Submitted:
10 April 2026
Posted:
14 April 2026
You are already at the latest version
Abstract
Oral drug delivery remains the most preferred route of administration; however, traditional oral dosage forms face several limitations, including low bioavailability, enzymatic degradation, poor permeability, and lack of site-specific drug release. Recent advances in nanotechnology have introduced nanoparticles as promising drug carriers capable of overcoming these challenges. Eudragit-based nanoparticles have demonstrated great potential in enhancing drug stability, controlling release profiles, and improving site-specific targeting in the gastrointestinal tract. These polymethacrylate copolymers exhibit pH-dependent solubility, mucoadhesive properties, and tuneable drug-loading capacities, making them highly suitable for advanced oral formulations. This review provides a comprehensive analysis of Eudragit®-based nanoparticulate systems for oral drug delivery, discussing their formulations, physicochemical properties, and mechanisms of controlled drug release. Emphasis is placed on controlled-release strategies, targeted delivery, and the impact of polymeric materials in optimising therapeutic outcomes. By exploring these aspects, this review aims to highlight the potential of Eudragit-based nanoparticles as a robust platform for improving oral drug bioavailability and efficacy.
Keywords:
controlled drug release
; eudragit
; nanoparticles
; oral drug delivery
; polymethacrylates
1. Introduction
Oral delivery is the most preferred route of drug delivery due to its high patient compliance, cost-effectiveness, non-invasiveness, ease of use, and safety [1,2,3]. It is also the most convenient way to treat chronic disorders. However, there have always been some concerns with this administration route as multiple biological barriers must be overcome to achieve the desired therapeutic outcomes. Ingestible dosage forms must be stable enough to withstand the conditions of the gastrointestinal tract and deliver drugs to a biological target within an adequate timeframe and dosage window, ensuring the appropriate concentration for the expected clinical outcomes [1,4]. To accomplish this, oral formulations must resist the harsh biochemical environment that characterises the GIT, effectively penetrate gastrointestinal tissues and enter the bloodstream [4]
Notwithstanding, traditional dosage forms still present significant limitations, including low bioavailability due to compromised stability under varying pH conditions, enzymatic degradation, poor permeability, low site-specific accumulation, and limited mucoadhesion [4,5]. Gastrointestinal barriers are important to protect the internal environment from potentially harmful external factors. Nonetheless, because oral drugs are externally administered, they undergo the same hydrolysis processes and structural disruptions as those potentially harmful agents. Additionally, the physicochemical and stability properties of the active pharmaceutical ingredient influence its absorption site, transport route, and, therefore, bioavailability. Thus, novel oral dosage forms aim to achieve site-specific targeting within the GIT while providing sustained and controlled release profiles [1].
Pharmaceutical formulations have evolved to better meet patient requirements and therapeutic expectations. Along with this change came drug carriers, which are defined as being a “system that can change the way the drug enters the body and its distribution in the body, control the release rate of the drug, and deliver the drug to the target organ” [6]. Among these, nanosized drug carriers are particularly advantageous as they enhance drug solubility due to the high surface area-to-volume ratio [2,7]. Nanotechnology has played a fundamental role in mitigating and overcoming some of the drawbacks associated with oral drug delivery and conventional treatments. It can be applied to diagnosing, preventing, and treating various diseases, enabling some formulations to be multipurpose [8]. Furthermore, nanoparticles (NPs) can be coated with biomaterials to design systems that protect the API from hydrolysis in the stomach, enabling it to be released in a sustained way at a targeted site [9].
One example of such polymer-based drug delivery systems involves the use of polymethacrylates, which are copolymers derived from acrylic or methacrylic esters and methacrylic acid, with variations that include anionic, cationic, and neutral copolymers [10]. Although there are several commercially available polymethacrylates, Eudragit® polymers are widely utilised due to their pH-dependent solubility, controlled-release properties, and ability to prolong drug loading [10]. Their versatility makes them valuable in developing advanced oral formulations capable of enhancing drug stability and therapeutic efficacy[10].
This review aims to provide a comprehensive analysis of Eudragit-based NPs for oral drug delivery, focusing on different formulations and evaluating their specific applications. The objective is to highlight both the potential and limitations of these types of polymeric drug delivery systems intended for oral administration. To achieve this, the review first examines the role of nanotechnology in enhancing oral drug delivery, with particular emphasis on controlled-release systems and targeted drug delivery mechanisms. Moreover, a detailed description of the distinct types of polymethacrylate copolymers and their specific properties will be provided to elucidate their suitability for optimising oral drug release profiles. This discussion will be supported by an analysis of multiple drug delivery systems incorporating Eudragit, with a focus on the APIs used, target sites, and therapeutic approaches.
2. Controlled Drug Release and Targeted Drug Delivery
When discussing oral drug formulations, it is crucial to differentiate between immediate-release and controlled-release systems. Immediate-release dosage forms are defined as “a solid, semi-solid, solution or suspension that is designed to release its active and/or inert ingredient(s) upon administration with no enhanced, delayed or extended-release effect”[11]. According to the Pharmaceutical Quality/Chemistry, Manufacturing and Controls (PQ/CMC) terminology, an immediate-release dosage form can also be described as a formulation in which no deliberate modifications have been made to alter the release rate of the API [12]
In contrast, controlled release dosage forms are a “solid, semi-solid, solution or suspension designed to release active and/or inert ingredient(s) at a controlled rate”[13]. These formulations belong to the broader category of modified-release dosage forms, as they exhibit an intentionally altered release profile of their active and/or inert ingredients [14].
The capability of extending drug activity through formulation strategies was first observed in the early 1950s. These advances led to the introduction of the concept of “drug delivery systems (DDS)”, which, although often used interchangeably with “dosage form”, represents a distinct idea. A DDS implies that there is a technology controlling the release rate of the drug [9,15]. Over the years, DDS have evolved significantly, allowing for precise targeting of drug concentrations within specific tissues and organs. This results in drug accumulation in the desired target sites and minimisation of off-target distribution [9,15].
These controlled-release systems offer multiple advantages, not only for patients but also for oral drug design. They help maintain optimal drug concentration and extend the duration of the therapeutic effect with lower drug doses. In the absence of periods where drug levels in the bloodstream are either sub- or supratherapeutic, patient convenience is enhanced, and adverse effects are minimised, resulting in increased safety [15]. These systems are used to improve drug pharmacokinetics, including controlling drug release kinetics, improving solubility, enhancing drug stability, overcoming biological barriers, and targeting drug delivery [9].
2.1. Kinetics of Controlled Drug Release
Controlling the drug release rate in a system may be achieved through several mechanisms (9). Concentration gradients play a significant role in determining the release rate across most controlled-release delivery systems. Diffusion, dissolution, swelling, affinity, and ion exchange are the primary mechanisms behind drug release control. Moreover, most controlled-release formulations are influenced by multiple mechanisms simultaneously [15].
The most observed release kinetics are first-order and/or zero-order releases. Zero-order release kinetics, characterised by a constant drug release rate following an initial equilibration phase, are particularly desirable in controlled-release systems. This is because they maintain drug concentrations within the therapeutic window for an extended period, minimising side effects and maximising efficacy, while also reducing dosing frequency [16]. On the other hand, first-order release kinetics involve a progressively decreasing release rate [9,15].
When designing controlled-release oral dosage forms, it is essential to consider the physiological constraints of the GIT, where drug transit typically lasts approximately 24 h. Therefore, most orally controlled-release formulations must be engineered to release the full drug dose within 12 to 18 h to ensure effective absorption and therapeutic efficacy [15]. Release profiles can generally be split into three phases: the initial burst or lag phase, followed by the controlled release phase, ending with drug depletion and release rate tail-off [17].
In matrix-type systems, the drug molecules are uniformly dispersed or dissolved within the polymeric material forming the matrix, whereas in reservoir-type systems, the drug is surrounded by a permeable, porous or non-porous membrane layer [9,15]. Diffusion-controlled formulations tend to be matrix-based or reservoir-based, where drug release is governed by Fick´s law. This release mechanism refers to nonswelling, noneroding drug-containing matrices in which there is, primarily, a concentration-driven release and where the rate-limiting step is drug diffusion [17]. With matrix systems, it is common to have an initial burst release of the drug due to the rapid diffusion of surface-localised drug particles, and first-order kinetics are inherently followed. On the other hand, reservoir systems can present zero-order release kinetics. However, if membrane failure occurs, then dose dumping is observed [9,15].
In this case, the system´s properties control drug release. So, characteristics like pore size, bond types, hydrophobicity, and thickness of the membrane/matrix will directly influence the dissolution rate, which influences drug release. This means that drug release will decrease with time throughout the dissolution process because the matrix size will also decrease. For this reason, these systems tend to present a non-zero-order release [9,15]. When the drug is dispersed in the matrix, the release process involves a dissolution step before diffusion can occur, which alters the release time dependency. In many controlled release systems, dissolution and diffusion are both responsible for drug release. Additionally, the dissolution step depends on how the drug is dispersed in the polymeric matrix. If it is molecularly dispersed, dissolution is minimal; if the drug is present in a solid or crystalline form, dissolution-diffusion occurs [15,17].
When referring to swelling as a controlled release mechanism, the materials forming the matrix/reservoir need to have some water-absorbing capacity. This way, they can expand and release the drug. In other words, they need to be hydrophilic on some level. Matrix systems based on hydrophilic polymers will have a drug release dependent on the rate at which water can effectively penetrate the matrix. If the water penetration rate is quicker when compared to polymer erosion, an increased degree of swelling will increase the drug release rate. On the other hand, water penetration into the matrix can depend on the time it takes for the polymeric network to relax. In this case, the drug release rate depends on the hydration rate of the polymer, making polymer relaxation and swelling the main forces for drug release. Finally, there can be cases where both swelling, and diffusion are present [17].
Affinity-based systems leverage specific interactions between drug molecules and carrier materials to modulate the release kinetics. Such interactions are noncovalent, including electrostatic, van der Waals, hydrophobic, and hydrogen-bond interactions. For this reason, the drug's release rate is dependent on the association constant of drug-ligand interactions. These release rates are, therefore, tuneable [9,17].
Ion exchange systems rely on an ionic environment to obtain controlled release. The charged drug molecules are loaded into a resin, which is typically inert and can pass safely through the GIT. This system is especially suitable for enteral delivery [9].
2.2. Targeted Drug Delivery
Targeted delivery consists of selectively transporting a particular drug to a specific site, enabling therapeutic action to be secluded from other body areas and minimising potential adverse effects [18]. Targeted delivery can be attained passively or actively, by passive transport principles, the development of stimuli-responsive DDS, and via surface modification with specific targeting molecules. These stimuli-responsive systems are specifically engineered to selectively respond to internal or external stimuli, such as pH, temperature, biologically active molecules, heat, light, and ultrasound. Intrinsic stimuli are typically associated with pathological processes and can vary depending on the condition. The distinctive changes observed in pathological regions set them apart from normal healthy tissues. Consequently, a better understanding of local microenvironmental alterations in these areas has facilitated the development of advanced DDS capable of reacting to abnormal conditions, thus targeting drug delivery [19].
Passive targeting relies on the inherent properties of the DDS (shape, size, charge, and surface modifications), the physiological characteristics and pathological changes of the target tissue. A careful balance of these parameters is required to design a DDS capable of interacting with biological barriers and tissues, directing the drug to diseased areas without the need for specific ligands or active recognition mechanisms. Particle size (PS) plays a central role in circulation kinetics and tissue penetration. Systems below 10 nm are rapidly eliminated by renal filtration, while those exceeding 200 nm are more prone to uptake by the mononuclear phagocyte system (MPS). This uptake can be potentially beneficial in different contexts: if the therapeutic strategy is to target macrophages, such as in inflammatory or infectious conditions, the enhanced MPS uptake is advantageous. For oral nanocarriers, an intermediate range (50 – 200 nm) is considered optimal, as it balances uptake across the intestinal mucosa, avoidance of rapid clearance, and sufficient stability to exploit passive targeting mechanisms after absorption. Morphology also influences passive targeting efficiency. While spherical NPs are most common, non-spherical morphologies have been shown to display distinct hydrodynamic and cellular interaction profiles, while also exhibiting reduced phagocytic uptake and longer circulation times. Optimisation studies of NP size and morphology can be conducted to evaluate the impact of independent formulation variables on these properties. Surface charge determines how the DDS interacts with physiological components, with cationic systems displaying enhanced interaction with negatively charged cellular membranes, promoting uptake; whereas negatively charged systems are subject to serum protein adsorption [20,21,22].
Solid tumours provide an example of how pathological physiology can be exploited in the context of passive targeting. Their vasculature is often disorganised, presenting irregular branching, heterogeneous perfusion, and extensive fenestrations. In parallel, inefficient lymphatic drainage hinders interstitial fluid clearance. Together, these alterations create a microenvironment that favours the passive accumulation of nanosized DDS. This phenomenon, commonly known as enhanced permeability and retention (EPR) effect, represents a passive targeting mechanism solely derived from the pathophysiological changes in tumour environment [9].
However, this rather straightforward approach has its downsides. For one, the EPR effect is predominantly applied to tissues with ongoing inflammatory processes. Adding to this, extravasation increases interstitial pressure, which will paradoxically disfavour the EPR effect [23,24].
In active targeting, the DDS can interact with the desired target(s) due to the use of an affinity-based recognition sequence. The target can be a cell, tissue, or molecule, which makes site-specific interactions extremely relevant. Targeting ligands can be antibodies, proteins, peptides, aptamers, carbohydrates, or small molecules [9,17].
3. Polymethacrylates and Oral Drug Delivery
When choosing a material to be applied to a DDS for an intended administration route and site of action, biocompatibility, hydrophilicity, physical and protein binding properties, stability, degradability, pH and temperature behaviours must be evaluated. Biomaterials are “materials destined to interface with biological systems to evaluate, treat, augment, or replace any tissue, organ, or function in the body” [9,25].
Polymers, with tuneable chemistry, controllable and responsive properties, flexibility in conjugation and drug incorporation, and a wide variety of bulk compositions and physical properties, are the most widely used materials for DDS. Adding to this, they can be designed with different specifications, such as molecular weights, degradable linkages, crosslinking modes and block structures. They can be categorised as synthetic or natural; however, synthetics are more commonly used due to their predictable structure-function relationships, resulting in minimised batch-to-batch variations [9,26].
Polymer chemical structural analysis is useful for predicting possible degradation mechanisms, which primarily occur through hydrolysis, oxidation, photolysis, and proteolysis. However, the biological environment can also modulate polymer behaviour and for this reason, degradation can occur intentionally or unintentionally. Surface properties are also critical and lead to different levels of protein adsorption, influencing organic reactions and controlled drug release [27].
Considering a DDS, the polymer’s molecular weight, the presence of end functional groups (such as ester or carboxyl) and the nature of interactions between the polymer and drug influence the drug´s solubility and consequently, the drug loading and encapsulation efficiency [28,29,30].
When developing stimuli-responsive DDS, stimuli-responsive biomaterials are applied. When a change occurs in the environment, polymer behaviour will be affected by this alteration. These so-called “smart polymers” can respond to external or internal, physical or chemical stimuli (28). pH-sensitive polymers contain ionisable functional groups, either acidic or basic, that can accept or donate protons in response to pH fluctuations. The structural changes of these polymers are primarily governed by the presence of these groups, with the polymer's pKa value serving as a key parameter in defining the pH threshold at which ionisation occurs. At this critical pH, electrostatic repulsion within the polymer matrix leads to molecular expansion, which is accompanied by an increase in net surface charge, thereby enhancing the polymer’s hydrophilicity and solubility. Common protonatable moieties include amino, imidazolyl, and carboxyl groups. When incorporated into a DDS, these polymers must effectively prevent premature drug release at non-targeted sites. Their responsiveness arises from the presence of ionisable groups that undergo protonation or deprotonation depending on the pH variations encountered along physiological pathways [19,27].
Polymers containing weak acidic groups are polyacids, and at acidic pH values, such as those present in the gastric milieu (approximately pH 1.2), their carboxylic groups become protonated, stabilising the polymer matrix and restricting non-specific drug release. As the environmental pH transitions to neutral and alkaline conditions, carboxyl groups lose protons and become ionised, leading to modifications within the polymer matrix, promoting swelling and drug release [31]. An illustrative example includes methacrylic acid, which is structurally present in several pH-sensitive polymers. Conversely, polymers containing weak basic groups in their structure, such as amines or imidazole, have a low pH threshold for accepting protons, becoming protonated at low pH [31,32].
