclear-receptor model set built with DIGITS, indicating that the system migration introduced a performance trade-off that required further optimization [15]. Parallel comparisons on individual endpoints showed that DIGITS outperformed the TensorFlow/Keras system in mean AUC across all three endpoints tested, though systematic per-endpoint optimization of learning rate and batch size substantially improved the TensorFlow/Keras results [27].
Background Color Optimization
Background color was explored as an additional rendering variable. In the first-generation pipeline, six colors (white, red, yellow, green, blue, black) were tested for the PR endpoint; white and black produced significantly lower performance at the 360-degree single-image condition, while all colors performed comparably at 300 degrees [25]. The second-generation system expanded this search to more than 20 colors. For the PPARgamma agonist endpoint, wheat and Grey90 emerged as the best standard and pastel colors on the test set (ROC AUC 0.931 and 0.935, respectively), while for the aromatase antagonist endpoint, tv_orange yielded a test ROC AUC of 0.905 [15]. The authors noted that background color effects may relate to the colors present in the ImageNet pretraining data [15].
Learning Rate, Batch Size, and Epoch Selection
Learning rate and batch size required per-endpoint optimization. For the AhR activation endpoint, the NAG solver with a learning rate of 0.0025 and batch size of 37 achieved the lowest validation loss of 0.1466 [26]. For PR antagonist prediction, the learning rate range 0.01 to 0.001 was optimal, and performance generally decreased with increasing batch size; among six solver types tested, RMSprop performed significantly best and AdaDelta significantly worst [25]. In the second-generation system, even wider hyperparameter sweeps were conducted: for the GR antagonist endpoint, 39 learning rates and 84 batch sizes were evaluated, ultimately achieving a test ROC AUC of 0.983 at batch size 125 [27]. Epoch selection across all studies relied on the lowest validation loss criterion, with 30 epochs as the standard training duration and early stopping introduced in the TensorFlow/Keras system [15,27].
Aggregation, Cut-Off Determination, and Model Validation
Because DeepSnap generates multiple images per molecule, predictions must be aggregated across viewing angles. Across all studies, the median of per-image predicted probabilities was adopted as the representative prediction value for each molecule [21,23,25,26]. For binary classification, the optimal probability cut-off was determined using the Youden index [15,26,27].
Model validity was assessed through permutation tests in which class labels were randomly shuffled before training. For the CAR endpoint, the permutation test yielded a mean AUC of 0.553 (standard deviation 0.007, ten repetitions), far below the true-label AUC of 0.764, confirming that the model captured genuine structure-activity relationships rather than noise [21]. Similar random-label controls for the PR antagonist endpoint produced permutation AUC values of 0.519 to 0.527 [25], and for the AhR endpoint a permutation AUC of 0.539 [26]. The consistently near-chance permutation AUC values across multiple endpoints provide important evidence that DeepSnap-DL models are learning meaningful chemical features from the molecular images.
4. Applications to Toxicological Targets
The primary testing ground for DeepSnap-DL has been the Tox21 10K compound library, a publicly available collection of approximately 7,000–10,000 chemicals screened across multiple in vitro bioassays under the interagency Tox21 program. Throughout the DeepSnap series, binary activity was defined using a PubChem activity score threshold of 40 or above as active, with scores below 40 classified as inactive [14,23]. This standardized criterion replaced the more permissive threshold of score greater than zero used in the earliest parameter optimization study [21]. The Tox21 datasets are characterized by pronounced class imbalance, with active compound fractions typically ranging from below 1% to approximately 20% across different endpoints [14,15].
The first application of DeepSnap targeted mitochondrial membrane potential (MMP) disruption, one of the Tox21 Data Challenge 2014 endpoints. Using AlexNet with 512 images per molecule at 45-degree rotation increments, the model achieved an AUC of 0.921 on the external validation set of 647 compounds [13]. This result compared favorably with descriptor-based alternatives tested on the same data: a deep learning model using ECFP fingerprints yielded an AUC of 0.888, and random forest models with ECFP or 3D MOE descriptors achieved AUCs of 0.901 and 0.907, respectively [13]. However, the Tox21 Data Challenge 2014 winning model for this endpoint had achieved an AUC of 0.95, indicating a gap between the initial DeepSnap implementation and state-of-the-art competition entries [13].
Subsequent studies applied DeepSnap-DL to individual nuclear receptor targets from the Tox21 library, progressively refining the methodology and improving performance. For the constitutive androstane receptor (CAR) agonist endpoint, the initial parameter optimization study using AlexNet achieved an AUC of 0.791 on 9,523 compounds [21]. A follow-up study on the same CAR endpoint introduced GoogLeNet, optimized the 3D conformational sampling with CORINA, and applied a corrected activity threshold (score 40 or above), which together yielded an AUC of 0.999 on 7,141 compounds with a 4:4:1 train-validation-test split [23]. This represented a substantial improvement and far exceeded the performance of RF (AUC 0.884) and XGBoost (AUC 0.889) baselines trained on MORDRED descriptors from the same dataset, though these ML baselines were evaluated using a different split protocol [23]. For the progesterone receptor (PR) antagonist endpoint, DeepSnap-DL achieved an AUC of 0.999 on 7,582 compounds with 10-fold cross-validation, again outperforming five conventional machine learning methods, the best of which (CatBoost) reached an AUC of 0.894 [25].
A notable departure from the Tox21 data occurred with the prediction of aryl hydrocarbon receptor (AhR) activation using a small in-house dataset of 201 compounds measured by a reporter gene assay [26]. Despite the limited dataset size, DeepSnap-DL achieved an AUC of 0.959 at the optimal activity threshold, substantially exceeding the best conventional machine learning comparator (XGBoost, AUC 0.724) tested on the same data [26]. This result demonstrated that the molecular image approach could be effective beyond large public screening libraries, although the small sample size warrants cautious interpretation.
The scope of DeepSnap-DL was then expanded to 35 nuclear receptor agonist and antagonist endpoints from the Tox21 library in a single comprehensive study [14]. Using GoogLeNet with the DIGITS framework, a 176-degree angle yielding 27 images per compound, and a uniform set of hyperparameters across all endpoints, the models achieved a mean AUC of 0.884 across the 35 targets [14]. Individual model performance varied considerably; the best-performing model (TSHR2 agonist) achieved an AUC of 0.9994, while the overall mean reflected contributions from more challenging endpoints [14]. In a comparison with the Tox21 Data Challenge 2014 results for four overlapping endpoints, using BAC as the shared metric, DeepSnap-DL outperformed the Challenge winners in three (AR full agonist BAC 0.836 vs 0.650; ER-alpha LBD agonist BAC 0.820 vs 0.715; PPAR-gamma agonist BAC 0.849 vs 0.785) but fell short for AhR agonist (BAC 0.779 vs 0.853), noting that the evaluation protocols differed between the two studies [14]. The strong class imbalance across these endpoints (mean active fraction 3.7%) was reflected in the threshold-dependent classification metrics, with a mean F-measure of 0.309 and a mean MCC of 0.354 [14].
The transition to an improved system based on TensorFlow and Keras, with PyMOL replacing Jmol for rendering and a new 3D structure generation pipeline (SMILES_TO_SDF), enabled construction of 59 molecular initiating event (MIE) models encompassing both nuclear receptor and stress response pathway targets [15]. The mean AUC across the 59 models was 0.818 on the validation set and 0.803 on the test set [15]. These values were lower than the 0.884 achieved by the DIGITS-based system on 35 NR models, a difference that the authors attributed to the system migration rather than endpoint difficulty and noted as having potential for improvement through detailed per-endpoint optimization [15]. Separately, MORDRED descriptor-based XGBoost models covering 58 of the 59 MIE endpoints (VDR_ago excluded due to insufficient active compounds under criteria 40) achieved a mean AUC of 0.817 [28].
