Submitted:
21 April 2026
Posted:
22 April 2026
You are already at the latest version
Abstract
Cellular stress signaling conveys vital messages to the cell’s machinery to respond dynamically to internal and external environmental cues. One prevailing hypothesis for such signaling is to bring about crucial downstream functional changes in the cell’s ability to withstand intra- and extra-cellular stress, as exemplified by the NRF2-ARE pathway. Reactive electrophilic metabolites (REMs) are often generated from membrane lipids or respiratory metabolites in inflammatory and stress signaling contexts. REMs harbour an innate ability to irreversibly bind protein-cysteines, and form DNA and RNA adducts. Our work has led us to propose a new hypothesis regarding the role of locale-specific REM build-up in stress signalling; whereby they can label both the majority stable protein residents in a given locale, as well as minority guests transiently existing within a subcellular compartment, at a kinetically significant rate to trigger a “gain of function” signalling cascade. Much like the NRF2 signalling pathway, such downstream signalling messages could assist the cell’s ability to survive against microenvironment-specific stress and adapt on-demand. As REMs accumulate in several disease-specific cells, especially age-related disorders such as cancer and neurodegeneration, understanding the functional signal propagation mechanisms shaped by specific REMs engaging with specific biomolecular targets will prove vital for future therapeutic interventions with enhanced precision and context.
Keywords:
electrophile signaling
; gain of function signaling
; 4-hydroxynonenal
; reactive electrophilic metabolite sensor
Introduction
Reactive electrophilic metabolites (REMs) are often formed during essential metabolic processes [1]. These are typically housed in an enzyme active site, or channeled between active sites through complex associations and tunnels. Despite the huge efficiency and elegance of biological signaling and synthesis pathways, inevitably, the odd REM leaks out from enzyme active sites [2]. Due to their reactivity, these liberated REMs have the potential to wreak some level of havoc. The havoc caused, at least under transient exposure conditions, typically manifests itself through covalent labeling of amino acid side chains of proteins, which are more abundant and intrinsically more reactive to many reactive molecules than RNA and DNA [3]. Of course many REMs do react with DNA, and such events are also damaging and mutagenic. These are dealt with by DNA damage and repair enzymes [4] and are not discussed further here. Unsurprisingly, in healthy cells, levels of REMs are typically “low”. Although the precise concentrations estimated for each REM vary, the implication is that at resting levels, each REM does not obtain a concentration sufficient to allow modification of proteins significantly. Perhaps unsurprisingly, levels of REMs in cells are typically inversely proportional to individual REMs’ inherent chemical reactivity [2]. This situation effectively leads to a “healthy equilibrium” where proteins go about their business, and there is not sufficient of each REM to interfere with cellular processes.
Stress Upregulates a Host of REMs, Many of Which Can Covalently Modify Proteins
During cellular stress, it is established that concentrations of REMs can increase. This can be caused by an increase in various forms of reactive oxygen species that can themselves modulate proteins and also spawn REMs through oxidation of membrane components [5]. REMs can also be formed enzymatically upon stress (Figure 1) [2,3]. Despite the plethora of REMs and the ways they can arise in cells, we will discuss REMs mostly exemplified by lipid-derived REMs and mitochondria-derived (mt)-REMs [6]. There are many reasons why we favor these. Briefly, these are carbon-based REMs that are ostensibly bench stable in a laboratory, making them easy to study in cells and in vitro. Perhaps more importantly, they also are chemically reminiscent of covalent pharmacophores prevalent in drug discovery and approved drugs. For instance, aspects of these molecules can be found in Tecfidera [7] and Vumerity [8], as well as ibrutinib [9], neratinib [10], and sotorasib [11]. Thus, their interactions with proteins mimic protein–covalent drug interactions. Indeed, we have derived isoform-selective inhibitors of specific kinase isoforms by modification of endogenous REMs [12,13]. Therefore, we see particular translational relevance to understanding these interactions.
Reactive Carbon-Based REMs React with Proteins, but Not Sufficiently Enough to Achieve High Occupancy
Using the protypical REM, 4-hydroxynonenal (HNE), as an exemplar, we see that this REM, despite being stable in water and inert buffers, is inherently reactive with several amino acids [1]. HNE shows a second order rate of reactivity of ~1 M-1s-1 with cysteine, forming a covalent bond between the sulfur atom and the β-carbon of the enal system, a process often referred to as Michael or conjugate addition. HNE also shows measurable reactivity with histidine (at least one order of magnitude slower than with cysteine), and lysine (although this rate is very slow), again reacting through Michael/conjugate addition [2]. Although these rates are calculated for specific amino acids, it turns out that many generic proteins show similar levels of reactivity with HNE; indeed it is well established that protein reactivity to HNE is dominated by cysteine. Thus these rates seem to be a good model for how generic proteins interact with HNE. Nonetheless, even the rate of reaction between a free cysteine and HNE is overall disappointing. Assuming a constant cellular concentration of HNE 1 µM, the half-life of covalent labeling of a specific normal cysteine in a cell is 8 days [Ln2/(1 * 0.000001)]. For comparison, the cut-off second order rate constant (kcat/Km) for an enzymatic reaction to be considered biologically relevant is typically 100-1000 M-1s-1. The average (kcat/Km) for all known enzymes is ~100 000 M-1s-1. Thus, HNE reacting with a free cysteine is a very slow process under normal physiological conditions.
Of course, a constant generation of HNE and slow reactivity may still be expected to lead to augmentation of HNE and other REM concentrations over time. However, there is a counterbalance, performed by detoxification enzymes that deactivate REMs . Several classes of enzyme are involved in this process, including, glutathione-S-transferases (GST), that mark hydrophobic molecules with glutathione for expulsion and react with HNE with second order rate constants at minimally 103 M-1s-1 [14], and oxidoreductases, that convert HNE to either the acid or alcohol [15]; the alcohol cannot perform covalent chemistry, and the acid, not as effectively as HNE. Notably, different biological contexts, particularly, transformed versus non-transformed cells, manifest different detoxification potencies [16,17]. Different isoforms of GSTs are expressed at differing ratios in different types of cancer cells [17]. Different oxidoreductases can also be expressed. Interestingly, despite oxidoreductase chemistry lowering covalent reactivity of HNE and most other REMs in vitro, the products of these reactions can still serve as biological signals. For instance, we demonstrated a signaling role for reduced HNE in a recent paper [15]. Although being very interesting, the biological roles of these non-covalent secondary/tertiary metabolites are not within the scope of this piece and will thus be ignored. The bottom line is that REM stability, and half-lives of different REMs relative to each other likely change as a function of cell types and biological contexts. Thus, REM concentrations are a balance between all these complex, localized regulation factors, making each system essentially unique.
Extent of HNEylation Can Be Increased in Cells
Fortunately, the above analysis does not tell the full story. There are two principal mitigating factors that change the situation considerably: HNE concentration can significantly increase due to stress; and specific protein-cysteines manifest unexpectedly high reactivities to HNE (Figure 1)[2]. We have referred to such protein-cysteines as first responders or kinetically privileged sensors (KPS). We will deal with each consideration in turn. Increases in HNE concentration will naturally decrease the time taken to HNEylate all proteins. Assuming that HNE concentration can increase to 10 µM, half-life of covalently labeling generic cysteines (which we term by-stander cysteines) drops to about one day. Obviously, upon increase to 100 µM, half-life will drop further (~2 h). In fact, several of these timeframes indicate that during a normal cell cycle, when a cell has to remake 50% of its proteome, a significant amount of free cysteines or protein-bound by-stander cysteines could be modified by HNE.