When pH-sensitive polymers are assembled into DDS membranes, their behaviour becomes even more versatile. These pH-sensitive membranes respond to environmental pH by undergoing physical or chemical changes that directly affect their permeability and drug release performance [33]. Two main mechanisms drive this response: pH-dependent ion transport and molecular recognition [31,34,35]. The first process involves alterations in the protonation state of ionisable groups, which in turn modulate the membrane´s charge density, leading to altered ion selectivity and permeability. The subsequent mechanism relies on pH-dependent affinity between functional groups on the polymer chains forming the membrane, and interacting molecules, such as drugs, ligands, or biomolecules. Variations in acidity or alkalinity modify the ionisation state of these groups, which in turn affects specific, non-covalent interactions between them and other molecules. In this way, pH fluctuations regulate how strongly the membrane binds or releases specific molecules [31,32]. pH responsiveness offers numerous advantages compared to other stimuli-responsive mechanisms, primarily due to its specificity. pH is a parameter that indicates the acidity or alkalinity of an environment and directly influences a molecule's proton concentration. This characteristic enables precise control and prediction of polymer behaviour in various pH environments, enhancing these systems´ specificity [31].
In the pharmaceutical field, polymethacrylates are synthetic methacrylate-based polymers, which may consist of dimethyl aminoethyl methacrylates, methacrylic acid, and various methacrylic acid esters [36]. These polymers are monographed in several Pharmacopoeia, including the European Pharmacopoeia, and the USP/NF. According to the USP32-NF27, methacrylic acid copolymer is described as a fully polymerised copolymer of methacrylic acid and an acrylic or methacrylic ester [37]. They are commonly referred to as Eudragit, although various companies produce them under different names. For instance, Evonik Industries markets them as Eudragit, Colorcon calls them Acryl-EZE, Eastman Chemical refers to them as Eastacryl 30D, and BASF Fine Chemicals sells them under the name Kollicoat MAE [38]. Their physicochemical properties are determined by the nature of their functional groups, such as methacrylic acid moieties, carboxylic acid groups or quaternary ammonium groups; and these play a crucial role in modulating how and where drug release will occur [10,32]. Polymethacrylates classified as soluble facilitate pH-dependent drug release through the formation of salts, whereas their insoluble counterparts regulate release primarily through polymer swelling and diffusion-driven mechanisms [32].
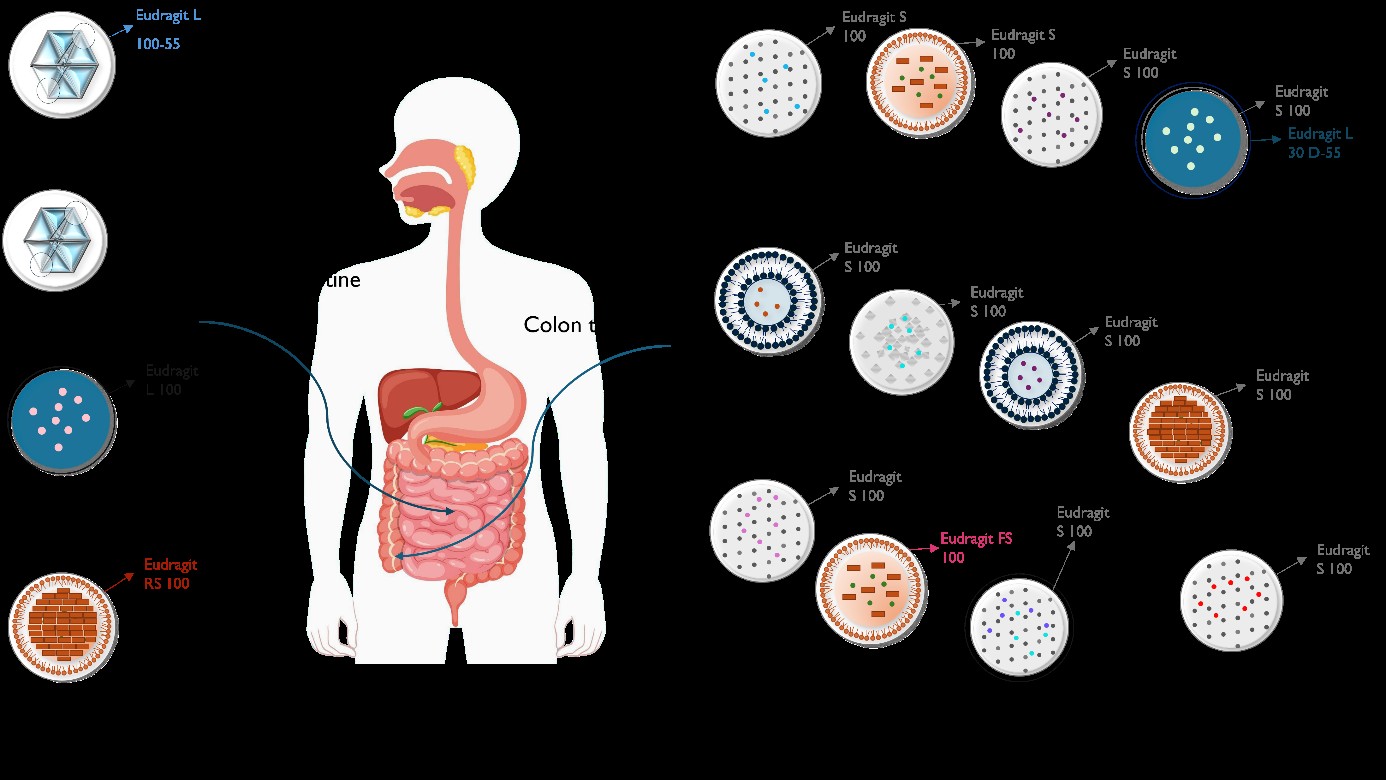
Eudragit polymers are available in multiple grades, each distinguished by specific compositions, chemical properties, and pH-dependent solubility profiles, as outlined in Table 1. The differing ratios of methacrylates, methacrylic acid, and/or methacrylic acid esters in each polymer´s composition determine whether they exhibit cationic, anionic, or neutral behaviour [10]. Among the cationic variants, Eudragit E is based on dimethyl aminoethyl methacrylate and other neutral methacrylic acid esters, such as butyl methacrylate and methyl methacrylate [36,38]. This polymer is commercially available in three different grades: Eudragit E100, Eudragit E 12,5 and Eudragit E PO. These grades are characterised by favourable properties such as strong adhesion, low viscosity, high pigment-binding capacity, and minimal polymer weight gain. The primary distinctions among them lie in their physical forms: Eudragit E100 is supplied as granules, Eudragit E12.5 as an organic solution, and Eudragit E PO as a fine powder [36,39]. These polymers are typically employed to enable immediate gastric drug release and taste-masking, since they are insoluble in pH values greater than 5. The taste-masking property is derived from the polymer´s insolubility in the neutral oral cavity pH, preventing drug release in the mouth [40]. Thus, these polymeric grades can increase the solubility and bioavailability of enterically insoluble compounds with unpleasant taste. For example, Deng et al. employed Eudragit E PO in a NP-based, orodispersible, paediatric-friendly formulation loaded with lopinavir and ritonavir, to improve palatability, dose flexibility, solubility, and oral bioavailability [41]. Regarding anionic Eudragit polymers, Eudragit L and S are copolymerisation products of methacrylic acid and methyl methacrylate [38]. The Eudragit L series includes five commercial grades: L100, L100-55, L12.5, L12.5 P, and L30D-55, while the Eudragit S series comprises S12.5, S12.5P, and S100 [10]. Eudragit L100 (EL100) is soluble in pH 6, which corresponds to the intestinal physiological pH. EL100 remains insoluble in gastric pH, enabling indirect targeted drug delivery to the upper small intestine. Due to its commercially available physical form (dry powder granules), it is commonly used for film coating and as a matrix former in controlled release and gastro-resistant dosage forms [42,43,44,45]. On the other hand, Eudragit S100 (ES100) is designed for enteric coating applications aimed at the colon (pH 7), remaining insoluble in gastric and upper intestinal fluids. When this polymer dissolves in the GIT, it exposes the positive charges, encouraging adhesion to negatively charged cell membranes and improving NP retention. This property makes ES100 the preferred coating polymer for indirect colon-targeted delivery [42,43,46]. Eudragit FS (grades FS30D and FS100) is also an anionic copolymer based on methyl acrylate, methyl methacrylate, and methacrylic acid. Their pH solubility properties are similar to ES100; however, their glass transition temperatures are significantly different. This makes Eudragit FS30D (FS30D) and Eudragit FS100 (FS100) more suitable carriers for hot melt extrusion processes [47].
In the Handbook of Pharmaceutical Excipients, Eudragit RL and RS are classified as neutral polymers, and the USP32 classifies them as ammonium methacrylate copolymers. Both consist of acrylic acid and methacrylic acid esters, but according to USP32-NF27, the key distinction between them lies in the ratio of ammonium methacrylate units [37,38]. The quaternary ammonium groups present in their backbone remain permanently ionised, therefore conferring a weak cationic charge to the matrix. Since Eudragit RL has a higher proportion of these functional groups than RS, it exhibits greater aqueous permeability. On the contrary, Eudragit RS, which contains fewer ammonium groups, is comparatively less permeable. Nonetheless, both polymers are pH-independent and remain insoluble across the physiological pH spectrum, exhibiting a controlled swelling behaviour that allows gradual fluid penetration and, subsequently, sustained drug release. Optimisation of controlled release profiles can be attained using these two polymers in different ratios [48]. Similarly, Eudragit NE and NM represent neutral, swellable, pH-independent, and permeable copolymers. According to The Handbook of Pharmaceutical Excipients, Eudragit NE 30 D and NE 40 D consist of poly methacrylic acid esters and form water-insoluble but swellable films. On the other hand, Eudragit NM 30 D is composed of ethyl acrylate and methyl methacrylate units and likewise exhibits swellable and permeable behaviour [10,38].
Several in vitro studies have suggested that certain Eudragit grades (ES100, L100, RL100) may indirectly modulate P-glycoprotein (P-gp) activity. These polymers, however, cannot be regarded as classical P-gp inhibitors, as there is no evidence of molecular downregulation at the protein or gene expression level. Notwithstanding, studies in Caco-2 cell monolayer models have reported reduced drug efflux ratios in formulations coated with these Eudragit grades, when compared with their uncoated counterparts. The exact mechanism underlying this effect remains under debate, as most of the evidence points to functional inhibition or circumvention of P-gp-mediated efflux. Importantly, such findings are limited to in vitro studies, and it remains uncertain whether similar behaviour would be observed in vivo [49,50,51,52].
Thiolated derivatives of Eudragit have been increasingly employed to overcome the intrinsic limitations of the unmodified polymer in oral nanoparticulate delivery, namely premature drug leakage and insufficient mucoadhesion. The introduction of free thiol groups into the polymer backbone can promote disulphide crosslinking with other thiolated polymers, enhancing their stability in gastric conditions and enabling selective redox-responsive cleavage in physiologically reducing environments, such as the colon, tumour tissues, or intracellular compartments. This chemical modification has proven particularly effective in combination systems, where thiolated Eudragit interacts with polymers such as sodium alginate, pectin, or chitosan [53,54,55,56]. In practical applications, thiolated sodium alginate/Eudragit RS100 NPs have been designed for Paclitaxel delivery, achieving enhanced colonic targeting while improving solubility, intestinal permeability, and oral bioavailability [55]. Similarly, NPs composed of thiolated chitosan and Eudragit RS100 loaded with moxifloxacin provided high drug loading and improved pharmacokinetic profiles in vivo [57], whereas poly(allylamine)/thiolated ES100 NPs enabled dual encapsulation of DOX and curcumin with high efficiency, releasing almost completely under reductive conditions and remaining stable in non-reducing media [53]. In targeted applications, thiolated pectin/Eudragit RL100 nanocarriers enhanced colonic delivery of aspirin and metformin, and thiolated EL100 reinforced with glycyrrhetinic acid improved liver accumulation and pharmacodynamic effects of 5-fluorouracil [58,59]. Collectively, these outcomes illustrate that thiolation of Eudragit not only enhances entrapment and stability compared with the unmodified polymer, but also imparts selective, redox-sensitive release profiles and superior oral bioavailability.
4. Nanoparticles Containing Eudragit for Oral Drug Delivery
According to the Data Bridge Market Research report, the “oral delivery market is expected to witness market growth at a rate of 7.60% in the forecast period of 2022 to 2029” [64]. Nanotechnology has emerged as a transformative field in the realm of medicine and drug design, particularly in oral drug delivery, playing a vital role in addressing the associated challenges. It has revolutionised the approach to oral drugs, facilitating the incorporation of increasingly complex APIs at a reduced dimensional scale [65]. PS is a critical factor in various stages of drug formulation development and delivery. It will directly influence stability, formulation feasibility, and the ability to overcome biological barriers for absorption. Reducing PS increases surface area, thereby enhancing interfacial solubility, adhesion, and interaction with cell membranes [66]. Consequently, nanoscale size reduction is very advantageous for oral delivery systems. Beyond this, nanotechnology further enhances therapeutic efficacy by increasing bioavailability, enabling extended and controlled drug release, protecting active compounds from degradation, and facilitating site-specific targeting [65].
To ensure optimal performance, effectiveness, and broad applicability, nanocarriers must possess specific properties. Such properties include high traceability and imaging capability, dispersibility, specificity and selective cellular binding, substantial cargo-loading capacity, biocompatibility, and minimal toxicity [67]. The following section provides a detailed explanation of Eudragit-based NPs, and Table 2 summarizes studies on their application.
4.1. Inorganic Nanoparticles
Inorganic nanoparticles (INPs) constitute a varied class of nanomaterials, including metal, metal oxide, and silica-based systems. Their nanoscale dimensions impart an abundance of surface atoms, resulting in distinct electronic, magnetic, catalytic, and optical properties that are highly sensitive to size, morphology, and crystal structure. These NPs can function as antibacterial or anticancer agents, biosensors, and regenerative or imaging tools. However, they present potential toxicity, urging the need for precise inorganic nanoparticulate systems to optimise safety and efficacy [83].
Mesoporous silica nanoparticles (MSNPs) belong to the class of INPS and exhibit distinctive attributes that make them stand out among other nanoparticulate drug delivery systems. In addition to being biocompatible and exhibiting low toxicity, these NPs also present voluminous pores, high internal surface area, flexible mesoporous structure, and tailorable pore dimensions. They are easily adsorbed and can be loaded with a diverse range of therapeutic agents, including low molecular weight drugs, genetic material, peptides, and fluorescent dyes [68,84,85]. MSNPs provide a high loading capacity of both hydrophilic and hydrophobic drugs, while also shielding them from degradation and immune system recognition [84]. Furthermore, these NPs can be chemically functionalized, both internally and/or externally, making it possible to have sustained drug release profiles, pH stimuli-responsive behaviour, and cell targeting. These capabilities stem primarily from the presence of free and germinal silanol groups of porous silica materials, which are chemically accessible and amenable to modification [85]. At physiological pH, these silanol groups undergo deprotonation, culminating in electrostatic interactions with positively charged moieties [67]. Amino modification of MSNPs is very commonly observed, as it introduces a basic character to the particle, allowing for the formation of amide bonds with other functional groups, such as, for example, carboxylic acids. Surface-functionalised MSNPs can also provide higher drug loading and controlled drug release [86,87,88].
Kassem et al. [68] developed catechin-loaded MSNPs (Figure 1A–F), with the objective of enhancing the solubility of this flavonoid and improving its oral bioavailability [89]. The authors prepared colon-targeted, catechin-loaded amino-functionalised MSNPs coated with ES100
(NH2- MSNPs/CHT@EUS-100) to enable pH-controlled, site-specific release. The ES100 coating was additionally intended to prolong the residence time of the drug at the colonic site. It was observed that amino grafting of MSNPs led to an increase in zeta potential (ZP) (Figure 1G), rendering the particles more positively charged. This favoured electrostatic attraction with the negatively charged ES100, thereby facilitating the coating process and enhancing its stability. In addition, amino groups also acted as additional binding sites for CHT, both through hydrogen bonding with its acceptor groups and electrostatic interactions with the phenolic hydroxyl moieties, resulting in higher drug loading and adsorption.
The release behaviour of unencapsulated CHT, catechin-loaded MNSPs (MSNPs/CHT ) , catechin-loaded NH2-MNSPs (NH2-MSNPs/CHT) and NH2- MSNPs/CHT@EUS-100 was assessed (Figure 1H), under a simulated gastrointestinal pH gradient, progressing from gastric (pH 1.9) to intestinal (pH 5.5) and colonic (pH 7.4) conditions, at 2- and 5-h intervals. Free CHT rapidly dissolved, with nearly 95% of the compound being released within the first 2 h, whereas CHT encapsulated in MSNPs demonstrated a slower, more controlled release, achieving approximately 79% within the same timeframe. In contrast, NH2-MSNPs/CHT@EUS-100 exhibited the most pronounced drug retention, with negligible release during the gastric stage. This stronger retention was attributed to the presence of aminated material, which possesses reduced pore dimensions and volume, when compared with pristine MSNPs, and to the stronger interactions with CHT. An initial burst release was noted in both CAT-loaded MSNPs and NH2-MSNPs/CHT@EUS-100 at pH 1.2, potentially due to surface-localised CHT. This was followed by a slower, diffusion-driven release attributed to the presence of adsorbed CHT molecules into the internal silica network. Importantly, the ES100 coating imparted pronounced pH-responsiveness, with less than 2% of CHT being released under acidic conditions, increasing only slightly to 4% at pH 5.5. On the other hand, a sharp release of approximately 90% occurred under colonic pH after 5 h. This lag time indicated that ES100 successfully shielded the drug from premature release in the upper GIT, thereby ensuring colonic targeting [68].
