Submitted:
21 April 2026
Posted:
22 April 2026
You are already at the latest version
Abstract
The synthesis of hydroxamic acids from sterically hindered substrates, such as abietane-type resin acids, remains a significant challenge due to the extreme congestion of the tricyclic skeleton. This study reports an efficient one-pot protocol for the direct conversion of abietic and dehydroabietic acids into their corresponding hydroxamic derivatives, achieving 65% and 74% isolated yields, respectively. Systematic screening of activating agents identified diethyl chlorophosphate (DCP) as the superior reagent for the hydroxy-amidation. A critical finding of this work is that the optimization of the isolation process specifically minimizing the water amount during aqueous work-up was essential to recover these polar products and prevent significant yield loss.
The reaction proceeds through diethyl phosphate mixed anhydride intermediate, which was successfully isolated, providing direct experimental evidence of the activation pathway. The reaction mechanism was further elucidated using density functional theory (DFT) calculations at the M062X/6-31G** level, identifying a concerted transition state for the simultaneous addition of hydroxylamine and expulsion of the phosphate group. Furthermore, the study rationalizes the observed chemoselectivity: although the ester is the more stable thermodynamic product, the formation of the N-hydroxy amide is kinetically favored through a substantially lower activation barrier. This combined experimental and theoretical approach establishes a robust and scalable methodology for the functionaliza-tion of abundant similar natural terpenoids.
Keywords:
abietane
; abietic acid
; dehydroabietic acid
; synthesis
; hydroxamic acid
1. Introduction
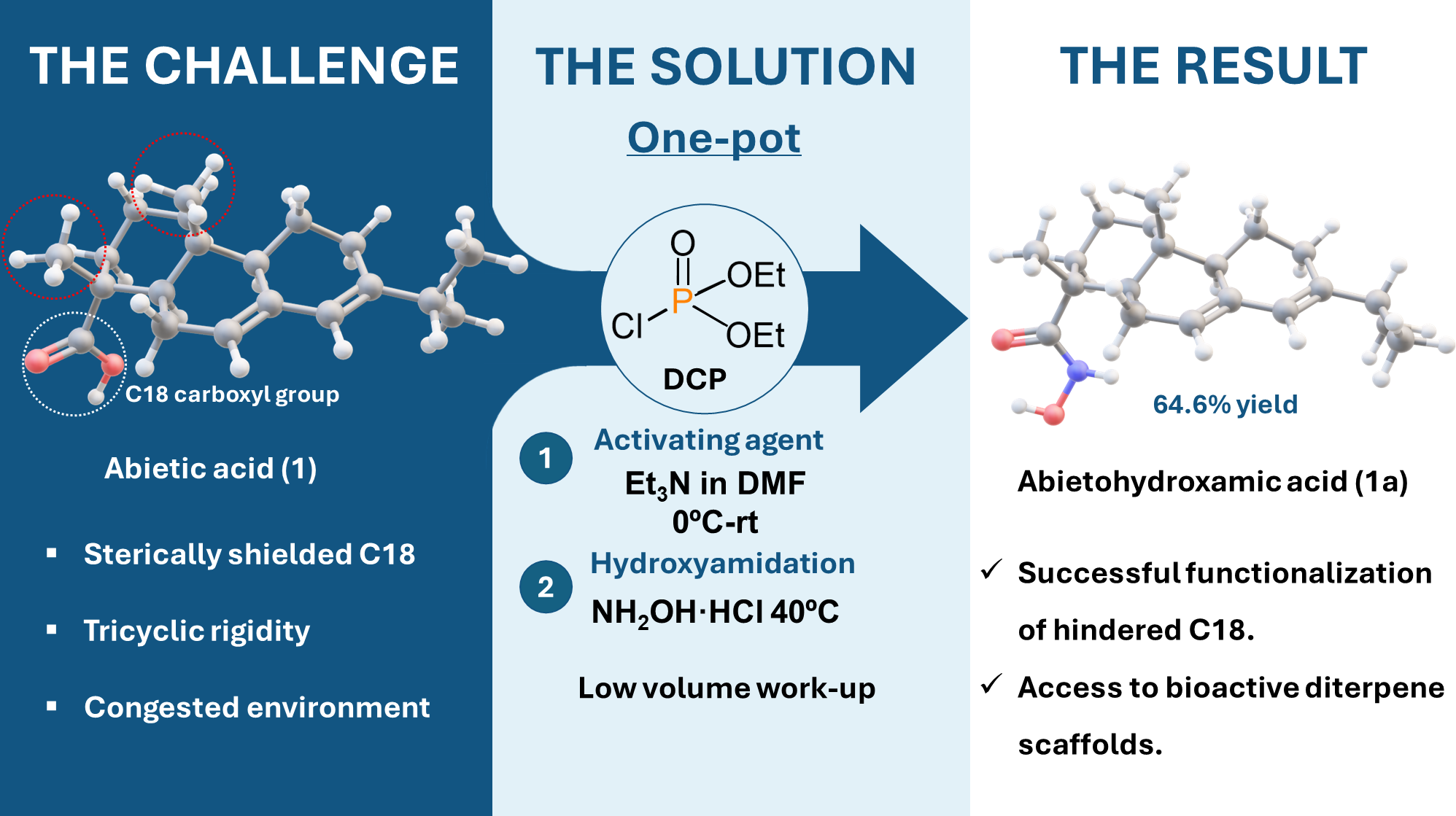
Abietane-type diterpenoids, such as abietic acid (1, Figure 1) and dehydroabietic acid (2, Figure 1), represent a class of tricyclic natural products characterized by significant structural diversity and widespread occurrence in coniferous resins [1,2]. These compounds have long served as versatile chiral scaffolds in organic synthesis for the preparation of bioactive derivatives and the semisynthesis of other complex natural products [3,4]. However, the functionalization of the C18 carboxyl group in the abietane skeleton remains a formidable synthetic challenge. This difficulty arises from the severe steric hindrance imposed by the rigid tricyclic system and the proximity of the methyl groups at the C4 and C10 positions [5].