Per-endpoint hyperparameter optimization of the TensorFlow/Keras system was subsequently demonstrated for three MIE targets [27]. Systematic tuning of learning rate, batch size, angle, and data split ratio yielded AUCs of 0.983 for GR antagonist, 0.934 for TRHR agonist, and 0.925 for TGF-beta antagonist on the test set [27]. These results substantially exceeded the mean performance of the initial 59-model run and demonstrated that the TensorFlow/Keras system could match or approach DIGITS-level performance when optimized on a per-endpoint basis (Table 1).
Beyond the Tox21 in vitro assay framework, DeepSnap-DL was applied to the prediction of acute oral toxicity using the CATMoS dataset of 11,886 compounds, with the binary endpoint defined as LD50 of 50 mg/kg or below [17]. DeepSnap-DL alone achieved an AUC of 0.887 and a BAC of 0.818, while the descriptor-based model yielded an AUC of 0.931 [17]. An ensemble combining both approaches by averaging predicted probabilities reached an AUC of 0.942 [17]. A consensus model, which retained only compounds where both methods agreed on classification, achieved a BAC of 0.916, exceeding the CATMoS collaborative consensus BAC of 0.87 [29] despite using only two model types compared to 32 participating organizations [17].
The DeepSnap approach was also extended to the S-COPHY model developed with Shiseido [18]. This application classified compounds as cosmetics or pharmaceuticals based on structural similarity, using a single molecular image per compound selected from 27 candidates by maximum pixel occupation, with AlexNet implemented in MATLAB [18]. The model achieved an AUC of 0.935 on the internal validation set of 689 compounds [18]. S-COPHY provided a peer-reviewed example of Grad-CAM visualization for a DeepSnap-type model, showing that the CNN focused on pharmaceutical-like structural motifs within the molecular image [18].
A recurring challenge across all DeepSnap-DL toxicological applications has been class imbalance. In the 59-MIE study, active compound fractions ranged from 0.08% (TGF-beta agonist) to 21.6% (PXR agonist), with a mean of 4.79% [15]. Threshold-dependent metrics such as MCC and F-measure reflected the difficulty of classifying the minority active class. For example, the 35-NR study reported a mean F-measure of 0.309 and a mean MCC of 0.354 across endpoints with a mean active fraction of only 3.7% [14]. Similarly, the TRHR agonist endpoint (0.87% active compounds) yielded an MCC of 0.200 after optimization [27]. These observations underscore that threshold-dependent classification of minority-class compounds remains challenging in the Tox21 context, a limitation shared with other modeling approaches applied to these datasets.

Table 1.
Chronological summary of DeepSnap-based QSAR studies. Abbreviations: CB, CatBoost; CL, clearance; DIGITS, NVIDIA Deep Learning GPU Training System; DL, deep learning; ens., ensemble; cons., consensus; LGBM, LightGBM; MD, molecular descriptors; RF, Random Forest; TF, TensorFlow; XGB, XGBoost.
Table 1.
Chronological summary of DeepSnap-based QSAR studies. Abbreviations: CB, CatBoost; CL, clearance; DIGITS, NVIDIA Deep Learning GPU Training System; DL, deep learning; ens., ensemble; cons., consensus; LGBM, LightGBM; MD, molecular descriptors; RF, Random Forest; TF, TensorFlow; XGB, XGBoost.
| Year | Ref. | Endpoint | Dataset (N) | Pipeline | Best Result | Comparator | Key Limitation |
|---|---|---|---|---|---|---|---|
| 2018 | [13] | MMP disruption | Tox21 (7967) | AlexNet / DIGITS | AUC 0.921 | RF+3D desc 0.907 | Single endpoint; no HP tuning |
| 2019 | [21] | CAR param. optim. | Tox21 (9523) | AlexNet / DIGITS | AUC 0.791 | RF+MOE 0.749 | Non-standard threshold; single split |
| 2019 | [23] | CAR agonist | Tox21 (7141) | GoogLeNet / DIGITS | AUC 0.999 | XGB 0.889; RF 0.884 | Near-ceiling AUC; class imbalance |
| 2020 | [25] | PR antagonist | Tox21 (7582) | GoogLeNet / DIGITS | AUC 0.999 | CB 0.894; LGBM 0.893 | Wash varies by target; imbalance |
| 2020 | [26] | AhR (in-house) | In-house (201) | GoogLeNet / DIGITS | AUC 0.959 | XGB 0.724; RF 0.716 | Small N; no wash optimization |
| 2020 | [14] | 35 NR models | Tox21 (mean 7262) | GoogLeNet / DIGITS | Mean AUC 0.884 | Tox21 Challenge (3/4 exceeded) | n = 2 replicates; class imbalance |
| 2021 | [15] | 59 MIE models | Tox21 (mean 9699) | GoogLeNet / TF-Keras | Mean AUC 0.818 | Prior DIGITS system | Underperforms DIGITS; NFkB failed |
| 2021 | [16] | Rat CL classif. | In-house (1545) | GoogLeNet / DIGITS + ens. | Ens. AUC 0.943 | RF+MD 0.883 | Consensus coverage 69% |
| 2022 | [27] | GR/TRHR/TGFb | Tox21 (7537–7662) | GoogLeNet / TF-Keras | GR AUC 0.983 | DIGITS GR 0.910 | TRHR MCC 0.200; 3 endpoints only |
| 2022 | [30] | Rat CL regression | In-house (1545) | DL prob. + AutoML | R² 0.736 | MD-only R² 0.649 | DL alone R² 0.359; private data |
| 2023 | [17] | LD50/BBBP/CL path. | CATMoS (11886) etc. | GoogLeNet / DIGITS + ens. | Cons. BAC 0.916 | CATMoS 32 orgs 0.87 | Coverage 77–86%; DL < CATMoS |
| 2024 | [18] | Cosmetics vs. pharma | PubChem etc. (2754) | AlexNet / MATLAB | AUC 0.935 | None | Ext. pred. rate 46%; regulatory |
5. Applications to ADME Parameters
While the initial applications of DeepSnap-DL focused on toxicological targets from the Tox21 library and related assays, a parallel line of investigation extended the approach to absorption, distribution, metabolism, and excretion (ADME) and pharmacokinetic (PK) parameters. This expansion was motivated by the practical importance of early PK prediction in drug discovery and by the hypothesis that molecular image-based features might capture structural information relevant to processes governed by drug-transporter interactions and metabolic clearance, not solely receptor binding.
The first ADME application of DeepSnap-DL targeted the binary classification of rat clearance (CL), a parameter that had historically proven difficult to predict with conventional machine learning methods [16]. Using an in-house dataset of 1,545 compounds from Japan Tobacco Inc., with CL measured after intravenous administration and classified at a threshold of 1 L/h/kg (approximately 30% of hepatic blood flow in rats), the study constructed two independent modeling pipelines [16]. The DeepSnap-DL pipeline employed GoogLeNet on 3D molecular images rendered at 145-degree rotation increments, while the conventional pipeline used a random forest model selected by DataRobot from over 40 candidate algorithms operating on 100 molecular descriptors derived from an initial pool of 4,795 [16]. On the external test set of 309 compounds, DeepSnap-DL achieved an AUC of 0.905, modestly exceeding the descriptor-based random forest AUC of 0.883 [16].
The more consequential finding was that combining the two approaches produced substantial improvements beyond either method alone. An ensemble model, which averaged the predicted probabilities from DeepSnap-DL and the descriptor-based model, achieved an AUC of 0.943 on the same test set [16]. A consensus model, which retained predictions only when both methods agreed on the class assignment, achieved an accuracy of 0.959 and an MCC of 0.915 [16]. However, this consensus approach reduced the number of evaluable compounds from 309 to 214, representing a trade-off between prediction confidence and coverage [16]. The authors attributed the synergy to complementary information capture, noting that molecular recognition in the image space and in the descriptor space appeared to be fundamentally different [16]. This interpretation is supported by the observation that the two methods exhibited different error profiles in their confusion matrices: DeepSnap-DL showed higher sensitivity (0.843 vs 0.772) while the descriptor-based model showed higher specificity (0.863 vs 0.824) [16].