However, it is worth asking, how long can the cell maintain high concentrations of HNE? Although this is unknown, it is unlikely that the cell can sustain high HNE concentrations for long periods of time. Indeed, elevated HNE levels are an indication of extreme stress that are known triggers for numerous cell death pathways[18]. Elevated external concentrations of HNE (>100 µM) are toxic. It is known that the external and internal concentration of HNE are not the same, with the internal concentrations always being lower, due to detoxification processes; for the reactive oxygen species, peroxide, the difference between the external and internal concentrations can reach up to 500-1000 fold, although this difference decreases as peroxide concentration rises [14,19]. Thus we can conclude that much lower than 100 µM HNE internal concentrations of HNE are toxic. This discussion leads to the conclusion that not insignificant labeling occupancy of HNE of by-stander protein-cysteines could be achievable by increasing HNE concentrations, providing it could be sustained for relatively long periods. Unfortunately, with this model, selective labeling is not possible, and as discussed, potentially toxic HNE concentrations would be needed.
This brings us onto the second possibility to increase labeling of specific proteins. This can actually be achieved in two, potentially synergistic ways. The first is through localized buildup of HNE in specific regions. In this case, potentially very high HNE concentrations are possible [20], allowing relatively high occupancy of proximal proteins to occur without affecting other proteins that are further away from the localized site of HNE generation. This localized generation also eschews HNE-induced toxicity (total cellular HNE concentration remains tolerable). Biochemically, localized buildup of HNE is very much possible because it is produced from lipid membranes. If we assume that we can achieve a transient bust of HNE 100 µM, in a specific locale, perhaps for 1-2 h, we could quite easily achieve high (~50%) HNE occupancy on proximal proteins. We will set this as a threshold value to achieve a biological phenotype through inhibition, which we will initially assume REMs do to their target. 50% inhibition mimics a heterozygotic state, which for a not insignificant number of proteins can lead to phenotypic changes [21,22]. This relatively high threshold also allows for the fact that HNEylation intrinsically cannot be 100% selective.
Such considerations address locale selectivity, although we need to remember that numerous proteins exist in each cellular (micro)environment, and each protein has numerous nucleophilic residues (for instance cysteine occurs around 2.3% of amino acids [23]). Therefore for selective HNEylation to occur, we really need to have specifically KPS proteins that show residue-specific HNEylation due to kinetic elevation of intrinsic reactivity. Several proteins have now been shown to exhibit such KPS properties[12,24,25]. This phenomenon has been demonstrated using numerous different, orthogonal methods, all of which are designed to pick out specific KPS. Methods include quantitative activity-based reactivity profiling [26], direct detection[27], and REX-technologies [28]. We will not go into the details of each method, as we have discussed them previously. We will only mention that all have relative strengths and weaknesses, but they provide a universal affirmation that some specific protein cysteines have enhanced reactivities. Consistent with a complex mechanism of activation, proteins that have elevated reactivity for one specific REM do not always show elevated selectivity for other REMs, despite them having similar reactive functional groups[29]. This finding and many others, discussed below hint that KPS’s may have enzyme-like properties. Thus, a confluence of elevated HNE concentrations, and apposite HNEylation kinetics can lead to preferential of specific proteins at a rate that is potentially biologically relevant; as a reminder we have set this to 50% occupancy, based on the idea that REMs inhibit their target and that some genes are haploinsufficient.
Kinetic Privilege: Context and Considerations
The above analysis outlines that several intrinsic and extrinsic factors give way to a protein’s proclivity to be HNEylaed in cells. Modification occurs at specific cysteines that have properties that promote reaction with specific REMs. This sort of reasoning hints that KPS’s are not all made equal and may not always show kinetically privileged modification factors generically. Indeed, proteins can undergo numerous changes in association, posttranslational modification (e.g. phosphorylation and ubiquitination), and locale (which can lead to changes in concentration and pH, etc). Our REX-technologies are one of the few that allow examination of proclivity of specific proteins to be covalently modified by specific REMs in defined locales/contexts. Using these technologies, we have indeed outlined many examples of context-dependent HNEylation that depends on specific protein-protein association, locales, and other aspects in cells. Doubtless many more are there to be discovered. Thus, kinetic privilege, in cells, in not an absolute or even intrinsic quality, in many cases. There are proteins that have emerged to be very good KPS’s; Keap1 is a very good example of such proteins. Keap1 has numerous kinetically-privileged cysteines that are adducted by different REMs triggering downstream responses[25]. A counter example is CDK9, a protein that senses HNE in the cytosol but signals in the nucleus. In the nucleus, due to association with a canonical binding partner, CDK9 cannot bind HNE at all. Knockdown of the binding partner, restores CDK9-HNEylation in the nucleus [30]. Nonetheless, to be a genuinely REM-responsive functional protein, the protein of interest in one state or another at least must be able to engage with REMs at an elevated rate.
Signaling Modes
The above discussion has dwelt on the first essential step in REM signaling, covalent interaction with a target protein. We have established that a confluence of localized REM generation and rapid reaction kinetics can lead to a signaling event able to achieve a putative level of occupancy necessary for signaling, on a biologically relevant timescale. However, modifiation / ligand binding alone may not trigger phenotypes. Indeed, many researchers think that a lack of response is the default situation; we will address this with specific case studies, below. We realized that orthogonal to increasing propensity to be HNEylated, a way to increase proclivity to REM-signal is to lower the threshold needed to observe phenotypes when a protein is HNEylated. Extending our discussion of how using a model where REMs inhibit their targets naturally leads to a threshold of 50% occupancy to achieve a phenotype, we postulated that REM-signaling may involve gain of function (GOF) signaling. This GOF scenario would enable tripping of signaling pathways at low modification stoichiometry (i.e., ligand occupancy). In an ideal world, GOF induction could synergistically act with activity elevation to ensure the correct signaling pathway is flipped. We have discussed how privilege of modification (i.e., REM sensing) and REM signaling could be related through binding sites, for instance.
Using REX-technologies, we have uncovered numerous GOF signaling pathways in REM-signaling [12,24,30]. Most of these pathways we have uncovered are dominant negative signaling events. In this signaling mode, the inhibited protein does not just lose function, but it gains the ability to inhibit other similar proteins. This can happen through (dis)aggregation, abortive complex formation, or sequestering of essential components, amongst others. One of our early examples sets out many of the concepts that we have discussed above. The isoforms of AKT are expressed from independent genetic loci, and have overall high identity [31]. One of the largest areas of divergence is a linker region between the two principal AKT domains, the N-terminal plextrin homology domain and the kinase domain[32]. We discovered that AKT3 reacts rapidly with the electrophile HNE through a unique cysteine located within this domain (Figure 2A-B). AKT2 also reacted via a non-analogous cysteine within this domain, although it was markedly slower than for AKT3. AKT1 did not react at all, and it has no cysteine within this linker region. When we examined how labeling affected activity of the different AKT isoforms, we found that only AKT3 activity was affected by HNE labeling (Figure 2B-C). As we could assess HNEylation efficiency using T-REX, and could assess maximal inhibition using activity assays, we correlated how much inhibition we observed relative to ligand occupancy [12].
