Submitted:
24 April 2026
Posted:
27 April 2026
You are already at the latest version
Abstract
This review summarizes mechanisms regulating mRNA translation under cellular stress and highlights design strategies to improve translation efficiency and stability in the gene therapy of human diseases. mRNA-based therapeutics are emerging as a versatile gene therapy platform enabling transient and controllable expression of therapeutic for the treatment of cancer, genetic disorders, and inflammatory diseases. The efficacy of mRNA-based gene therapy is strongly influenced by sequence design, chemical modifications, and structural features. Evidence shows that rational mRNA engineering can significantly enhance translation efficiency even under stress conditions that impair canonical protein synthesis, as observed in many pathological states. Cellular stress activates regulatory pathways that suppress global translation; however, optimized mRNA constructs can partially bypass these inhibitory mechanisms, enabling sustained protein expression. By improving mRNA stability and resistance to stress-responsive translational control, robust therapeutic protein production can be achieved even in challenging cellular environments. These advances position mRNA engineering as a promising component of next-generation gene therapy, offering new opportunities for effective treatments of human diseases.
Keywords:
mRNA therapeutics
; cellular stress
; alternative translation
; mRNA design
; innovative therapy
1. Introduction
Over the past decade, owing to technological innovations and research investments, mRNA has emerged as a promising therapeutic tool in the field of vaccine development and replenishment of missing proteins. As this approach allows for fast and efficient optimization of mRNA sequences, these medicines have the potential to revolutionize both the prevention and treatment of diseases. The use of mRNA has several advantages over killed and live attenuated viruses used in traditional vaccines [1] First, safety: because mRNA is a non-infectious molecule that does not integrate into genomic DNA, there is no risk of potential infection or insertional mutagenesis [2]. Furthermore, mRNA is degraded by physiological cellular processes; its half-life can be regulated by the structure of the coding sequence of the mRNA itself, as well as by delivery methods [3]. The inherent immunogenicity of mRNA can be reduced to further improve its safety profile [4]. Second, efficacy: various modifications stabilize the molecule and ensure high translation. In vivo delivery can be achieved by formulating mRNA in the carrier molecules, enabling rapid uptake and mRNA expression in the cytoplasm [5]. Third, mRNA vaccines can be manufactured rapidly, inexpensively, and scalably [6].
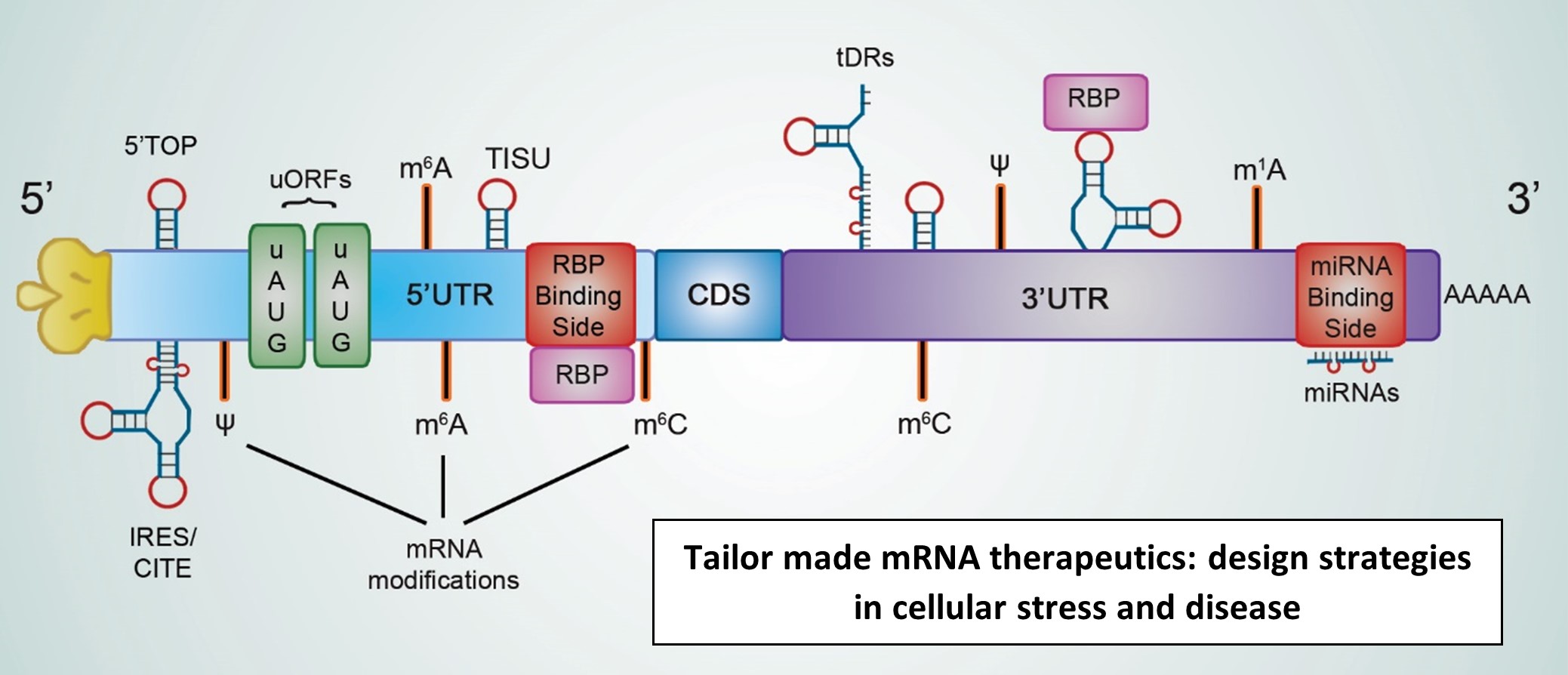
The mRNA sequence consists of five key elements. First, a protective molecule at the 5’end called cap, which consists of an atypical nucleoside, 7-methylguanosine, joined by a 5’,5’-triphosphate linkage to the 5’end of the mRNA chain, protects the mRNA from premature degradation by 5’exonuclease [7]. The cap is involved in many other biological functions including maturation, transport, and translation initiation. The 5′untranslated region (5’UTR) plays a crucial role in the regulation of translation processes, as it has an impact on its stability, localization, and translation initiation by interacting with ribosomes and initiation factors [8,9,10]. Immediately after the 5’UTR, a protein-coding sequence is located, and the 3’ untranslated region (3’UTR) is placed behind the stop codon. The 3’UTR is typically longer than the 5’UTR; it influences mRNA stability, translation efficiency, and cellular localization as it interacts with RNA binding proteins and microRNAs [11,12]. The last element of the mRNA sequence is a 3’poly-(A) tail that contributes to the mRNA translational status and decay as a result of interaction with poly(A)-binding proteins and deadenylases [13,14].
It might seem that designing an mRNA therapeutic is conceptually simple; however, in reality, it requires meeting several different, often contradictory, requirements to create a highly effective sequence that fits the desired application. Maximizing protein expression from delivered mRNA molecules depends on its half-life in the target tissue and translational potential, which is related to the number of ribosomes scanning and transiting mRNA [15]. However, ribosome overload leads to translation mRNA decay, lowering total protein yield and eliciting toxic effects; thus, appropriate ribosome loading is highly needed [16]. Other factors include ease and consistency of manufacturing, as well as regulation of mRNA shelf life, both unformulated and in its delivery vehicle [17]. Because mRNA molecules are inherently unstable and susceptible to degradation, extending mRNA stability is one of the key requirements for this group of therapeutics.
The central design consideration in mRNA medicine is its nucleotide sequence, which affects the structure, function, and stability of mRNA. The interplay between selected codon triplets, RNA secondary and tertiary structures, and nucleotide modification is crucial for the fulfillment of the target therapeutic profile [18]. For all organisms and even tissues, codon biases exist; more frequently used ones correlate with the abundance of tRNAs [19]. Mammalian mRNA sequences are generally GC-rich, with a strong emphasis on C as the third nucleotide in the codon. This phenomenon leads to increased mRNA stability and secondary structure formation [20]. Next, the spatial structure of the mRNA can positively or negatively influence the entire translation process. mRNA forms complex secondary structures as a result of intramolecular base pairing, which enables the binding of protein complexes and influences storage longevity [3,18].