In the context of IBD, application of Eudragit polymers can play an important role, contributing to the localised action and higher drug concentrations in colonic tissue. Li et al. [69] synthesised NPs that were loaded with budesonide (BUD) and coated with ES100 (Figure 2A). Three different shapes of MSNPs were produced: spherical, rod-shaped, and dendritic (Figure 2B to 2D). The spherical MSNPs were obtained using the cetyl trimethyl ammonium bromide (CTAB)-templated and base-catalysed methods, whereas the rod-like MSNPs were prepared using CTAB and tetraethoxysilane (TEOS). Dendritic MSNPs were synthesised using cetyl trimethyl ammonium tosylate, triethanolamine, and TEOS, with a final lyophilisation step, common in all 3 MSNPs. To study the release behaviour of BUD from the NPs, dissolution studies were conducted in simulated gastrointestinal pH conditions (pH 1.0, pH 4.5, pH 6.8, pH 7.8) (Figure 2E to 2J). At pH 1.0, drug release was minimal (less than 10%), while in alkaline conditions, rapid release was observed, exceeding 80% within 30 minutes. Among the three morphologies, dendritic MSNPs showed the highest cumulative release across the tested pH range, while rod-shaped particles consistently exhibited the lowest. Morphological analysis revealed that rod-like MSNPs had an elongated morphology with fewer pores, which contributed to a slower drug diffusion rate. In contrast, dendritic MSNPs featured an extensive porous network with a large surface area, facilitating faster dissolution and more efficient drug release. After the oral administration of the MSNPs suspensions in a murine model of IBD, the colonic tissue was analysed using in vivo imaging systems. It was concluded that after degradation of the Eudragit layer, dendritic MSNPs achieved the highest accumulation in the inflamed colon, followed by rod-shaped and spherical particles. This was attributed to the dendritic architecture, which has a branched, fibrous surface that enhances mucoadhesion, allowing for more efficient penetration through the dense mucous barrier, contributing to prolonged mucosal retention, and slowing mucosal clearance. Pharmacokinetic results (Figure 2K) indicated that low systemic absorption of BUD was observed after administration of both coated and uncoated MSNPs, supporting the hypothesis of a localised therapeutic effect. It was observed that MSNPs coated with Eudragit elicited a significantly higher anti-inflammatory response compared to free BUD, while uncoated MSNPs did not show comparable therapeutic benefits. The highest effectiveness of dendritic MSNPs with Eudragit is likely due to their enhanced accumulation at the target site, sustained drug release profile, and improved colonic retention, all of which resulted in better therapeutic outcomes, with reduced adverse effects. Dendritic MSNPs without the Eudragit coating failed to demonstrate therapeutic efficacy, likely because the absence of a protective polymer led to premature release of BUD from the mesopores, reducing the drug's availability at the inflammation site [69].
Raza et al. [52] developed pH-responsive MSPNPs loaded with meropenem (MER). MER, a broad-spectrum carbapenem antibiotic, is usually administered parenterally, either through intermittent bolus or continuous infusion, to prevent degradation in aqueous media, and maximise therapeutic plasma concentrations. However, bolus infusion often leads to sub-therapeutic levels, falling below the minimum inhibitory concentration. Additionally, due to its short half-life and rapid renal clearance, MER requires frequent dosing, typically three times daily, which imposes a significant burden, both because of the need for repeated drug preparation and reconstitution, and also since more frequent dosing is often linked to lower patient compliance. Furthermore, owing to its hydrophilic nature, MER has limited oral bioavailability. Although prodrugs have been proposed to overcome this limitation, economic and manufacturing challenges make this approach commercially unfeasible. In this context, MSNPs were chosen as carriers for MER´s oral delivery, given their ability to enhance the stability of labile compounds in the GIT, improve thermostability, and facilitate the oral drug absorption. However, because MER is thermolabile, loading methods involving heat, such as solvent evaporation and hot-melt extrusion, are unsuitable. To address this, the authors used liquid CO₂ as an alternative for drug incorporation. ES100 served as a coating agent, shielding the drug from acidic gastric conditions and aiming at targeted release in the intestinal environment. The MSNPs were successfully synthesised and functionalised with either phosphonate (MER-MCM-PO3) or amine terminal groups (MER-MCM-NH2 ), followed by MER loading (Figure 3A). Although MER-MCM-PO3 exhibited a higher drug loading capacity, this surface modification did not support subsequent coating, as it failed to confer MSNPs’ a positive surface charge. Therefore, despite their comparatively lower loading capacity, MER-MCM-NH2 were selected for further coating with ES100. In vitro drug release studies (Figure 3B and 3C) demonstrated that Eud-MER-MCM-NH2 – MER exhibited pH-responsive release behaviour, with no drug release at gastric pH, and sustained release under intestinal conditions, effectively protecting MER from gastric degradation, and enabling controlled drug release at simulated intestinal pH. In vitro permeability assays (Figure 3D and 3E) were performed using the Caco-2 monolayer culture model, and evidenced that both MER-MCM-NH2 and Eud-MER-MCM-NH2 significantly enhanced MER´s absorptive transport. The improved permeation was higher for Eud-MER-MCM-NH2 (around 2.4-fold) than for MER-MCM-NH2 (1.9-fold) [52].
Nanodiamond-based NPs (NDNPs) present good biocompatibility and lower toxicity when compared with other carbon-based nanomaterials, such as carbon black, carbon nanotubes, or quantum dots. Their characteristic sharp edges facilitate endosomal membranes´ penetration, benefiting the cytosolic delivery of several drugs, including antitumour ones. Nonetheless, the strong covalent bonding within the diamond framework renders NDNPs chemically inert, which limits the possibility for subsequent modifications, requiring more specialised approaches [72,90].
Su et al. [72] developed NDNPs designed for the targeted delivery of DOX to colorectal tumours (Figure 4). Due to the strong covalent bonds between carbon atoms of NDNPs, the authors incorporated polydopamine as a nanocarrier. Its structure, rich in aromatic rings, allows efficient adsorption of anthraquinone-based antitumor agents on its surface, and it also offers notable photothermal conversion abilities. This dual functionality creates a synergistic effect between chemotherapy and photothermal therapy. For better targeting and adhesion to the colon, the researchers used diamino polyethylene glycol as a linker (NH2-PEG-NH2) and triphenylphosphonium (TPP) as a cationic surface charger with high lipophilicity, which aids in mitochondrial targeting. To protect the nanocarrier from degradation in the stomach, prevent adhesion and absorption in the small intestine, allow dissolution in the colonic environment, and hide its positive charge (Figure 5B), ES100 was used as a protective coating. Once inside colon cancer cells, these NPs can be directed to the mitochondria because of the presence of TPP. The activation of photothermal effects via Near-Infrared (NIR) laser irradiation can then boost drug release and cause localised cytotoxic heating, which can lead to mitochondrial dysfunction, and greatly enhance the cytotoxic effects against cancer cells. Figure 4 schematically illustrates the rationale behind these NPs’ formulation and assembly.
In vitro release assays (Figure 5A) of uncoated (PND-PEG-TPP/DOX) and coated (ES@PND-PEG-TPP/DOX) nanodiamonds were performed using simulated gastrointestinal fluids [gastric (SGF), intestinal (SIF), and colonic (SCF)]. The cumulative drug release of PND-PEG-TPP/DOX and ES@PND-PEG-TPP/DOXwas 55.09% and 40.34%, respectively. Under laser irradiation, drug release increased to 75.31% for PND-PEG-TPP/DOX and 51.99% for ES@PND-PEG-TPP/DOX. These results suggest that the ES100 coating effectively reduced premature release in the upper GIT, and that NIR laser irradiation can promote enhanced drug release. The colonic delivery of DOX was indicated by PS modification: in SGF and SIF, ES@PND-PEG-TPP/DOXshowed no significant changes in PS, whereas in SCF, a clear PS reduction was observed, indicating ES100 dissolution and DOX release. In vitro fluorescence imaging revealed minimal fluorescence signals for free DOX and PND-PEG-TPP/DOX, predominantly in the liver. On the other hand, ES@PND-PEG-TPP/DOX exhibited an even significantly lower fluorescence, compared to the former, which was suggestive of effective mitigation of DOX´s release prior to reaching the colon, reducing intestinal absorption, and thereby decreasing systemic side effects. Regarding retention in the GIT, ES@PND-PEG-TPP/DOX exhibited stronger fluorescence signals in the small intestine and colon, when compared with free DOX and PND-PEG-TPP/DOX. Furthermore, in vivo tumour therapy efficacy studies (Figure 5C to 5F), performed on Balb/c mice bearing colon cancer tumours, highlighted that ES@PND-PEG-TPP/DOX combined with NIR laser radiation had the strongest growth inhibition rate on tumours [72].
4.2. Nanocrystals
Nanocrystals (NCs) are crystalline particles with dimensions below 100 nm, comprising a carrier-free nanotechnology, as the drug itself forms the matrix, thereby eliminating the need for large amounts of excipients. NCs improve the bioavailability of poorly water-soluble drugs, as they enhance dissolution velocity and saturation solubility. Stabilisers and polymers are usually required to prevent particle agglomeration and recrystallisation, ensuring physical stability during storage and administration. They are versatile NPs that can encompass semiconductor systems, displaying size-tunable fluorescence, photostability, and energy transfer efficiency, rendering them useful in fields other than nanomedicine [91]
Lopez-Vidal et al. [73] reported the development of pH-sensitive NCs coated with Eudragit L100-50 (EL100-55), using ivermectin (IVM) as the model drug. The design rationale aimed to enhance IVM´s solubility, a Biopharmaceutical Classification System (BCS) Class II compound, with variable bioavailability, and significant inter- and intra-individual variabilities. The choice of EL100-55 was based on its capacity to confer delayed-release properties, while protecting the drug from premature degradation. The effect of varying polymer concentrations (10%, 25%, 40%) on the NC´s PS, PDI, yield, and encapsulation efficiency was investigated. All formulations presented PS lower than 500 nm, while the ones containing 25% and 40% of EL100-55 exhibited a more homogeneous PS distribution, with a PDI lower than 0.3. An increase in polymer content was also associated with higher encapsulation efficiency. Encapsulation stability was monitored for 112 days, under moisture-protected conditions, and results showed practically no alterations for all three samples, underscoring the technique´s high efficiency in obtaining efficiently coated particles, therefore achieving pH-dependent release. Additional stability studies were conducted for 3 months to evaluate whether the NCs underwent significant changes in PS and PDI during storage periods without protection from moisture or light. The sample with 25% polymer content exhibited a PS increase of approximately 30 nm, with no registered changes for PDI. On the other hand, the 40% formulation experienced an enlargement of around 75 nm. Notwithstanding, the altered values still remained within an acceptable range. In vitro dissolution studies were conducted, simulating gastrointestinal pH shifts to study the influence of polymer coating, and comparing NCs containing 25% polymer with their physical mixture. In the first 2 h, both systems demonstrated less than 10% release (pH 1.2). As pH increased (pH 6.8), IVM´s release from the NCs rapidly occurred, whereas the physical mixture continued to show poor dissolution, with still less than 10% released even after pH adjustment. This disparity between systems confirmed that the polymer coating effectively shielded the drug from gastric acidity, enabling indirect selective delivery to the intestine due to EL100-55 ionization. A second dissolution test (pH 7) was performed, adding sodium lauryl sulphate (SLS) to both formulations, in order to enhance IVM´s solubility and prevent medium saturation. In this case, the dissolution profiles of the coated NCs and the physical mixture did not differ so markedly. For the first 90 minutes, both formulations behaved similarly, gradually increasing drug release. For the following duration of the study, which was of 180 minutes, the NCs were able to increase drug release when compared with the physical mixture. These results show that differences in surface tension between media with and without SLS further contributed to enhanced drug solubility, increasing released drug quantities. For the coated NCs, dissolution remained dependent on polymer disintegration prior to drug release, confirming the system´s pH-responsive behaviour. In spite of this, it was observed that the encapsulation resulting from Eudragit led to a controlled drug release. All in all, the NC´s solubility increased by 47-fold in water and 4.8-fold in simulated intestinal conditions (pH 6.8) compared to pure IVM [73].
Che et al. [74] developed nintedanib (BIBF) nanocrystals (BIBF-NCs) (Figure 6A to 6C) to lower the solubility of the drug in the stomach, while maintaining drug supersaturation in the intestine, thereby improving oral bioavailability. BIBF’s pH-dependent solubility, combined with drug recrystallisation in the intestinal lumen and P-gp mediated efflux, contributes to its low oral bioavailability. NCs were prepared by using a hydrochloric acid solution at pH 1.2 as solvent, avoiding the use of organic solvents typically required for BIBF solubilisation. These NCs, in which the drug is maintained in a supersaturated state to prevent precipitation and improve dissolution, were coated with EL100, aiming to guarantee their stability. To obtain coated particles, freeze drying and spray drying were evaluated, using hydroxypropyl methylcellulose E5 (HPMC-E5) at different ratios, as a protective agent to prevent aggregation. At HPMC-E5: drug ratios of 1:1 and 2:1, PS was preserved. However, excess HPMC-E5 slowed redispersion due to matrix formation around the crystals, physically blocking aggregation. Therefore, the chosen HPMC-E5: drug ratio to prepare coated BIBF-NC was 1:1. Dissolution assays demonstrated that uncoated BIBF-NC were completely dissolved at pH 1.2 within 2 h, whereas coated NCs with different polymer ratios avoided unwanted and early onset disintegration. The higher the polymer: drug ratio, the lesser quantity of BIBF-NC dissolved in pH 1.2.
Continuous increase in polymer concentration was responsible for the downward trend in NCs dissolution and drug release. A polymer: drug ratio of 5:1 enabled the reduction of drug release more significantly than all other ratios employed. No further ratios were tested because, although increasing polymer quantity reduces BIBF´s gastric dissolution, drug concentration would need to be decreased in order to maintain PS. The used drying technique also influenced NCs solubilisation, since at the same amount of Eudragit, spray-dried crystals presented a more significant acid-resistance, with lower drug release quantities. For this reason, the spray drying method, with a 5:1 ratio of EL100 to BIBF-NC, was selected as the final formulation process. PS stability of both coated and non-coated BIBF-NC was evaluated as well, under simulated physiological conditions, incubating them in pH 6.8 and 7.4 for 10 h (Figure 6D and 6E). Uncoated NCs showed an increase in PS, which may be related to the pH and ionic strength sensitivity of SDS. For the coated crystals, HPMC-E5 played an important role in spatial stabilisation, maintaining the nanocrystalline PS. In vitro dissolution studies were performed for BIBF soft capsules, uncoated BIBF-NC, coated BIBF-NC (BIBF-NCs@L100), and their physical mixture, at pH 1.0 to pH 6.8, for 2 h (Figure 6F). Due to BIBF´s pH-dependent solubility, results showed dissolution of BIBF in the stomach without the enteric material, with posterior precipitation in less acidic pH values. The presence of EL100 enabled a significant reduction in BIBF dissolution in the gastric environment. However, BIBF-NCs@L100showed a cumulative dissolution of 36.43 +/- 3.50%, while their physical mixture showed a value of 36.43 +/- 4.97%. This indicates that the polymer does not interact with the drug and that the pH-dependent solubility is due to the spray-dried coating layer. An in vivo pharmacokinetic study was also performed on rats and included the utilisation of diluted powders with a BIBF concentration of 16.5 mg/mL. The formulations included uncoated BIBF-NC, its coated version, and BIBF soft capsules. The results showed that BIBF-NCs@L100achieved the highest plasma concentrations compared with uncoated NC´s and BIBF soft capsules. This enhancement was attributed to reduced PS, pH-dependent protection from gastric release, and maintenance of supersaturations, ultimately leading to superior oral bioavailability [74].