Turn on GOF Signaling
Recently, we discovered a GOF turn-on signaling pathway orchestrated by a nuclear-specific HNE sensor, NCBP1[24] (Figure 3A). The human protein has 19 cysteines (Figure 3B). Individual cysteine mutants did not affect HNEylation efficiency, indicating that many individual cysteines were KPS sites. Using a phenotype screen, we linked NCBP1 single cysteine site-specific HNEylation to inhibition of translation (Figure 3C). When we examined individual cysteine point mutants for hypomorphism in inhibition of translation, noting that no individual cysteine mutant could suppress HNEylation of the protein; remarkably, only one cysteine (C436) mutant prevented translation inhibition. Indeed, when we mutated all cysteines to alanine, bar the proposed functionally-privileged cysteine, HNEylation extent of NCBP1 was maintained, and crucially, dominant inhibition of translation upon HNEylation was retained. So, we arrived at a situation where likely several cysteines were KPS’s, but only one was functionally privileged (Figure 3C). This is perhaps the best example that functionally privileged cysteines are a subset of KPS cysteines. Further biochemical analysis uncovered that HNEylation of NCBP1 via the specific functionally privileged site led to a change in splicing creating an uncharacterized isoform of the translational regulator S6K that is dominant negative for translation when independently expressed.
General Similarities of GOF Signaling
The key thread linking the above GOF pathways is that the labeled protein itself contributes to the function of the reactive molecule. Unlike inhibition, where a dead end is reached, a new protein state is obtained that has its own biological activity. Unsurprisingly, as the modified state is essential to biological activity of the signaling molecule, the persistence of the protein-ligand complex is critical for determining how signaling manifests. It is thus perhaps not unsurprising that these signaling modes are common for irreversible protein binders, as irreversible labeling confers apposite properties to the labeled state, that is not present to reversible binders. This for instance, endows cells with a memory for transient stress upregulation, that could be useful to prepare for potentially dangerous levels of stress in the future. For instance, cells treated with a minor amount of a stressor (preconditioned) tend to be more robust to subsequent stresses; a similar concept can be derived from hormesis, the idea that low concentration of toxins are health promoting, but high concentrations are deleterious [33].
Factoring in Protein Stability on Labeling and Sensing
As discussed above, the half-life for protein cysteine labeling in cells is around 8 days. Despite being slow, this could readily lead to the conclusion that as cells are maintained in culture oftentimes for many months, all proteins should be HNE labelled. However, in cultured cells, division rates are usually once per ~18 h. Given that most cultured cells divide once per 18-24 h and half-life of labeling is 8 days, buildup of HNEylated proteins is therefore low (around 5% over 18 h, for 100 nM protein and 1 µM HNE, with second order rate constant of 1 M-1s-1). For shorter lived proteins, for instance those that are cell cycle-regulated and rapidly degraded (t1/2 ~1-2 h), fractional occupancy is considerably lower (< 1%), all other aspects being equal. Indeed, simple simulations the authors have run, assuming the same conditions as above, show that the amount of protein labeled at the half-life for labeling decreases markedly as the rate of reversion of the HNEylated protein to the unlabeled protein (equivalent to degradation and resynthesis for irreversible protein modifications) increases. Even very slow reversion k = 0.000002 s-1 (t1/2 ~1 day), leads to 50% or more reduction in labeling for 100 nM protein and 1 µM HNE, with second order rate constant of 1 M-1s-1. With a second order rate constant of 10 M-1s-1, a four-fold higher rate of reversion is needed to reduce labeling by 50%. Generalizing this concept, a significant perturbation for labeling will occur whenever the net rate of destruction of the labeled protein gives a half-life similar to the half-life of labeling. Although we are attuned now to believe that irreversible binding trumps reversible binding every time, it should be noted that this analysis does not apply to reversible binders, and so slowly metabolized, reversible binders are actually “better” than irreversible ones for rapidly turned over proteins.
At the other extent of the spectrum, for slower growing cells, where protein degradation rates may be negligible, HNE occupancy may indeed be higher. For instance, in vivo, differentiated cells have relatively slow protein turnover (several days), which may allow buildup of more HNEylated proteins, although there could also be lower/altered metabolism in many of these cells that could lower HNE concentrations [34]. Nonetheless, for very long-lived proteins, such as crystallin in the eye (a protein that remains in the lens throughout life without being replaced) [35], there is some evidence that slow irreversible labeling events can have biological relevance.
With the tools to understand the complex kinetics of protein labeling in hand, we can now look back on our discussion and ask how protein stability should fit into this conundrum. The conclusion is interesting- depending on how we chose to define kinetic privilege, this may not actually be a fixed quantity. Thinking back to our example of crystallins and very stable proteins that may exist for 60 or more years, labeling could be very slow, but we could consider this as a kinetically privileged sensor if a threshold labeling could be attained on this timescale. On the other hand, for a reasonably short-lived protein, such as p53 (t1/2 20-60 min) [36] this requires a much higher labeling rate to attain a threshold 50% occupancy (>200 M-1s-1). Some of the shortest-lived proteins are MYC (20-30 min) [37], and CDC25A (10-20 min) [38]. These would require rate constants of ~600 and ~1300 M-1s-1 to achieve 50% occupancy respectively.
Obviously, this topic is very nuanced, and context-dependent, but there is a clear correlation between protein half-life and its modification[39]. Indeed, this sort of logic permeates all irreversible protein-ligand/inhibitor interactions. Thus, from a labeling and occupancy standpoint, we can conclude that that reactive molecule signaling is perhaps more likely to function on stable proteins, as proteins that are unstable would need a higher reactivity than those that are long lived to obtain a threshold value. There would also be a lower bang for the buck in terms of each signaling event and signals would have less duration-effectively the cellular memory would be wiped too early.
Protein Half-Lives Set Lower Limits on Reactivity Needed to Be Sensors
As we mentioned, a significant portion of the phenotypically relevant outputs of REM signaling that we have studied have been derived by virtue of gain of function signaling. We explained why irreversible labeling assisted this mechanism as it effectively bolts the irreversible ligand onto the target, permanently turning that protein’s hidden function on. Aside from deHNEylation, a process that is unknown in cells, the only way to turn off this function is through protein degradation. Indeed, one of the early concepts for why irreversible drug binding was beneficial over reversible drug binding was the concept that new protein synthesis was needed to reinitialize protein activity for irreversible binding, but not for reversible binding [40]. This was postulated to put a larger burden on resources of cells addicted to the inhibited protein than that of non-addicted cells. Of course, this covalent burden is effectively alleviated if the target protein has a low half-life, as the cell is obligated to remake the protein anyway-effectively counterbalancing the win from covalent binding.
Although in the drug development field these concepts have subsided in favor of pharmacokinetic arguments, arguments centered around degradation are highly applicable to GOF-signaling. This is because in GOF signaling the labeled protein itself is the principal causative agent of phenotypically relevant responses. The system is no longer limited by need to achieve a high threshold occupancy, nor is it subject to overexpression of the target overwhelming the inhibited state [40]. Thus, to reinstate the default cellular state, degradation is the only simple way. This logic once again indicates that transient bursts of a specific REM can exert a prolonged effect on signaling outputs, potentially extending the cellular memory beyond the duration of the cell cycle. In modern pharmaceutical terms, this goes beyond occupancy-driven pharmacology to persistent “event-driven” behaviors [41]. However, it also indicates that the strength and persistence of REM signaling is strongly dictated by the half-life of the labelled protein of interest. Assuming that REM signaling is a network of cellular stress sensors that seeks to prepare cells to cope with future problems, it makes sense that reactive molecule signaling would occur on stable proteins. Thus from both angles, that of labeling and that of signaling, one may predict that reactive signal sensing will occur on stable proteins.