Hydroxamic acids, or N-hydroxy amides, are of particular interest in organic and medicinal chemistry due to their unique ability to form stable chelates with transition metal ions and their prevalence in enzyme inhibitors [6,7]. While the incorporation of a hydroxamic acid moiety has been shown to enhance the properties of various terpenoids, including derivatives of betulin, oleanolic acid, and glycyrrhetinic acid [8,9,10], reports on abietane-derived hydroxamates are remarkably scarce. A landmark study by Bardyshev reported the preparation of abietohydroxamic acid (1a, Figure 1) through the formation of intermediate anhydrides [11]. Despite this early precedent, the method lacked optimization for highly hindered substrates, and a detailed understanding of the factors governing the competitive N- versus O-attack of hydroxylamine on such congested centers was not established.
Traditional methods for hydroxamic acid synthesis often involve the reaction of esters or acid chlorides with hydroxylamine [12,13]. Nevertheless, these procedures frequently fail or require harsh conditions when applied to the sterically shielded environment of resin acids. In this context, the development of stepwise activation strategies is essential to channel reactivity in a controlled manner. Diethyl chlorophosphate (DCP) has emerged as an effective reagent for the in-situ generation of reactive mixed anhydride intermediates, yet its application to the synthesis of hindered abietane hydroxamates has not been systematically explored until now.
In this work, we describe a robust and optimized one-pot protocol for the direct conversion of abietic and dehydroabietic acids into their corresponding hydroxamic derivatives using DCP as the activating agent. Beyond the synthetic optimization, we provide the first comprehensive mechanistic rationalization of the observed chemoselectivity through Density Functional Theory (DFT) calculations. This work expands upon our preliminary findings reported at the 29th International Electronic Conference on Synthetic Organic Chemistry [14]. By establishing a unified energy reference point, we demonstrate how the reaction is governed by strict kinetic control. This control favors the formation of the N-hydroxy amide over the hydroximic ester, which is the thermodynamically more stable product, successfully overcoming the steric demand of the diterpene skeleton.
2. Results and Discussion
2.1. Screening of Activation Methods and Mechanistic Discovery
The synthesis of hydroxamic acids derived from abietane-type resin acids represents a formidable synthetic challenge that has long hindered the functionalization of these natural products. This difficulty arises primarily from the extreme steric hindrance surrounding the C18 carboxyl group. This specific carbon center is effectively shielded by the rigid tricyclic framework of the diterpene skeleton and is further congested by the presence of axial methyl groups located at the C4 and C10 positions. Such a dense chemical environment precludes the possibility of direct functionalization of the carboxylic acid, thereby necessitating an indirect activation pathway via a relay-activation mechanism to overcome the high organizational and steric barriers of the system. This general activation approach, designed to bypass the steric congestion of the resin acid, is illustrated in Scheme 1.
Attempts to facilitate this transformation using ethyl chloroformate and triethylamine to form a classic mixed anhydride resulted in the complete recovery of the starting material. We subsequently studied a more aggressive activation via the acyl chloride using thionyl chloride (SOCl2) and pyridine. However, the used conditions favored the formation of the symmetric anhydride of dehydroabietic acid over the desired attack by the nitrogen nucleophile. Furthermore, modern coupling reagents such as EDC/DMAP were evaluated, but these yielded highly complex mixtures with low selectivity, making the purification of the target hydroxamic acid practically impossible.
Before arriving at the optimized phosphorus-mediated route, we systematically evaluated several conventional and modern carboxyl activation strategies to benchmark the reactivity of the C18 center. We initially explored the use of propylphosphonic anhydride (PPAA) in acetonitrile at room temperature [15]. For abietic acid (1), this reagent resulted in the complete recovery of the starting material (Table 1, Entry 1). Interestingly, when PPAA was applied to dehydroabietic acid (2), the results were inconsistent; specific experiments yielded the desired hydroxamic acid with a modest yield of approximately 20%, but the reactions were characterized by poor selectivity and the formation of unknown side products (Table 1, Entry 2).