Having established the value of the combination approach for CL classification, a subsequent study addressed the regression prediction of continuous CL values using the same 1,545-compound dataset [30]. Rather than averaging probabilities as in the ensemble classification model, this study introduced a conceptually distinct strategy: the DeepSnap-DL classification probability was appended as an additional descriptor to the conventional molecular descriptor set, and the augmented feature matrix was used to train regression models via DataRobot [30]. This probability-as-descriptor approach was validated using a five-pattern cross-dataset design, in which the compounds were divided into five equal subsets and each pattern used four subsets for training and one for testing [30]. The combination model achieved a mean test R-squared of 0.736 with a mean RMSE of 0.265, compared with R-squared of 0.649 and RMSE of 0.306 for the descriptor-only regression [30]. The improvement was consistent across all five data partitions, with test R-squared values ranging from 0.710 to 0.769 for the combination model versus 0.625 to 0.669 for the descriptor-only model [30].
Feature importance analysis of the combination regression model revealed the dominance of the image-derived probability feature. The DeepSnap-DL prediction probability had an average effect of 1.000 (normalized to the top feature), while the next most important descriptor (BCUT_SLOGP_0, a lipophilicity-related descriptor) had an average effect of only 0.107 [30]. This pronounced gap indicated that the image-based classification probability encoded structural information largely orthogonal to conventional physicochemical descriptors. Notably, the DeepSnap-DL probability alone explained only R-squared of 0.359 of the variance in log(CL), suggesting that its value lies primarily in its complementarity with descriptor-based features rather than in its standalone predictive power [30].
The generalizability of the ensemble and consensus combination strategies was subsequently tested on two additional ADME-related endpoints alongside the LD50 toxicity target [17]. For blood-brain barrier penetration (BBBP), using the MoleculeNet dataset of 2,049 compounds classified by a log BB threshold of negative one, the ensemble model achieved an AUC of 0.936 on the test set of 409 compounds, surpassing both DeepSnap-DL alone (AUC 0.893) and the descriptor-based model (AUC 0.919) [17]. For clearance pathway classification (hepatic metabolism versus renal elimination), using a dataset of 636 compounds, the ensemble model achieved an AUC of 0.908, again exceeding both individual methods (DeepSnap-DL AUC 0.883, descriptor-based AUC 0.900) [17]. The consensus models further improved balanced accuracy at the cost of reduced coverage: for BBBP, the consensus BAC was 0.918 with 328 to 351 evaluable compounds out of 409, and for CL pathway, the consensus BAC was 0.847 with 100 to 110 evaluable compounds out of 127 [17].
The robustness of ESOL (estimated aqueous solubility) prediction was also examined using the probability-as-descriptor approach on 1,128 compounds from the MoleculeNet benchmark [30]. Here the improvement was modest: the combination model achieved a mean R-squared of 0.950 versus 0.943 for the descriptor-only model [30]. This smaller gain, compared with the larger improvement observed for rat CL, suggests that the combination approach provides the greatest benefit when conventional descriptors alone are insufficient to capture the relevant structure-property relationships. When descriptor-based models already perform well, as with ESOL, the marginal contribution of image-derived features diminishes.
Across all tested ADME and PK endpoints, a consistent pattern emerged: the ensemble model outperformed either individual method in AUC for every endpoint examined [16,17,30]. This held for both classification tasks (rat CL, BBBP, CL pathway) and, in terms of R-squared, for regression tasks (rat CL, ESOL). However, an important observation is that, unlike the earlier Tox21-based toxicity applications where DeepSnap-DL sometimes outperformed descriptor-based models, the descriptor-based method yielded a higher AUC than DeepSnap-DL alone for BBBP and CL pathway [17]. Only for rat CL classification did DeepSnap-DL modestly exceed the descriptor-based model in AUC (0.905 vs 0.883) [16]. This suggests that the primary value of DeepSnap-DL in the ADME domain lies not in standalone superiority but in contributing complementary structural information that strengthens combination models.
Several limitations should be noted. The rat CL studies relied on a proprietary dataset from a single pharmaceutical company, preventing external validation or direct comparison with published benchmarks from other groups [16,30]. The CL pathway dataset contained only 636 compounds with a test set of 127, limiting the statistical confidence of the results [17]. The consensus model’s requirement for agreement between methods introduces a coverage-accuracy trade-off that may constrain its applicability in early drug screening, where comprehensive evaluation of all candidate compounds is often preferred [16]. Furthermore, the DeepSnap-DL angle was fixed at 145 degrees for all ADME applications based on the initial CL study, without per-endpoint optimization, which may have reduced the image-based model’s standalone performance [17]. Despite these caveats, the ADME studies collectively establish that the complementarity between molecular image features and conventional descriptors is not limited to toxicological endpoints but extends to pharmacokinetic and physicochemical property prediction.
6. Technical Evolution and Variants
Since its introduction in 2018, the DeepSnap-DL system has undergone substantial technical changes affecting the deep learning framework, the 3D structure generation pipeline, the molecular rendering software, and the strategies used for combining predictions across models. These modifications can be organized into two broad pipeline generations, a single-image variant (S-COPHY), combination strategies that integrate image-based and descriptor-based predictions, and emerging work on model interpretability (Figure 3).
The first generation of DeepSnap-DL (2018–2020) was built on the NVIDIA Deep Learning GPU Training System (DIGITS) with Caffe as the underlying framework, running on four Tesla V100 GPUs [23,25]. Three-dimensional molecular structures were generated through a pipeline in which SMILES strings were processed by MOE (Chemical Computing Group) for protonation and desalting, then optimized by CORINA Classic to produce SDF files [23]. The open-source Java-based viewer Jmol rendered these structures as ball-and-stick images at 256 by 256 pixel resolution [21]. The CNN architecture transitioned from AlexNet in the earliest study [13] to GoogLeNet beginning with the 2019 CAR agonist work, which demonstrated that GoogLeNet outperformed AlexNet on the same dataset (AUC 0.886 versus 0.857) [23]. Transfer learning from ImageNet (ILSVRC 2012) pretrained weights was employed in the first-generation DIGITS-based studies [21,22]. This first-generation system produced the highest reported performances in the DeepSnap series, including AUC values of 0.999 for CAR agonist [23] and PR antagonist [25], and a mean AUC of 0.884 across 35 nuclear receptor models [14]. However, the pipeline comprised four separate steps that were not automated, limiting throughput [15].
The second generation, introduced in 2021, migrated the deep learning framework from DIGITS/Caffe to TensorFlow and Keras, replaced Jmol with the Python-based renderer PyMOL, and substituted the MOE/CORINA workflow with the SMILES_TO_SDF program for 3D structure generation [15]. Critically, these components were integrated into a one-step sequential pipeline that automatically executed 3D generation, snapshot rendering, model training, and statistical evaluation [15]. The GoogLeNet architecture was retained, and early stopping based on validation loss was introduced as a regularization strategy [15]. This redesign prioritized throughput and reproducibility over peak accuracy. When applied to 59 molecular initiating event (MIE) models from the Tox21 library, the second-generation system achieved a mean validation AUC of 0.818, compared with the mean AUC of 0.884 reported for 35 nuclear receptor models in the first-generation system [14,15]. Direct parallel comparisons on individual endpoints showed that DIGITS consistently outperformed TensorFlow/Keras in mean AUC across all angles and splits tested [27]. For example, on the glucocorticoid receptor antagonist endpoint, the DIGITS system achieved a mean validation AUC of 0.856 versus 0.832 for the Python system [27]. However, with systematic per-endpoint optimization of learning rates and batch sizes, the TensorFlow/Keras system could reach high individual-endpoint performance, as demonstrated by a test AUC of 0.983 for GR antagonist after batch size optimization [27].