4.3. Lipid Nanoparticles
Lipid nanoparticles (LNPs) are widely utilised for vehiculation of poorly water-soluble and highly lipophilic drugs, as their solubilization is a major challenge [92]. These NPs are composed of ionisable lipids, which are highly compatible with cell membranes, adopting a positive charge at low pH and remaining predominantly neutral at physiological pH values around 7.4. This charge modulation enhances drug encapsulation, controlled release, and interaction with biologic membranes [93]. Due to their nanoscale size and unique physicochemical properties, LNPs exhibit efficient cellular uptake, making them highly effective for safely delivering genetic material, as well as small-molecule drugs and other biologically active compounds. Additionally, LNPs can be functionalized with targeting ligands, stimuli-responsive components, or polymeric coatings, to achieve greater precision in drug delivery, enabling the development of more sophisticated and efficient therapeutic systems [94]
4.3.1. Liposomes
Liposomes (LPs) are spherical vesicles composed of lipids arranged in a unique bilayer structure, forming their outer layer, and surrounding an aqueous internal compartment. These NPs can carry both hydrophilic and hydrophobic drugs, while keeping a structural similarity with living cells, making them a promising DDS for a variety of therapeutic agents [95]. LPs are composed of single or multiple lipidic bilayers, derived from natural or synthetic phospholipids, and may contain unsaturated lipids, sphingolipids, steroids, glycosphingolipids, polymeric materials, or charge-inducing lipids [96]. LPs have already been studied for enteric drug delivery, mainly in the colonic environment. They have effectively interacted with both normal and inflamed colonic tissues in laboratory settings, strongly suggesting their potential as carriers for colon-targeted drug delivery [75].
A study conducted by Alghurabi et al. [75] sought to further assess Eudragit-coated bile salt-containing LPs (ES-SG/LP) (Figure 7C), through a combination of in vitro and in vivo experiments. 5-aminosalicylic acid (5-ASA), a compound characterised by low water solubility and limited permeability, primarily indicated to treat IBD, was selected as a model drug [97,98]. Bile salts (specifically sodium glycocholate) were incorporated into the liposomal bilayer for two main reasons: to promote membrane destabilisation upon contact with the intestinal epithelium, thereby facilitating vesicle uptake; and to stabilise the liposomal membrane against the disruptive effects of endogenous physiological bile salts. The inclusion of bile salts decreased PS and EE, with no effect on ZP. The reduction in EE was attributed either to competition between the anionic sodium glycocholate and 5-ASA for binding sites on the cationic lipid, or to destabilisation of the bilayer caused by the hydrophilic nature of sodium glycocholate, which may induce pore formation and facilitate drug leakage into the aqueous medium.
LPs with varying bile salt concentrations (0.25, 0.5, 1 mg/mL) were subsequently coated with ES100. The coating process resulted in increased NP’s PS values and negative ZP values, indicating reversal of the liposomal surface charge. Additionally, a slight increase in entrapment efficiency (EE) was also observed, which was attributed to the possible adsorption of 5-ASA beneath the polymeric layer. The influence of bile salt concentration on the dissolution profile of ES100-coated LPs was assessed at pH 5, in a fed state simulating medium (FeSSIF). At a concentration of 1mg/mL, the presence of bile salts resulted in a 17% release of 5-ASA, whereas coated LPs without bile salts released 37% of the drug under the same conditions.
Drug release behaviour (Figure 7A) was then evaluated for several liposomal formulations [LPs without bile salts, bile salt-containing LPs (SG/LP), and ES-SG/LP], under simulated gastrointestinal conditions, using three sequential biorelevant media, with pH values of 1.2, 5.0, and 7.4, thereby reproducing the physiological progression along the GIT. To exclude artefacts due to drug-dialysis membrane interactions, a 5-ASA solution was used as a control, and results confirmed that 80% of the drug remained within the dialysis bag after 1 h, indicating negligible drug-membrane binding. On the other hand, within the first 2 h, the LPs without bile salts had released approximately 60% of their 5-ASA content, whereas ES-SG/LP released only around 20% of 5-ASA, with the majority of drug release happening at pH 7.4. SG/LP showed slightly modified but significant release profiles compared to LPs without bile salts. After 5 h, ES-SG/LP showed a drug release of approximately 40%, confirming that the ES100 coating successfully inhibited release in an acidic environment, hence ensuring a more targeted delivery to the colon. As the therapeutic efficacy of 5-ASA is strongly dependent on the concentrations reached at the target site (100), the authors further examined cellular uptake as a determinant of local in vitro drug accumulation (Figure 7B). LPs without bile salts, SG/LP, and ES-SG/LP were marked with fluorescein isothiocyanate, and all exhibited significantly improved cellular uptake than a fluorescein solution at pH 7.4, evidencing efficient cell membrane permeation and intracellular transport. Moreover, a pharmacokinetic assay, performed on male Wistar rats, demonstrated that ES-SG/LP reduced Cmax by about 37% compared with LPs without bile salts, while maintaining elevated plasma concentrations for up to 6 h. Furthermore, when comparing SG/LP and ES-SG/LP, the coated preparation reduced Cmax by approximately 15% comparatively to the uncoated one, underscoring the role of Eudragit in further delaying systemic absorption, favouring higher colonic fluorescein delivery [75].
The same authors developed a similar colon-targeted delivery system using liposomal carriers loaded with BUD and coated with ES100 (BUD-E-BSLPs). As before, the LPs were conjugated with bile salts to enhance stability and facilitate epithelial uptake. However, stearylamine was additionally included to impart a positive surface charge, enabling electrostatic attraction with ES100, and improving coating efficiency. To investigate coating conditions, the authors varied several parameters, including liposomal concentration (2, 4, 5, 7, 10 mM), ES100 concentration (0.25, 0.5, 1, 2, 2.5 mg/mL in 100 mM phosphate buffer, pH 8.0), and liposome/polymer volume ratio (1:1, 1:2, 1:3). The impact of these variables on PS, PDI, and ZP was evaluated. Two interactions between the LPs and ES100 occurred simultaneously: particle re-stabilisation, where charge reversal occurred, and full coating was achieved; or aggregation, which resulted from partially coated LPs interacting with uncoated ones, thereby increasing PS. A lower LP/ES100 ratio (1:1) was associated with a lower PS, owing to reduced lipid concentrations. Conversely, at a ratio of 1:3, an excess of ES100 was available, which led to an increased coating rate and minimal collisions between LPs. Similarly, enhanced polymer availability also promoted reduced PS by preventing aggregation and favouring re-stabilisation. Regarding the process, the rate of LP addition also affected coating success, since a slower addition allowed partially coated, negatively charged LPs to interact more efficiently with subsequently added cationic LPs. In contrast, faster addition rates led to simultaneous charge reversal across all particles, maintaining repulsive interactions between dispersed particles and reducing coating efficiency.
Drug release studies were performed as well, for LPs without bile salts, SG/LP, BUD-E-LPs, and Bud-E-BSLPs, in media simulating different GIT regions: simulated gastric fluid pH 1.2 (SGF), fasted-state pH 6.5 (FaSSIF) and fed-state (FeSSIF) simulated intestinal fluids, and phosphate buffer saline pH 7.4, representing the colonic conditions. In SGF, BUD-E-BSLPs inhibited approximately 85% of drug release within 2 h, compared with 65% of drug release from BS-LPs. In FaSSIF, around 60% of the drug was released from SG/LP after 4 h, whereas only 20% was released from BUD-E-BSLPs in the same timeframe. Both ES100 coated formulations, with and without bile salts (BUD-E-LPs and BUD-E-BSLPs), showed stability in FeSSIF, maintaining liposomal structure and size distribution. In this medium, BUD-E-BSLPs showed only 15% drug release after a 4 h incubation, while BUD-E-LPs released about 30%. Due to the lack of bile salts in its liposomal structure, BUD-E-LPs doubled drug release when compared to BUD-E-BSLPs. At pH 7.4, the two formulations showed similar release patterns, where about 65-85% was released within 24 h. Collectively, this data confirmed that drug release from BUD-E-BSLPs was pH-dependent, with minimal release under gastric and upper intestinal conditions, and marked increase at higher pH values, consistent with colon targeting. In a simulated colonic environment, the majority of the drug was released within 1 h.
Stability studies were performed as well, for 3 days and 5 weeks, at 25 and 4ºC, respectively, and further demonstrated that BUD-E-BSLPs exhibited a better capacity to prevent liposomal aggregation and destabilisation. The negative surface charge imparted by bile salts promoted electrostatic repulsion, which prevented liposomal aggregation during storage. Hence, overall, BUD-E-BSLPs showed better performance and stability across all in vitro studies [76].
4.3.2. Solid Lipid Nanoparticles
Solid lipid nanoparticles (SLNPs) are nanoscale carriers built from a are composed of a solid lipid matrix with a surfactant in the outer coat layer. The physical and chemical properties of these nanostructures are largely dictated by their constituent components, which in turn determine their biological behaviour. By adjusting their composition, these systems can fine-tune drug release patterns and influence the way a drug is distributed within the body, leading to improvements in solubility and overall bioavailability. SLNPs provide an attractive method for delivering drugs with precise control, longer bloodstream residency times, and reduced toxicity [99].
With the purpose of improving saxagliptin´s (SAX) oral bioavailability, Alhamhoom et al. [77] developed RS100-coated SLNPs loaded with SAX. This antidiabetic drug is characterised by limited membrane permeability, low aqueous solubility, and short elimination half-life (4 - 6 h), all of which contribute to its poor bioavailability. The use of SLNPs as a carrier allows bypassing hepatic first-pass metabolism and promotes lymphatic drug transport, thereby enhancing oral bioavailability. For successful encapsulation, SAX´s solubility in different lipids was first evaluated. Among the tested lipids, stearic acid exhibited the highest solubility and was therefore chosen for NP preparation. The optimised SLNPs were obtained using Poloxamer188 as an emulsifier and polyvinyl alcohol as a stabiliser, excipients that contributed to preventing particle aggregation and improving their stability. A Quality by Design approach, employing a Central Composite Design, was applied to optimise the formulation. In vitro diffusion studies were performed for pure drug and optimised SLNPs at pH 7.4. The SLNPs exhibited a slower, controlled release profile relative to the pure drug, which was attributed to the presence of RS100. The reduced RS100´s permeability limits the penetration of the dissolution medium into the NPs, thereby delaying drug dissolution and diffusion across the polymeric coating layer. Subsequently, in vivo pharmacokinetic studies were performed in male albino Wistar rats, comparing pure SAX with the RS100 SLNPs. Results showed improved Cmax, Tmax, and overall bioavailability for the coated SLNPs formulation, compared with pure SAX. In addition, the RS100 SLNPs exhibited elimination rate constants, confirming a more sustained drug release. Hence, RS100 enabled a controlled release profile due to low polymer permeability and, consequently, slow polymer swelling, which had a direct impact on the NPs’ biological performance [77].
Colonic drug delivery can also be attained using SLNPs. Using oxaliplatin (OXA) as the model drug, Golla et al. [78] developed ES100-coated SLNPs (OXA-ES100 SLNPs) for colorectal cancer treatment. Conventional OXA-based chemotherapy presents off-target toxicity on healthy cells and low bioavailability, therefore justifying the need for advanced delivery systems. Similar to what was done in the previous study, an experimental design methodology was applied to optimise OXA SLNPs. A Box-Behnken design was employed to investigate the influence of lipid, surfactant, and co-surfactant concentrations on PS and EE. These SLNPs were subsequently lyophilised and processed into pellets using an extrusion spheronization technique, after which they were spray-coated with ES100. In vitro drug release was evaluated for two different pH values: gastric pH 1.2 and colonic pH 7.0. The release profiles demonstrated that OXA-ES100 SLNPs began releasing OXA after 4 h at neutral pH. At colonic pH, the percentage of OXA released from the coated NPs was significantly higher compared to the uncoated formulation. Since these two dissolution assays were performed separately, the reason for this difference lies in ES100´s insolubility at acidic pH, resulting in lower percentages of released OXA. These findings confirmed that OXA-ES100 SLNPs could successfully deliver OXA to the colon, indirectly achieving organ targeting through the pH-dependent solubility of ES100. Furthermore, the dissolution data suggest that drug release followed a controlled-release mechanism, where the polymer´s solubility properties regulated and sustained OXA´s release [78].
4.3.3. Nanostructured Lipid Carriers
Nanostructured lipid carriers (NLCs) are advanced NPs built from a mixture of solid and liquid lipidic components. At physiological temperature, the lipids exist in either a solid or liquid state, creating a partially disordered internal structure that provides spaces within the lipid core, facilitating drug incorporation. The ratio and nature of the lipids forming NLCs will influence their configuration. Their structural irregularities reduce the risk of drug displacement, enabling higher drug loading compared with other nanoparticulate systems. These nanocarriers are transformed into chylomicrons following intestinal absorption, entering the lymphatic system, and promoting systemic delivery while bypassing first-pass metabolism, similarly to SLNPs [79]. Moreover, NLCs have demonstrated enhanced particle mucoadhesion, contributing to a prolonged residence time at the target site and, consequently, improved therapeutic efficacy [100,101].
Tacrolimus (TAC) is a calcineurin inhibitor used to manage refractory IBD. It is a significantly potent, water-insoluble drug that undergoes extensive first-pass metabolism. Aiming to achieve targeted colonic delivery, tacrolimus-loaded NLCs (TAC-NLCs) coated with FS100 (TAC-NLCs/E FS100) were designed (Figure 8A). NLCs were obtained using the solid lipid compritol, the surfactant CTAB, and TAC as the model drug. FS100 was employed because it enables colon-targeted drug release and prevents premature degradation at gastric pH. When it reaches neutral pH, polymer swelling and dissolution occur, resulting in immediate drug release at the desired target site. Optimisation of both TAC-NLCs and TAC-NLCs/E FS100 was also performed using the Box-Behnken design (Figure 8B). Different NLC: FS100 ratios were studied to assess their impact on PS, ZP, and EE. The optimized formulation, containing a 1:2 ratio of NLCs to FS100, presented an increased PS and ZP, but a reduced drug release at pH 1.2, likely due to limited penetration of the dissolution medium. A decrease in EE was also observed, potentially caused by drug migration from the lipid core to the outer aqueous phase, during the coating stage.
In vitro drug release of TAC-NLCs/E FS100 and TAC-NLCs, performed at pH 1.2, 4.5, and 7.4, using the dialysis bag diffusion method, revealed that TAC-NLCs/E FS100 exhibited controlled drug release over a 72 h period, with maximal release happening under colonic pH. Release kinetics analysis indicated that uncoated NLCs followed Fickian diffusion, explaining the initially observed burst release. Conversely, TAC-NLCs/E FS100 demonstrated a release mechanism governed by polymer swelling and drug diffusion, consistent with the controlled release properties of RS100, and thus regulating burst release.
The in vitro cytotoxicity of TAC, TAC-NLCs, and TAC-NLCs/E FS100 (25, 50, and 100 µg/mL) was evaluated on thioglycolate elicited macrophages and colon cells, using the MTT assay (Figure 8C). After 24 h of incubation, the viability of both cell types remained above 80% for all tested formulations, compared with the negative control Triton-X. Additionally, the results indicated that TAC-NLCs/E FS100 exhibited a favourable safety profile at all tested concentrations, with cell viability remaining independent of the applied concentration.
In vivo studies were performed as well, using a DSS-induced colitis rat model (the same model used by Raza et al.) [52]. Treatment groups received TAC, TAC-NLCs, or TAC-NLCs/E FS100. Among these, TAC-NLCs/E FS100 demonstrated superior therapeutic outcomes, with significantly greater body weight recovery and an improvement in survival rate of up to 80%. Biodistribution studies confirmed the presence and accumulation of the developed NLCs in the stomach, small intestine, and colon, with TAC-NLCs/E FS100 achieving markedly higher colonic drug concentrations. In contrast, and due to the initial burst release, TAC-NLCs exhibited increased drug content in the stomach [79].
Another colon targeting strategy was proposed by Borderwala et al. [80], using 5-fluorouracil (5-FUO) as the model drug. The clinical use of 5-FUO is limited by its short plasma half-life, and uncontrolled systemic concentrations, resulting in lack of therapeutic efficacy, and the need for frequent dose administration to maintain drug concentrations above the minimum effective level. These problems contribute to poor patient compliance, resistance development, systemic side effects, and drug distribution in high amounts to normal tissues. Intravenous administration often results in plasma concentrations above the maximum safe level, while oral administration is conditioned by first-pass hepatic and intestinal metabolisms, leading to unpredictable toxicity and efficacy. To overcome these limitations, 5-FUO-loaded NLC coated with ES100 (5-FUO-E-NLCs) were formulated. Regarding the optimized formulations, it was observed that the PS of 5-FUO-E-NLCs (154 nm) was higher than the PS of uncoated NLC (102 nm), and the ZP of uncoated NLC ( -8.19) was higher than that of coated NLC (-21.7), attributable to the acrylic acids from the coating polymer. The coating process enhanced the EE of NLCs, with coated NLCs reaching 89.8%, compared to 83.5% for the uncoated NLCs The 5-FUO release from a 5-FUO solution, 5-FUO NLCs, and 5-FUO-E-NLCs was evaluated in vitro, using pH variations to mimic the physiological GI transit. The 5-FUO solution burst-released the drug in the first 2 h at pH 1.2, while 5-FUO NLCs showed maximum release at pH 4.5, suggesting indirect intestinal targeting. The presence of ES100 effectively controlled drug release, being in line with providing the colon with the largest drug concentrations, and achieving a spatial and temporal release pattern.