Following these trials, we explored the use of diethyl phosphorocyanidate (DEPC) [16]. When the reaction was performed by mixing all reagents simultaneously with substrate 1, we observed a low yield of 13.7% (Table 1, Entry 3). However, these results were fundamental in unraveling the reaction mechanism. By employing dehydroabietic acid (2), we successfully isolated and characterized the corresponding stable diethyl phosphate mixed anhydride intermediate (Int2, Scheme 2) (Table 1, Entry 4). The subsequent transformation of this isolated intermediate into the desired hydroxamic acid 2a was achieved in a separate step by reaction with hydroxylamine in DMF (Table 1, Entry 5).
The outcomes of Entry 3 and Entry 4 provided the critical mechanistic validation needed to move forward. These results confirmed that phosphorus-mediated activation was the correct chemical strategy. Therefore, it was envisaged that phosphorylation with diethyl chlorophosphate (DCP) would operate via a nearly identical activation pathway, but as a more robust and cost-effective phosphorous source. To conclude this screening phase, we transitioned to a one-pot protocol using DCP and DMF at 40 °C. While this confirmed the chemical viability of the method, the initial yields obtained using standard isolation procedures remained disappointing: 17% starting from abietic acid (1) and 34% from dehydroabietic acid (2) (Table 1, Entry 6 and Entry 7, respectively).
2.2. Optimization One-Pot Synthesis
The focus of the investigation then shifted to the optimization of the isolation process (work-up). Our findings revealed that the true bottleneck was not the chemical reactivity, but rather the loss of product during extraction. The resin acid derivatives studied here possess an amphiphilic nature, which causes a significant portion of the hydroxamic acid to partition back into the aqueous phase during the traditional washing steps.
The impact of this optimization is most evident when comparing the physical state and purity of the crude products. In the initial trials using a standard volume of 20 mL of water (Table 1, Entries 6), the resulting crude was a dry yellow foam. However, 1H NMR analysis revealed a discouraging 1:1 ratio between the hydroxamic acid and the starting carboxylic acid, confirming significant product loss.
To address this, we implemented a modified work-up by drastically reducing the water volume to 5 mL. This change resulted in a crude product that appeared as a yellowish solution in residual DMF. Crucially, the 1H NMR purity improved to a 3:1 ratio (product to starting acid). This decision represented a strategic trade-off: we deliberately sacrificed the total removal of DMF in the crude stage to ensure the maximum recovery of hydroxamic acid. While residual DMF can sometimes complicate purification, we found that the remaining volume was sufficiently small to not interfere with the subsequent flash column chromatography fractions.
By applying this optimized lower water amount work-up, the isolated yields increased up to 64.6% for abietic acid (Table 1, Entry 8) and 73.8% for dehydroabietic acid (Table 1, Entry 9). The finalized optimized one-pot protocol is illustrated in Scheme 2.
Finally, the standardized heating to 40 °C was validated as essential for the one-pot protocol, ensuring the full solubility of the hydroxylamine hydrochloride in DMF. As demonstrated by our computational analysis, this energy input allows the system to overcome easily the calculated activation barrier of 13.3 kcal/mol (Figure 2), facilitating access to the transition state without compromising the stability of the activated intermediates.
2.3. Mechanistic Rationalization via DFT Studies
To gain deeper mechanistic insight into the observed chemoselectivity of the hydroxyamidation reaction, we investigated the competitive nucleophilic acyl substitution pathways occurring in the second stage of the one-pot process. While the global reaction starts from abietic acid (1), the computational study specifically evaluated the energy profiles for both routes N-attack and O-attack starting from the activated phosphate mixed anhydride as the common reactive precursor (Figure 2). This intermediate is formed in situ during the first activation step and serves as the branching point for the subsequent nucleophilic attack by hydroxylamine.
For the hydroxamic amide pathway, the calculated energy variation (∆E) for the RC (Reactant Complex), TS (Transition State), and PC (Product Complex) presents a profile of 0.0, +10.8, and -10.0 kcal/mol relative to its initial association complex, as detailed in Figure S19. This activation barrier of 10.8 kcal/mol is relatively low. While the inclusion of entropic factors (∆S) will expectedly increase this value, the overall exothermic nature of the process suggests that amide formation is unlikely to be reversible under the employed experimental conditions. Conversely, the route leading to the amino ester via oxygen attack exhibits a profile of 0.0, +20.2, and -9.4 kcal/mol relative to its own initial complex, as illustrated in Figure S23.
However, to provide a comprehensive and comparable view of the chemoselectivity during this second step, a common energy reference point must be established, considering that both initial reactant complexes exist in a rapid exchange equilibrium. A comparison of the total electronic energies (HF) reveals that the ester-type reactant complex is lower in energy (HF = –1708.1497 a.u., see Table S24) than its amide-type counterpart (HF = –1708.1449 a.u., see Table S20). Since the ester-type complex is the more stable of the two, it was selected as the global zero-energy reference for Figure 2. Under this unified framework, the profile for amide formation is adjusted to 0.0, +13.3, and -7.0 kcal/mol, while the ester pathway remains at 0.0, +20.2, and -9.4 kcal/mol.