An intriguing phenomenon observed in the second-generation system was a two-peak pattern in performance as a function of rotation angle: TensorFlow/Keras models showed performance peaks at approximately 180 degrees and 360 degrees, a pattern not observed in DIGITS models [15]. This observation was reproduced across multiple endpoints [27] and may reflect differences in how the two frameworks handle image preprocessing or weight initialization, though the precise cause remains unidentified. An important caveat in interpreting the performance gap between generations is that the system migration changed multiple components simultaneously—the DL framework, the 3D structure generator, and the renderer—making it impossible to isolate the contribution of any single factor to the observed decrease in mean performance [15].
The S-COPHY variant, developed with Shiseido, departed from the multi-angle snapshot paradigm [18]. Instead of generating multiple images per compound and aggregating predictions across angles, S-COPHY generated 27 candidate images by rotating the 3D structure at 120-degree increments on each axis and selected a single image per compound based on the maximum percentage of pixels occupied by the molecular structure [18]. The implementation used AlexNet rather than GoogLeNet, MATLAB R2021a as the deep learning framework, and Marvin (ChemAxon) for 3D structure generation [18]. Applied to the classification of compounds as cosmetics versus pharmaceuticals (2,754 compounds), S-COPHY achieved an AUC of 0.935 on the internal validation set [18]. Although the task differs from Tox21-based activity prediction, this result demonstrated that a single well-chosen molecular image can carry sufficient structural information for classification, substantially reducing the computational cost of the multi-angle approach.
Beyond modifications to the image generation and classification pipeline, the DeepSnap series introduced strategies for combining image-based predictions with conventional descriptor-based machine learning. For classification tasks, an ensemble model that averaged the predicted probabilities from DeepSnap-DL and a descriptor-based random forest was shown to improve AUC from 0.905 (DeepSnap-DL alone) to 0.943 on a rat clearance dataset [16]. A consensus model, which retained predictions only when both methods agreed, achieved higher accuracy (0.959) but at the cost of reducing the evaluable compounds from 309 to approximately 69–71% of the test set [16]. For regression tasks, a conceptually distinct probability-as-descriptor strategy was introduced, in which the DeepSnap-DL classification probability was appended as a feature to the conventional descriptor set and used to train regression models [30]. This image-derived probability emerged as the single most important feature in the regression model, with a normalized importance of 1.000 compared with 0.107 for the next most important conventional descriptor [30]. These combination strategies, framed within an ensemble learning conceptual framework [31], represent a shift from treating DeepSnap-DL as a standalone classifier to positioning it as a complementary component within hybrid prediction systems.
Efforts toward interpreting DeepSnap-DL predictions have advanced through multiple channels, though the area remains at an early stage. The S-COPHY study provided Grad-CAM [32] visualizations for a molecular image-based CNN, demonstrating that the model’s attention concentrated on specific pharmacophoric substructures (such as beta-lactam rings and pyrazolin-5-one motifs) in compounds predicted as pharmaceuticals [18]. In contrast, for compounds predicted as cosmetics, the attention maps highlighted surrounding background areas rather than specific structural motifs [18]. Within the main DeepSnap series, Grad-CAM analysis was reported to confirm that the CNN detected chemical structure features within the molecular images, although the results were not shown [15]. In the METI AI-SHIPS project, DeepSnap was implemented in the integrated toxicity prediction system and equipped with image feature-region visualization functionality [33,34]. In this system, seven CYP inhibition prediction models were constructed, and CAM-based highlighting of molecular regions contributing to predicted activity was achieved; ScoreCAM and GuidedGradCAM were also made available as visualization options for DeepSnap-based predictions [33,34]. A book chapter discussed the applicability of additional explainable AI techniques—including LIME, SHAP, Anchor, and integrated gradients—to molecular image-based models [2], In the DeepSnap publications and AI-SHIPS project materials covered in this review, the documented explainability implementations are CAM-family methods, whereas practical applications of LIME, SHAP, Anchor, and integrated gradients to DeepSnap molecular images have not yet been described. The interpretability of DeepSnap-DL thus remains a developing area, identified across the series as a priority for future investigation [2,22].
7. Limitations and Unresolved Issues
Despite the strong prediction performance reported across multiple endpoints, the DeepSnap-DL approach exhibits several methodological limitations and unresolved issues that warrant critical examination. These can be organized into five thematic areas: parameter sensitivity and generalizability, statistical evaluation practices, system migration challenges, interpretability constraints, and practical deployment barriers.
Parameter Sensitivity and the Absence of Universal Settings
A persistent finding throughout the DeepSnap literature is that optimal rendering and training parameters are endpoint-dependent and cannot be transferred across targets. Matsuzaka and Uesawa systematically demonstrated that six rendering parameters (molecules per SDF, zoom factor, atom size, bond radius, minimum bond distance, and bond tolerance) each followed a quadratic relationship with validation loss for the CAR agonist endpoint, with R-squared values exceeding 0.90 [21]. However, the optimal angle differed substantially between endpoints: 280 degrees for CAR agonist [23], 176 degrees for the 35 nuclear receptor models [14], and 145 degrees for rat clearance [16]. Similarly, optimal learning rates and batch sizes varied: 0.0001 and batch size 15 for PPARgamma agonist versus 0.0008 and batch size 65 for aromatase antagonist, both evaluated within the same study [15]. Background color preferences also proved endpoint-specific, with white and wheat tones performing well for PPARgamma agonist but orange-family tones preferred for aromatase antagonist [15]. The dependence of optimal background color on the pretrained CNN model was noted explicitly, suggesting that the ImageNet-derived feature representations interact with rendering choices in ways that remain poorly understood [15]. No study has proposed a principled method for selecting parameters a priori for a new endpoint, and the full grid search required for each target incurs substantial computational cost. Mamada et al. reduced the search space from 100 conditions to 25 per endpoint in later work [17], but the need for per-endpoint tuning remains a practical barrier to high-throughput deployment.
Statistical Evaluation Practices and Class Imbalance
Several aspects of the evaluation methodology across DeepSnap studies limit confidence in the reported performance estimates. The most pervasive issue is class imbalance in the Tox21 datasets: the mean active fraction across 35 nuclear receptor models was only 3.72% [14], and the 59 MIE models had a mean active percentage of 4.79% [15]. Under this severe class imbalance, threshold-dependent classification metrics remained modest; the 35 NR models had a mean F-measure of 0.309 and a mean MCC of 0.354 [14], and the LD50 model (positive-class fraction 8.3%) yielded an MCC of 0.393 [17]. These values indicate that effective threshold-dependent classification of minority-class compounds remains challenging in practical screening scenarios.
Validation design also varied across studies. The 35 nuclear receptor models used only two test replicates per endpoint [14], providing limited statistical power for estimating performance variance. Some studies relied on a single data split without cross-validation, as in the rat clearance study where primary results derived from a single 4:1 train-test partition [16]. No DeepSnap study has reported an applicability domain analysis to delineate the chemical space within which predictions are reliable. Additionally, a numerical discrepancy exists in the rat clearance consensus model: Table 2 of that study reported the evaluable compound count as 214, but the confusion matrix in Table 4 summed to 221 [16]. While this does not invalidate the overall findings, it introduces uncertainty regarding the exact consensus coverage.
Reproducibility and Data Availability
Several DeepSnap studies relied on proprietary, non-public datasets that preclude independent reproduction. The AhR activation study used 201 in-house compounds from an XRE reporter assay [26], and the rat clearance studies employed 1,545 in-house compounds from Japan Tobacco Inc. [16]. While the Tox21-based studies drew on publicly available data, the 3D structure generation pipelines (MOE washing, CORINA Classic conformer generation) relied on commercial software whose exact behavior may vary across versions. Later studies partially addressed this by adopting open-source tools such as RDKit with the MMFF force field [17], but the comparability of structures generated by different pipelines has not been formally evaluated.