Exceptions could come about from extremely reactive proteins (k2 >> 1000 M-1s-1) that attain high REM-occupancy rapidly, regardless of known protein half-lives; proteins that trigger exaggerated GOF signaling mechanisms, i.e., that need exceptionally low occupancy; or potentially REM modification events that themselves stabilize target proteins (itself effectively a GOF phenotype). The last group could also be applicable to proteins that are, for instance, transient cell cycle drivers that are converted to dominant negative inhibitors upon REM labeling (this would naturally prevent degradation of the target protein). Conversely, following our proposed link between REM binding affinity, rapid labeling and GOF-signaling, and that ligand binding stabilizes targets as a function of Kd, all the latter examples would very much fit into a scenario where some super REM-sensors may have all the above attributes. These have yet to be found. Moreover, although it goes against some of the published data and dogma, it is plausible that some REM-sensing events are not intended to have prolonged effects on cells. These events would be expected to occur on proteins with short half-lives and could constitute a different class of REM sensors to what we have discovered to date, perhaps those involved in nuanced, transient signaling pathways (as opposed to translation, transcription, one carbon metabolism, etc). It is likely that our current detection methods are not apposite for detection of such species, as they are often performed on transfected cells, or lysates. Nonetheless, it is likely that REM sensors that are short lived proteins (or derived from proteins that exist in transient complexes) are particularly interesting.
Basal Levels of Electrophiles Set Upper Limits on Reactivity
One final thing to consider is that reactive molecules are part of all of us. For reactive molecules to function as signals it is paramount that redox signaling switches are not triggered all the time. For instance, we calculated that at 10 µM HNE, half-life of labeling of generic protein cysteines approaches the maximal division rate of cancer cells. This could reflect a threshold moment where the cellular defenses are intrinsically overwhelmed that is defined by maximal cellular division rates. As mentioned earlier, there is indeed a correlation between oxidative stress levels, HNE and cellular division rates [1], which means that less proliferative cells naturally lower HNE levels, making basal labeling rates intrinsically lower, potentially accounting for longer half-lives. Our model would require this sort of relationship, in the absence of other compensatory factors for removal of damaged proteins. Thus, it is clear that sensing trigger mechanisms cannot be too sensitive to react with stray HNE over a reasonable course of time.
This logic indicates that, at least for stable proteins, we are unlikely to find proteins in typical cellular compartments that can sense HNE at the diffusion limited (approximately 10^8-10^9 M-1s-1). Such a rate would lead to a half-life of labeling of less than a second at our standard conditions. Indeed, with a second order rate of labeling of 10000 M-1s-1 half-life of labeling for a stable protein is only ~1 min. Of course, some subcellular compartments may have particularly low HNE concentrations, or the cellular concentration of HNE itself may be very much overestimated. Without (locale-specific) sensors we cannot be sure about these ideas. Nonetheless, it is very unlikely concentrations are overestimated by more than 10-100 fold.
Fortunately, we can use data from T-REX (Figure 2A) to assess these assumptions. T-REX releases a defined quantity of HNE from a specific point source. This is achieved by expressing Halo protein that binds to a photocaged HNE irreversible. Photouncaging rates are known[42]. The expression of Halo for most proteins is similar, and for Halo, it has been calculated to be around 5 µM[43]. Using metabolic rates for HNE, half-life of photouncaging, and concentration of HALO, we have modelled how HNE accumulates in cells as a consequence of T-REX, peaking around 1 µM. In T-REX, HALO is usually fused to a target protein, e.g. KEAP1. For KEAP1, the amount of HNE that labels KEAP1 upon photocaging is 20-40%. However, we have also performed controls in which KEAP1 and HALO are expressed separately; this is often referred to as the split system. Under these conditions KEAP1 was not labelled effectively (<3% labeling) [25]. Modeling in the concentrations of KEAP1 and varying second order reaction with KEAP1 and HNE we measured that if KEAP1 were to react with HNE at a rate >20000 M-1s-1 then >85% Keap1 would have been labelled under these conditions. In fact, based on the data we collected the second order rate constant between KEAP1 and HNE is likely < 500 M-1s-1. Although we could easily be a factor of 10-100 out in these calculations, it is clear that KEAP1 does not react with HNE near the diffusion limit, as we predicted above. Nor does it need to obtain high labeling in cells. Indeed, under our standard conditions, a second order rate of reaction of 500 M-1s-1 would require around 30 min to achieve 50% labeling, which may still be too rapid, at least for a concentration of HNE 1 µM.
Two Different Paradigms of REM Sensors
These lines of reasoning argue that standard REM sensing proteins are stable and likely walk a fine line between premature triggering and inability to respond. Nonetheless, from several different angles, the sensors that we have discovered seem to fit into this paradigm. However our discussion also indicates that there may be other classes of sensors that are even more reactive, and have more diverse mechanistic repertoires that are cell cycle regulated or involved in transient complexes/signalosomes. Such proteins would require similar properties to already characterized privileged sensors but these would have to be particularly exaggerated. Such proteins would be particularly relevant to drug design and discovery.
Comparison with Current GOF Drugs
GOF drugs are entering the pharmaceutical canon. Several serendipitous GOF-drugs already exist; these include SERDs that cause degradation of the estrogen receptor[44,45]. They also extend to molecules that rewire the substrate spectrum of specific enzymes, such as thalidomide [46]; often referred to as a “molecular glue”, although we find this term confusing as it is unlikely thalidomide itself is able to directly “glue” two molecules together. We thus prefer small molecule neo-functionalizer. A new class of GOF drugs are also poised to enter the clinic, PROTACs (Figure 4). These are complex bifunctional molecules that seek to create new target protein-E3 interactions to usher target protein degradation [47]. At least for neofunctionalizers and PROTACs, there are numerous parallels with REM signaling. All three work through event-driven pharmacology. Initially focusing on PROTACs, these like REMs, also show prolonged efficacy on cells, and are often more effective than simple inhibition alone[48]. The last point derives from the fact that all functions of the target are ablated rather than simply the enzymatic function. However, there are several interesting differences between PROTACS and REMs that deserve discussion.
Firstly, whereas natural REM signals (and indeed neofunctionalizers like thalidomide) minimally elicit turn on function of a specific protein through a binary interaction, PROTACS (and neofunctionalizers that behave like a true “glue”) by definition function through a ternary complex. This means that for a PROTAC, or other two-headed GOF-inducer, both targeted proteins must be present, and able to engage with the specific poles of the bifunctional molecule.
A large amount of effort has been expended into optimizing every aspect of PROTACs in terms of recruited protein [49]. These considerations lead to differences in the way PROTACs and REM-based drugs may work.