These results reveal a clear dichotomy between kinetic and thermodynamic control in the functionalization of the abietane skeleton. From a thermodynamic perspective, the hydroximic ester product is more stable than the hydroxy amide by 2.4 kcal/mol. Nevertheless, the reaction is strictly governed by kinetic control, as the activation barrier for N-attack is 6.9 kcal/mol lower than that required for O-attack. At room temperature (298.15 K), this energetic difference favors amide formation by approximately five orders of magnitude in the rate constant ratio. Such a significant kinetic preference provides a compelling rationalization for the exclusive formation of the hydroxamic acid observed experimentally.
The second stage of the reaction was conducted at 40 °C to ensure the complete solubility of the hydroxylamine hydrochloride in DMF. This temperature ensures a high reaction rate while remaining sufficiently low to prevent the thermal decomposition of the sensitive activated phosphate intermediate.
Finally, Intrinsic Reaction Coordinate (IRC) calculations confirmed that the transition state for the amide pathway connects the activated phosphate reactant specifically to the desired hydroxamic product. These findings validate a mechanistic framework where the steric hindrance of the abietane skeleton is overcome by the superior kinetic accessibility of the N-attack route.
3. Materials and Methods
3.1. Materials and Equipment
All reagents were obtained from commercial sources and used without further purification unless otherwise noted. Abietic acid (95% purity) was purchased from Biosynth. (Staad, Switzerland). Despite dehydroabietic acid (DHA) is commercially available, it was synthesized under similar conditions of the procedure reported by Halbrook and Lawrence [17], from an old aged sample of abietic acid (ca. 50-60% purity) we had available in the laboratory, by disproportionation. To this end, 1% (w/w) of 10% Pd on carbon (300 mg) (Merck Life Science S.L.U., St. Louis, MO, USA) were added to 30 g of impure abietic acid and heated at 240 ˚C for 4 h without a stream of nitrogen gas. It was cooled to around 50 ˚C and diluted with 50 mL of ethyl acetate and filtered through a paper filter. After removal of the solvent the crude product (26 g) was chromatographed on silica eluting with hexane-ethyl acetate 7:3 to give 9.6 g of DHA which contained ca. 10% of an impurity. 6,7-dehydrodehydroabietic acid. It was attempted further purification by making the 2-aminoethanol salt and crystallization [17]. For this purpose, 9.6 g of impure DHA were dissolved in 5 mL of 96% ethanol at 80 ˚C with continuous stirring and 1.8 mL of ethanolamine (Merck Life Science S.L.U., St. Louis, MO, USA) and 5 mL of pre-heated water were added. Without cooling, it was extracted once with 7 mL of isooctane (Merck Life Science S.L.U., St. Louis, MO, USA) but we were unable to do more hot extractions with isooctane like Halbrook and Lawerence since the product rapidly precipitated. The salt was collected and washed with cold 1:1 (v/v) ethanol-water (50 mL) and dried to the air overnight. The resulting salt (9.8 g) was recrystallized once dissolving with 20 mL of 96% ethanol at 70 ˚C and 21 mL of water used as anti-solvent, allowing to cool down to rt with a crystal seed. It was obtained 5 g of salt as a white solid with trace brownish powder. This salt was dissolved in 10 mL of 96% ethanol at 70 ˚C and acidified to pH=1 by dropwise addition of 1 ml of conc. aqueous HCl. The resulting white precipitate, after cooling to rt, was filtered off and washed with water and dried to the air at rt overnight to afford 3.7 g of DHA which still contained the ca. 10% impurity of the double bond between C6-C7. Finally, 3.2 g of this impure DHA was hydrogenated using 10% Pd/C (210 mg) in MeOH (60 mL) under 1 atm of hydrogen for 20 h. After filtration through a pad of celite under reduced pressure and washing with 40 mL of MeOH, the clear filtrate was concentrated to give 3.04 g of pure DHA as a white solid.
Diethyl chlorophosphate (DCP, 97%), triethylamine (Et3N, 99%), and hydroxylamine hydrochloride (NH2OH·HCl, 99%) were obtained from Merck Life Science (St Louis, MO, USA). Anhydrous N,N-dimethylformamide (DMF) was purchased from ACROS Organics (Geel, Belgium) (extra dry over molecular sieves, AcroSeal). All moisture-sensitive reactions were performed under an argon atmosphere using an argon-filled balloon.