On the other hand, the performed ex vivo studies showed only partial and slow release of 5-FUO from 5-FUO solution, which was likely attributed to P-gp presence, which leads to drug efflux from the intestinal epithelium [102,103]. The release pattern of 5-FUO-E-NLCs in the performed ex vivo study was 7.40 ± 2.19%, 26.5 ± 4.45%, and 100.90 ± 3.96% after 2, 6, and 24 h, respectively. In vitro cytotoxicity of blank ES100-NLCs, 5-FUO NLCs, and 5-FUO-E-NLCs was carried out by MTT assay on Caco-2 cells. Blank ES100-NLCs led to a cell viability of 99%, indicating excipient safety. For the other formulations, cytotoxicity was dose-dependent, and cell viability was inversely proportional to 5-FUO concentration. 5-FUO NLCs and 5-FUO-E-NLCs exhibited higher cytotoxic activity, which could be associated with enhanced permeation and retention effect.
Pharmacokinetic studies, in albino Wistar rats, further demonstrated the advantages of the ES100 coating. 5-FUO was immediately released from the solution, represented by low Tmax, whereas both coated and uncoated formulations significantly prolonged 5-FUO´s Tmax, indicating their capacity to delay drug release. The relative bioavailability of 5-FUO NLCs and 5-FUO-E-NLCs were found to increase by 5 and 11-fold, respectively, compared to the 5-FUO solution [80].
4.4. Polymeric Nanoparticles
Polymeric nanoparticles (PNPs) have become a cornerstone in modern drug delivery, primarily due to their customizable architecture and physicochemical adaptability. Their size ranges between 1 to 1000 nm, and they can be loaded with active compounds, entrapped within or surface-adsorbed to the polymeric core. Their advantages include controlled drug release, drug protection from the biological environment, and improved bioavailability and therapeutic efficacy. Frequently used polymeric materials include PLGA, PEG, PMMA, and Eudragit, which offer tuneable surface chemistry and mechanical properties, enabling precise modulation of drug release profiles [104,105].
In the context of ulcerative colitis therapy, Gao et al. [81] developed iridoid glycoside (IG)-loaded, dual-Eudragit-coated polymeric NPs (PLGA-E-PNPs), to improve IG´s oral bioavailability. The PNPs were designed using poly (lactic-co-glycolic acid) (PLGA) as a sustained release polymer, and ES100 and Eudragit L30-D 55 (EL30) as coating agents to overcome the limitations of single-triggered release systems. The aim of this dual-Eudragit formulation was to optimise colonic drug distribution by choosing the most advantageous EL30:ES100 ratio. These polymers were meant to prevent gastric drug release, minimising drug loss, and leading to higher colonic drug concentrations. The in vitro drug release of IG from PLGA-E-PNPs was evaluated in simulated physiological conditions along the GIT, along with two other nanoparticulate systems: IG-loaded pH-sensitive NPs (E-PNPs), containing EL30 and ES100, and IG-loaded PLGA NPs (TDNPs). In the first 6 h, less than 20% of IG had been released from E-PNPs and PLGA-E-PNPs, corresponding to inhibited drug release in the simulated stomach and intestinal fluids. TDNPs exhibited a burst drug release profile during the first 6 h, due to PLGA´s pH-independent properties, which corresponded to 70% of IG release. On the other hand, E-PNPs showed 100% burst release at pH 7.4, owing to the complete dissolution of ES100 and EL30D at pH > 7, whereas PLGA-E-PNPs maintained their intact NP morphology, suggesting no immediate colonic drug release. This was due to the presence of PLGA, which was able to sustain drug release after polymer dissolution when reaching pH 7.4.
After in vivo therapeutic efficacy assessment of orally administered PLGA-E-NPs in a DSS-induced colitis rat model, the results showed a longer Tmax for PLGA-E-NPs than for free IG. Notwithstanding, free IG achieved higher Cmax and AUC0-48h values than the controlled targeted delivery NPs. The reason for this difference was the higher IG release and absorption, which occurred in the upper GIT. However, after 8 h, the drug´s poor stability and short half-life rendered it undetectable. On the contrary, PLGA-E-NPs exhibited a longer residence time, reaching their Cmax after 10 h, and maintaining high plasmatic concentrations during a 16 h period. By studying the different IG containing formulations, the authors were able to assess that the dual polymeric strategy resulted in better performance outcomes than the other employed formulations. Burst release inhibition, increased colonic drug accumulation, and improved bioavailability of PLGA-E-NPs were some important results to encourage dual-targeting PNPs [81].
Similar strategies were reported by Naeem et al. [106], who developed dual-targeting NPs destined for ulcerative colitis, using FS30D and PLGA as polymers to deliver cyclosporine A. For comparison, NPs containing only PLGA and only FS30 D were produced. All formulations exhibited equivalent PS (< 300 nm), PDI (< 0.1), and encapsulation efficiency (> 50%). Additionally, it was observed that particles maintained their size across different pH media mimicking the gastrointestinal tract. Regarding drug release at pH 1.2 and 6.8, formulations containing either FS30D alone or a mixture of PLGA and FS30D showed a similar profile, with less than 20% of drug release. In contrast, formulations with only PLGA released approximately 70% of the drug after 6 hours. At pH 7.4 (simulating the ileum), differences emerged between the particles containing a mixture of polymers and NPs containing only FSD30D. Although this system employed only one Eudragit grade, the results were comparable: FS30D (as well as ES100 and EL30) controlled drug release time and location, while PLGA prevented burst release and provided sustained drug release after reaching pH 7.4, promoting higher drug concentrations in the desired targeted location, the inflamed colon. In vivo studies in mice with colitis demonstrated the superiority of NPs containing both polymers [106].
Together, these studies underscore the complementary role of Eudragit coatings in controlling release location and PLGA in prolonging drug release.
Beyond inflammatory bowel disease, PNPs have also been employed to enhance the oral delivery of anticancer drugs. G. et al. [82] developed PNPs loaded with Dasatinib (DTN). DTN is a second-generation tyrosine kinase inhibitor, efficacious in targeting multiple tyrosine kinases associated with tumour cell proliferation. This drug´s oral bioavailability ranges from 14 to 34%, primarily due to its poor water solubility, short half-life of 3 - 5h, and pH susceptibility, which complicate its effective delivery. Because of its weak basic nature, at a pH greater than 4, DTN´s solubility declines, meaning that DTN is more soluble in acidic pH [107]. This can lead to high gastric drug concentrations, which subsequently can result in gastric irritation and restricted drug absorption in the intestinal environment. DTN-loaded PNPs coated with EL100 (DTN-E-PNPs) were designed to achieve better tumour selectivity, minimise toxicity, control drug release, and increase DTN´s oral bioavailability. To optimise the synthesis of the PNPs, a 23 factorial design was employed. EL100 concentration, Poloxamer 407 concentration, and sonication power were the studied independent variables, whereas PS, ZP, and EE were the dependent ones. Among the several prepared batches, the optimised batch of DTN-E-PNPs included a drug polymer ratio of 1:2, sonication power of 30 mV, and a Poloxamer 407 concentration of 1%. The in vitro drug release study was conducted for the optimised DTN-E-PNPs, and pure DTN, in two media (pH 1.2 and 6.8). Single DTN exhibited the lowest release profile, whereas DTN-E-PNPs significantly improved drug release. An initial burst release was registered for both formulations in the first hour. However, sustained drug release profiles were maintained throughout the following duration of the assay. Additionally, the release kinetics model that best fitted DTN-E-PNPs’ release profile suggested a diffusion-controlled mechanism. The cytotoxicity of single DTN and DTN-E-PNPs was evaluated as wel, in vitro, in MCF-7, MDA-MB-231, and 4T1 cell lines, using the MTT assay. The used negative control was DOX, and the incubation period was of 24 h. The results showed higher cytotoxic effects for DTN-E-PNPs when compared with DOX and DTN, with lower IC50 values, indicating a more potent anticancer activity. In vivo pharmacokinetics and biodistribution studies, in Wistar albino rats, confirmed the superiority of the nanoparticulate system, compared to the DTN. DTN-E-PNPs exhibited a Cmax of 12.60 ng/mL at 2 h (Tmax), an AUC0-t of 71.23 ng/mL.h, and an elimination half-life of 6.15 h, whereas for single DTN´s Cmax was 8.50 ng/mL at 4 h (Tmax), AUC0-t was 66.06 ng/mL.h, and the elimination half-life was 3.97 h. Furthermore, biodistribution assays revealed that both formulations accumulated in the spleen and kidney, though DTN-E-PNPs showed slightly reduced levels (2.83 ng/10 mg for the spleen, and 2.87 ng/10 mg for the kidney). The mammary tissue was not actively targeted, which indicates that the only mechanism targeting cancer cells was the passive EPR effect, leaving the mammary tissue with lower drug concentrations for both formulations (1.11 ng/10 mg for the DTN-E-PNPs). The formulation´s performance was studied in a rat mammary carcinoma model, which was 7,12-Dimethylbenz[a]anthracene (DMBA)-induced. The results supported the pharmacokinetic findings, since DTN-E-PNPs significantly reduced tumour volumes, improved survival rates, preserved body weight, and maintained healthier haematological parameters, compared to DMBA-treated and single DTN groups. The PNP´s remained stable in terms of PS, ZP, and EE for 6 months at 4.0 ± 1.0 ºC, 25 ± 1.0 ºC, and 40 ± 1.0 ºC storage conditions. All in all, this work demonstrates that EL100 coated PNPs effectively supressed gastric burst release, redirected drug release to the small intestine, and provided sustained delivery, thereby enhancing DTN´s oral bioavailability, stability, tolerability, and antitumour activity [82].
5. Discussion
The development of oral DDS based on Eudragit-coated NPs represents a versatile strategy to address multiple limitations associated with conventional oral pharmaceuticals. The selection of a suitable Eudragit grade is a critical determinant in the rational design of oral nanoparticulate systems, as the polymer strongly influences the carrier´s physicochemical behaviour, as well as the site and kinetics of drug release. Across the diverse NPs considered in this work, Eudragit coatings were consistently employed to address solubility limitations of poorly water-soluble drugs, provide protection against the gastrointestinal environment, and introduce site-specific delivery. Beyond their traditional role as enteric or protective coatings, Eudragit polymers have also demonstrated the capacity to indirectly modulate biological barriers in vitro, such as P-gp-mediated efflux, thereby increasing intracellular drug retention. Moreover, with the opportunity to be chemically modified and crosslinked with other polymeric compounds, such as polymer thiolation and disulfide-bridge formation, their already favourable properties are further enhanced.
Different Eudragit grades provide unique functionalities that can be tailored to specific therapeutic goals. Soluble cationic grades, such as Eudragit E, have been employed for immediate release, where they also improve the palatability of bitter or weakly basic drugs. In contrast, other pH-dependent grades such as Eudragit L and S enable release in the small intestine and colon, respectively, and are often combined to broaden the solubility range and achieve precise pH-triggered targeting. ES100, in particular, was predominantly utilised for colon targeting, successfully enhancing oral bioavailability through targeted delivery, gastric degradability protection, and controlled drug release. pH-independent grades, Eudragit RS and RL, have been employed to achieve sustained release throughout the GIT, with drug release controlled by diffusion through the swollen polymer matrix. When using MSNPs as drug carriers, amino modification was often introduced, owing to this carrier´s anionic nature under physiological pH conditions. This functionalisation enhances coating stability, preventing premature release. Altogether, these diverse examples illustrate the adaptability of Eudragit polymers across nanoparticulate platforms. Their wide-ranging functionalities, spanning from protective effects to tailored release kinetics and biofunctionalization opportunities, establish them as a highly versatile and clinically relevant family of polymers for oral drug administration.
Eudragit polymers have a long record of safe use in conventional oral formulations, supported by more than 70 years of pharmaceutical application [108]. Their inclusion in pharmacopeial monographs further underscores their regulatory acceptance. For instance, EL30-55 has been incorporated into the FDA-approved Procysbi® for nephropatic cystinosis, where it functions as a protective coating. Toxicological evaluations of EL30-55, including 6-month and 1-year oral studies in rats and dogs, have demonstrated no significant adverse effects at doses up to 600 mg/kg/day and 80 mg/kg/day, respectively [109]. However, the application of these polymers in nanoparticulate systems necessitates additional safety evaluations due to the unique physicochemical properties of NPs. In vitro evaluations have shown that Eudragit-coated NPs generally maintain low toxicity at relevant concentrations, with surface charge and surfactant composition modulating cellular responses [110]. An in vitro comparative study assessing RL100, chitosan, PLGA, and poly-ε-caprolactone-based NPs revealed that RL100-based formulations displayed the highest cytotoxicity and haemolysis, effects attributed to the presence of quaternary amine groups, which can induce cytotoxicity, suppress DNA replication, and trigger inflammatory responses [111,112]. Another research study showed the implications of chronic exposure to different polymeric NPs on the biological function of Drosophila melanogaster. The studied polymers were polysorbate 80, PEG, chitosan, and RS100. When employing 500 µL doses, all polymers showed potential toxicity in several parameters. However, the RS100-based NPs caused the most changes, even at doses of 100 µL. Notably, cationic NPs have more extensive toxicity than anionic ones, due to electrostatic interactions with negatively charged cell membranes [113,114]. Hence, these findings emphasize the importance of systematic toxicological assessments, particularly given the need for chronic dosing in many of the therapeutic indications targeted by oral NPs.
Translation to clinical applications also requires consideration of manufacturing and purification processes. NP production at scale must preserve critical quality attributes, such as size, surface charge, and drug release kinetics. Purification strategies, including tangential flow filtration and solvent-exchange methods, are essential to remove residual solvents, unreacted polymer, and surfactants, which could otherwise impact safety and efficacy [115]. In this regard, alternative solvent free approaches can be investigated to enhance reproducibility and clinical feasibility. In this work, the use of a liquid CO2-assisted process was described as an appropriate alternative to formulate MSNPs loaded with MER and coated with ES100. This technique not only minimised the risk of residual organic solvents, but also improved drug loading, ensuring a scalable and environmentally friendly strategy for oral NP production [52].
The patent landscape reflects the growing pharmaceutical interest in Eudragit-coated NPs for oral drug delivery and highlights key priorities for clinical translation. Patents such as US 9,700,544 B2[116] and US 10,391,059 B2[117] from Rapamycin Holdings, disclose rapamycin NPs protected by ES100 for gastric stability and intestinal release. US 9,452,930 B2 [118], which describes a continuous polymer-coating process with supporting data on RL100-coated NPs, underscores the importance of scalable and reproducible manufacturing methods. Additional filings highlight therapeutic diversification. For example, US 20130034602 A1[119] discloses enteric-coated capsules containing insulin-loaded cationic NPs protected with Eudragit-based coatings. Similarly, CN 108379560 A [120]describes insulin NPs coated with Eudragit for enteric solubility control. More recent applications, such as US 2018/0318230 A1 [121], focus on blended systems, including PLGA-PEG/Eudragit NPs with pH-triggered oral insulin release. Collectively, these patents demonstrate the versatility of Eudragit across therapeutic areas, while also illustrating the dual emphasis on functional performance and scalable production technologies.
6. Conclusions
Altogether, the evidence indicates that Eudragit-coated NPs hold significant promise in the rational design of next-generation oral therapies. The small intestine and colon remain the primary target sites, which explains the frequent use of Eudragit L and S grades in formulations for IBD and colorectal cancer. Beyond site-specific targeting, these polymers are also employed to improve the solubility of poorly water-soluble drugs, thereby broadening their therapeutic applicability. Among the various delivery platforms, polymeric NPs have emerged as the most widely used carriers, underscoring their versatility and compatibility with Eudragit-based coatings.