The original PROTACs were non-covalent [47]. This makes sense as reversible binding to both target proteins allows release of the drug each time either of the two proteins bound are degradaed, endowing PROTACs with a catalytic mechanism. Covalent binding of PROTACS to the target protein has been tested, although this winnows the benefits of the PROTAC by preventing recycling[50,51]. Alternatively irreversible binding to specific E3s has also been tried [52]. Once again E3’s that trigger targeted degradation often have relatively low half-lives due to autoubiquitination functions
GOF Protein Sensors-Ideal for Drug Discovery
The above discussion demonstrates that there are many potential benefits of targeting natural GOF sensors for drug discovery. Their binary GOF mechanism immediately simplifies mechanistic analysis and minimizes complexity over two-headed strategies like PROTACs. Moreover, by screening for sentinel proteins regulating specific pathways, based on the discussion we are immediately wired in directly to proteins that have been selected to be apposite for orchestrating meaningful signaling processes (i.e. posses all the virtues described above, elevated kinetics, GOF functions, based around natural sentinel proteins that are designed to propagate signals over the long term). Of course, as we discussed, there may be no absolute definition of a privileged sensor protein-many proteins may be sensors under specific conditions. Nonetheless, proteins with GOF signaling that show elevated reaction kinetics usually come out of profiling screens, for instance Localis-REX (Figure 5), and these are a good start for drug discovery. By extension, identifying proteins that are sensors in specific disease conditions would be a good way to identify potential drug targets for a disease. Moreover, unlike artificial systems that co-opt regulatory functions of proteins and create new signaling pathways that are non-essential for the cell, targeting GOF protein sensors taps into natural signaling pathways that are likely evolutionarily conserved. Because REM signaling is likely more important in diseased states than in healthy cells, it is our opinion that mutagenesis to suppress these pathways would have significant negative effects on the diseased cells themselves. This means that gaining resistance to REM-signaling mimics may be overall disadvantageous to cells.
Identifying Sentinel Proteins in Unexpected Places
We have released numerous ways to identify sentinel proteins. Our current best strategy is called Localis-REX [15,30]. This method has allowed us to identify REM-sensitive proteins in more restricted environments. These include the cytosol, the nucleus, and specific tissues in C elegans. These have started to investigate more and more spatially restricted realms. However, based on the above discussion, questions remain if we should turn our attention to trying to investigate sensing of shorter-lived proteins. These may be missed from our profiling approaches as we used unsynchronized, transfected cells that typically have a very high G0 content. By changing strategies, we would readily investigate specific phases of the cell cycle, where many transiently expressed proteins have peak expression profiles. Perhaps using this sort of system, or by profiling other transient biological structures, such as signalosomes, we will be able to identify more rapid sensors. The applicability of these sensors to drug design may or may not be apposite, but it will still be interesting to measure kinetics of sensing in vitro, for instance, and see if we can extract general principles of sensing from hits derived from such systems.
Author contributions
Outline & content: M.J.C.L. and Y.A.; first draft: M.J.C.L.; figures and references: L.B.; editing & finalization: Y.A. All authors approve the final version of the article.
Acknowledgements
Research involving the development and applications of spatiofunctional omics and signaling interrogation technologies within the Aye laboratory are presently supported by the European Research Council Advanced Grant, Swiss National Science Foundation, UK Academy of Medical Sciences, and the Royal Society Wolfson Fellowship (Y.A.). Novartis Medical-Biological Research Foundation (M.J.C.L).
Conflict of interest
There is no conflict of interest to declare.
References
- Esterbauer, H.; Schaur, R. J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M. J. C.; Poganik, J. R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Schopfer, F. J.; Cipollina, C.; Freeman, B. A. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef]
- Blair, I. A. DNA adducts with lipid peroxidation products. J Biol Chem. 2008, 283, 15545–9. [Google Scholar] [CrossRef] [PubMed]
- Sakanyan, V. Reactive Chemicals and Electrophilic Stress in Cancer: A Minireview. High Throughput 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Huang, K. T.; Aye, Y. Toward decoding spatiotemporal signaling activities of reactive immunometabolites with precision immuno-chemical biology tools. Commun Chem. 2024, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Poganik, J. R.; Huang, K. T.; Parvez, S.; Zhao, Y.; Raja, S.; Long, M. J. C.; Aye, Y. Wdr1 and cofilin are necessary mediators of immune-cell-specific apoptosis triggered by Tecfidera. Nat Commun. 2021, 12, 5736. [Google Scholar] [CrossRef]
- Hauer, L.; Sellner, J. Diroximel Fumarate as a Novel Oral Immunomodulating Therapy for Relapsing Forms of Multiple Sclerosis: A Review on the Emerging Data. Drug Des Devel Ther. 2022, 16, 3915–3927. [Google Scholar] [CrossRef]
- Montano, J. L.; Wang, B. J.; Volk, R. F.; Warrington, S. E.; Garda, V. G.; Hofmann, K. L.; Chen, L. C.; Zaro, B. W. Improved Electrophile Design for Exquisite Covalent Molecule Selectivity. ACS Chem Biol. 2022, 17, 1440–1449. [Google Scholar] [CrossRef]
- Gu, T. J.; Cai, J.; Auster, A.; Torres, E.; Zhang, D.; Khojasteh, S. C.; Wang, S. In vitro metabolism of targeted covalent inhibitors and their thiol conjugates by gut microbiome from rats, mice, and humans. Drug Metab Dispos. 2025, 53, 100027. [Google Scholar] [CrossRef]
- Baird, L.; Zhang, L.; Hidaka, T.; Xi, L.; Wang, K.; Tateno, K.; Iso, T.; Suzuki, T.; Kumada, K.; Katsuoka, F.; Kinoshita, K.; Yamamoto, M. Systemic activation of NRF2 contributes to the therapeutic efficacy of clinically-approved KRAS-G12C anti-cancer drugs. Br J Cancer 2025, 133, 1377–1390. [Google Scholar] [CrossRef]