System Migration and Framework Confounding
The transition from the original DIGITS/Caffe/Jmol system to TensorFlow/Keras/PyMOL, documented in the 59 MIE model study, resulted in measurably lower prediction performance. The TF/Keras system achieved a mean test AUC of 0.803 across 59 MIE models, compared with a mean AUC of 0.884 for the previous DIGITS-based 35 nuclear receptor models [15]. Direct comparison on the PPARgamma agonist endpoint showed the DIGITS system achieving a best test AUC of 0.962 versus 0.934 for TF/Keras, and for aromatase antagonist the gap was 0.950 versus 0.893 [15]. The migration simultaneously changed multiple components — the DL framework, the molecular renderer (Jmol to PyMOL), and the 3D structure generator (MOE/CORINA to SMILES_TO_SDF) — making it impossible to isolate which change was responsible for the performance decrease [15]. This confounded migration complicates interpretation of subsequent results produced with the newer system.
Interpretability and Comparison with Alternative Architectures
Throughout most of the DeepSnap series, CNN-based prediction has operated as a black box. The interpretability gap was acknowledged explicitly in several publications [2,14,22]. Grad-CAM analysis was mentioned in the 59 MIE study, but results were described as “data not shown” [15]. The S-COPHY study by Hisaki et al. provided peer-reviewed Grad-CAM visualizations, demonstrating that the CNN focused on chemically meaningful structural motifs within pharmaceutical compounds [18]. However, S-COPHY used AlexNet with a single molecular image rather than the standard multi-angle GoogLeNet pipeline, limiting the transferability of its interpretability findings. In the AI-SHIPS project, CAM-family visualization including ScoreCAM and GuidedGradCAM was implemented for DeepSnap-based CYP inhibition models [33,34], representing a broader deployment of interpretability tools beyond the peer-reviewed S-COPHY example. Other XAI methods discussed in the literature—LIME, SHAP, Anchor, and integrated gradients [2]—have not yet been reported as applied to DeepSnap molecular images in the publications covered in this review.
Finally, no DeepSnap study has conducted a head-to-head comparison with graph neural network approaches (GCN, GNN, MPNN, or AttentiveFP) on the same datasets and splits. While Matsuzaka and Uesawa reviewed GNN architectures and positioned DeepSnap as providing richer 3D structural information than 2D graph-based methods [2,22], this claim remains untested empirically. The computational cost of multi-angle image generation and CNN training, which in the DIGITS-based studies required a four-GPU Tesla V100 system [23], further motivates such comparisons, as graph-based methods may offer more efficient alternatives for some prediction tasks.
8. Position Relative to External Methods
Positioning DeepSnap within the broader landscape of molecular property prediction methods requires comparison along several axes: input representation, predictive performance, data efficiency, interpretability, computational cost, and task fit. Because no published study has performed a head-to-head comparison between DeepSnap and any of the external methods discussed below using the same datasets and identical train–test splits, all contrasts presented in this section rely on results obtained under different experimental conditions. Differences in data preprocessing, splitting strategy, class balance, and evaluation metrics can substantially affect reported performance values, and apparent numerical advantages should not be interpreted as definitive evidence of methodological superiority.
Comparison with Tox21 Data Challenge 2014 winners (indirect comparison; different data splits and evaluation protocols). The earliest external benchmark for DeepSnap was the Tox21 Data Challenge 2014, whose overall winner, DeepTox, employed deep neural networks trained on extended connectivity fingerprints and toxicophore features [7]. When Matsuzaka et al. applied DeepSnap-DL to 35 nuclear receptor endpoints from the Tox21 10K library, the method outperformed the Challenge winners in three of four comparable endpoints on balanced accuracy: AR full agonist (BAC 0.836 vs. 0.650), ER-alpha LBD agonist (0.820 vs. 0.715), and PPAR-gamma agonist (0.849 vs. 0.785), while the Challenge winner retained superiority for AhR agonist (0.853 vs. 0.779) [14]. These comparisons, however, involved different data splits and evaluation protocols: the Challenge used a held-out test set provided by the organizers, whereas DeepSnap-DL used a 4:4:1 train-validation-test split generated internally. Moreover, the Challenge was conducted in 2014 with methods of that era, and the state of the art has advanced considerably since then. A separate descriptor-based baseline on the same Tox21 assays reported balanced accuracy values ranging from 0.58 to 0.82 using Random Forest and deep neural network classifiers with Dragon and SiRMS descriptors [35], which are generally lower than the DeepSnap-DL mean BAC of 0.847 across 35 NR endpoints [14], though these studies differed in the number of endpoints evaluated and the data preprocessing procedures. Given the absence of matched experimental conditions, these numerical differences should not be interpreted as definitive evidence of methodological superiority.
CATMoS acute oral toxicity (indirect comparison; same dataset but different modeling pipelines and coverage). For acute oral toxicity (LD50), Mamada et al. compared DeepSnap-based models against the Collaborative Acute Toxicity Modeling Suite (CATMoS) [29], a consensus model constructed by 32 organizations [17]. The DeepSnap-DL plus molecular descriptor consensus model achieved a BAC of 0.916, exceeding the CATMoS consensus BAC of 0.87 [17]. This result is noteworthy because it was achieved with only two model types (image-based and descriptor-based) compared with the 32 participating organizations in CATMoS. However, the consensus approach reduced the number of evaluable compounds from 2,894 to approximately 2,232–2,342, meaning that this higher accuracy applied only to compounds where the two models agreed [17]. The ensemble model, which retained full coverage, achieved a BAC of 0.842, comparable to the CATMoS weight-of-evidence BAC of 0.84 [17].
Blood-brain barrier penetration (indirect comparison; different data split strategies). For BBBP prediction, the DeepSnap ensemble model achieved an AUC of 0.936, higher than the MoleculeNet scaffold-split baseline of 0.729 reported by Wu et al. [36] and the value of 0.763 reported by Chen et al. [37]. However, the DeepSnap study used a random train-test split rather than the scaffold split employed by MoleculeNet, and scaffold splits generally yield lower performance estimates because they test generalization to novel chemical scaffolds [36,38]. The Directed Message Passing Neural Network (D-MPNN, Chemprop) reported a BBBP AUC of approximately 0.71 on scaffold split [9], which is again not directly comparable to the DeepSnap result obtained under different splitting conditions. In the absence of a matched-split comparison, it remains unclear whether the DeepSnap ensemble genuinely outperforms graph-based methods on this endpoint.
Clearance pathway prediction (indirect comparison; same compound set but independent evaluation). For binary classification of hepatic metabolism versus renal elimination, the DeepSnap ensemble achieved an AUC of 0.908 and an accuracy of 0.841, exceeding the AUC range of 0.776–0.870 and accuracy range of 0.72–0.77 reported by Kaboudi and Shayanfar [39] on the same compound set using random-split evaluation [17]. The consensus model further improved accuracy to 0.875, approaching the value of 0.88 reported by Lombardo et al. [40] for a restricted subset with excretion above 70% [17]. As these comparisons involve independently conducted evaluations rather than a controlled head-to-head design, the observed performance differences should be interpreted cautiously.