In addition to optimizing the protein-coding sequence, appropriate selection of UTRs modulate translation towards increasing or decreasing translation efficiency. Unstructured 5’UTRs, in general, ensure better translation initiation, [21] suggesting the simplest design of these elements; however, rapid initiation leads to faster mRNA decay, affecting total protein expression [22]. The selection of the appropriate cap analog is also important as the interaction of initiation factors with the 7-methylguanosine cap is one of the rate-limiting steps in the entire translation process [23]. The poly-(A) tail is another mRNA design that influences stability and translation efficacy.
Translation modulation can be achieved by engineering mRNA sequences adapted to the cell state. This is especially important in the case of mRNA medicines for the treatment of certain diseases, where mRNA is translated by cells under various stress conditions. As changes in translation are a key feature of many diseases such as cancer, viral infections, and myocardial hypertrophy, [24] designing mRNA therapeutics whose translation involves mechanisms induced by cellular stress may lead to an expression advantage when canonical translation is inhibited. [25] In this review, the principles of translation control under cellular stress are described, with an emphasis on mRNA design elements that can boost exogenous mRNA expression in the context of stress-induced changes in the translatome.
2. Canonical Mode of mRNA Translation
To better understand the mechanisms of translational adaptation to cellular stress, it is essential to summarize the processes underlying mRNA translation under normal conditions. Protein synthesis is a periodic phenomenon that can be divided into three main stages: initiation of mRNA translation, elongation of the polypeptide chain, and termination via ribosome recycling [25,26]. All steps are tightly regulated; however, most regulatory events occur at the initiation phase, as they require the coordinated action of multiple factors collectively called messenger ribonucleoproteins (eukaryotic initiation factors, ribosomes, mRNAs, and tRNAs). Translation initiation is also a rate-limiting phase [27]; when translation enters the elongation step, ribosomes are obliged to complete the synthesis of the developing protein to avoid the formation of the potentially dangerous, incomplete polypeptide.
Protein synthesis begins when 40S and 60S ribosomal subunits assemble as qualified 80S ribosomes on the start codon of mRNA. During this process, numerous steps occur that are mediated by multiple eukaryotic initiation factors (eIFs) and the initiator . The translation initiation begins with the formation of 43S pre-initiation complex (43S PIC). Additionally, eIF2B as a guanine nucleotide exchange factor (GEF), commands TC assembly by transforming eukaryotic initiation factor 2-GDP (eIF2–GDP) into the active eIF2–GTP complex before each round of translation. eIF4E directly binds the 5′-terminal m7G cap of the transcripts and activates an mRNA for translation, facilitating the interaction of the scaffold protein eIF4G, DEAD-box RNA helicase eIF4A, and helicase stimulator eIF4B to form the eIF4F complex [26,28]. The 43S PIC scans the 5’UTR of an mRNA in the 5’ to 3’ direction, searching for the start codon. In the canonical mode of translation initiation, translation begins with AUG codon surrounded by a specific Kozak sequence [29,30].
ATP hydrolysis is used by eIF4A to unroll the secondary structures of mRNA and move the ribosome in the 3’direction. 5’UTR scanning usually stops when the 43S PIC reaches the first AUG codon and efficient codon-anticodon pairing occurs. Eukaryotic cells require GTP hydrolysis by eIF2 and eIF5B to initiate translation. eIF5B-GTP binding to the 40S subunit stimulates the 60S ribosome subunit connection to form the 80S ribosome, ready for the next step of translation, decoding the message [31,32]. Figure 1 shows schematic representation of cannonical mode of translation initiation.
3. mRNA Translation Initiation in Cellular Stress and Disease
Inhospitable environmental conditions under which multicellular life evolves and persists require specialized mechanisms for the efficient repair of molecular damage. This pressure allows all cells to adapt to adverse environmental conditions. Upon exposure to physiological intra- and extracellular stress stimuli, eukaryotic cells activate an adaptive pathway called the integrated stress response (ISR), which reprograms cellular metabolism and focuses on maintaining homeostasis [33] ISR is activated under most types of stress stimuli, including endoplasmic reticulum (ER) stress (which induces the unfolded protein response [34]), hypoxia [35], nutrition deprivation [36] , heat shock [37], viral infection [38], oxidative stress [39], UV irradiation [40], and proteasome inhibition [41].
4. Cellular Pathways Involved in Stress Responses
4.1. Stress-Induced eIF2a Phosphorylation and Disease Development
Upon activation of the integrated stress response by numerous stimuli, eIF2α (a subunit of eIF2) is phosphorylated, leading to the inhibition of global mRNA translation and general protein synthesis shutdown. By lowering global translation rates, cells reorganize their proteomes to allow their death or survival. While the translation of most mRNAs is entirely inhibited (such as transcripts of housekeeping genes), eIF2α phosphorylation causes translation upregulation of certain mRNAs encoding pro-survival and stress-responsive proteins. Under conditions of prolonged cellular stress and irreparable damage, the ISR program, as a response to acute cellular stress, may transform into chronic stress, leading to many cell pathologies and diseases [34,35]. In mammalian cells, there are four different types of eIF2a kinases: general control non-derepressible-2 (GCN2/EIF2AK4) [42], PKR-like ER kinase (PERK/EIF2AK3) [43], protein kinase RNA (PKR/ EIF2AK2) [44], and heme-regulated inhibitor kinase (HRI/ EIF2AK1) [45] 5′UTR features, particularly upstream open reading frames (uORFs), critically regulate selective mRNA translation during cellular stress. Phosphorylation of eIF2α by stress-activated kinases globally represses translation while enabling preferential translation of uORF-containing transcripts through reinitiation mechanisms, exemplified by ATF4 and related stress-responsive mRNAs [26] A further 5′UTR-based mechanism involves internal ribosome entry sites (IRESs), which enable cap-independent translation initiation during stress. When eIF2α phosphorylation suppresses global cap-dependent translation, IRES elements recruit ribosomes independently of eIF4E, allowing continued synthesis of stress-response proteins such as BiP and Apaf-1 [46]. Another 5′UTR feature is the presence of RNA secondary structures and specific RNA-binding protein motifs. During eIF2α phosphorylation, stress-induced RBPs (e.g., HuR or TIA-1) remodel structured 5′UTRs to selectively promote or repress translation of target mRNAs, enabling stress-adaptive proteome reprogramming [47]. Figure 2 illustrates eIF2α phosphorylation and the activation of the integrated stress response (ISR) program.
4.2. Regulation of eIF4E Activity Under Cellular Stress
eIF4E plays an essential role in regulating the expression of proliferation and survival proteins; therefore, its action is strictly controlled. To date, several cellular mechanisms have been described in the context of eIF4E activity, including mammalian target of rapamycin (mTOR) signaling through eIF4E-binding proteins (4E-BPs), phosphorylation of eIF4E by Mnk, and stimulation of eIF4E transcription by Myc.
4.2.1. The mTOR Pathway
The activation or inhibition of eIF4E is led by proteins that compete for binding to the residues on the opposite side of its cap-binding pocket. The eIF4E dorsal surface can bind EIF4G to stimulate translation or eIF4E-binding proteins (4E-BPs) and inhibit translation. In mammalian cells, 4E-BP1, 2 and 3 abolish eIF4F complex formation by blocking eIF4G binding to eIF4E [48,49]. When the dorsal surface of eIF4E is blocked by 4E-BPs, eIF4G cannot induce structural changes in the loops near the cap-binding site; thus, eIF4E orientation is incapable of cap-binding [50].
Regulation of 4E-BPs activity in the cell is carried out by phosphorylation via the mechanistic target of rapamycin complex (mTORC) pathway. It consists of two different protein complexes, mTORC1 and mTORC2. The first one, under physiological conditions, phosphorylates 4E-BPs, blocking their binding to eIF4E [49]. However, in stress responses, such as hypoxia or low energy, mTORC1 is inactivated and 4E-BPs are dephosphorylated. The second class of mTORC1 target—S6 kinases (S6Ks) phosphorylate ribosomal protein S6 and eIF4B[51]. S6Ks upon activation by mTORC1 promote translation of mRNAs necessary for biosynthesis, such as ribosomal proteins, elongation factors and poly(A)-binding protein. Its role is also inhibition of eukaryotic translation elongation factor 2 kinase (eEF2K), which increase eEF2 activity and elongation during protein synthesis under external stimuli by growth factors [52].