- Long, M. J.; Parvez, S.; Zhao, Y.; Surya, S. L.; Wang, Y.; Zhang, S.; Aye, Y. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat Chem Biol. 2017, 13, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Long, M. J. C.; Hopkins, B. D.; Luo, C.; Wang, L.; Aye, Y. Precision Targeting of pten-Null Triple-Negative Breast Tumors Guided by Electrophilic Metabolite Sensing. ACS Cent Sci. 2020, 6, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Alnasser, S. M. The role of glutathione S-transferases in human disease pathogenesis and their current inhibitors. Genes Dis. 2025, 12, 101482. [Google Scholar] [CrossRef]
- Liu, J.; Kulkarni, A.; Gao, Y. Q.; Urul, D. A.; Hamelin, R.; Novotny, B. A.; Long, M. J. C.; Aye, Y. Organ-specific electrophile responsivity mapping in live C. elegans. Cell. 2024, 187, 7450–7469 e29. [Google Scholar] [CrossRef] [PubMed]
- Recktenwald, C. V.; Kellner, R.; Lichtenfels, R.; Seliger, B. Altered detoxification status and increased resistance to oxidative stress by K-ras transformation. Cancer Res. 2008, 68, 10086–93. [Google Scholar] [CrossRef]
- Singh, R. R.; Reindl, K. M. Glutathione S-Transferases in Cancer. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Gueraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–30. [Google Scholar] [CrossRef]
- Huang, B. K.; Sikes, H. D. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biol. 2014, 2, 955–62. [Google Scholar] [CrossRef]
- Gueraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P. M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010, 44, 1098–124. [Google Scholar] [CrossRef]
- Lam, M.; Mast, N.; Pikuleva, I. A. Drugs and Scaffold That Inhibit Cytochrome P450 27A1 In Vitro and In Vivo. Mol Pharmacol. 2018, 93, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Normandin, K.; Coulombe-Huntington, J.; St-Denis, C.; Bernard, A.; Bourouh, M.; Bertomeu, T.; Tyers, M.; Archambault, V. Genetic enhancers of partial PLK1 inhibition reveal hypersensitivity to kinetochore perturbations. PLoS Genet. 2023, 19, e1010903. [Google Scholar] [CrossRef] [PubMed]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000, 17, 1232–9. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Assari, M.; Suwathep, C.; Sappakhaw, K.; Uttamapinant, C.; Long, M. J. C.; Aye, Y. NCBP1 stress signaling drives alternative S6K1 splicing inhibiting translation. Nat Chem Biol. 2026. [Google Scholar] [CrossRef]
- Parvez, S.; Fu, Y.; Li, J.; Long, M. J.; Lin, H. Y.; Lee, D. K.; Hu, G. S.; Aye, Y. Substoichiometric hydroxynonenylation of a single protein recapitulates whole-cell-stimulated antioxidant response. J Am Chem Soc. 2015, 137, 10–3. [Google Scholar] [CrossRef]
- Zhou, Y. F.; Zhang, L.; Niu, Z. L.; Wang, Z. A. Targeting the Reactive Proteome: Recent Advances in Activity-Based Protein Profiling and Probe Design. Biomolecules 2025, 15. [Google Scholar] [CrossRef]
- Wall, S. B.; Smith, M. R.; Ricart, K.; Zhou, F.; Vayalil, P. K.; Oh, J. Y.; Landar, A. Detection of electrophile-sensitive proteins. Biochim Biophys Acta 2014, 1840, 913–22. [Google Scholar] [CrossRef]
- Long, M. J. C.; Urul, D. A.; Aye, Y. REX technologies for profiling and decoding the electrophile signaling axes mediated by Rosetta Stone proteins. Methods Enzymol. 2020, 633, 203–230. [Google Scholar]
- Liu, X.; Long, M. J. C.; Aye, Y. Proteomics and Beyond: Cell Decision-Making Shaped by Reactive Electrophiles. Trends Biochem Sci. 2019, 44, 75–89. [Google Scholar] [CrossRef]
- Zhao, Y.; Miranda Herrera, P. A.; Chang, D.; Hamelin, R.; Long, M. J. C.; Aye, Y. Function-guided proximity mapping unveils electrophilic-metabolite sensing by proteins not present in their canonical locales. Proc Natl Acad Sci U S A. 2022, 119. [Google Scholar] [CrossRef]
- Guerau-de-Arellano, M.; Piedra-Quintero, Z. L.; Tsichlis, P. N. Akt isoforms in the immune system. Front Immunol. 2022, 13, 990874. [Google Scholar] [CrossRef]
- Kumar, B. H.; Kabekkodu, S. P.; Pai, K. S. R. Structural insights of AKT and its activation mechanism for drug development. Mol Divers. 2025, 29, 5443–5463. [Google Scholar] [CrossRef]
- Calabrese, E. J.; Kozumbo, W. J. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol Res. 2021, 167, 105526. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G. L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. K.; Santhoshkumar, P. Lens aging: effects of crystallins. Biochim Biophys Acta 2009, 1790, 1095–108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Sheikh, M. S.; Huang, Y. Decision Making by p53: Life versus Death. Mol Cell Pharmacol. 2010, 2, 69–77. [Google Scholar]
- Gregory, M. A.; Hann, S. R. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol. 2000, 20, 2423–35. [Google Scholar] [CrossRef]
- Mailand, N.; Podtelejnikov, A. V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–20. [Google Scholar] [CrossRef]
- Lee, J. M.; Hammaren, H. M.; Savitski, M. M.; Baek, S. H. Control of protein stability by post-translational modifications. Nat Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Basu, R.; Fletcher, S. Protein structural dynamics in covalent drug design: insights from irreversible and reversible covalent inhibitors. RSC Chem Biol. 2026, 7, 376–399. [Google Scholar] [CrossRef]
- Lai, A. C.; Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M. J.; Lin, H. Y.; Zhao, Y.; Haegele, J. A.; Pham, V. N.; Lee, D. K.; Aye, Y. T-REX on-demand redox targeting in live cells. Nat Protoc. 2016, 11, 2328–2356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Long, M. J. C.; Wang, Y.; Zhang, S.; Aye, Y. Ube2V2 Is a Rosetta Stone Bridging Redox and Ubiquitin Codes, Coordinating DNA Damage Responses. ACS Central Science 2018, 4, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A. E.; Dukes, M.; Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991, 51, 3867–73. [Google Scholar] [PubMed]
- Wang, G. Fulvestrant as a reference antiestrogen and estrogen receptor (ER) degrader in preclinical studies: treatment dosage, efficacy, and implications on development of new ER-targeting agents. Transl Cancer Res. 2020, 9, 4464–4468. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–50. [Google Scholar] [CrossRef]