Author Contributions
Conceptualization, F.B., A.S., and A.V.; writing—original draft preparation, F.B.; writing—review and editing, P.P., F.V., A.S., A.V.; supervision, P.P, F.V., A.S, A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by National Funds through FCT/MCTES—Portuguese Foundation for Science and Technology within the scope of the project UIDB/50006/2025—https://doi.org/10.5 4499/UID/50006/2025.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing does not apply to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-ASA | 5-Aminosalicylic acid |
| 5-FUO | 5-Fluorouracil |
| 5-FUO-E-NLCs | 5-Fluorouracil loaded, Eudragit coated nanostructured lipid carriers |
| AUC | Area under the curve |
| AZM | Acetazolamide |
| AZM-SH | Acetazolamide derivative |
| BCS | Biopharmaceutical Classification System |
| SG/LP | Bile salt containing liposomes |
| BIBF | Nintedanib |
| BIBF-NCs | Nintedanib nanocrystals |
| BIBF-NCs@L100 | Eudragit-coated BIBF-NCs |
| BUD | Budenoside |
| BUD-A-MSNPs | Budesonide loaded, amino-functionalised MSNP |
| BUD-A-E-MSNPs | Budesonide loaded, Eudragit-coated, amino-functionalised MSNP |
| BUD-E-LPs | Budesonide-loaded, Eudragit-coated liposomes |
| BUD-E-BSLPs | Budesonide-loaded, bile salt-containing liposomes |
| CHT | Catechin |
| CTAB | Cetyl trimethyl ammonium bromide |
| DAI | Disease Activity Index |
| DMBA | 7,12-Dimethylbenz[a]anthracene |
| DOX | Doxorubicin hydrochloride |
| DDS | Drug delivery system |
| DSS | Dextran sulphate sodium |
| DTN | Dasatinib |
| DTN-E-PNPs | Dasatinib loaded, Eudragit coated polymeric nanoparticles |
| ES@PND-PEG-TPP/DOX | Eudragit coated PND-PEG-TPP/DOX |
| EE | Entrapment efficiency |
| EL100 | Eudragit L100 |
| EL100-55 | Eudragit L100-55 |
| EL30D | Eudragit L30D-55 |
| EGFR | Endothelial growth factor receptor |
| E-PNPs | Iridoid glycoside loaded pH-sensitive polymeric nanoparticles |
| EPR | Enhanced permeability and retention |
| ES100 | Eudragit S100 |
| ES-SG/LP | Eudragit coated, bile salt containing liposomes |
| E@MSND-BUD | Enteric-Coated MSNDs-BUD |
| Eud-MER-MCM-NH2 | Eudragit coated, MER-MCM-NH2 |
| FaSSIF | Fasted-state simulated intestinal fluid |
| FDA | Food and Drug Administration |
| FeSSIF | Fed-state simulated intestinal fluid |
| FS30D | Eudragit FS30D |
| FS100 | Eudragit FS100 |
| hCA IX | Human Carbonic Anhydrase IX |
| HPMC-E5 | Hydroxypropyl methylcellulose E5 |
| IBD | Inflammatory bowel disease |
| IG | Iridoid glycoside |
| INP | Inorganic nanoparticle |
| IVM | Ivermectin |
| LNP | Lipid nanoparticle |
| LP | Liposome |
| MCM | Mesoporous silica nanoparticles MCM 41 |
| MCM-NH2 | Amino-functionalised MCM |
| MER | Meropenem |
| MER-MCM | MER loaded-MCM |
| MER-MCM-NH2 | MER loaded-amino-functionalised MCM |
| MER-MCM-PO3 | MER loaded-phosphonate functionalised MCM |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MSNP | Mesoporous silica nanoparticle |
| MSND | Dendritic MSNP |
| MSND-BUD | BUD-Loaded MSND |
| NC | Nanocrystal |
| ND | Nanodiamond |
| NDNP | Nanodiamond-based nanoparticle |
| NH2-MNSPs | unloaded amino-functionalized MSNPs |
| NH2-MNSPs@EUS-100 | NH2-MNSPs coated with ES100 |
| NH2-MSNPs/CHT | Catechin-loaded NH2-MNSPs |
| NH2-MSNPs/CHT@EUS-100 | NH2-MNSPs/CHT coated with ES100 |
| NIR | Near Infrared |
| NLC | Nanostructured lipid carrier |
| NP | Nanoparticle |
| MSNPs/CHT | Catechin-loaded MNSPs |
| OFLO | Ofloxacin |
| OXA | Oxaliplatin |
| P-gp | P-glycoprotein |
| PCR | Polymerase chain reaction |
| PDI | Polydispersity index |
| PEG | Polyethylene glycol |
| PLGA | Poly (lactic-co-glycolic acid) |
| PLGA-E-PNPs | Iridoid glycoside loaded, Eudragit coated, PLGA-based polymeric NPs |
| PMMA | Poly (methyl methacrylate) |
| PM | Physical mixture |
| PND | ND coated with polydopamine |
| PND-PEG | PEG-Functionalized PND |
| PND-PEG-TPP | TPP-functionalized PND-PEG |
| PND-PEG-TPP/DOX | Doxorrubicin loaded- PND-PEG-TPP |
| PNP | Polymeric NP |
| Pred | Prednisolone |
| Pred-A-MSNPs | Prednisolone loaded, amino-functionalised mesoporous silica NPs |
| Pred-A-E-MSNPs | Prednisolone loaded, Eudragit coated, amino-functionalised mesoporous silica NPs |
| PS | Particle size |
| RS100 | Eudragit RS100 |
| SAX | Saxagliptin |
| SLN | Solid lipid NPs |
| SLS | Sodium lauryl sulphate |
| SGF | Simulated gastric fluid |
| TAC | Tacrolimus |
| TAC-NLCs | Tacrolimus-loaded nanostructured lipid carriers |
| TAC-NLCs/E FS100 | Tacrolimus-loaded, Eudragit-coated nanostructured lipid carriers |
| TDNPs | Iridoid glycoside loaded time-dependent polymeric NPs |
| TEOS | Tetraethoxysilane |
| TPP | Triphenylphosphonium |
| ZP | Zeta potential |
References
- Milián-Guimerá, C.; McCabe, R.; Thamdrup, L.H. E.; Ghavami, M.; Boisen, A. Smart pills and drug delivery devices enabling next generation oral dosage forms. Journal of Controlled Release 2023, 364, 227–245. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, Q.; Dong, Z.; Meng, T.; Hu, F.; Wang, J.; Yuan, H. Nanocarriers transport across the gastrointestinal barriers: The contribution to oral bioavailability via blood circulation and lymphatic pathway. Advanced Drug Delivery Reviews 2023, 203, 115130. [Google Scholar] [CrossRef] [PubMed]
- Sant, S.; Tao, S.L.; Fisher, O.Z.; Xu, Q.; Peppas, N.A.; Khademhosseini, A. Microfabrication technologies for oral drug delivery. Advanced Drug Delivery Reviews 2012, 64, 496–507. [Google Scholar] [CrossRef]
- Ahadian, S.; Finbloom, J.A.; Mofidfar, M.; Diltemiz, S.E.; Nasrollahi, F.; Davoodi, E.; Hosseini, V.; Mylonaki, I.; Sangabathuni, S.; Montazerian, H.; et al. Micro and nanoscale technologies in oral drug delivery. Advanced Drug Delivery Reviews 2020, 157, 37–62. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Duan, H.; Qin, Q.; Teng, Z.; Gan, F.; Zhou, X.; Zhou, X. Advances in Oral Drug Delivery Systems: Challenges and Opportunities. Pharmaceutics 2023, 15, 484. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xie, Z.; Li, D.; Kassymova, M.; Zang, S.Q.; Jiang, H.L. Nano-sized metal-organic frameworks: Synthesis and applications. Coordination Chemistry Reviews 2020, 417, 213366. [Google Scholar] [CrossRef]
- Johnsen, H.M.; Filtvedt, W.; Klaveness, J.; Hiorth, M. Nano-strategies for advancing oral drug delivery: Porous silicon particles and cyclodextrin encapsulation for enhanced dissolution of poorly soluble drugs. International journal of pharmaceutics 2024, 666, 124809. [Google Scholar] [CrossRef]
- Singh, K.; Singhal, S.; Pahwa, S.; Sethi, V.A.; Sharma, S.; Singh, P.; Kale, R.D.; Ali, S.W.; Sagadevan, S. Nanomedicine and drug delivery: A comprehensive review of applications and challenges. Nano-Structures and Nano-Objects 2024, 40, 101403. [Google Scholar] [CrossRef]
- Aggas, J.R.; Guiseppi-Elie, A. Responsive Polymers in the Fabrication of Enzyme-Based Biosensors. In Biomaterials Science: An Introduction to Materials in Medicine; Elsevier, 2020; pp. 1267–1286. [Google Scholar]
- Nikam, A.; Sahoo, P.R.; Musale, S.; Pagar, R.R.; Paiva-Santos, A.C.; Giram, P.S. A Systematic Overview of Eudragit® Based Copolymer for Smart Healthcare. Pharmaceutics 2023, 15, 587. [Google Scholar] [CrossRef]
- NCI, Immediate Release Dosage Form ( Code - C42669 ). 2026. Available online: https://evsexplore.semantics.cancer.gov/evsexplore/concept/ncit/C42669.
- Thesaurus, N.C.I. Pharmaceutical Quality/CMC Terminology Files in the National Cancer Institute Thesaurus (NCIt).
- 2026-02-23, N. T.-V. d. R. D., Controlled Release Dosage Form ( Code - C42731 ). Available online: https://evsexplore.semantics.cancer.gov/evsexplore/concept/ncit/C42731.
- 2026-02-23, N. T.-V. d. R. D., Modified Release Dosage Form ( Code - C42712 ). Available online: https://evsexplore.semantics.cancer.gov/evsexplore/concept/ncit/C42712.
- Hong Wen, K.P. Introduction And Overview Of Oral Controlled Release Formulation Design. In Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice; Hong Wen, K.P., Ed.; Wiley, 2010. [Google Scholar]
- Laracuente, M.L.; Yu, M.H.; McHugh, K.J. Zero-order drug delivery: State of the art and future prospects. Journal of Controlled Release 2020, 327, 834–856. [Google Scholar] [CrossRef]
- Bader, R.A.; Putnam, D.A. Engineering polymer systems for improved drug delivery; John Wiley & Sons, 2014; pp. 492–492. [Google Scholar]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. Journal of Pharmacy and Pharmacology 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Amiji, A.S.M.M. Stimuli-responsive Drug Delivery Systems 2018.
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nature Reviews Drug Discovery 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Frey, M.; Bobbala, S.; Karabin, N.; Scott, E. Influences of nanocarrier morphology on therapeutic immunomodulation. Nanomedicine 2018, 13, 1795–1795. [Google Scholar] [CrossRef]
- Garapaty, A.; Champion, J.A. Tunable particles alter macrophage uptake based on combinatorial effects of physical properties. Bioengineering & Translational Medicine 2017, 2, 92–92. [Google Scholar] [CrossRef] [PubMed]
- Huynh, E.; Zheng, G. Cancer nanomedicine: Addressing the dark side of the enhanced permeability and retention effect. Nanomedicine 2015, 10, 1993–1995. [Google Scholar] [CrossRef] [PubMed]
- Poole-Warren, L.; Martens, P.J.; Green, R.A. Biosynthetic polymers for medical applications; Woodhead Publishing, 2016. [Google Scholar]
- Fenton, O.S.; Olafson, K.N.; Pillai, P.S.; Mitchell, M.J.; Langer, R. Advances in Biomaterials for Drug Delivery. Advanced Materials 2018, 30. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: advances, insights and prospects. Nature Reviews Drug Discovery 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Hardenia, A.; Maheshwari, N.; Hardenia, S.S.; Dwivedi, S.K.; Maheshwari, R.; Tekade, R.K. Scientific rationale for designing controlled drug delivery systems. In Basic Fundamentals of Drug Delivery; Elsevier, 2018; pp. pp 1–28. [Google Scholar]
- Depan, D.; Shah, J.; Misra, R.D.K. Controlled release of drug from folate-decorated and graphene mediated drug delivery system: Synthesis, loading efficiency, and drug release response. Materials Science and Engineering: C 2011, 31, 1305–1312. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef]
- Pandi, J.S.; Pavadai, P.; Sundar, L.M.; Sankaranarayanan, M.; Panneerselvam, T.; Pandian, S.R.K.; Kunjiappan, S. Pharmacokinetics and Brain Tumor Delivery Studies of Thymoquinone-Encapsulated Eudragit L100-Coated Solid-Lipid Nanoparticles. Journal of Cluster Science 2025, 36. [Google Scholar] [CrossRef]
- Al-Shaeli, M.; Benkhaya, S.; Al-Juboori, R.A.; Koyuncu, I.; Vatanpour, V. pH-responsive membranes: Mechanisms, fabrications, and applications. Science of the Total Environment 2024, 946. [Google Scholar] [CrossRef]
- Huang, T.; Su, Z.; Hou, K.; Zeng, J.; Zhou, H.; Zhang, L.; Nunes, S.P. Advanced stimuli-responsive membranes for smart separation. Chemical Society Reviews 2023, 52, 4173–4207. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the development of amorphous solid dispersions: The role of polymeric carriers. Asian Journal of Pharmaceutical Sciences 2023, 18, 100834. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Zhao, N.; Chen, H.; Guo, G. Advancements in pH-responsive nanocarriers: enhancing drug delivery for tumor therapy. Expert Opinion on Drug Delivery 2023, 20, 1623–1642. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yue, R.; Guan, G.; Zhang, C.; Zhou, Y.; Song, G. Recent development of pH-responsive theranostic nanoplatforms for magnetic resonance imaging-guided cancer therapy. Exploration 2023, 3. [Google Scholar] [CrossRef]
- Patra, C.N.; Priya, R.; Swain, S.; Jena, G.K.; Panigrahi, K.C.; Ghose, D. Pharmaceutical significance of Eudragit: A review. Future Journal of Pharmaceutical Sciences 2017, 3, 33–45. [Google Scholar] [CrossRef]
- Convention, U.S.P. United States Pharmacopeia 32 - National Formulary 27 (USP32-NF27); United States Pharmacopeial Convention, 2009. [Google Scholar]
- Rowe, C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; RPS Publishing, 2009. [Google Scholar]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit®: A technology evaluation. Expert Opinion on Drug Delivery 2013, 10, 131–149. [Google Scholar] [CrossRef]
- Brewer, K.; Blencowe, A. Composition-Property Relationships of pH-Responsive Poly[(2-vinylpyridine)-co-(butyl methacrylate)] Copolymers for Reverse Enteric Coatings. 2023. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Shen, L.; Yang, Y.; Shen, J. Development of nanoparticle-based orodispersible palatable pediatric formulations. International journal of pharmaceutics 2021, 596, 120206. [Google Scholar] [CrossRef]
- EUDRAGIT® L 100 & S 100: Specs & Tests. 14 Aug 2025. Available online: https://studylib.net/doc/25298564/evonik-eudragit-l-100-and-eudragit-s-100-specificationsheet.
- The Global Market Leader for Enteric Release Coatings | EUDRAGIT® - Evonik Industries. 14 Aug 2025. Available online: https://healthcare.evonik.com/en/drugdelivery/oral-drug-delivery/oral-excipients/eudragitportfolio/delayed-release.
- EUDRAGIT® L 100. 14 Aug 2025. Available online: https://www.evonik.com/en/products/hc/pr_52000884.html.
- Evonik, Functional excipients to take control of your release profile Versatility and reliability for oral solid dosage forms. 13 Aug 2025. Available online: www.evonik.com/healthcare.
- EUDRAGIT® Functional Polymers for Sustained Release - Evonik Industries. In [Internet]. 14 Aug 2025. Available online: https://healthcare.evonik.com/en/drugdelivery/oral-drug-delivery/oral-excipients/eudragit-portfolio/sustained-release.