Analytical TLC was performed on silica gel 60 F254 plates, and column chromatography was carried out on silica gel 60 (0.040–0.063 mm) (Merck Life Science S.L.U., St. Louis, MO, USA). Melting points were determined on a Cole-Parmer MP-800D apparatus (Vernon Hills, IL, USA). Optical rotations were measured on a JASCO P2000 polarimeter (Tokyo, Japan). 1H and 13C NMR spectra were recorded on a Bruker Ascend 400 spectrometer (Billerica, MA, USA) at 400 and 100 MHz, respectively, using CDCl3 as solvent. Chemical shifts (δ) are reported in ppm relative to internal CDCl3 (δH 7.26 and δC 77.00). High-resolution mass spectra (HRMS) were obtained on an AB Sciex QTOF 6600+ mass spectrometer (Framingham, MA, USA). Elemental analyses (C, H, N) were performed with a Thermo Scientific™ Flash Smart™ analyzer (Waltham, USA), using sulfanilamide as reference. The UV-Vis measurements were performed in solid in a Cary 7000 spectrophotometer (Agilent, Santa Clara, CA, USA) in the range between 190 and 800 nm.
3.2. General Procedure for the Synthesis of N-Hydroxy-abieta-7,13-dien-18-amide (1a, Abietohydroxamic Acid)
To a cooled solution of abietic acid (93% purity estimated by 1H, 649 mg, 2.0 mmol), Et3N (2.54 mL, 18.0 mmol) in anhydrous DMF (12 mL) at 0 ˚C under Ar atmosphere, diethyl chlorophosphate (DCP) (595 µL, 4.0 mmol) was added dropwise allowing to warm to rt. After being stirred for 7 h, it was added NH2OH·HCl (702 mg, 10.0 mmol) in one portion. The resulting reaction mixture was heated at 40 ˚C overnight (16 h). Then, it was diluted with H2O (5 mL) and extracted with ethyl acetate (4 × 20 mL). The combined organic extracts were washed with 1N HCl (2 × 5 mL), H2O (5 mL), brine (5 mL), dried (Na2SO4) and concentrated. The crude residue (2.52 g, yellow solution, estimation by 1H NMR ca. 3.3:1 hydroxamic acid:carboxylic acid) was chromatographed on silica eluting with n-hexane- ethyl acetate (6:4) to give 101.7 mg of recovered abietic acid followed by 410 mg (64.6% yield, estimated 77.7% yield based on recovered starting material (brsm)) of abietohydroxamic acid 1a as a white solid: m.p. 125-128 °C (lit. [11], 127-131°C); = -72.9° (c 0.5, DCM)(lit. [11], = -78 °); UV-Vis λmax 226 nm; 1H NMR (CDCl3, 400 MHz) δH: 5.75 (1H, s), 5.32 (1H, br s), 2.21 (1H, sept., J = 6.8), 2.08-1.90 (4H, m), 1.88-1.77 (3H, m), 1.60-1.50 (4H, m), 1.23 (3H, s), 1.25-1.20 (3H, m), 1.00 (3H, d, J = 6.8), 0.99 (3H, d, J = 6.8), 0.82 (3H, s); 13C NMR (CDCl3, 100 MHz) δC: 176.6 (s), 145.3 (s), 135.5 (s), 122.3 (d), 120.1 (d), 50.9 (d), 45.2 (d), 45.1 (s), 38.1 (t), 37.2 (t), 34.9 (d), 34.6 (s), 27.4 (t), 25.2 (t), 22.4 (t), 21.4 (q), 20.8 (q), 17.9 (t), 15.8 (q), 14.2 (q); HRMS (ESI) m/z 318.2425 [M+ H]+, calcd for C20H32NO2: 318.2433; Anal. calcd. for C20H31NO2: C, 75.7; H, 9.8; N, 4.4 Found: C, 75.2; H, 10.1; N, 4.4.
3.3. Synthesis of N-Hydroxy-abieta-8,11,13-trien-18-amide (2a, Dehydroabietohydroxamic Acid)
Following the general procedure described in section 4.2, dehydroabietic acid (600 mg, 2.0 mmol) was treated with DCP and then with NH2OH·HCl. After stirring overnight (16 h) at 40 ˚C, The reaction was processed using the optimized low-volume work-up (5 mL H2O) to yield the crude residue (2.55 g, orangish solution, estimated 1H NMR ratio ca. 3.3:1 hydroxamic acid:carboxylic acid). Purification by column chromatography yielded 465.1 mg (73.8% yield, 93.5% brsm) of dehydroabietohydroxamic acid 2a as a white solid: mp 123-125 °C; = +38.2° (c 1.0, DCM); UV-Vis λmax 228 , 269, 277 nm; 1H NMR (CDCl3, 400 MHz) δH 7.15 (1H, d, J = 8.4), 6.99 (1H, d, J = 8.4, 2.4), 6.86 (1H, br s), 2.89-2.85 (2H, m), 2.82 (1H, sept., J = 6.8), 2.30 (1H, br d, J = 12.6), 2.21 (1H, dd, J = 12.6, 2.2), 1.89-1.69 (4H, m), 1.58-1.53 (1H, m), 1.51-1.45 (2H, m), 1.25 (3H, s), 1.22 (6H, d, J = 6.8), 1.22 (3H, s); 13C NMR (CDCl3, 100 MHz) δC 176.7 (s), 146.6 (s), 145.8 (s), 134.5 (s), 126.8 (d), 124.0 (d), 123.9 (d), 46.2 (s), 44.9 (d), 37.7 (t), 37.0 (s), 36.8 (t), 33.4 (d), 29.8 (t), 25.2 (q), 24.0 (q), 24.0 (q), 21.0 (t), 18.3 (t), 15.4 (q); HRMS (ESI) m/z 316.2264 [M+ H]+, calcd for C20H30NO2: 316.2277; Anal. calcd. for C20H29NO2: C, 76.2; H, 9.3; N, 4.4 Found: C, 75.7; H, 9.0; N, 4.2.