- Sakamoto, K. M.; Kim, K. B.; Kumagai, A.; Mercurio, F.; Crews, C. M.; Deshaies, R. J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001, 98, 8554–9. [Google Scholar] [CrossRef]
- Suo, Y.; Du, D.; Chen, C.; Zhu, H.; Wang, X.; Song, N.; Lu, D.; Yang, Y.; Li, J.; Wang, J.; Luo, Z.; Zhou, B.; Luo, C.; Zhou, H. Uncovering PROTAC Sensitivity and Efficacy by Multidimensional Proteome Profiling: A Case for STAT3. J Med Chem. 2024, 67, 4804–4818. [Google Scholar] [CrossRef]
- Pike, A.; Williamson, B.; Harlfinger, S.; Martin, S.; McGinnity, D. F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today 2020, 25, 1793–1800. [Google Scholar] [CrossRef]
- Tinworth, C. P.; Lithgow, H.; Dittus, L.; Bassi, Z. I.; Hughes, S. E.; Muelbaier, M.; Dai, H.; Smith, I. E. D.; Kerr, W. J.; Burley, G. A.; Bantscheff, M.; Harling, J. D. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem Biol. 2019, 14, 342–347. [Google Scholar] [CrossRef]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; Brandis, A.; Shulman, Z.; Katz, B. Z.; Herishanu, Y.; London, N. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J Am Chem Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V. M.; Wucherpfennig, T. G.; Dix, M. M.; Cravatt, B. F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
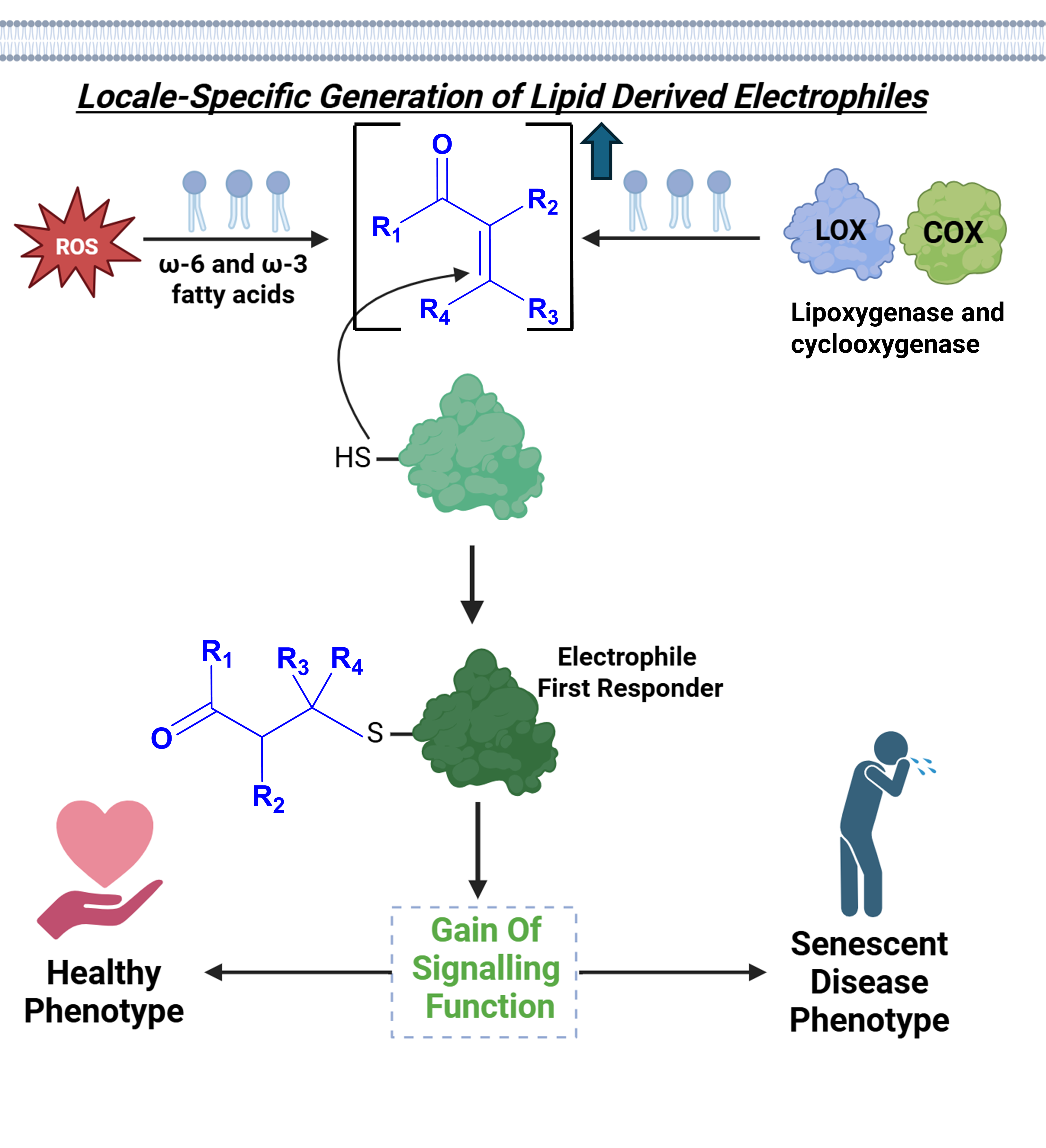
Figure 1.
Electrophilic Lipid Peroxidation products at the phospholipid bilayer and their downstream labelling mechanisms. (A) Generation of 4-Hydroxynonenal (HNE) occurs through both enzymatic and non-enzymatic pathways, from peroxidation of lipids residing in the phospholipid membrane. LOX and COX oxygenase enzymes convert Arachidonic acid starting material to Polyunsaturated fatty acids (PUFAs), of which many contain electrophilic moieties. Non-enzymatic lipid peroxidation follows a free-radical pathway with hydroxy radicals produced at the lipid membrane from Fenton-type chemistry, resulting in lipid peroxidation products such as 4-hydroxynonenal (HNE) [2]. (B) Lipid peroxidation product HNE at a cytosolic concentration of 1 µM at the phospholipid membrane can have three potential mechanisms of action. Firstly, kinetically privileged first responders will react quickly, second order rate k2 > 10 M-1s-1, with HNE at the cytosolic concentration, leading to a gained functional downstream response. Secondly, a small burst of locally-produced HNE, reaching up to 100 µM for 1 – 2 h, can lead to non-specific tagging of bystander protein cysteines at a second order rate k2 ~ 1 M-1s-1, triggering a new downstream function of the protein. The competition between the rate of tagging of HNE by nearby cysteines and the detoxification process by GSH controls the overall strength of the downstream signalling response triggered by the HNE modification. At the modelled cytosolic concentration of 1 µM HNE, the half-life of covalent non-specific tagging of bystander proteins is 8 days, therefore aforementioned “non-specific” covalent tagging of protein cysteines by HNE is likely only possible after a stress- or inflammation-induced increase in lipid peroxidation levels.
Figure 1.
Electrophilic Lipid Peroxidation products at the phospholipid bilayer and their downstream labelling mechanisms. (A) Generation of 4-Hydroxynonenal (HNE) occurs through both enzymatic and non-enzymatic pathways, from peroxidation of lipids residing in the phospholipid membrane. LOX and COX oxygenase enzymes convert Arachidonic acid starting material to Polyunsaturated fatty acids (PUFAs), of which many contain electrophilic moieties. Non-enzymatic lipid peroxidation follows a free-radical pathway with hydroxy radicals produced at the lipid membrane from Fenton-type chemistry, resulting in lipid peroxidation products such as 4-hydroxynonenal (HNE) [2]. (B) Lipid peroxidation product HNE at a cytosolic concentration of 1 µM at the phospholipid membrane can have three potential mechanisms of action. Firstly, kinetically privileged first responders will react quickly, second order rate k2 > 10 M-1s-1, with HNE at the cytosolic concentration, leading to a gained functional downstream response. Secondly, a small burst of locally-produced HNE, reaching up to 100 µM for 1 – 2 h, can lead to non-specific tagging of bystander protein cysteines at a second order rate k2 ~ 1 M-1s-1, triggering a new downstream function of the protein. The competition between the rate of tagging of HNE by nearby cysteines and the detoxification process by GSH controls the overall strength of the downstream signalling response triggered by the HNE modification. At the modelled cytosolic concentration of 1 µM HNE, the half-life of covalent non-specific tagging of bystander proteins is 8 days, therefore aforementioned “non-specific” covalent tagging of protein cysteines by HNE is likely only possible after a stress- or inflammation-induced increase in lipid peroxidation levels.

Figure 2.
Experimental workflow to identify and validate kinetically-privileged first responders to specific REMs with spatiotemporal precision in living subjects. (A) To first validate a set protein of interest (POI) and confirm its REM-sensing ability using T-REX: Halo, a protein engineered to irreversibly bind the cell/animal permeable and biocompatible photocaged-REM probe, is fused to the POI and, once the REM is rapidly uncaged by UV irradiation (365 nm, 2-5 mW/cm2, t1/2 photouncaging <1 min), any POI with inherent REM-sensing ability will bind the probe, and be identified in the subsequent pulldown and proteomics steps [42] . Two isoforms of Protein Kinase B; Akt2 and Akt3, were identified as electrophile sensors using such tools [12]. (B) Diagram of FRET-based reporter to identify the changes in phosphorylation activity of Akt3 before (left) and after (right) release of electrophiles, and the subsequent hydroxynonenalation of the reactive cysteine residue, C119. (C) T-REX experimental procedure in HEK293T cells expressing Halo-Akt3 fusion protein. Results validate the downstream functional effects of the electrophile-sensing Cysteine 119 in Akt3, located in the linker region between the N-terminal plextrin homology (NTPH) and kinase domains when compared to the respective C119S mutant. Subsequent activation of downstream protein target of Akt3, FOXO, was monitored using a luciferase reporter assay, confirming perturbation of Akt3 phosphorylation activity in response to electrophile release [12].
Figure 2.