Upstream regulator of mTORC1 activity is class I phosphoinositide 3-kinase (PI3K), a key signaling enzyme activated by secreted ligands including hormones (such as insulin), growth factors, cytokines, and chemokines [53]. Another signaling pathway controlled by mTORC1 involves 5′-adenosine monophosphate-activated protein kinase (AMPK), which is essential for cellular metabolism and growth [54].
mRNAs most sensitive to eIF4E availability upon mTOR belong to specific classes defined by distinct 5′UTR features. Canonical examples include 5′terminal oligopyrimidine (5′TOP) and TOP-like mRNAs encoding ribosomal proteins and translation factors, whose translation is acutely suppressed when mTOR–4E-BP signaling is inactive [55]. In addition, mRNAs with long, GC-rich, highly structured 5′UTRs, such as MYC, CCND1, VEGFA, and ODC1, exhibit strong dependence on eIF4E and are preferentially downregulated when cap-dependent initiation is limited [10]. These findings underscore that mTOR signaling selectively controls translation of defined mRNA subsets rather than uniformly regulating protein synthesis. In a Figure 3, the eIF4E activity regulation in stress response is presented.
5. mRNA Specific Regulation
5.1. Trans-Acting Factors Regulate mRNA Fate in Cellular Stress Response
Trans-acting factors are proteins and RNAs that recognize specific elements in mRNA molecules and facilitate rapid changes in gene expression in response to cellular stress. Diverse mechanisms are involved in the action of RNA-binding proteins and non-coding regulatory RNAs such as microRNAs and tRNA-derived RNAs [56,57,58,59].
5.1.1. RNA-Binding Proteins (RBPs)
RBPs play an important role in the mRNA life cycle [60]. This is implemented by interacting with sequence elements in the 5′ and 3′ untranslated regions and relies on the recruitment of mRNAs to the ribosome and regulation of protein synthesis or, alternatively, repression of mRNA translation by regulating mRNA instability and decay [61]. Under oxidative stress, there is a strong association between transcripts that are translationally repressed and those associated with the RNA-binding protein PUF3p, a well-known regulator of proteins targeted to mitochondria. PUF-3p–responsive elements are short UGUA-containing motifs in the 3′UTR that confer stress-dependent regulation of mRNA stability and translation; avoiding these elements can therefore allow therapeutic mRNAs to escape oxidative stress–induced translational shutdown [62]. In turn, upon DNA damage, RBP HuR/ELAV1 reduces its binding to mRNA partners MDM2 and BAX, which enhances cell survival [63]. Given that HuR recognizes AU-rich elements (AREs) in 3’UTR, insertion of these motifs into therapeutic mRNA may enhanced translation compared to non-ARE mRNAs [64].
5.1.2. MicroRNAs
MicroRNAs are short RNAs consisting of around 22 nucleotides that modulate the translational potential of their target mRNA [65]. Mature miRNAs bind to the 3’UTR via the seed region. The degree of complementarity determines cleavage (exact matching) or translation repression (partial matching). Multiple forms of stress, including hypoxia, DNA damage, ER stress, and inflammatory signaling, alter miRNA transcription, processing, and target engagement [58,66]. For therapeutic mRNAs delivered into stressed tissues, these dynamics present an opportunity: rather than attempting to override stress-induced translational repression globally, mRNA constructs can be designed to cooperate with endogenous miRNA stress responses. One of the most consistently observed features of cellular stress is the selective downregulation of specific miRNAs. In cancer and ischemic tissues, hypoxia can reduce global miRNA biogenesis through inhibition of Drosha and Dicer, leading to broad attenuation of miRNA-mediated repression [67]. From a therapeutic design perspective, inclusion of target sites for stress-downregulated miRNAs in the 3′UTR of a therapeutic mRNA can create a conditional expression switch. Under stress, loss of the miRNA relieves repression, resulting in relative translational enhancement [68]. Although miRNAs are most often associated with repression, several studies have demonstrated that miRNAs can activate translation in specific cellular states. For example, miR-369-3 directs translational activation of TNF-α mRNA in quiescent cells by recruiting AGO2 and FXR1 to AU-rich elements [69].
5.1.3. tRFs and tiRNAs
Other non-protein-coding RNA are tRNA fragments that serve as precursors for subsequent cleaving agents, generating two classes of functional RNA fragments: 17–26-nucleotide long tRNA-derived RNA fragments (tRFs) and tRNA-derived stress-induced RNAs (tiRNAs) [70]. It has been previously reported that ANG cleaves tRNAs nonspecifically during cellular stress, such as UV irradiation, heat shock, arsenite treatment, nutrition deficiency, hypoxia, and hypothermia [71,72]. While many tiRNAs repress global cap-dependent translation, specific tRFs modulate translation in a sequence-, context-, and RBP-dependent manner, including interactions with Argonaute proteins and stress-response RBPs [71,72] Mechanistic studies have demonstrated that tRNA-derived fragments can selectively repress translation of mRNAs containing specific UTR motifs by displacing RNA-binding proteins such as YBX1 under stress conditions, suggesting that incorporation of tRF- or tiRNA-responsive elements into therapeutic mRNA UTRs could be exploited to modulate translation during stress adaptation [73].
5.2. Specific cis-Acting Features of mRNA Control the Rate and Mode of Translation During Stress
Cells engage in a variety of mechanisms to achieve selective translation, involving special elements on mRNA. The majority of cis-elements are sequence fragments in the untranslated regions of mRNA and mainly consist of 5ʹcap, 5ʹUTR and 3ʹUTR. Together, these fragments influenced both the expression level and degradation of mRNA [74].
5.2.1. Stress-Induced mRNA Modification
mRNA can be covalently modified during post-transcriptional processes. Together with their coordinators, writers, erasers, and readers, these modifications serve as signaling machines that regulate cellular stress responses and play important roles in disease pathogenesis, including metabolic diseases, cancer, and neurological disorders [75]. m6A modifications on 5′UTRs or coding sequences can recruit the m6A “reader” YTHDF1, enhancing cap-independent translation. Incorporating strategic m6A consensus motifs (DRACH) into the 5′UTR or coding sequence of therapeutic mRNAs can promote preferential translation under stress, when eIF4E-dependent cap-mediated initiation is suppressed, such as during inflammation or viral infection [76]. In line, site-specific 2′-O-methyl modifications in UTRs or coding regions of therapeutic mRNAs, may protect from degradation and improving ribosome upon oxidative stress and UV damage [77].
5.2.2. Upstream Open Reading Frames (uORFs)
Upstream open reading frames (uORFs) and upstream AUGs (uAUGs) are present in the 5’ UTR of mRNAs in approximately half of the human and mouse transcripts that encode regulatory proteins. The vast majority of uORFs serve as regulatory cis-elements, usually reducing translation of the main coding region under physiological conditions [78]. One of the best-known transcripts containing uORFs in mammalian cells are ATF4, ATF5, CHOP, CEBPB, ATG5, and BiP [79,80,81,82,83]. By incorporating certain uORFs (e.g., in ATF4 mRNA) we allow reinitiation at the main coding sequence when initiation is limited due to eIF2α is phosphorylated during stress [84]. Additionally, engineering short, weakly conserved uORFs upstream of therapeutic ORFs can permit partial ribosome bypass (leaky scanning). Under stress conditions such as heat shock or oxidative stress, slowed ribosome scanning increases the likelihood of initiation at the main ORF [85]. The use of multiple uORFs establishes a tiered translational control mechanism during the integrated stress response, ensuring efficient main ORF translation only at defined stress thresholds and restricting expression under non-stress conditions [86].