- Zhang, F. Melt-Extruded Eudragit® FS-Based Granules for Colonic Drug Delivery. AAPS PharmSciTech 2015, 17, 56. [Google Scholar] [CrossRef]
- Cardoso, A.M. L.; Oliveira, E.E.; Machado, B.A.S.; Marcelino, H.R. Eudragit®-based nanoparticles for controlled release through topical use. Journal of Nanoparticle Research 2023, 25. [Google Scholar] [CrossRef]
- Nassar, T.; Rom, A.; Nyska, A.; Benita, S. Novel double coated nanocapsules for intestinal delivery and enhanced oral bioavailability of tacrolimus, a P-gp substrate drug. Journal of Controlled Release 2009, 133, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.P.; Sun, S.P.; Cheng, S.H.; Lee, C.H.; Wu, C.Y.; Yang, C.S.; Lo, L.W.; Lai, Y.K. Enhanced chemotherapy of cancer using pH-sensitive mesoporous silica nanoparticles to antagonize P-glycoprotein-mediated drug resistance. Molecular Cancer Therapeutics 2011, 10, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, R.; Baradaran, B.; Valizadeh, H.; Yousefi, B.; Zakeri-Milani, P. Reduced ABCB1 Expression and Activity in the Presence of Acrylic Copolymers. Advanced Pharmaceutical Bulletin 2014, 4, 219–219. [Google Scholar] [CrossRef]
- Raza, A.; Sime, F.B.; Cabot, P.J.; Roberts, J.A.; Falconer, J.R.; Kumeria, T.; Popat, A. Liquid CO2 formulated mesoporous silica nanoparticles for pH-responsive oral delivery of meropenem. ACS Biomaterials Science and Engineering 2021, 7, 1836–1853. [Google Scholar] [CrossRef]
- Gupta, A.; Sood, A.; Dhiman, A.; Shrimali, N.; Singhmar, R.; Guchhait, P.; Agrawal, G. Redox responsive poly(allylamine)/eudragit S-100 nanoparticles for dual drug delivery in colorectal cancer. Biomaterials Advances 2022, 143, 213184. [Google Scholar] [CrossRef]
- Gupta, A.; Dhiman, A.; Sood, A.; Bharadwaj, R.; Silverman, N.; Agrawal, G. Dextran/eudragit S-100 based redox sensitive nanoparticles for colorectal cancer therapy. Nanoscale 2023, 15, 3273–3283. [Google Scholar] [CrossRef]
- Khalid, H.M. B.; Rasul, A.; Shah, S.; Abbas, G.; Mahmood, A. Disulfide Bridged Nanoparticles of Thiolated Sodium Alginate and Eudragit RS100 for Oral Delivery of Paclitaxel: In Vitro and In Vivo Evaluation. ACS Omega 2023, 8, 9662–9672. [Google Scholar] [CrossRef]
- Leichner, C.; Jelkmann, M.; Bernkop-Schnürch, A. Thiolated polymers: Bioinspired polymers utilizing one of the most important bridging structures in nature. Advanced Drug Delivery Reviews 2019, 151-152, 191–221. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, O.; Shah, S.; Abbas, G.; Rasul, A.; Hanif, M.; Ashfaq, M.; Afzal, Z. Moxifloxacin loaded nanoparticles of disulfide bridged thiolated chitosan-eudragit RS100 for controlled drug delivery. International journal of biological macromolecules 2021, 182, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.S.; Mukherjee, D.; Sukharamwala, P.; Dehuri, R.; Murali, A.; Teja, B.V. Thiolated polymer nanocarrier reinforced with glycyrrhetinic acid for targeted delivery of 5-fluorouracil in hepatocellular carcinoma. Drug delivery and translational research 2021, 11, 2252–2269. [Google Scholar] [CrossRef]
- Abbas, N.; Rasul, A.; Abbas, G.; Shah, S.; Hanif, M. Targeted delivery of aspirin and metformin to colorectal cancer using disulfide bridged nanoparticles of thiolated pectin and thiolated Eudragit RL100. Materials Today Communications 2023, 35, 105586. [Google Scholar] [CrossRef]
- EUDRAGIT® FS 100 - Pharma Excipients. 16 Aug 2025. Available online: https://www.pharmaexcipients.com/product/eudragit-fs-100/.
- EUDRAGIT®, E.P.O. 29 Aug 2025. Available online: https://www.evonik.com/en/products/hc/pr_52011259.html.
- EUDRAGIT® E 100. 29 Aug 2025. Available online: https://www.evonik.com/en/products/hc/pr_52000110.html.
- Balogh, A.; Farkas, B.; Domokos, A.; Farkas, A.; Démuth, B.; Borbás, E.; Nagy, B.; Marosi, G.; Nagy, Z.K. Controlled-release solid dispersions of Eudragit® FS 100 and poorly soluble spironolactone prepared by electrospinning and melt extrusion. European Polymer Journal 2017, 95, 406–417. [Google Scholar] [CrossRef]
- Oral Drug Delivery Market Size & Industry Growth Forecast By 2029. 19 Feb 2025. Available online: https://www.databridgemarketresearch.com/reports/global-oral-drug-delivery-market.
- Sharma, A.; Jangra, N.; Dheer, D.; Jha, S.K.; Gupta, G.; Puri, V.; Kesharwani, P. Understanding the journey of biopolymeric nanoformulations for oral drug delivery: Conventional to advanced treatment approaches. European Polymer Journal 2024, 218, 113338–113338. [Google Scholar] [CrossRef]
- Mok, Z.H. The effect of particle size on drug bioavailability in various parts of the body. Pharmaceutical Science Advances 2024, 2, 100031–100031. [Google Scholar] [CrossRef]
- Tarn, D.; Ashley, C.E.; Xue, M.; Carnes, E.C.; Zink, J.I.; Brinker, C.J. Mesoporous silica nanoparticle nanocarriers: Biofunctionality and biocompatibility. Accounts of Chemical Research 2013, 46, 792–801. [Google Scholar] [CrossRef]
- Kassem, A.M.; Almukainzi, M.; Faris, T.M.; Ibrahim, A.H.; Anwar, W.; Elbahwy, I.A.; El-Gamal, F.R.; Zidan, M.F.; Akl, M.A.; Abd-ElGawad, A.M.; et al. A pH-sensitive silica nanoparticles for colon-specific delivery and controlled release of catechin: Optimization of loading efficiency and in vitro release kinetics. European Journal of Pharmaceutical Sciences 2024, 192, 106652. [Google Scholar] [CrossRef]
- Li, Q.; Liu, W.; Liu, K.; Dong, Z.; Kong, W.; Lu, X.; Wei, Y.; Wu, W.; Yang, J.; Qi, J. The Role of Nanoparticle Morphology on Enhancing Delivery of Budesonide for Treatment of Inflammatory Bowel Disease. ACS Applied Materials and Interfaces 2024, 16, 33081–33092. [Google Scholar] [CrossRef]
- Qu, Z.; Wong, K.Y.; Moniruzzaman, M.; Begun, J.; Santos, H.A.; Hasnain, S.Z.; Kumeria, T.; McGuckin, M.A.; Popat, A. One-Pot Synthesis of pH-Responsive Eudragit-Mesoporous Silica Nanocomposites Enable Colonic Delivery of Glucocorticoids for the Treatment of Inflammatory Bowel Disease. Advanced Therapeutics 2021, 4. [Google Scholar] [CrossRef]
- Marcelo, G.A.; Montpeyó, D.; Galhano, J.; Martínez-Máñez, R.; Capelo-Martínez, J.L.; Lorenzo, J.; Lodeiro, C.; Oliveira, E. Development of New Targeted Nanotherapy Combined with Magneto-Fluorescent Nanoparticles against Colorectal Cancer. International journal of molecular sciences 2023, 24, 6612. [Google Scholar] [CrossRef]
- Su, Y.; Pan, H.; Wang, J.; Liu, D.; Pan, W. Eudragit S100 coated nanodiamond-based nanoparticles as an oral chemo-photothermal delivery system for local treatment of colon cancer. Colloids and Surfaces B: Biointerfaces 2024, 237. [Google Scholar] [CrossRef]
- Lopez-Vidal, L.; Parodi, P.; Actis, M.R.; Camacho, N.; Real, D.A.; Paredes, A.J.; Irazoqui, F.J.; Real, J.P.; Palma, S.D. Formulation and optimization of pH-sensitive nanocrystals for improved oral delivery. Drug delivery and translational research 2024, 14, 1301–1318. [Google Scholar] [CrossRef]
- Che, J.; Fu, Y.; Li, Y.; Zhang, Y.; Yin, T.; Gou, J.; Tang, X.; Wang, Y.; He, H. Eudragit L100-coated nintedanib nanocrystals improve oral bioavailability by reducing drug particle size and maintaining drug supersaturation. International journal of pharmaceutics 2024, 658, 124196. [Google Scholar] [CrossRef]
- Alghurabi, H.; Muhammad, H.J.; Tagami, T.; Ogawa, K.; Ozeki, T. Optimization, cellular uptake, and in vivo evaluation of Eudragit S100-coated bile salt-containing liposomes for oral colonic delivery of 5-aminosalicylic acid. International journal of pharmaceutics 2023, 648, 123597. [Google Scholar] [CrossRef] [PubMed]
- Alghurabi, H.; Tagami, T.; Ogawa, K.; Ozeki, T. Preparation, Characterization, and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide. Polymers 2022, 14, 2693. [Google Scholar] [CrossRef] [PubMed]
- Alhamhoom, Y.; Ravi, G.; Osmani, R.A. M.; Hani, U.; Prakash, G.M. Formulation, Characterization, and Evaluation of Eudragit-Coated Saxagliptin Nanoparticles Using 3 Factorial Design Modules. Molecules 2022, 27, 7510. [Google Scholar] [CrossRef] [PubMed]
- Golla, V.S. K.; Boddu, P.; Nageswara, S. Statistical Experimental Approach in Designing pH-Sensitive Oxaliplatin Lipid Nanoparticles for Application in Colorectal Cancer Therapy. BioNanoScience 2023, 13, 1100–1109. [Google Scholar] [CrossRef]
- Altaf, S.; Zeeshan, M.; Ali, H.; Zeb, A.; Afzal, I.; Imran, A.; Mazhar, D.; Khan, S.; Khan, F.A. pH-Sensitive Tacrolimus loaded nanostructured lipid carriers for the treatment of inflammatory bowel disease. European Journal of Pharmaceutics and Biopharmaceutics 2024, 204, 114461. [Google Scholar] [CrossRef]
- Borderwala, K.; Rathod, S.; Yadav, S.; Vyas, B.; Shah, P. Eudragit S-100 Surface Engineered Nanostructured Lipid Carriers for Colon Targeting of 5-Fluorouracil: Optimization and In Vitro and In Vivo Characterization. AAPS PharmSciTech 2021, 22. [Google Scholar] [CrossRef]
- Gao, C.; Yu, S.; Zhang, X.; Dang, Y.; Han, D.D.; Liu, X.; Han, J.; Hui, M. Dual functional eudragit® s100/l30d-55 and plga colon-targeted nanoparticles of iridoid glycoside for improved treatment of induced ulcerative colitis. International journal of nanomedicine 2021, 16, 1405–1422. [Google Scholar] [CrossRef]
- G, H.; Patil, A.; Mg, H.; Redhwan, M.A. M.; Guha, S. Development, optimization, and characterization of Eudragit-based nanoparticles for Dasatinib delivery. Journal of Biomaterials Science, Polymer Edition 2024. [Google Scholar] [CrossRef]
- Yaqoob, A.A.; Ahmad, H.; Parveen, T.; Ahmad, A.; Oves, M.; Ismail, I.M.I.; Qari, H.A.; Umar, K.; Ibrahim, M.N.M. Recent Advances in Metal Decorated Nanomaterials and Their Various Biological Applications: A Review. Frontiers in Chemistry 2020, 8, 528583–528583. [Google Scholar] [CrossRef]
- Noreen, S.; Maqbool, A.; Maqbool, I.; Shafique, A.; Khan, M.M.; Junejo, Y.; Ahmed, B.; Anwar, M.; Majeed, A.; Abbas, M.; et al. Multifunctional mesoporous silica-based nanocomposites: Synthesis and biomedical applications. Materials Chemistry and Physics 2022, 285, 126132. [Google Scholar] [CrossRef]
- Elmowafy, M.; Alruwaili, N.K.; Ahmad, N.; Kassem, A.M.; Ibrahim, M.F. Quercetin-Loaded Mesoporous Silica Nanoparticle–Based Lyophilized Tablets for Enhanced Physicochemical Features and Dissolution Rate: Formulation, Optimization, and In Vitro Evaluation. AAPS PharmSciTech 2023, 24. [Google Scholar] [CrossRef]
- Juère, E.; Favero, G.D.; Masse, F.; Marko, D.; Popat, A.; Florek, J.; Caillard, R.; Kleitz, F. Gastro-protective protein-silica nanoparticles formulation for oral drug delivery: In vitro release, cytotoxicity and mitochondrial activity. European Journal of Pharmaceutics and Biopharmaceutics 2020, 151, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, Z.; Subramaniam, S.; Khan, G.M.; Abeer, M.M.; Qu, Z.; Janjua, T.; Kumeria, T.; Batra, J.; Popat, A. Encapsulation and Controlled Release of Resveratrol Within Functionalized Mesoporous Silica Nanoparticles for Prostate Cancer Therapy. Frontiers in Bioengineering and Biotechnology 2019, 7, 482266–482266. [Google Scholar] [CrossRef]
- Estevão, B.M.; Miletto, I.; Hioka, N.; Marchese, L.; Gianotti, E. Mesoporous Silica Nanoparticles Functionalized with Amino Groups for Biomedical Applications. ChemistryOpen 2021, 10, 1251–1259. [Google Scholar] [CrossRef]
- Catechin | C15H14O6 | CID 9064 - PubChem.
- Reina, G.; Zhao, L.; Bianco, A.; Komatsu, N. Chemical Functionalization of Nanodiamonds: Opportunities and Challenges Ahead. Angewandte Chemie - International Edition 2019, 58, 17918–17929. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, A.K.; John, M.; Misra, M.; Menezes, P.L. Nanocrystalline Materials: Synthesis, Characterization, Properties, and Applications. Crystals 2021, 11, 1317–1317. [Google Scholar] [CrossRef]
- How Lipid Nanoparticles Overcome Solubility Challenges For Oral And Injectable Formulations. 27 Feb 2025. Available online: https://www.pharmaceuticalonline.com/doc/how-lipid-nanoparticles-overcome-solubility-challenges-for-oral-and-injectable-formulations-0001.
- Exploring Lipid Nanoparticles For Drug Delivery. 25 Feb 2025. Available online: https://www.pharmaceuticalonline.com/doc/exploring-lipid-nanoparticles-for-drug-delivery-0001.
- Lipid Nanoparticles Are Having A Breakout Moment. 4 Mar 2025. Available online: https://www.pharmaceuticalonline.com/doc/lipid-nanoparticles-are-having-a-breakout-moment-0001.
- Liposomes Challenges And Opportunities. 27 Feb 2025. Available online: https://www.pharmaceuticalonline.com/doc/liposomes-challenges-and-opportunities-0001.
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Advanced Pharmaceutical Bulletin 2017, 7, 3–9. [Google Scholar] [CrossRef]
- Berends, S.E.; Strik, A.S.; Löwenberg, M.; D’Haens, G.R.; Mathôt, R.A.A. Clinical Pharmacokinetic and Pharmacodynamic Considerations in the Treatment of Ulcerative Colitis. Clinical Pharmacokinetics 2018, 58, 15–37. [Google Scholar] [CrossRef]
- Nakashima, J.; Patel, P.; Preuss, C.V. Mesalamine (USAN). StatPearls 2024. [Google Scholar]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N. A brief review on solid lipid nanoparticles: part and parcel of contemporary drug delivery systems. RSC advances 2020, 10, 26777–26791. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, M.; Al-Sanea, M.M. Nanostructured lipid carriers (NLCs) as drug delivery platform: Advances in formulation and delivery strategies. Saudi Pharmaceutical Journal SPJ 2021, 29, 999–999. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.Á; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine: Nanotechnology, Biology, and Medicine 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Gurjar, R.; Chan, C.Y. S.; Curley, P.; Sharp, J.; Chiong, J.; Rannard, S.; Siccardi, M.; Owen, A. Inhibitory Effects of Commonly Used Excipients on P-Glycoprotein in Vitro. Molecular pharmaceutics 2018, 15, 4835–4842. [Google Scholar] [CrossRef]
- Mady, O.Y.; Khaled, S.; Hedaya, A.A.; Abdine, N.; Haggag, Y. Formulation and characterization of novel oral pH-sensitive electrospun nanofibers for boosting dissolution and penetration of model class IV drug. Pharmaceutical Development and Technology 2025, 30, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Sánchez-López, E.; Ettcheto, M.; López-Machado, A.; Espina, M.; Souto, E.B.; Galindo, R.; Camins, A.; García, M.L.; Turowski, P. Current advances in the development of novel polymeric nanoparticles for the treatment of neurodegenerative diseases. Nanomedicine 2020, 15, 1239–1261. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M.; et al. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25, 3731–3731. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Bae, J.; Oshi, M.A.; Kim, M.S.; Moon, H.R.; Lee, B.L.; Im, E.; Jung, Y.; Yoo, J.W. Colon-targeted delivery of cyclosporine A using dual-functional eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis. International journal of nanomedicine 2018, 13, 1225–1240. [Google Scholar] [CrossRef]
- Hassouneh, W.B.; Al-Ghazawi, M.A.; Saleh, M.I.; Najib, N. Population Pharmacokinetics of Dasatinib in Healthy Subjects. Pharmaceuticals 2024, 17, 671–671. [Google Scholar] [CrossRef]
- 70 years of EUDRAGIT - Coatings for targeted drug delivery. 8 Sep 2025. Available online: https://www.pharmaexcipients.com/news/70-years-eudragit/?utm_source=chatgpt.com.
- Center For Drug Evaluation And Research Application Number: 203389Orig1s000 PHARMACOLOGY REVIEW(S).