3.4. Isolation and Characterization of the Diethyl Phosphate Mixed Anhydride Intermediate of Dehydroabietic Acid (Int2)
Following the experimental conditions described in Table 1 (Entry 4), the intermediate was isolated for mechanistic validation.
Diethyl (abieta-8,11,13-trien-18-oyl) phosphate (Int2, Figure 3). Colorless oil; 1H NMR (CDCl3, 400 MHz) δH: 7.16 (1H, d, J = 8.0), 7.01 (1H, d, J = 8.4, 2.0), 6.89 (1H, br s), 4.30–4.20 (4H, m), 2.95–2.87 (2H, m), 2.82 (1H, sept., J = 6.8), 2.34–2.29 (1H, m), 2.19 (1H, dd, J = 12.4, 2.0), 1.92–1.71 (5H, m), 1.58–1.46 (2H, m), 1.35 (6H, tt, J = 7.2, 0.8), 1.31 (3H, s), 1.22 (6H, d, J = 7.2), 1.21 (3H, s); 13C NMR (CDCl3, 100 MHz) δC: 172.9 (d, JC-P = 10.8), 146.3 (s), 145.9 (s), 134.5 (s), 126.6 (d), 124.1 (d), 124.0 (d), 65.0 (d, JC-P = 5.7), 64.9 (d, JC-P = 5.7), 48.8 (d, JC-P = 5.7), 44.7 (d), 37.7 (t), 36.9 (s), 35.8 (t), 33.4 (q), 29.9 (t), 25.1 (d), 23.9 (q), 23.9 (q), 21.6 (t), 18.3 (t), 16.4 (q), 16.1 (q), 16.0 (q).
3.5. Computational Methods
All quantum-chemical calculations were performed using the Gaussian 09 software package (Revision D.01). Geometry optimizations and frequency calculations were carried out at the M06-2X/6-31G** level of theory, which is widely validated for describing thermochemistry and activation barriers in organic reactions. Solvent effects were incorporated implicitly through the Polarizable Continuum Model (PCM) using N,N-dimethylformamide (DMF, ε = 37.22) as the solvent, consistent with the experimental reaction conditions.
To reduce computational costs while preserving the essential stereoelectronic features of the sterically congested abietane skeleton, a simplified model system was employed. In this model, dimethoxyphosphate groups replaced the full diethyl phosphate substituents, yielding a system that preserves the reactive environment at the C-18 center.
Stationary points on the potential energy surface were located using analytical gradient optimization. Transition state (TS) structures were obtained using the Berny algorithm and were initially identified through relaxed potential energy surface (PES) scans along the forming N–C(=O) or O–C(=O) bond coordinates. The nature of all stationary points was confirmed by harmonic frequency analysis computed analytically at the same level of theory. Local minima displayed zero imaginary frequencies, while each transition state exhibited exactly one imaginary frequency corresponding to the normal mode of the bond-forming event.
Intrinsic Reaction Coordinate (IRC) calculations were performed for each TS to rigorously confirm the connectivity between the transition states and the corresponding reactants and products. Electronic energy variations (∆E) are reported for the individual pathways relative to their respective reactant complexes to describe the intrinsic barriers. For the global mechanistic discussion and the rationalization of chemoselectivity, all relative energies are referenced to the most stable ester-type reactant complex (R) set to 0.0 kcal/mol.
4. Conclusions
We have developed a robust and efficient single-vessel protocol for the synthesis of abietane-type hydroxamic acids. This methodology successfully overcomes the extreme steric hindrance associated with the C18 carboxyl group. The use of diethyl chlorophosphate (DCP) as an activating agent proved superior to traditional strategies, as it facilitates the formation of reactive diethyl phosphate mixed anhydride intermediates. These species were successfully isolated and characterized, providing direct experimental evidence of the activation pathway.
A fundamental contribution of this study is the optimization of the isolation process to handle the amphiphilic nature of the target products. By reducing the aqueous volume to 5 mL (for 2 mmol of starting acid) during the work-up, we effectively prevented product loss into the aqueous phase. This modification allowed isolated yields to increase from initial values of 17.3–34.2% to 64.6% from abietic acid and 73.8% from dehydroabietic acid. We concluded that the deliberate retention of a small amount of residual DMF in the crude material was a necessary strategic decision to ensure maximum product recovery. Crucially, this residual solvent did not interfere with the subsequent flash chromatography purification.