5.2.3. 5’Terminal OligoPyrimidines (5’TOP) Motifs
The 5’TOP motif is a regulatory element of mRNA that begins immediately after the m7G cap containing 5’cytidine, followed by a continuous tract of 4-15 pyrimidines, together with a subsequent G-rich region [87]. The core set of the 5’TOP motif has been shown to be constitutively expressed in 16 human tissues [88] and is found in all human ribosomal proteins, together with non-ribosomal translation regulators such as eIF3, eIF4A, eEF2, and PABP (poly(A)-binding protein). Translation of 5′TOP mRNAs is controlled by mTOR signaling: active mTOR promotes translation, while stress-induced mTOR inhibition reduces translation thus incorporating 5′TOP motifs upstream of therapeutic ORFs enable coupling translation with mTOR activity: under moderate stress with partial mTOR activity, translation of therapeutic mRNAs can be preferentially maintained, mimicking endogenous ribosomal protein mRNA behavior [89,90]
5.2.4. Translation Initiator of Short 5′UTR (TISU)
TISU, or Translation Initiator of Short 5’UTR, is a regulatory element that controls the translation of mRNAs with very short 5’UTRs [91]. It is located downstream of the transcription start site (TSS) and contains a fixed ATG sequence that serves as the translation initiation codon in approximately two-thirds of the genes bearing it [92]. Engineering therapeutic mRNAs with a TISU element immediately downstream of the cap enable may enable robust translation even when global initiation factors are limited, especially in energy-stress–adaptive conditions [93].
5.2.5. Internal Ribosome Entry Site (IRES)
One of the best characterized mechanisms of cap-independent translation is the use of IRES elements. It relies on the binding of the 40S ribosomal subunit directly to the start codon through an IRES element in the 5’UTR region [94]. Approximately 15% of the total cell mRNA can be translated via the IRES mechanism; however, only about 100 transcripts contain IRES elements, suggesting that IRES-containing mRNAs are usually translated in a cap-dependent mechanism and switch to an IRES-dependent mechanism under stress [95]. By incorporating viral or cellular IRES upstream of the therapeutic mRNA ORF, the translation continues even in stress conditions such as hypoxia, viral infection or oxidative stress [96]. Certain IRES elements (e.g., VEGF, HIF-1α, c-Myc) are naturally more active under specific stresses, thus usage of stress-responsive cellular IRES allow control of therapeutic mRNA translation selectively in stressed cells [97]. By using IRES elements, a second ORF can be translated independently of cap-dependent initiation, permitting co-expression of a therapeutic protein alongside a reporter or chaperone from a single mRNA. This strategy is particularly useful for proteins that demand improved folding or modulation of immune responses [98].
5.2.6. Cap-independent translation enhancers (CITE)
CITE are RNA sequence elements, stimulating translation initiation without the need for the 5′cap. CITE contacts the 5’UTR of the same mRNA and recruits initiation complexes to the uncapped 5’UTR. Unlike IRESes, they require a free 5’end directing translation initiation from the 5’UTR in a cap-dependent manner and employ general ribosomal scanning to find the start codon, even if they bypass the direct participation of the eIF4F complex [99]. 5’CITE-like structures can bind eIF4F in a cap-independent manner, providing a moderate dependence on the 5’cap and making their translation, at least partially, resistant to stress-induced inactivation [100]. Viral or synthetic CITE-like RNA structures utilization in therapeutic mRNAs enable recruitment of initiation factors (e.g., eIF4G, eIF4A) directly to the mRNA, ensuring sustained translation despite global repression of cap-dependent initiation [101]. By embedding 3′UTR CITE motifs downstream of therapeutic coding sequences, especially in oxidative stress, viral infection or inflammation, translation may persists when 5′end–dependent initiation is blocked [102].
5.2.7. Alternative Cap-Dependent Mechanism in Stress Response
In addition to the non-canonical, cap-independent mechanism of translation regulation, translation of multiple capped mRNAs remains unaffected or even increases during cellular stress despite inactivation of eIF4E, suggesting the existence of other non-canonical but cap-dependent translation initiation mechanisms [103]. Specific subunits of eIF3, an initiation factor participating in nearly every step of canonical translation, have been shown to have specific non-canonical functions [104]. eIF3 binds to specific subsets of mRNAs involved in cell growth, differentiation, and apoptosis using different modes of 5’UTR stem-loop binding to activate or repress mRNA translation [105]. In this context, eIF3d is of particular interest due to its cap-binding capability. eIF3d has been reported to become activated under metabolic stress, such as glucose deprivation; accordingly, engineering specialized 5′cap structures and 5′UTR sequences that preferentially recruit eIF3 may enable therapeutic mRNA expression during stress-induced translatome reprogramming [106].
6. Conclusions
Balancing multiple design considerations in therapeutic mRNA optimization is one of the main challenges in the development of these drugs. An extremely important, yet difficult, design decision is the selection of appropriate elements before and after the coding sequence. Recent advances in deep-learning methods have enabled the design of high-performance UTRs based on certain cell types [107,108]. The main obstacle in such estimations is the prediction of generated UTRs for in vitro and in vivo mRNA performance. Cis-regulatory elements of mRNA affect multiple cellular processes, such as interactions with RBPs [109] or other RNA forms [110], and determine alternative modes of translation, depending on the cell state [47]. The cellular context and biology of the target ORF influence the general stability and expression of mRNA. Thus, the use of one universal UTR to enhance protein production from exogenous mRNA across different organs and cell types may not be optimal.
Designing therapeutic mRNA sequences to treat diseases should take into account additional elements to overcome: on the one hand—the general inhibition of translation associated with the transition of cells into a stress state and, on the other hand—utilize altered endogenous translation agents to engineer cell state-dependent mRNA therapeutics. Selection of mRNA elements: A 5’cap, 5’ and 3’UTRs, codon selection, and poly(A)-tail should be preceded by an in-depth analysis of the cell state and expression mechanisms to which the delivered therapeutic mRNA is subject. For example, in cancer, a subset of mRNA involved in cell survival, angiogenesis, and cell progression is more efficiently translated, and these mRNA contain long and structured 5’UTRs[111]. Oncogenic transformation also leads to the overexpression of mRNA containing purine-rich motifs in the 5’UTR, which activates eIF4A1 [109]. In squamous cell carcinomas, genes with alternative 5’UTR isoforms containing TOP motifs show increased translational efficiency [112]. Designing mRNAs capable of interacting with different trans-acting factors involved in cellular stress conditions (specific RNA-binding proteins or small regulatory RNAs), as well as the implementation of cis-elements in mRNA sequences, could tremendously change the therapeutic efficacy of mRNA. It has been reported that 2′-O-methylation of the second transcribed nucleotide in the 5’cap contributes to higher mRNA expression in IFN-α-treated cells [113]. Pseudouridine, another common mRNA modification, enhances RNA stability and optimizes secondary structure to facilitate ribosome scanning and translation, making it a standard feature of therapeutic mRNAs and helping mitigate cellular stress through reduced innate immune activation [114]. In addition, mRNA containing N6-benzyl analog of the m6Am-cap has higher translation potential in human dendritic cells compared to the m6Am-capped mRNA, however, the effect was even more pronounced when the 5’UTR promoting an alternative translation pathway was used [115]. Another example is the use of a specific 5’UTR from the fatty acid metabolism gene carboxylesterase 1D (Ces1d), which improved mRNA translation by 2-fold in the heart post- myocardial infarction (MI) [116]. As mechanistic understanding deepens and delivery technologies mature, stress-aware mRNA design is poised to enable therapies that are not only efficient but also context-sensitive and self-regulating. Future studies combining ribosome profiling, RNA structure modeling, and in vivo stress mapping, alongside computational analyses that integrate these features, will further optimize these strategies, extending mRNA therapeutics from constitutive expression toward truly adaptive, stress-responsive gene medicines.
Author Contributions
I hereby declare that Edyta Trepkowska-Mejer is the sole author of this publication. All contributions related to the conception, design, methodology, data collection, analysis, interpretation, writing, and revision of the manuscript were carried out exclusively by Edyta Trepkowska-Mejer. No other individuals meet the criteria for authorship.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no data were created or analyzed in this study.