Descriptor-based machine learning (internal comparisons within the same studies). Within the DeepSnap literature, several studies included internal comparisons against conventional descriptor-based machine learning methods. For PR antagonist prediction, DeepSnap-DL achieved an AUC of 0.999, exceeding CatBoost (AUC 0.894), LightGBM (0.893), XGBoost (0.889), neural network (0.842), and Random Forest (0.821), all trained on MORDRED descriptors with the same 10-fold cross-validation splits [25]. Similarly, for AhR activation prediction on a small in-house dataset of 201 compounds, DeepSnap-DL (AUC 0.959) exceeded XGBoost (0.724), CatBoost (0.719), LightGBM (0.715), and Random Forest (0.716), representing a performance gap of approximately 0.24 AUC units [26]. However, for the three drug screening endpoints examined by Mamada et al. (LD50, BBBP, CL pathway), the descriptor-based models actually exceeded DeepSnap-DL alone in AUC across all three endpoints, with the image-based method contributing complementary rather than superior information [17]. Although the number of studies is limited, the available evidence suggests a tentative pattern in which DeepSnap-DL alone may be particularly competitive for toxicological endpoints involving nuclear receptor interactions, whereas descriptor-based methods may retain an advantage for certain ADME and pharmacokinetic properties. Further comparisons across a wider range of endpoints would be needed to confirm whether this pattern is general.
Graph neural networks (no shared benchmark; no direct comparison available). Graph neural networks represent the most prominent alternative paradigm in molecular property prediction. The foundational MPNN framework unified message passing over molecular graphs for property prediction [8], and subsequent developments such as D-MPNN (Chemprop) [9] and AttentiveFP [10] have been extensively benchmarked on public datasets. No study in the DeepSnap literature has performed a direct head-to-head comparison with any GNN architecture using the same data splits and evaluation protocols. Matsuzaka and Uesawa discussed GNN and GCN methods as alternatives to image-based approaches, noting that graph-based models operate on two-dimensional molecular topology and therefore do not incorporate three-dimensional conformational information [22]. Uesawa further stated that DeepSnap can provide more structural information than GCN because it uses three-dimensional structure as input data [2]. However, this theoretical advantage has not been empirically validated through controlled comparison, and whether three-dimensional image encoding provides measurable improvements over two-dimensional graph representations for specific endpoints remains an open question.
Other image-based approaches (no shared benchmark; no direct comparison available). DeepSnap is not the only method to use molecular images as CNN input. Chemception applied an Inception-ResNet architecture to two-dimensional chemical structure drawings, achieving performance comparable to expert-developed QSAR models on toxicity and activity prediction tasks [11]. More recently, ImageMol employed self-supervised pretraining on 10 million two-dimensional molecular images using a ResNet-18 backbone, achieving competitive results across 10 benchmark datasets including BBBP and toxicity endpoints [12]. The critical distinction between these approaches and DeepSnap is the dimensionality of structural information: Chemception and ImageMol use two-dimensional structural drawings that lack conformational and stereochemical detail, whereas DeepSnap renders three-dimensional ball-and-stick models from multiple viewing angles, thereby encoding three-dimensional shape and spatial relationships into the pixel representation [22]. No published study has directly compared DeepSnap with either Chemception or ImageMol on the same dataset and under matched conditions.
Voxel-based three-dimensional CNNs (different task domain; no direct comparison applicable). A distinct class of three-dimensional deep learning methods employs voxelized grids of protein-ligand complexes. AtomNet [41] and the Gaussian atom density approach of Ragoza et al. [42] demonstrated that three-dimensional CNNs can learn binding features directly from structural data. However, these methods address a fundamentally different task domain: structure-based binding prediction requiring protein target structures, whereas DeepSnap performs ligand-based QSAR requiring only molecular structure. Additionally, three-dimensional convolutions scale cubically with spatial resolution, creating substantial computational costs that DeepSnap avoids through its two-dimensional image projection strategy [22]. The major characteristics of these molecular property prediction approaches are compared in Table 2 in terms of input representation, use of three-dimensional information, interpretability, computational cost, and benchmark relationship to DeepSnap.
Table 2.
Comparison of molecular property prediction approaches. Benchmark relation: indirect = same dataset, different split/protocol; none = no overlapping benchmark with DeepSnap.
Table 2.
Comparison of molecular property prediction approaches. Benchmark relation: indirect = same dataset, different split/protocol; none = no overlapping benchmark with DeepSnap.
| Method Family | Example | Representation | 3D Info | Interpret. | Cost | Benchmark Relation |
|---|---|---|---|---|---|---|
| Descriptor ML | RF/DNN + ECFP [35] | Descriptors, fingerprints | Partial | Medium | Low | Indirect: Tox21 (BA 0.58–0.82) |
| Descriptor DL | DeepTox [7] | ECFP + toxicophores | No | Low | Medium | Indirect: Tox21 Challenge winner |
| GNN | MPNN [8] | Molecular graph | No | Low | Medium | None: QM9 only |
| GNN | D-MPNN [9] | Directed graph + RDKit | No | Low | Medium | Indirect: MoleculeNet scaffold split |
| GNN | AttentiveFP [10] | Graph + attention | No | High | Medium | None: not on DeepSnap endpoints |
| 2D Image | Chemception [11] | 2D structure drawings | No | Low | Low | Indirect: Tox21 different splits |
| 2D Image | ImageMol [12] | 2D images (ResNet-18) | No | Medium | High | Indirect: BBBP overlap |
| 3D Voxel | AtomNet [41] | Voxelized complex | Yes | Low | High | None: structure-based task |
| 3D Voxel | Ragoza et al. [42] | Voxelized densities | Yes | Low | High | None: structure-based task |
| DeepSnap | DeepSnap-DL [13,14] | 3D molecular images | Yes | Low | Medium | Reference method |
| DeepSnap Ens. | DeepSnap + ML [17] | Hybrid: images + descriptors | Yes | Medium | Medium | Reference method |
These comparisons highlight several strengths and weaknesses of DeepSnap relative to other molecular property prediction approaches. Along the axis of input representation, DeepSnap occupies a distinctive position: it requires no explicit feature engineering (unlike descriptor-based methods), encodes three-dimensional conformational information (unlike two-dimensional graph or two-dimensional image methods), and avoids the cubic memory scaling of voxel-based approaches. On data efficiency, DeepSnap has been applied to datasets ranging from 201 to approximately 12,000 compounds [17,26], which is modest compared with the 10-million-molecule pretraining corpus used by ImageMol [12]. With respect to interpretability, graph attention methods such as AttentiveFP provide atom-level attention maps that directly identify substructural contributions [10], whereas DeepSnap has explored CAM-family visualization in S-COPHY [18] and the AI-SHIPS platform [33,34], atom-level attribution from pixel-space gradients remains undeveloped. The fixed image resolution of DeepSnap may also result in information loss at the pixel level for large or complex molecules, and the method does not currently support atom-level gradient analysis in the way that graph-based approaches do. In terms of computational cost, DeepSnap requires generation of multiple images per molecule and training of a large CNN, which is more expensive than descriptor-based ML but less expensive than three-dimensional voxel approaches. Based on the currently available evidence, the image-based approach appears to perform well for toxicological endpoints, though the number of cross-method comparisons remains too small to draw general conclusions about task-specific advantages. What can be stated with greater confidence is that DeepSnap provides complementary information to descriptor-based methods, as demonstrated by the consistent improvement observed in ensemble and consensus models across all tested endpoints [17].
9. Future Directions
The body of work reviewed in the preceding sections has established the DeepSnap-DL approach as a viable molecular-image-based QSAR methodology with demonstrated utility for toxicological endpoints and selected ADME/PK parameters. However, several limitations identified throughout this series of studies point toward concrete areas where further development could substantially strengthen the approach.
Expanding the Endpoint Repertoire
DeepSnap-DL has been applied primarily to nuclear receptor and stress-response endpoints from the Tox21 10K library [14,15], rat clearance [16,30], and a limited set of additional drug screening endpoints [17]. Many ADME and toxicological endpoints of pharmaceutical relevance remain untested, including metabolic stability, plasma protein binding, hERG channel inhibition, hepatotoxicity, and genotoxicity. The S-COPHY model demonstrated that the molecular image concept could be adapted to chemical categorization tasks beyond toxicological activity [18], and its authors noted the potential for expanding the model to include additional compound categories [18].