Furthermore, DFT calculations provided a comprehensive rationalization for the exclusive formation of N-hydroxy amides. The process is governed by strict kinetic control. Although the hydroximic ester is the thermodynamically more stable product, the formation of the hydroxamic amide is favored by an activation barrier that is 6.9 kcal/mol lower than the oxygen-attack route. The application of 40 °C ensured the solubility of reagents and the stability of the activated intermediates. This integrated approach establishes a reliable and scalable methodology for the chemical valorization of abundant natural terpenoids into potential bioactive scaffolds.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1–S11: copies of 1H, 13C, DEPT NMR spectra of 1a, 2a and int2; Figures S12–S15: copies of HRMS and UV spectra of 1a and 2a; Figures S16, S19 and S23: Optimized structures and relative energies for the species; Tables S17-S18, S20-S22, and S24-S26: Cartesian coordinates; Video animation of reaction path.
Author Contributions
Conceptualization, M.A.G.-C.; methodology, M.A.G.-C., W.E.M.-H., U.D. and R.J.Z.; validation, M.A.G.-C. and R.J.Z.; formal analysis, M.A.G.-C. and W.E.M.-H.; investigation, M.A.G.-C., W.E.M.-H., U.D. and R.J.Z.; resources, M.A.G.-C. and R.J.Z.; writing—original draft preparation, W.E.M.-H.; writing—review and editing, M.A.G.-C. and W.E.M.-H.; funding acquisition, U.D. and M.A.G.-C. All authors have read and agreed to the published version of the manuscript.
Funding
The authors thank the Spanish Government (Projects PID2023-146114NB-C21 and CEX2021-001230-S funded by MCIN/AEI /10.13039/501100011033 /FEDER, UE) for financial support. This research was also supported by the regional government “Generalitat Valenciana, Conselleria de Educación, Universidades y Empleo”, grant number CIAICO/2022/220 to M.A.G.-C. W.E.M.-H. also thanks the regional government “Generalitat Valenciana” for a PhD contract (grant number CIACIF/2024/510).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data and sample Availability Statement
The data is available in the manuscript and further details or samples of 1a or 2a upon request.
Acknowledgments
We acknowledge the support given by the characterization department of the “Instituto de Tecnologia Quimica, ITQ” for providing the NMR, elemental composition and high-resolution mass spectra (through Universitat de Valencia) of the samples.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Talapatra, S.K.; Talapatra, B. Diterpenoids (C20). In Chemistry of Plant Natural Products; Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; pp. 469–510. [CrossRef]
- Sargazifar, Z.; Ghorbanian Charami, D.; Esmaeilzadeh Kashi, M.; Asili, J.; Shakeri, A. Abietane-Type Diterpenoids: Insights into Structural Diversity and Therapeutic Potential. Chemistry & Biodiversity 2024, 21, e202400808. [CrossRef]
- González, M.A. Aromatic Abietane Diterpenoids: Total Syntheses and Synthetic Studies. Tetrahedron 2015, 71, 1883–1908. [CrossRef]
- Antoniuk, O.; Maranha, A.; Salvador, J.A.R.; Empadinhas, N.; Moreira, V.M. Bi- and Tricyclic Diterpenoids: Landmarks from a Decade (2013–2023) in Search of Leads against Infectious Diseases. Nat. Prod. Rep. 2024, 41, 1858–1894. [CrossRef]
- Hamulić, D.; Stadler, M.; Hering, S.; Padrón, J.M.; Bassett, R.; Rivas, F.; Loza-Mejía, M.A.; Dea-Ayuela, M.A.; González-Cardenete, M.A. Synthesis and Biological Studies of (+)-Liquiditerpenoic Acid A (Abietopinoic Acid) and Representative Analogues: SAR Studies. J. Nat. Prod. 2019, 82, 823–831. [CrossRef]
- Citarella, A.; Moi, D.; Pinzi, L.; Bonanni, D.; Rastelli, G. Hydroxamic Acid Derivatives: From Synthetic Strategies to Medicinal Chemistry Applications. ACS Omega 2021, 6, 21843–21849. [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; De La Torre, B.G.; Albericio, F. Hydroxamate Siderophores: Natural Occurrence, Chemical Synthesis, Iron Binding Affinity and Use as Trojan Horses against Pathogens. Eur. J. Med. Chem. 2020, 208, 112791. [CrossRef]
- Stanetty, C.; Czollner, L.; Koller, I.; Shah, P.; Gaware, R.; Cunha, T.D.; Odermatt, A.; Jordis, U.; Kosma, P.; Claßen-Houben, D. Synthesis of Novel 3-Amino and 29-Hydroxamic Acid Derivatives of Glycyrrhetinic Acid as Selective 11β-Hydroxysteroid Dehydrogenase 2 Inhibitors. Bioorg. Med. Chem. 2010, 18, 7522–7541. [CrossRef]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic Acid Derived Hydroxamates and Betulin Derived Carbamates Are Interesting Scaffolds for the Synthesis of Novel Cytotoxic Compounds. Eur. J. Med. Chem. 2015, 106, 194–210. [CrossRef]
- Wiemann, J.; Heller, L.; Csuk, R. Targeting Cancer Cells with Oleanolic and Ursolic Acid Derived Hydroxamates. Bioorg. Med. Chem. Lett. 2016, 26, 907–909. [CrossRef]
- Bardyshev, I.I. Diterpenoid Carboxylic Acid Anhydrides of the Abietane, Pimarane, and Isopimarane Series. Russ. J. Org. Chem. 1999, 35, 41–55.