Conflicts of Interest
The author declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| mRNA | Messenger RNA |
| 5’UTR | 5’ Untranslated Region |
| 3’UTR | 3’ Untranslated Region |
| tRNA | Transfer RNA |
| 43S PIC | 43S pre-initiation complex |
| eIF | Eukaryotic Translation Initiation Factor |
| GEF | Guanine Nucleotide Exchange Factor |
| eIF2–GDP | Eukaryotic Initiation Factor 2-Guanosine Diphosphate |
| eIF2–GTP | Eukaryotic Initiation Factor 2-Guanosine Triphosphate |
| eIF4 | Eukaryotic Translation Initiation Factor 4 |
| eEF2K | Eukaryotic Elongation Factor 2 Kinase |
| TC | Ternary Complex |
| GTP | Guanosine triphosphate |
| ISR | Integrated Stress Response |
| GCN2 | General Control Non-Derepressible-2 |
| PERK | PKR-like ER Kinase |
| PKR | Protein Kinase RNA |
| HRI | Heme-Regulated Inhibitor Kinase |
| uORFs | Upstream Open Reading Frames |
| ATF4 | Activating Transcription Factor 4 |
| IRESs | Internal Ribosome Entry Sites |
| BiP | Binding Immunoglobulin Protein |
| APAF-1 | Apoptotic Protease Activating Factor-1 |
| RBPs | RNA-Binding Protein |
| HuR | Human Antigen R |
| TIA-1 | T-cell intracellular antigen-1 |
| mTOR | Mammalian Target of Rapamycin |
| 4E-BPs | eIF4E-Binding Proteins |
| PI3K | Class I Phosphoinositide 3-Kinase |
| AMPK | 5′-Adenosine Monophosphate-Activated Protein Kinase |
| 5′TOP | 5′Terminal Oligopyrimidine |
| CCND1 | Cyclin D1 |
| VEGFA | Vascular Endothelial Growth Factor A |
| ODC1 | Ornithine Decarboxylase |
| MNKs | Mitogen-Activated Protein Kinase (MAPK)-Interacting Kinases |
| TSC | hamartin |
| MDM2 | Mouse Double Minute 2 |
| BAX | Bcl-2-Associated X Protein |
| AGO2 | Argonaute 2 |
| FXR1 | Fragile X-Related Protein 1 |
| tRFs | tRNA-Derived RNA Fragments |
| tiRNAs | tRNA-Derived Stress-Induced RNAs |
| ANG | Angiogenin |
| YTHDF1 | YTH N6-Methyladenosine RNA Binding Protein 1 |
| CHOP | DNA Damage-Inducible Transcript 3 |
| ATG5 | Autophagy Related 5 |
| 5’TOP | 5’Terminal OligoPyrimidines |
| PABP | Poly(A)-Binding Protein |
| TISU | Translation Initiator of Short 5′ UTR |
| TSS | Transcription Start Site |
| IRES | Internal Ribosome Entry Site |
| VEGF | Vascular Endothelial Growth |
| HIF-1α | Hypoxia-Inducible Factor 1-alpha |
| CITE | Cap-Independent Translation Enhancers |
References
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the Promise of MRNA Therapeutics. Nat. Biotechnol. 2022, 40. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug. Discov. 2021, 20. [Google Scholar] [CrossRef]
- Mauger, D.M.; Joseph Cabral, B.; Presnyak, V.; Su, S. V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. MRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. U. S. A. 2019, 116. [Google Scholar] [CrossRef]
- Lee, J.; Woodruff, M.C.; Kim, E.H.; Nam, J.H. Knife’s Edge: Balancing Immunogenicity and Reactogenicity in MRNA Vaccines. Exp. Mol. Med. 2023, 55. [Google Scholar] [CrossRef]
- Berger, S.; Lächelt, U.; Wagner, E. Dynamic Carriers for Therapeutic RNA Delivery. Proc. Natl. Acad. Sci. U. S. A. 2024, 121. [Google Scholar] [CrossRef]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. MRNA Vaccines Manufacturing: Challenges and Bottlenecks. Vaccine 2021, 39. [Google Scholar] [CrossRef]
- Potužník, J.F.; Cahova, H. If the 5’ Cap Fits (Wear It)–Non-Canonical RNA Capping. RNA Biol. 2024, 21, 1–13. [Google Scholar] [CrossRef]
- Chu, Y.; Yu, D.; Li, Y.; Huang, K.; Shen, Y.; Cong, L.; Zhang, J.; Wang, M. A 5′ UTR Language Model for Decoding Untranslated Regions of MRNA and Function Predictions. Nat. Mach. Intell. 2024, 6. [Google Scholar] [CrossRef]
- Li, X.; Kazan, H.; Lipshitz, H.D.; Morris, Q.D. Finding the Target Sites of RNA-Binding Proteins. Wiley Interdiscip. Rev. RNA 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Ryczek, N.; Łyś, A.; Makałowska, I. The Functional Meaning of 5′UTR in Protein-Coding Genes. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. What Are 3′ Utrs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Hong, D.; Jeong, S. 3’UTR Diversity: Expanding Repertoire of RNA Alterations in Human MRNAs. Mol. Cells 2023, 46. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.A.; Coller, J. Roles of MRNA Poly(A) Tails in Regulation of Eukaryotic Gene Expression. Nat. Rev. Mol. Cell. Biol. 2022, 23. [Google Scholar] [CrossRef]
- Harnisch, C.; Moritz, B.; Rammelt, C.; Temme, C.; Wahle, E. Chapter Nine—Activity and Function of Deadenylases. In The Enzymes; Chanfreau, G.F., Tamanoi, F., Eds.; Academic Press, 2012; Vol. 31, pp. 181–211. ISSN ISBN 1874-6047. [Google Scholar]
- Vavilis, T.; Stamoula, E.; Ainatzoglou, A.; Sachinidis, A.; Lamprinou, M.; Dardalas, I.; Vizirianakis, I.S. MRNA in the Context of Protein Replacement Therapy. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Bicknell, A.A.; Reid, D.W.; Licata, M.C.; Jones, A.K.; Cheng, Y.M.; Li, M.; Hsiao, C.J.; Pepin, C.S.; Metkar, M.; Levdansky, Y.; et al. Attenuating Ribosome Load Improves Protein Output from MRNA by Limiting Translation-Dependent MRNA Decay. Cell. Rep. 2024, 43. [Google Scholar] [CrossRef]
- Reinhart, A.G.; Osterwald, A.; Ringler, P.; Leiser, Y.; Lauer, M.E.; Martin, R.E.; Ullmer, C.; Schumacher, F.; Korn, C.; Keller, M. Investigations into MRNA Lipid Nanoparticles Shelf-Life Stability under Nonfrozen Conditions. Mol. Pharm. 2023, 20. [Google Scholar] [CrossRef] [PubMed]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V. V.; Choe, C.; Rothschild, D.; et al. Combinatorial Optimization of MRNA Structure, Stability, and Translation for RNA-Based Therapeutics. Nat. Commun. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Alhusaini, N.; Morris, N.; Sweet, T.; Coller, J. Translation Elongation and MRNA Stability Are Coupled through the Ribosomal A-Site. RNA 2018, 24. [Google Scholar] [CrossRef]
- Courel, M.; Clément, Y.; Bossevain, C.; Foretek, D.; Cruchez, O.V.; Yi, Z.; Bénard, M.; Benassy, M.N.; Kress, M.; Vindry, C.; et al. Gc Content Shapes MRNA Storage and Decay in Human Cells. Elife 2019, 8. [Google Scholar] [CrossRef]
- Wieder, N.; D’Souza, E.N.; Martin-Geary, A.C.; Lassen, F.H.; Talbot-Martin, J.; Fernandes, M.; Chothani, S.P.; Rackham, O.J.L.; Schafer, S.; Aspden, J.L.; et al. Differences in 5’untranslated Regions Highlight the Importance of Translational Regulation of Dosage Sensitive Genes. Genome Biol. 2024, 25. [Google Scholar] [CrossRef]
- Dave, P.; Roth, G.; Griesbach, E.; Mateju, D.; Hochstoeger, T.; Chao, J.A. Single-Molecule Imaging Reveals Translation-Dependent Destabilization of MRNAs. Mol. Cell. 2023, 83. [Google Scholar] [CrossRef]
- Lindqvist, L.; Imataka, H.; Pelletier, J. Cap-Dependent Eukaryotic Initiation Factor-MRNA Interactions Probed by Cross-Linking. RNA 2008, 14. [Google Scholar] [CrossRef]
- Jia, X.; He, X.; Huang, C.; Li, J.; Dong, Z.; Liu, K. Protein Translation: Biological Processes and Therapeutic Strategies for Human Diseases. Signal Transduct. Target. Ther. 2024, 9. [Google Scholar] [CrossRef]