Hybrid Image-Graph Models
Graph neural networks operate on 2D molecular graph representations [22], whereas DeepSnap encodes 3D structural information that graph-based methods typically lack. The ensemble strategy already demonstrated in the DeepSnap series [16,17] provides a precedent for multi-representation integration that could be extended to incorporate graph-derived features. Whether a tighter integration at the feature level, analogous to the probability-as-descriptor approach used for clearance regression [30], could yield further improvements when graph features replace or supplement conventional descriptors remains an open question.
Systematic Interpretability Analysis
The interpretability of DeepSnap-DL models has been identified as a persistent limitation [2,22]. The S-COPHY study provided peer-reviewed Grad-CAM visualizations for a DeepSnap-type model [18], and in the AI-SHIPS project, ScoreCAM and GuidedGradCAM were implemented for CYP inhibition models [33,34]. However, these examples remain limited to specific endpoints and architectures. Future work should extend explainability analyses beyond the currently documented CAM-family visualizations and apply additional techniques discussed by Uesawa—including LIME, SHAP, Anchor, and integrated gradients [2]—to DeepSnap molecular images across a wider range of endpoints and CNN architectures.
Reducing Hyperparameter Sensitivity
The DeepSnap-DL pipeline requires optimization of numerous parameters, including six rendering parameters, viewing angles, learning rate, batch size, and epoch count [21,22]. The requirement for per-endpoint tuning remains a practical barrier to high-throughput application. Automated hyperparameter optimization approaches could systematically identify effective parameter configurations while reducing the manual tuning burden. The quadratic relationship between rendering parameters and validation loss [21] suggests that the parameter landscape may be sufficiently smooth for such automated approaches to be effective.
Regression Without Classification Intermediary
The regression extension for clearance prediction relied on a two-stage procedure in which a classification model first generated prediction probabilities that were then used as features in a separate regression model [30]. Direct regression from molecular images, without the classification intermediary, would simplify the pipeline and eliminate the threshold dependency, though it would require modifications to the CNN output layer and training procedure.
Multi-Task Learning
The current DeepSnap-DL framework constructs independent models for each endpoint. Given that many Tox21 endpoints share molecular determinants, multi-task learning architectures that predict multiple endpoints simultaneously could improve prediction efficiency and potentially benefit data-scarce endpoints through shared representation learning. The 59 MIE models [15] and the 35 nuclear receptor models [14] represent natural candidates for multi-task reformulation, where a shared CNN backbone could learn general molecular image features while endpoint-specific heads capture target-dependent patterns.
10. Conclusions
DeepSnap is a descriptor-free QSAR approach that converts three-dimensional molecular structures into image representations and feeds them into convolutional neural networks for biological activity and property prediction. Since its introduction in 2018, the method has been systematically developed through a series of studies that optimized rendering parameters, CNN architectures, and combination strategies across toxicological and pharmacokinetic endpoints.
The key quantitative achievements of the DeepSnap series include a mean AUC of 0.884 across 35 nuclear receptor agonist and antagonist endpoints from the Tox21 10K library [14], individual endpoint AUCs exceeding 0.99 for CAR agonist [23] and PR antagonist [25], and ensemble AUCs of 0.943 for rat hepatic clearance classification [16] and 0.936 for blood–brain barrier penetration [17]. A recurring and arguably the most important finding is the complementarity between molecular image-based and descriptor-based predictions: ensemble models that combine both approaches have consistently outperformed either method alone across all tested endpoints [16,17,30], and the image-derived prediction probability emerged as the single most important feature in a clearance regression model [30]. This complementarity supports the view that three-dimensional molecular images encode structural information that is at least partially orthogonal to conventional physicochemical descriptors.
Several limitations constrain the current evidence base. No head-to-head comparison with graph neural network architectures on the same datasets and identical splits has been reported, leaving the relative merits of image-based and graph-based molecular representations empirically unresolved [22]. The interpretability of DeepSnap-DL models is developing, with peer-reviewed Grad-CAM visualizations reported in S-COPHY [18] and CAM-family visualization implemented in the AI-SHIPS platform [33,34], though systematic application across endpoints remains pending. The requirement for per-endpoint hyperparameter optimization, including six rendering parameters and multiple training hyperparameters [21], presents a practical barrier to high-throughput deployment. Class imbalance in the Tox21 datasets substantially affects threshold-dependent classification metrics [14], and several studies relied on proprietary datasets that preclude independent reproduction [16,26].
Future priorities for DeepSnap development include systematic application of explainable AI methods across endpoints and architectures, controlled benchmarking against graph neural networks on standardized datasets [36], automated hyperparameter optimization to reduce per-endpoint tuning burden, and expansion to pharmacological and toxicological endpoints not yet examined. The consistent benefit of ensemble strategies suggests that the greatest practical impact of DeepSnap may lie not as a standalone method but as a complementary component within multi-representation prediction systems.
Author Contributions
Conceptualization, literature review, writing—original draft preparation, writing—review and editing, Y.U. The author has read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated in this review. Data and performance values discussed herein are available in the cited publications.
Acknowledgments
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
Generative AI Disclosure
During the preparation of this manuscript, the author used Claude Opus 4.7 (Anthropic) for assistance in creating and refining parts of the figures and the graphical abstract. The author reviewed, edited, and verified all AI-assisted graphical outputs, including scientific content, chemical representations, labels, and numerical values, and takes full responsibility for the content of this publication.
References
- Uesawa, Y. AI-based QSAR Modeling for Prediction of Active Compounds in MIE/AOP. Yakugaku Zasshi 2020, 140, 499–505. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Deep learning using molecular image of chemical structure. In Cheminformatics, QSAR and Machine Learning Applications for Novel Drug Development; Roy, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2023; Chapter 18, pp. 473–501. [CrossRef]
- Huang, R.; Xia, M.; Nguyen, D.-T.; Zhao, T.; Sakamuru, S.; Zhao, J.; Shahane, S.A.; Rossoshek, A.; Simeonov, A. Tox21 Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci. 2016, 3, 85. [CrossRef]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; Serrano, J.A.; Tietge, J.E.; Villeneuve, D.L. Adverse Outcome Pathways: A Conceptual Framework to Support Ecotoxicology Research and Risk Assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; Consonni, V.; Kuz’min, V.E.; Cramer, R.; Benigni, R.; Yang, C.; Rathman, J.; Terfloth, L.; Gasteiger, J.; Richard, A.; Tropsha, A. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. In Proceedings of the 34th International Conference on Machine Learning (ICML 2017), PMLR 70:1263–1272, 2017. arXiv:1704.01212.
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; Palmer, A.; Settels, V.; Jaakkola, T.; Jensen, K.; Barzilay, R. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [CrossRef]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; Zheng, M. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2020, 63, 8749–8760. [CrossRef]
- Goh, G.B.; Hodas, N.O.; Siegel, C.; Vishnu, A. Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models. arXiv preprint 2017, arXiv:1706.06689.