Experimental workflow to identify and validate kinetically-privileged first responders to specific REMs with spatiotemporal precision in living subjects. (A) To first validate a set protein of interest (POI) and confirm its REM-sensing ability using T-REX: Halo, a protein engineered to irreversibly bind the cell/animal permeable and biocompatible photocaged-REM probe, is fused to the POI and, once the REM is rapidly uncaged by UV irradiation (365 nm, 2-5 mW/cm2, t1/2 photouncaging <1 min), any POI with inherent REM-sensing ability will bind the probe, and be identified in the subsequent pulldown and proteomics steps [42] . Two isoforms of Protein Kinase B; Akt2 and Akt3, were identified as electrophile sensors using such tools [12]. (B) Diagram of FRET-based reporter to identify the changes in phosphorylation activity of Akt3 before (left) and after (right) release of electrophiles, and the subsequent hydroxynonenalation of the reactive cysteine residue, C119. (C) T-REX experimental procedure in HEK293T cells expressing Halo-Akt3 fusion protein. Results validate the downstream functional effects of the electrophile-sensing Cysteine 119 in Akt3, located in the linker region between the N-terminal plextrin homology (NTPH) and kinase domains when compared to the respective C119S mutant. Subsequent activation of downstream protein target of Akt3, FOXO, was monitored using a luciferase reporter assay, confirming perturbation of Akt3 phosphorylation activity in response to electrophile release [12].

Figure 3.
Identification of “Turn on Signalling” in response to nuclear-specific HNEylation of Kinetically Privileged First Responder (KPS) Nuclear Cap Binding Protein Subunit 1 (NCBP1). (A) REX-based technologies, including Localis-REX and T-REX, were employed to identify potential kinetically priviledged first responders, of which NCBP1 was a hit [24]. Site-directed mutagenesis of each 19 cysteines of NCBP1 individually identified Cys 436 as a residue with a functional response to the photouncaged HNE. Results demonstrated that HNEylation of Cys 436 leads to an inhibition of translation of several genes relating to regulation of transcription, translation and the stress response. A GCE reporting system was used to quantify rates of protein synthesis, whereby site-specific integration of non-canonical amino acid bicyclononynelysine was integrated into a synthetic HA-tagged actin. The rate of protein synthesis was measured by western blot using anti-HA, and by in-gel fluorescence, using the intensity of the fluorescence yielded from Diels-Alder coupling to Cy5 azide. Both methods confirm a decline in protein synthesis signal is observed only after the cells are introduced to the photouncaged electrophile probe. (B ) Left: X – ray Crystal structure of NCBP1 (blue) – NCBP2 (green) heterodimer (PDB ID: 1H6K, 2.00 Å), with 18 cysteine residues of NCBP1 highlighted in yellow, and the functionally priviledged electrophilic sensor cysteine 436 highlighted in red. Right: Expansion of the 4-hydroxynonenal binding site at Cysteine 436, with nearby residues PHE 448 and TYR 399 highlighted in orange and pink respectively. (C) The proposed gain of function signalling function of post-translational HNE tagging of NCBP1 at Cysteine 436. HNE binding leads to the disassociation of binding partner SF3A1, consequently triggering the alternative splicing of several gene products. One such alternatively spliced gene product, S6K1-X, was identified as a negative regulator for translation, halting global protein production in response to electrophilic tagging of NCBP1.
Figure 3.
Identification of “Turn on Signalling” in response to nuclear-specific HNEylation of Kinetically Privileged First Responder (KPS) Nuclear Cap Binding Protein Subunit 1 (NCBP1). (A) REX-based technologies, including Localis-REX and T-REX, were employed to identify potential kinetically priviledged first responders, of which NCBP1 was a hit [24]. Site-directed mutagenesis of each 19 cysteines of NCBP1 individually identified Cys 436 as a residue with a functional response to the photouncaged HNE. Results demonstrated that HNEylation of Cys 436 leads to an inhibition of translation of several genes relating to regulation of transcription, translation and the stress response. A GCE reporting system was used to quantify rates of protein synthesis, whereby site-specific integration of non-canonical amino acid bicyclononynelysine was integrated into a synthetic HA-tagged actin. The rate of protein synthesis was measured by western blot using anti-HA, and by in-gel fluorescence, using the intensity of the fluorescence yielded from Diels-Alder coupling to Cy5 azide. Both methods confirm a decline in protein synthesis signal is observed only after the cells are introduced to the photouncaged electrophile probe. (B ) Left: X – ray Crystal structure of NCBP1 (blue) – NCBP2 (green) heterodimer (PDB ID: 1H6K, 2.00 Å), with 18 cysteine residues of NCBP1 highlighted in yellow, and the functionally priviledged electrophilic sensor cysteine 436 highlighted in red. Right: Expansion of the 4-hydroxynonenal binding site at Cysteine 436, with nearby residues PHE 448 and TYR 399 highlighted in orange and pink respectively. (C) The proposed gain of function signalling function of post-translational HNE tagging of NCBP1 at Cysteine 436. HNE binding leads to the disassociation of binding partner SF3A1, consequently triggering the alternative splicing of several gene products. One such alternatively spliced gene product, S6K1-X, was identified as a negative regulator for translation, halting global protein production in response to electrophilic tagging of NCBP1.

Figure 4.
Schematic Diagram of the design of non-covalent small molecule PROteolysis TArgeting Chimeras (PROTACs) used to drug protein targets. The reversible binding of PROTACs to both the protein of interest (POI) and an E3 ligase in close proximity triggers selective polyubiquitination, and downstream degradation by the proteasome [47]. This could lead to a haploinsufficient phenotype of the cell, whereby at least 50% of the disease-causing phenotype can be lost by degradation of the POI. Such techniques therefore depend on the endogenous E3 ligase and proteasome systems of the diseased cell to effectively destroy the disease-causing protein and restore the “healthy” phenotype.
Figure 4.
Schematic Diagram of the design of non-covalent small molecule PROteolysis TArgeting Chimeras (PROTACs) used to drug protein targets. The reversible binding of PROTACs to both the protein of interest (POI) and an E3 ligase in close proximity triggers selective polyubiquitination, and downstream degradation by the proteasome [47]. This could lead to a haploinsufficient phenotype of the cell, whereby at least 50% of the disease-causing phenotype can be lost by degradation of the POI. Such techniques therefore depend on the endogenous E3 ligase and proteasome systems of the diseased cell to effectively destroy the disease-causing protein and restore the “healthy” phenotype.

Figure 5.
Schematic Diagram of the Electrophilic drug design process guided by REX technologies. Locale-specific first responders (POI) are identified in response to the UV-triggered release of an uncaged derivative of an electrophile, such as 4-HNE [30]. Hits from initial testing are then validated using T-REX and phenotypic assays in disease relevant cell lines to assess the signalling role of the “gain of function” post-translational modification on the Kinetically Privileged Sensor (sensor) [42]. Subsequent screening of drug libraries to identify electrophilic small molecules either already in the market, or commercially available is used to assist the drug design process. Such electrophilic drugs, once administered into diseased cells, will trigger a downstream signalling function of the disease-implicated POI, leading to an alleviated or disease-free phenotype.
Figure 5.
Schematic Diagram of the Electrophilic drug design process guided by REX technologies. Locale-specific first responders (POI) are identified in response to the UV-triggered release of an uncaged derivative of an electrophile, such as 4-HNE [30]. Hits from initial testing are then validated using T-REX and phenotypic assays in disease relevant cell lines to assess the signalling role of the “gain of function” post-translational modification on the Kinetically Privileged Sensor (sensor) [42]. Subsequent screening of drug libraries to identify electrophilic small molecules either already in the market, or commercially available is used to assist the drug design process. Such electrophilic drugs, once administered into diseased cells, will trigger a downstream signalling function of the disease-implicated POI, leading to an alleviated or disease-free phenotype.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