- Advani, V.M.; Ivanov, P. Translational Control under Stress: Reshaping the Translatome. BioEssays 2019, 41. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sormanni, P.; Ahmed, N.; Ciryam, P.; Friedrich, U.A.; Kramer, G.; O’Brien, E.P. A Chemical Kinetic Basis for Measuring Translation Initiation and Elongation Rates from Ribosome Profiling Data. PLoS Comput. Biol. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Lorsch, J.R. The Mechanism of Eukaryotic Translation Initiation: New Insights and Challenges. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Pushing the Limits of the Scanning Mechanism for Initiation of Translation. Gene 2002, 299. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Niimura, Y.; Gojobori, T.; Tanaka, H.; Miura, K. ichiro Diversity of Preferred Nucleotide Sequences around the Translation Initiation Codon in Eukaryote Genomes. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef]
- Lee, J.H.; Pestovat, T. V.; Shin, B.S.; Cao, C.; Choi, S.K.; Dever, T.E. Initiation Factor EIF5B Catalyzes Second GTP-Dependent Step in Eukaryotic Translation Initiation. Proc. Natl. Acad. Sci. U. S. A. 2002, 99. [Google Scholar] [CrossRef]
- Huang, B.Y.; Fernández, I.S. Long-Range Interdomain Communications in EIF5B Regulate GTP Hydrolysis and Translation Initiation. Proc. Natl. Acad. Sci. U. S. A. 2020, 117. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368. [Google Scholar] [CrossRef]
- Guan, B.J.; van Hoef, V.; Jobava, R.; Elroy-Stein, O.; Valasek, L.S.; Cargnello, M.; Gao, X.H.; Krokowski, D.; Merrick, W.C.; Kimball, S.R.; et al. A Unique ISR Program Determines Cellular Responses to Chronic Stress. Mol. Cell. 2017, 68. [Google Scholar] [CrossRef]
- Chee, N.T.; Lohse, I.; Brothers, S.P. MRNA-to-Protein Translation in Hypoxia. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Shu, X.E.; Swanda, R. V.; Qian, S.B. Nutrient Control of MRNA Translation. Annu. Rev. Nutr. 2020, 40. [Google Scholar] [CrossRef]
- Panniers, R. Translational Control during Heat Shock. Biochimie 1994, 76. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Thompson, S.R.; Mathews, M.B.; Mohr, I. Translational Control in Virus-Infected Cells. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Ghosh, A.; Shcherbik, N. Effects of Oxidative Stress on Protein Translation: Implications for Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Hu, Y.; Wang, J.L.; Chatterjee, M.; Shi, Y.; Kaufman, R.J. Ultraviolet Light Inhibits Translation through Activation of the Unfolded Protein Response Kinase PERK in the Lumen of the Endoplasmic Reticulum. J. Biol. Chem. 2002, 277. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the Ubiquitin-Proteasome System Induces Stress Granule Formation. Mol. Biol. Cell. 2007, 18. [Google Scholar] [CrossRef]
- Castilho, B.A.; Shanmugam, R.; Silva, R.C.; Ramesh, R.; Himme, B.M.; Sattlegger, E. Keeping the EIF2 Alpha Kinase Gcn2 in Check. Biochim. Biophys. Acta Mol. Cell. Res. 2014, 1843. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein Kinase R-like ER Kinase and Its Role in Endoplasmic Reticulum Stress-Decided Cell Fate. Cell. Death Dis. 2015, 6. [Google Scholar] [CrossRef]
- Williams, B.R.G. PKR; A Sentinel Kinase for Cellular Stress. Oncogene 1999, 18. [Google Scholar] [CrossRef]
- Girardin, S.E.; Cuziol, C.; Philpott, D.J.; Arnoult, D. The EIF2α Kinase HRI in Innate Immunity, Proteostasis, and Mitochondrial Stress. FEBS J. 2021, 288. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-Mediated Translation: The War of ITAFs in Pathophysiological States. Cell. Cycle 2011, 10. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational Regulation of Gene Expression during Conditions of Cell Stress. Mol. Cell. 2010, 40. [Google Scholar] [CrossRef]
- Topisirovic, I.; Svitkin, Y. V.; Sonenberg, N.; Shatkin, A.J. Cap and Cap-Binding Proteins in the Control of Gene Expression. Wiley Interdiscip. Rev. RNA 2011, 2. [Google Scholar] [CrossRef]
- Igreja, C.; Peter, D.; Weiler, C.; Izaurralde, E. 4E-BPs Require Non-Canonical 4E-Binding Motifs and a Lateral Surface of EIF4E to Repress Translation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, A.; Marcotrigiano, J.; Stepinski, J.; Jankowska-Anyszka, M.; Wyslouch-Cieszynska, A.; Dadlez, M.; Gingras, A.C.; Mak, P.; Darzynkiewicz, E.; Sonenberg, N.; et al. Biophysical Studies of EIF4E Cap-Binding Protein: Recognition of MRNA 5′ Cap Structure and Synthetic Fragments of EIF4G and 4E-BP1 Proteins. J. Mol. Biol. 2002, 319. [Google Scholar] [CrossRef] [PubMed]
- Volarević, S.; Thomas, G. Role of S6 Phosphorylation and S6 Kinase in Cell Growth. Prog. Nucleic Acid. Res. Mol. Biol. 2000, 65. [Google Scholar]
- Ballard, D.J.; Peng, H.Y.; Das, J.K.; Kumar, A.; Wang, L.; Ren, Y.; Xiong, X.; Ren, X.; Yang, J.M.; Song, J. Insights Into the Pathologic Roles and Regulation of Eukaryotic Elongation Factor-2 Kinase. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of MTORC1 by PI3K Signaling. Trends Cell. Biol. 2015, 25. [Google Scholar] [CrossRef]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. MTORC1 Directly Inhibits AMPK to Promote Cell Proliferation under Nutrient Stress. Nat. Metab. 2020, 2. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The Translational Landscape of MTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, X.; Chen, J. RNA-Binding Proteins in Regulating MRNA Stability and Translation: Roles and Mechanisms in Cancer. Semin. Cancer Biol. 2022, 86. [Google Scholar] [CrossRef]
- Vind, A.C.; Genzor, A.V.; Bekker-Jensen, S. Ribosomal Stress-Surveillance: Three Pathways Is a Magic Number. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L.; Sharp, P.A. MicroRNA Functions in Stress Responses. Mol. Cell. 2010, 40. [Google Scholar] [CrossRef] [PubMed]
- Gebetsberger, J.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A TRNA-Derived Fragment Competes with MRNA for Ribosome Binding and Regulates Translation during Stress. RNA Biol. 2017, 14. [Google Scholar] [CrossRef]
- Harvey, R.F.; Smith, T.S.; Mulroney, T.; Queiroz, R.M.L.; Pizzinga, M.; Dezi, V.; Villenueva, E.; Ramakrishna, M.; Lilley, K.S.; Willis, A.E. Trans-Acting Translational Regulatory RNA Binding Proteins. Wiley Interdiscip. Rev. RNA 2018, 9. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A Large-Scale Binding and Functional Map of Human RNA-Binding Proteins. Nature 2020, 583. [Google Scholar] [CrossRef]
- Rowe, W.; Kershaw, C.J.; Castelli, L.M.; Costello, J.L.; Ashe, M.P.; Grant, C.M.; Sims, P.F.G.; Pavitt, G.D.; Hubbard, S.J. Puf3p Induces Translational Repression of Genes Linked to Oxidative Stress. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global Dissociation of HuR-MRNA Complexes Promotes Cell Survival after Ionizing Radiation. EMBO J. 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, S.; Fan, B.; Jin, D.; Miao, L.; Liu, L.; Du, S.; Lin, J. Enhancing MRNA Translation Efficiency by Introducing Sequence Optimized AU-Rich Elements in 3’ UTR via HuR Anchorage. Mol. Ther. Nucleic Acids 2025, 36. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. . 2018, 9. [Google Scholar] [CrossRef]