- Ganeshpurkar, A.; Kumar, D.; Singh, S.K. Strategies for the Synthesis of Hydroxamic Acids. Curr. Org. Synth. 2018, 15, 154–165. [CrossRef]
- Alam, M.A. Methods for Hydroxamic Acid Synthesis. Curr. Org. Chem. 2019, 23, 978–993. [CrossRef]
- Mendoza-Hernández, W.E.; Zaragozá, R.J.; González-Cardenete, M.A. Study and Development on the Hydroxamation of Natural Resinic Acids: Synthesis and Computational Studies. In Proceedings of the ECSOC 2025; MDPI, November 12 2025; p. 81. [CrossRef]
- Ech-Chahad, A.; Minassi, A.; Berton, L.; Appendino, G. An Expeditious Hydroxyamidation of Carboxylic Acids. Tetrahedron Lett. 2005, 46, 5113–5115. [CrossRef]
- Harusawa, S.; Shioiri, T. Diethyl Phosphorocyanidate (DEPC): A Versatile Reagent for Organic Synthesis. Tetrahedron 2016, 72, 8125–8200. [CrossRef]
- Halbrook, N.J.; Lawrence, R.V. The Isolation of Dehydroabietic Acid from Disproportionated Rosin. J. Org. Chem. 1966, 31, 4246–4247. [CrossRef]
Figure 1.
Structures of starting resin acids (1, 2) and their corresponding hydroxamic acid derivatives (1a, 2a).
Figure 1.
Structures of starting resin acids (1, 2) and their corresponding hydroxamic acid derivatives (1a, 2a).

Scheme 1.
General relay-activation strategy for the synthesis of hindered abietane hydroxamic acids.
Scheme 1.
General relay-activation strategy for the synthesis of hindered abietane hydroxamic acids.

Scheme 2.
Optimized one-pot synthesis of abietohydroxamic acid (1a) and dehydroabietohydroxamic acid (2a) via an in situ generated diethyl phosphate mixed anhydride intermediate.
Scheme 2.
Optimized one-pot synthesis of abietohydroxamic acid (1a) and dehydroabietohydroxamic acid (2a) via an in situ generated diethyl phosphate mixed anhydride intermediate.

Figure 2.
Computed free-energy profile (M06-2X/6-31G**/PCM(DMF)) for the competitive N-attack versus O-attack of hydroxylamine on the activated phosphate of abietic acid (1).
Figure 2.
Computed free-energy profile (M06-2X/6-31G**/PCM(DMF)) for the competitive N-attack versus O-attack of hydroxylamine on the activated phosphate of abietic acid (1).


Table 1.
Optimization of the activation and hydroxyamidation conditions for abietic and dehydroabietic acids (1 and 2).
Table 1.
Optimization of the activation and hydroxyamidation conditions for abietic and dehydroabietic acids (1 and 2).
| Entry | Substrate | Reaction conditions | Results |
|---|---|---|---|
| 1 | 1 | PPAA, Et3N, NH2OH· HCl in MeCN rt | SM recovered |
| 2 | 2 | PPAA, Et3N, NH2OH· HCl in MeCN rt | 20.1% yield of 2a |
| 3 | 1 | DEPC, Et3N, NH2OH/Et3N in THF rt-40 °C | 13.7% yield of 1a |
| 4 | 2 | DEPC, Et3N, NH2OH/Et3N in THF rt | Phosphate intermediate isolated (Int2) |
| 5 | Int2 | NH2OH· HCl, Et3N in DMF | Confirmed formation of 2a |
| 6 | 1 | 1) DCP, Et3N 0 °C-rt 2) NH2OH· HCl 40 °C in DMF one pot |
17.3% yield of 1a |
| 7 | 2 | 1) DCP, Et3N 0 °C-rt 2) NH2OH· HCl rt in DMF one pot |
34.2% yield of 2a |
| 81 | 1 | 1) DCP, Et3N 0 °C-rt 2) NH2OH· HCl 40 °C in DMF one pot |
64.6% yield of 1a |
| 91 | 2 | 1)DCP, Et3N 0 °C-rt 2)NH2OH· HCl 40 °C in DMF one pot |
73.8% yield of 2a |
1Optimized work-up utilizing a minimized aqueous volume for both the initial quenching and each subsequent washing step of the organic phase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