- Zeng, X.; Xiang, H.; Yu, L.; Wang, J.; Li, K.; Nussinov, R.; Cheng, F. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 2022, 4, 1004–1016. [CrossRef]
- Uesawa, Y. Quantitative structure-activity relationship analysis using deep learning based on a novel molecular image input technique. Bioorg. Med. Chem. Lett. 2018, 28, 3400–3403. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Molecular Image-Based Prediction Models of Nuclear Receptor Agonists and Antagonists Using the DeepSnap-Deep Learning Approach with the Tox21 10K Library. Molecules 2020, 25, 2764. [CrossRef]
- Matsuzaka, Y.; Totoki, S.; Handa, K.; Shiota, T.; Kurosaki, K.; Uesawa, Y. Prediction Models for Agonists and Antagonists of Molecular Initiation Events for Toxicity Pathways Using an Improved Deep-Learning-Based Quantitative Structure-Activity Relationship System. Int. J. Mol. Sci. 2021, 22, 10821. [CrossRef]
- Mamada, H.; Nomura, Y.; Uesawa, Y. Prediction Model of Clearance by a Novel Quantitative Structure-Activity Relationship Approach, Combination DeepSnap-Deep Learning and Conventional Machine Learning. ACS Omega 2021, 6, 23570–23577. [CrossRef]
- Mamada, H.; Takahashi, M.; Ogino, M.; Nomura, Y.; Uesawa, Y. Predictive Models Based on Molecular Images and Molecular Descriptors for Drug Screening. ACS Omega 2023, 8, 37186–37195. [CrossRef]
- Hisaki, T.; Yoshida, K.; Nukaga, T.; Iwanaga, S.; Mori, M.; Uesawa, Y.; Sekine, S.; Tamura, A. S-COPHY: A deep learning model for predicting the chemical class of compounds as cosmetics or pharmaceuticals based on single 3D molecular images. Comput. Toxicol. 2024, 30, 100311. [CrossRef]
- Uesawa, Y. A Novel Molecular Information Input Method for Deep Learning: Deep Snap. CICSJ Bull. 2018, 36, 51. [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. In Advances in Neural Information Processing Systems 25 (NeurIPS 2012); Pereira, F., Burges, C.J.C., Bottou, L., Weinberger, K.Q., Eds.; Curran Associates: Red Hook, NY, 2012; pp. 1097–1105.
- Matsuzaka, Y.; Uesawa, Y. Optimization of a Deep-Learning Method Based on the Classification of Images Generated by Parameterized Deep Snap a Novel Molecular-Image-Input Technique for Quantitative Structure-Activity Relationship (QSAR) Analysis. Front. Bioeng. Biotechnol. 2019, 7, 65. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Computational Models That Use a Quantitative Structure-Activity Relationship Approach Based on Deep Learning. Processes 2023, 11, 1296. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. Int. J. Mol. Sci. 2019, 20, 4855. [CrossRef]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going Deeper with Convolutions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015; pp. 1–9. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. DeepSnap-Deep Learning Approach Predicts Progesterone Receptor Antagonist Activity With High Performance. Front. Bioeng. Biotechnol. 2020, 7, 485. [CrossRef]
- Matsuzaka, Y.; Hosaka, T.; Ogaito, A.; Yoshinari, K.; Uesawa, Y. Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap-Deep Learning. Molecules 2020, 25, 1317. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. A Deep Learning-Based Quantitative Structure-Activity Relationship System Construct Prediction Model of Agonist and Antagonist with High Performance. Int. J. Mol. Sci. 2022, 23, 2141. [CrossRef]
- Kurosaki, K.; Wu, R.; Uesawa, Y. A Toxicity Prediction Tool for Potential Agonist/Antagonist Activities in Molecular Initiating Events Based on Chemical Structures. Int. J. Mol. Sci. 2020, 21, 7853. [CrossRef]
- Mansouri, K.; Karmaus, A.; Fitzpatrick, J.; Patlewicz, G.; Pradeep, P.; et al. CATMoS: Collaborative Acute Toxicity Modeling Suite. Environ. Health Perspect. 2021, 129, 47013. [CrossRef]
- Mamada, H.; Nomura, Y.; Uesawa, Y. Novel QSAR Approach for a Regression Model of Clearance That Combines DeepSnap-Deep Learning and Conventional Machine Learning. ACS Omega 2022, 7, 17055–17062. [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Ensemble Learning, Deep Learning-Based and Molecular Descriptor-Based Quantitative Structure-Activity Relationships. Molecules 2023, 28, 2410. [CrossRef]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. In Proceedings of the IEEE International Conference on Computer Vision (ICCV), Venice, Italy, 22–29 October 2017; pp. 618–626. [CrossRef]
- Ministry of Economy, Trade and Industry (METI). Technology Evaluation Report (Final Evaluation) for the Project on the Development of Evaluation Technologies for Energy-Saving Electronic Device Materials (Development of High-Speed, High-Efficiency Safety Evaluation Technologies Supporting the Social Implementation of Functional Materials) [in Japanese]; METI: Tokyo, Japan, 2023. Available online: https://www.meti.go.jp/policy/tech_evaluation/e00/03/r04/600.pdf (accessed on 6 April 2026).
- Ministry of Economy, Trade and Industry (METI). Supplementary Explanatory Materials for the Final Evaluation of the Project on the Development of Evaluation Technologies for Energy-Saving Electronic Device Materials (Development of High-Speed, High-Efficiency Safety Evaluation Technologies Supporting the Social Implementation of Functional Materials) [in Japanese]; METI: Tokyo, Japan, 2023. Available online: https://www.meti.go.jp/shingikai/sankoshin/sangyo_gijutsu/kenkyu_innovation/hyoka_wg/pdf/064_h02_00.pdf (accessed on 6 April 2026).
- Capuzzi, S.J.; Politi, R.; Isayev, O.; Farag, S.; Tropsha, A. QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci. 2016, 4, 3. [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [CrossRef]
- Chen, D.; Gao, K.; Nguyen, D.D.; Chen, X.; Jiang, Y.; Wei, G.-W.; Pan, F. Algebraic Graph-Assisted Bidirectional Transformers for Molecular Property Prediction. Nat. Commun. 2021, 12, 3521. [CrossRef]
- Sheridan, R.P. Time-Split Cross-Validation as a Method for Estimating the Goodness of Prospective Prediction. J. Chem. Inf. Model. 2013, 53, 783–790. [CrossRef]
- Kaboudi, N.; Shayanfar, A. Predicting the Drug Clearance Pathway with Structural Descriptors. Eur. J. Drug Metab. Pharmacokinet. 2022, 47, 363–369. [CrossRef]
- Lombardo, F.; Obach, R.S.; Varma, M.V.; Stringer, R.; Berellini, G. Clearance Mechanism Assignment and Total Clearance Prediction in Human Based upon in Silico Models. J. Med. Chem. 2014, 57, 4397–4405. [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv preprint 2015, arXiv:1510.02855.
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [CrossRef]
Figure 1.
Conceptual illustration of multi-angle snapshot acquisition in DeepSnap. A single three-dimensional molecular conformer is viewed from multiple directions, producing distinct two-dimensional projected molecular images. These orientation-dependent snapshots collectively provide the convolutional neural network with structurally diverse visual inputs derived from the same molecule.
Figure 1.
Conceptual illustration of multi-angle snapshot acquisition in DeepSnap. A single three-dimensional molecular conformer is viewed from multiple directions, producing distinct two-dimensional projected molecular images. These orientation-dependent snapshots collectively provide the convolutional neural network with structurally diverse visual inputs derived from the same molecule.

Figure 2.
Schematic of the DeepSnap pipeline. SMILES strings are converted to three-dimensional conformers, rendered as color-coded ball-and-stick images from multiple viewing angles, and classified by a CNN with transfer learning from ImageNet. Adapted from [21].
Figure 2.
Schematic of the DeepSnap pipeline. SMILES strings are converted to three-dimensional conformers, rendered as color-coded ball-and-stick images from multiple viewing angles, and classified by a CNN with transfer learning from ImageNet. Adapted from [21].

Figure 3.
Timeline of DeepSnap development (2018–2024). Blue: first-generation DIGITS/Caffe/Jmol pipeline; orange: second-generation TensorFlow/Keras/PyMOL pipeline; green: S-COPHY industrial variant. Milestone annotations indicate key publications and methodological advances.
Figure 3.
Timeline of DeepSnap development (2018–2024). Blue: first-generation DIGITS/Caffe/Jmol pipeline; orange: second-generation TensorFlow/Keras/PyMOL pipeline; green: S-COPHY industrial variant. Milestone annotations indicate key publications and methodological advances.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