- Varghese, J.; Lim, S.F.; Cohen, S.M. Drosophila MiR-14 Regulates Insulin Production and Metabolism through Its Target, Sugarbabe. Genes. Dev. 2010, 24. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Wu, S.Y.; Pradeep, S.; Ivan, C.; Pecot, C. V.; Gharpure, K.M.; Nagaraja, A.S.; Armaiz-Pena, G.N.; McGuire, M.; Zand, B.; et al. Hypoxia-Mediated Downregulation of MiRNA Biogenesis Promotes Tumour Progression. Nat. Commun. 2014, 5, 5202. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Lopez-Berestein, G.; Sood, A. MicroRNA Therapeutics: Principles, Expectations, and Challenges. Chin. J. Cancer 2011, 30, 368–370. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J. Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2008, 318, 1931–1934. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A Novel Class of Small RNAs: TRNA-Derived RNA Fragments (TRFs). Genes. Dev. 2009, 23. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Feng, J.; Liu, Q.; Sun, F.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Stress Induces TRNA Cleavage by Angiogenin in Mammalian Cells. FEBS Lett. 2009, 583. [Google Scholar] [CrossRef]
- Yamasaki, S.; Ivanov, P.; Hu, G.F.; Anderson, P. Angiogenin Cleaves TRNA and Promotes Stress-Induced Translational Repression. J. Cell. Biol. 2009, 185. [Google Scholar] [CrossRef]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.B.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous TRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 2015, 161. [Google Scholar] [CrossRef] [PubMed]
- Medina-Muñoz, S.G.; Kushawah, G.; Castellano, L.A.; Diez, M.; DeVore, M.L.; Salazar, M.J.B.; Bazzini, A.A. Crosstalk between Codon Optimality and Cis-Regulatory Elements Dictates MRNA Stability. Genome Biol. 2021, 22. [Google Scholar] [CrossRef]
- Delaunay, S.; Helm, M.; Frye, M. RNA Modifications in Physiology and Disease: Towards Clinical Applications. Nat. Rev. Genet. 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5’ UTR M6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yi, Y.; Gao, X.; Wang, X.; Zhao, D.; Wang, R.; Zhang, L.-S.; Gao, B.; Zhang, Y.; Zhang, L.; et al. 2-O-Methylation at Internal Sites on MRNA Promotes MRNA Stability. Mol. Cell. 2024, 84, 2320–2336.e6. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream Open Reading Frames Cause Widespread Reduction of Protein Expression and Are Polymorphic among Humans. Proc. Natl. Acad. Sci. U. S. A. 2009, 106. [Google Scholar] [CrossRef]
- Xiao, W.; Sun, Y.; Xu, J.; Zhang, N.; Dong, L. UORF-Mediated Translational Regulation of ATF4 Serves as an Evolutionarily Conserved Mechanism Contributing to Non-Small-Cell Lung Cancer (NSCLC) and Stress Response. J. Mol. Evol. 2022, 90. [Google Scholar] [CrossRef]
- Jousse, C.; Bruhat, A.; Carraro, V.; Urano, F.; Ferrara, M.; Ron, D.; Fafournoux, P. Inhibition of CHOP Translation by a Peptide Encoded by an Open Reading Frame Localized in the Chop 5′UTR. Nucleic Acids Res. 2001, 29. [Google Scholar] [CrossRef]
- Chen, H.H.; Tarn, W.Y. UORF-Mediated Translational Control: Recently Elucidated Mechanisms and Implications in Cancer. RNA Biol. 2019, 16. [Google Scholar] [CrossRef]
- Yang, Y.; Gatica, D.; Liu, X.; Wu, R.; Kang, R.; Tang, D.; Klionsky, D.J. Upstream Open Reading Frames Mediate Autophagy-Related Protein Translation. Autophagy 2023, 19. [Google Scholar] [CrossRef]
- Russo, G.L.; Stampone, E.; Cervellera, C.; Borriello, A. Regulation of P27kip1 and P57kip2 Functions by Natural Polyphenols. Biomolecules 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liang, J. Activating Transcription Factor 4: A Regulator of Stress Response in Human Cancers. Front. Cell. Dev. Biol. 2024, 12, 1370012. [Google Scholar] [CrossRef]
- Somers, J.; Pöyry, T.; Willis, A.E. A Perspective on Mammalian Upstream Open Reading Frame Function. Int. J. Biochem. Cell. Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef]
- Young-Baird, S.; Wek, R. Upstream Open Reading Frames Differentially Regulate Gene-Specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, jbc.R116.733899. [Google Scholar] [CrossRef]
- Cockman, E.; Anderson, P.; Ivanov, P. Top Mrnps: Molecular Mechanisms and Principles of Regulation. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Philippe, L.; van den Elzen, A.M.G.; Watson, M.J.; Thoreen, C.C. Global Analysis of LARP1 Translation Targets Reveals Tunable and Dynamic Features of 5′ TOP Motifs. Proc. Natl. Acad. Sci. U. S. A. 2020, 117. [Google Scholar] [CrossRef] [PubMed]
- Tuxworth, W.J.; Shiraishi, H.; Moschella, P.C.; Yamane, K.; McDermott, P.J.; Kuppuswamy, D. Translational Activation of 5′-TOP MRNA in Pressure Overload Myocardium. Basic Res. Cardiol. 2008, 103, 41–53. [Google Scholar] [CrossRef]
- Jefferies, H.B.J.; Fumagalli, S.; Dennis, P.B.; Reinhard, C.; Pearson, R.B.; Thomas, G. Rapamycin Suppresses 5′TOP MRNA Translation through Inhibition of P70s6k. EMBO J. 1997, 16, 3693–3704. [Google Scholar] [CrossRef]
- Elfakess, R.; Sinvani, H.; Haimov, O.; Svitkin, Y.; Sonenberg, N.; Dikstein, R. Unique Translation Initiation of MRNAs-Containing TISU Element. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Elfakess, R.; Dikstein, R. A Translation Initiation Element Specific to MRNAs with Very Short 5′UTR That Also Regulates Transcription. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Sinvani, H.; Haimov, O.; Svitkin, Y.; Sonenberg, N.; Tamarkin-Ben-Harush, A.; Viollet, B.; Dikstein, R. Translational Tolerance of Mitochondrial Genes to Metabolic Energy Stress Involves TISU and EIF1-EIF4GI Cooperation in Start Codon Selection. Cell. Metab. 2015, 21. [Google Scholar] [CrossRef]
- Terenin, I.M.; Smirnova, V. V.; Andreev, D.E.; Dmitriev, S.E.; Shatsky, I.N. A Researcher’s Guide to the Galaxy of IRESs. Cell. Mol. Life Sci. 2017, 74. [Google Scholar] [CrossRef]
- Mokrejš, M.; Mašek, T.; Vopálenský, V.; Hlubuček, P.; Delbos, P.; Pospíšek, M. IRESite A Tool for the Examination of Viral and Cellular Internal Ribosome Entry Sites. Nucleic Acids Res. 2009, 38. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Internal Ribosome Entry Sites in Cellular MRNAs: Mystery of Their Existence. J. Biol. Chem. 2005, 280, 23425–23428. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational Control in Stress and Apoptosis. Nat. Rev. Mol. Cell. Biol. 2005, 6. [Google Scholar] [CrossRef]
- Gao, X.; Wu, Z. IRES-Mediated Translation: Expanding the Toolkits of RNA Therapy. Int. J. Mol. Sci. 2025, 26, 10542. [Google Scholar] [CrossRef]
- Sorokin, I.I.; Vassilenko, K.S.; Terenin, I.M.; Kalinina, N.O.; Agol, V.I.; Dmitriev, S.E. Non-Canonical Translation Initiation Mechanisms Employed by Eukaryotic Viral MRNAs. Biochemistry 2021, 86. [Google Scholar] [CrossRef]
- Andreev, D.E.; Dmitriev, S.E.; Terenin, I.M.; Prassolov, V.S.; Merrick, W.C.; Shatsky, I.N. Differential Contribution of the M7G-Cap to the 5′ End-Dependent Translation Initiation of Mammalian MRNAs. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef]
- Shatsky, I.N.; Terenin, I.M.; Smirnova, V. V.; Andreev, D.E. Cap-Independent Translation: What’s in a Name? Trends Biochem. Sci. 2018, 43, 882–895. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, J.; Hoffman, A.R.; Hu, J.-F. Eukaryotic Translation Initiation Factor EIF4G2 Opens Novel Paths for Protein Synthesis in Development, Apoptosis and Cell Differentiation. Cell. Prolif. 2023, 56, e13367. [Google Scholar] [CrossRef]
- Roiuk, M.; Neff, M.; Teleman, A.A. EIF4E-Independent Translation Is Largely EIF3d-Dependent. Nat. Commun. 2024, 15, 6692. [Google Scholar] [CrossRef]
- Lee, A.S.Y.; Kranzusch, P.J.; Cate, J.H.D. EIF3 Targets Cell-Proliferation Messenger RNAs for Translational Activation or Repression. Nature 2015, 522. [Google Scholar] [CrossRef]
- Hayek, H.; Gross, L.; Janvier, A.; Schaeffer, L.; Martin, F.; Eriani, G.; Allmang, C. EIF3 Interacts with Histone H4 Messenger RNA to Regulate Its Translation. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- Lamper, A.M.; Fleming, R.H.; Ladd, K.M.; Lee, A.S.Y. A Phosphorylation-Regulated EIF3d Translation Switch Mediates Cellular Adaptation to Metabolic Stress. Science (1979) . 2020, 370. [Google Scholar] [CrossRef]
- Castillo-Hair, S.; Fedak, S.; Wang, B.; Linder, J.; Havens, K.; Certo, M.; Seelig, G. Optimizing 5’UTRs for MRNA-Delivered Gene Editing Using Deep Learning. Nat. Commun. 2024, 15. [Google Scholar] [CrossRef]
- Castillo-Hair, S.M.; Seelig, G. Machine Learning for Designing Next-Generation MRNA Therapeutics. Acc. Chem. Res. 2022, 55. [Google Scholar] [CrossRef]
- Koletsou, E.; Huppertz, I. RNA-Binding Proteins as Versatile Metabolic Regulators. Npj Metab. Health Dis. 2025, 3, 1. [Google Scholar] [CrossRef]
- Singh, S.; Shyamal, S.; Das, A.; Panda, A.C. Global Identification of MRNA-Interacting Circular RNAs by CLiPPR-Seq. Nucleic Acids Res. 2024, 52. [Google Scholar] [CrossRef]
- Raza, F.; Waldron, J.A.; Le Quesne, J. Translational Dysregulation in Cancer: EIF4A Isoforms and Sequence Determinants of EIF4A Dependence. Biochem. Soc. Trans. 2015, 43. [Google Scholar] [CrossRef]
- Weber, R.; Ghoshdastider, U.; Spies, D.; Duré, C.; Valdivia-Francia, F.; Forny, M.; Ormiston, M.; Renz, P.F.; Taborsky, D.; Yigit, M.; et al. Monitoring the 5′UTR Landscape Reveals Isoform Switches to Drive Translational Efficiencies in Cancer. Oncogene 2023, 42. [Google Scholar] [CrossRef] [PubMed]
- Drazkowska, K.; Tomecki, R.; Warminski, M.; Baran, N.; Cysewski, D.; Depaix, A.; Kasprzyk, R.; Kowalska, J.; Jemielity, J.; Sikorski, P.J. 2′-O-Methylation of the Second Transcribed Nucleotide within the MRNA 5′ Cap Impacts the Protein Production Level in a Cell-Specific Manner and Contributes to RNA Immune Evasion. Nucleic Acids Res. 2022, 50. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of Pseudouridine into MRNA Enhances Translation by Diminishing PKR Activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef]
- Warminski, M.; Trepkowska, E.; Smietanski, M.; Sikorski, P.J.; Baranowski, M.R.; Bednarczyk, M.; Kedzierska, H.; Majewski, B.; Mamot, A.; Papiernik, D.; et al. Trinucleotide MRNA Cap Analogue N6-Benzylated at the Site of Posttranscriptional M6Am Mark Facilitates MRNA Purification and Confers Superior Translational Properties In Vitro and In Vivo. J. Am. Chem. Soc. 2024, 146. [Google Scholar] [CrossRef]
- Sultana, N.; Hadas, Y.; Sharkar, M.; Kaur, K.; Magadum, A.; Kurian, A.; Hossain, N.; Alburquerque, B.; Ahmed, S.; Chepurko, E.; et al. Optimization of 5’ Untranslated Region of Modified MRNA for Use in Cardiac or Hepatic Ischemic Injury. Mol. Ther. Methods Clin. Dev. 2020, 17. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of cannonical mode of translation initiation. At the beginning, a small ribosome subunit (40S) binds with translation initiation factors: 3, 1, 1A and 5. Also, eukaryotic translation initiation factor 2 (eIF2) and GTP associate with methionyl-transfer RNA, leading to ternary complex formation. Next, 43S pre-initiation complex formation occurs as a result of ternary complex binding to 40S ribosome subunit. The cap-binding complex, consisting of eIF4A, eIF4E and eIF4G, binds to the 5’cap of the mRNA; eIF4G also associate with the poly(A)-binding protein (PABP). The activated mRNA, complexed with initiation factors, binds to the 43S pre-initiation complex and scanning of the mRNA occurs to find the AUG start codon. In the last step, GTP is hydrolyzed by eIF2, which enable the dissociation of the initiation factors from the 48S complex, thereby allowing binding of the large 60S ribosomal subunit, resulting in the formation of the 80S ribosome, ready for translation elongation and protein synthesis.
Figure 1.
Schematic representation of cannonical mode of translation initiation. At the beginning, a small ribosome subunit (40S) binds with translation initiation factors: 3, 1, 1A and 5. Also, eukaryotic translation initiation factor 2 (eIF2) and GTP associate with methionyl-transfer RNA, leading to ternary complex formation. Next, 43S pre-initiation complex formation occurs as a result of ternary complex binding to 40S ribosome subunit. The cap-binding complex, consisting of eIF4A, eIF4E and eIF4G, binds to the 5’cap of the mRNA; eIF4G also associate with the poly(A)-binding protein (PABP). The activated mRNA, complexed with initiation factors, binds to the 43S pre-initiation complex and scanning of the mRNA occurs to find the AUG start codon. In the last step, GTP is hydrolyzed by eIF2, which enable the dissociation of the initiation factors from the 48S complex, thereby allowing binding of the large 60S ribosomal subunit, resulting in the formation of the 80S ribosome, ready for translation elongation and protein synthesis.

Figure 2.
eIF2α phosphorylation and ISR program. Upon various different stimuli, kinases GCN2 (amino acid starvation and UV radiation), HRI (oxidative stress and heat shock), PKR (viral infection and double-stranded RNA) and PERK (ER stress, hypoxia and proteostasis) phosphorylate eIF2 at Ser-51 of its α-subunit, preventing GDP to GTP exchange by eIF2B, thus the ternary complex cannot be regenerated. eIF2α phosphorylation leads to global cap-dependent translation inhibition and promotes selective, stress responsive mRNA translation.
Figure 2.
eIF2α phosphorylation and ISR program. Upon various different stimuli, kinases GCN2 (amino acid starvation and UV radiation), HRI (oxidative stress and heat shock), PKR (viral infection and double-stranded RNA) and PERK (ER stress, hypoxia and proteostasis) phosphorylate eIF2 at Ser-51 of its α-subunit, preventing GDP to GTP exchange by eIF2B, thus the ternary complex cannot be regenerated. eIF2α phosphorylation leads to global cap-dependent translation inhibition and promotes selective, stress responsive mRNA translation.

Figure 3.
eIF4E activity regulation in stress response. Activation of extracellular signal-regulated kinases results in the mitogen-activated protein kinase (MAPK)-interacting kinases (MNKs) phosphorylation, leading to eIF4E phosphorylation and enabling mRNA translation initiation. Growth factors binds to the receptors and activates phosphatidylinositol 3-kinase (PI3K) and AKT, resulting in dissociation of the TSC1/ TSC2 complex and activation of mTORC1 pathway: mTORC1 hyper-phosphorylates 4EBP, enabling mRNA translation and turning on S6Ks for further elongation facilitation by eukaryotic translation elongation factor 2 (eEF2).
Figure 3.
eIF4E activity regulation in stress response. Activation of extracellular signal-regulated kinases results in the mitogen-activated protein kinase (MAPK)-interacting kinases (MNKs) phosphorylation, leading to eIF4E phosphorylation and enabling mRNA translation initiation. Growth factors binds to the receptors and activates phosphatidylinositol 3-kinase (PI3K) and AKT, resulting in dissociation of the TSC1/ TSC2 complex and activation of mTORC1 pathway: mTORC1 hyper-phosphorylates 4EBP, enabling mRNA translation and turning on S6Ks for further elongation facilitation by eukaryotic translation elongation factor 2 (eEF2).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