- Eisele, J.; Haynes, G.; Rosamilia, T. Characterisation and toxicological behaviour of Basic Methacrylate Copolymer for GRAS evaluation. Regulatory Toxicology and Pharmacology 2011, 61, 32–43. [Google Scholar] [CrossRef]
- Esporrín-Ubieto, D.; Sonzogni, A.S.; Fernández, M.; Acera, A.; Matxinandiarena, E.; Cadavid-Vargas, J.F.; Calafel, I.; Schmarsow, R.N.; Müller, A.J.; Larrañaga, A.; et al. The role of Eudragit® as a component of hydrogel formulations for medical devices. Journal of Materials Chemistry B 2023, 11, 9276–9289. [Google Scholar] [CrossRef]
- Mathes, D.; Macedo, L.B.; Pieta, T.B.; Maia, B.C.; Rolim, C.M.B.; Nogueira-Librelotto, D.R. The role of polymer type and surfactant composition on the toxicological profile of nanoparticles: an in vitro comparative study. Journal of Biomaterials Science, Polymer Edition 2025. [Google Scholar] [CrossRef]
- Ronzani, C.; Belle, C.V.; Didier, P.; Spiegelhalter, C.; Pierrat, P.; Lebeau, L.; Pons, F. Lysosome mediates toxicological effects of polyethyleneimine-based cationic carbon dots. Journal of Nanoparticle Research 2019, 21, 1–17. [Google Scholar] [CrossRef]
- Machado, F.R.; Bortolotto, V.C.; Araujo, S.M.; Dahleh, M.M.M.; Fernandes, E.J.; Musachio, E.A.S.; Funguetto-Ribeiro, A.C.; Haas, S.E.; Guerra, G.P.; Prigol, M.; et al. Toxicological analysis of chronic exposure to polymeric nanocapsules with different coatings in Drosophila melanogaster. Comparative Biochemistry and Physiology Part - C: Toxicology and Pharmacology 2024, 283. [Google Scholar] [CrossRef]
- Tehrani, S.F.; Bharadwaj, P.; Chain, J.L.; Roullin, V.G. Purification processes of polymeric nanoparticles: How to improve their clinical translation? Journal of Controlled Release 2023, 360, 591–612. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, N.K.V.M. Oral rapamycin nanoparticle preparations. 2014. [Google Scholar]
- Vaughn, N.K.V.M. Oral rapamycin nanoparticle preparations and use. 2014.
- Sirkar, Kamalesh; Singh, R.P. Dhananjay; Chen, Dengyue. System and method for continuous polymer coating of particles. 2014. [Google Scholar]
- Yu Qian, L. J. Z., Zhi Min WU, Li Ying ZHOU, Wei Jiang, Li Ling, Qian Luo, Xin Dong GUO Enteric-coated capsule containing cationic nanoparticles for oral insulin delivery. Enteric-coated capsule containing cationic nanoparticles for oral insulin delivery 2012.
- A kind of enteric solubility nano-particle of load insulin and its preparation method and application. 2018.
- Chopra, Sunandini; Wang, R.K. Amy; Farokhzad, Omid C.; Zhang, Xue-Qing. Nanoparticles with pH Triggered Drug Release. 2016. [Google Scholar]
Figure 1.
A – Transmission electron microscopy image of the developed MSNPs; B – Transmission electron microscopy image of NH2MSNPs/CHT@EUS-100) C – Magnification of transmission electron microscopy images of NH2MSNPs/CHT@EUS-100 D - Scanning electron microscopy image of the developed MSNPs; E – Scanning electron microscopy image NH2MSNPs/CHT@EUS-100; F – Magnification of a scanning electron microscopy image of NH2MSNPs/CHT@EUS-100; G – Particle size and ZP of the developed NP; CHT: Catechin; MSNPs: Mesoporous silica nanoparticles; MSNPs/CHT: catechin-loaded MNSPs; NH2-MNSPs: unloaded amino-functionalized MSNPs; NH2-MNSPs@EUS-100: NH2-MNSPs coated with ES100; NH2-MSNPs/CHT: catechin-loaded NH2-MNSPs; NH2MSNPs/CHT@EUS-100: NH2-MNSPs/CHT coated with ES100; NP: nanoparticle; ZP: Zeta potential; adapted from Kassem et al. [68].
Figure 1.
A – Transmission electron microscopy image of the developed MSNPs; B – Transmission electron microscopy image of NH2MSNPs/CHT@EUS-100) C – Magnification of transmission electron microscopy images of NH2MSNPs/CHT@EUS-100 D - Scanning electron microscopy image of the developed MSNPs; E – Scanning electron microscopy image NH2MSNPs/CHT@EUS-100; F – Magnification of a scanning electron microscopy image of NH2MSNPs/CHT@EUS-100; G – Particle size and ZP of the developed NP; CHT: Catechin; MSNPs: Mesoporous silica nanoparticles; MSNPs/CHT: catechin-loaded MNSPs; NH2-MNSPs: unloaded amino-functionalized MSNPs; NH2-MNSPs@EUS-100: NH2-MNSPs coated with ES100; NH2-MSNPs/CHT: catechin-loaded NH2-MNSPs; NH2MSNPs/CHT@EUS-100: NH2-MNSPs/CHT coated with ES100; NP: nanoparticle; ZP: Zeta potential; adapted from Kassem et al. [68].

Figure 2.
A – Schematic representation of the developed NP systems, including the intended behaviour under colonic conditions; B – Scanning electron microscopy image of the developed spherical MSNPs; C – Scanning electron microscopy image of the developed rod-shaped MSNPs; D – Scanning electron microscopy image of the developed dendritic MSNPs; E – In vitro drug release profiles of the developed spherical MSNPs; F – In vitro drug release profiles of the developed rod-shaped MSNPs; G – In vitro drug release profiles of the developed MSND; H – Transmission electron microscopy image of the residue of the developed spherical MSNPs after in vitro drug release assessment at pH 7.8; I – Transmission electron microscopy image of the residue of the developed rod-shaped MSNPs after in vitro drug release assessment at pH 7.8; J – Transmission electron microscopy image of the residue of the developed MSNDsafter in vitro drug release assessment at pH 7.8; K – In vivo pharmacokinetic study results (left – plasma; right – colon); MSND-BUD ; E@MSND-BUD - Enteric-Coated MSNDs-BUD; BUD; BUD: budenoside; E@MSND-BUD: Enteric-Coated MSNDs-BUD; MSNDs: Dendritic MSNPs; MSND-BUD: BUD-loaded MSNDs; MSNPs - mesoporous silica nanoparticles; NP – nanoparticle; ; adapted from Li et al. [69]Also for the treatment of IBD, Qu et al. [70] aimed to achieve localised, targeted delivery with minimal systemic absorption of glucocorticoids. These drugs are therapeutically used to treat IBD, but they often present challenges, such as undesirable systemic side effects due to upper GIT absorption. Furthermore, conventional pH-responsive formulations can be quickly expelled from the body, reducing treatment effectiveness. To address these issues, the authors developed pH-responsive, amino-modified MSNPs highly loaded with prednisone (Pred) and BUD, respectively. Unmodified MSNPs initially displayed a negative ZP, which shifted to positive values after functionalisation with amino groups. Subsequent coating with ES100 effectively masked the surface charge, leading to an overall negative ZP, demonstrating that ES100 was successfully deposited onto the surface of amino-functionalized MSNPs. Using the same method employed by Kassem et. al.(69), in vitro drug release results showed that both free Pred and amino-modified MSNPs containing Pred (Pred-A-MSNPs) demonstrated a rapid dissolution profile, with approximately 86% of the drug being released within the first 2 h under acidic conditions. In contrast, Eudragit-coated MSNPs (Pred-A-E-MSNPs) displayed a more controlled release, with only 18% of prednisolone being released at pH 1.9 during the initial 2 h. Then, at pH 7.4, the NPs showed a cumulative release of 78% was observed over 24 h, indicating a pH-sensitive release mechanism inherent to the formulation. For BUD, the non-encapsulated drug displayed a 56% release within the first 2 h, and reached 71% after 24 h. Its limited release rate is attributed to its poor aqueous solubility. The release profile of amino-modified MSNPs loaded with BUD (BUD-A-MSNPs) resembled that of the free drug, with an overall release of approximately 80% within 5 h. Conversely, Eudragit-coated MSNPs (BUD-A-E-MSNPs) demonstrated pH-dependent behaviour: only 12% of the drug was released at pH 1.9 during the first 2 h, followed by an additional 7% at pH 5.5. Upon elevation of the pH to 7.4, around 60% of BUD was released, reflecting a release pattern analogous to that observed with Pre-A-E-MSNPs. In vivo experiments were conducted, using a murine model of colitis induced by DSS, to investigate the therapeutic potential of BUD-loaded NPs. This study was not carried out with prednisolone, since it, according to the authors, was not able to reverse inflammation using the same model. All treatment groups, including free Bud, BUD-A-MSNPs, and BUD-A-E-MSNPs, showed improvements in Disease Activity Index (DAI) scores compared to the untreated DSS group, with the nanoparticulate formulations having the most impactful results. In the proximal colon, no significant differences in therapeutic outcomes were noted between BUD-A-MSNPs and BUD-A-E-MSNPs compared to the untreated DSS colitis group. In the distal colon, however, both groups treated with MSNPs exhibited improved drug delivery efficiency, resulting in less tissue damage compared to the untreated DSS group and the free drug treatment. The authors noted that incomplete dissociation of Eudragit, possibly due to the lower pH from intestinal inflammation, may have limited drug release. To confirm targeted delivery and its biological effects at the molecular level, quantitative reverse transcription PCR analysis was conducted on colonic tissues. The results showed that treatments with BUD-A-MSNPs and BUD-A-E-MSNPs modulated gene expression related to inflammation and significantly boosted the anti-inflammatory effects of BUD, compared with the free drug. These findings indicate that NP-based delivery systems, whether coated or uncoated, enhance drug targeting and therapeutic effectiveness in colitis models. However, the presence of Eudragit may further contribute to stability, site-specific release, and pharmacokinetic advantages [70].
Figure 2.
A – Schematic representation of the developed NP systems, including the intended behaviour under colonic conditions; B – Scanning electron microscopy image of the developed spherical MSNPs; C – Scanning electron microscopy image of the developed rod-shaped MSNPs; D – Scanning electron microscopy image of the developed dendritic MSNPs; E – In vitro drug release profiles of the developed spherical MSNPs; F – In vitro drug release profiles of the developed rod-shaped MSNPs; G – In vitro drug release profiles of the developed MSND; H – Transmission electron microscopy image of the residue of the developed spherical MSNPs after in vitro drug release assessment at pH 7.8; I – Transmission electron microscopy image of the residue of the developed rod-shaped MSNPs after in vitro drug release assessment at pH 7.8; J – Transmission electron microscopy image of the residue of the developed MSNDsafter in vitro drug release assessment at pH 7.8; K – In vivo pharmacokinetic study results (left – plasma; right – colon); MSND-BUD ; E@MSND-BUD - Enteric-Coated MSNDs-BUD; BUD; BUD: budenoside; E@MSND-BUD: Enteric-Coated MSNDs-BUD; MSNDs: Dendritic MSNPs; MSND-BUD: BUD-loaded MSNDs; MSNPs - mesoporous silica nanoparticles; NP – nanoparticle; ; adapted from Li et al. [69]Also for the treatment of IBD, Qu et al. [70] aimed to achieve localised, targeted delivery with minimal systemic absorption of glucocorticoids. These drugs are therapeutically used to treat IBD, but they often present challenges, such as undesirable systemic side effects due to upper GIT absorption. Furthermore, conventional pH-responsive formulations can be quickly expelled from the body, reducing treatment effectiveness. To address these issues, the authors developed pH-responsive, amino-modified MSNPs highly loaded with prednisone (Pred) and BUD, respectively. Unmodified MSNPs initially displayed a negative ZP, which shifted to positive values after functionalisation with amino groups. Subsequent coating with ES100 effectively masked the surface charge, leading to an overall negative ZP, demonstrating that ES100 was successfully deposited onto the surface of amino-functionalized MSNPs. Using the same method employed by Kassem et. al.(69), in vitro drug release results showed that both free Pred and amino-modified MSNPs containing Pred (Pred-A-MSNPs) demonstrated a rapid dissolution profile, with approximately 86% of the drug being released within the first 2 h under acidic conditions. In contrast, Eudragit-coated MSNPs (Pred-A-E-MSNPs) displayed a more controlled release, with only 18% of prednisolone being released at pH 1.9 during the initial 2 h. Then, at pH 7.4, the NPs showed a cumulative release of 78% was observed over 24 h, indicating a pH-sensitive release mechanism inherent to the formulation. For BUD, the non-encapsulated drug displayed a 56% release within the first 2 h, and reached 71% after 24 h. Its limited release rate is attributed to its poor aqueous solubility. The release profile of amino-modified MSNPs loaded with BUD (BUD-A-MSNPs) resembled that of the free drug, with an overall release of approximately 80% within 5 h. Conversely, Eudragit-coated MSNPs (BUD-A-E-MSNPs) demonstrated pH-dependent behaviour: only 12% of the drug was released at pH 1.9 during the first 2 h, followed by an additional 7% at pH 5.5. Upon elevation of the pH to 7.4, around 60% of BUD was released, reflecting a release pattern analogous to that observed with Pre-A-E-MSNPs. In vivo experiments were conducted, using a murine model of colitis induced by DSS, to investigate the therapeutic potential of BUD-loaded NPs. This study was not carried out with prednisolone, since it, according to the authors, was not able to reverse inflammation using the same model. All treatment groups, including free Bud, BUD-A-MSNPs, and BUD-A-E-MSNPs, showed improvements in Disease Activity Index (DAI) scores compared to the untreated DSS group, with the nanoparticulate formulations having the most impactful results. In the proximal colon, no significant differences in therapeutic outcomes were noted between BUD-A-MSNPs and BUD-A-E-MSNPs compared to the untreated DSS colitis group. In the distal colon, however, both groups treated with MSNPs exhibited improved drug delivery efficiency, resulting in less tissue damage compared to the untreated DSS group and the free drug treatment. The authors noted that incomplete dissociation of Eudragit, possibly due to the lower pH from intestinal inflammation, may have limited drug release. To confirm targeted delivery and its biological effects at the molecular level, quantitative reverse transcription PCR analysis was conducted on colonic tissues. The results showed that treatments with BUD-A-MSNPs and BUD-A-E-MSNPs modulated gene expression related to inflammation and significantly boosted the anti-inflammatory effects of BUD, compared with the free drug. These findings indicate that NP-based delivery systems, whether coated or uncoated, enhance drug targeting and therapeutic effectiveness in colitis models. However, the presence of Eudragit may further contribute to stability, site-specific release, and pharmacokinetic advantages [70].

Figure 3.
A – Schematic representation of the developed nanosystem’s composition, preparation method, and performed assays; B – In vitro drug release results from the different developed formulations (MER-MCM; MER-MCM-NH2;MER-MCM-PO3) , at different pH levels; C – In vitro drug release results from Eud-MER-MCM-NH2 NPs in universal buffer, first at gastric pH for 2 h, and then at intestinal pH for 6 h; D – In vitro drug permeation results, showing the cumulative amount of MER transported from A to B (drug absorption study); E – Apparent permeability coefficient of the developed formulations; Eud-MER-MCM-NH2: Eudragit coated MER-MCM-NH2; MCM - mesoporous silica nanoparticles MCM 41; MER: Meropenem; MER-MCM: MER loaded-MCM; MER-MCM-NH2:: MER-loaded-amino-functionalised MCM; MER-MCM-PO3: MER-loaded-phosphonate-functionalised MCM; adapted from Raza et al. [52].
Figure 3.
A – Schematic representation of the developed nanosystem’s composition, preparation method, and performed assays; B – In vitro drug release results from the different developed formulations (MER-MCM; MER-MCM-NH2;MER-MCM-PO3) , at different pH levels; C – In vitro drug release results from Eud-MER-MCM-NH2 NPs in universal buffer, first at gastric pH for 2 h, and then at intestinal pH for 6 h; D – In vitro drug permeation results, showing the cumulative amount of MER transported from A to B (drug absorption study); E – Apparent permeability coefficient of the developed formulations; Eud-MER-MCM-NH2: Eudragit coated MER-MCM-NH2; MCM - mesoporous silica nanoparticles MCM 41; MER: Meropenem; MER-MCM: MER loaded-MCM; MER-MCM-NH2:: MER-loaded-amino-functionalised MCM; MER-MCM-PO3: MER-loaded-phosphonate-functionalised MCM; adapted from Raza et al. [52].

Figure 4.
Schematic representation of Doxorubicin-loaded NDNP coated with Eudragit S100, including the intended anticancer mechanism of action. Eudragit S100 coated nanodiamond-based NPs as an oral chemo-photothermal delivery system for local treatment of colon cancer. DOX: Doxorrubicin hydrochloride; ES@PND-PEG-TPP/DOX: Eudragit coated PND-PEG-TPP/DOX; ND: nanodiamond;PEG: Polyethylene glycol; PND: ND coated with polydopamine; PND-PEG: PEG-Functionalized PND; PND-PEG-TPP: TPP-functionalized PND-PEG; PND-PEG-TPP/DOX: Doxorrubicin-loaded- PND-PEG-TPP. Developed using PowerPoint, adapted from [72].
Figure 4.
Schematic representation of Doxorubicin-loaded NDNP coated with Eudragit S100, including the intended anticancer mechanism of action. Eudragit S100 coated nanodiamond-based NPs as an oral chemo-photothermal delivery system for local treatment of colon cancer. DOX: Doxorrubicin hydrochloride; ES@PND-PEG-TPP/DOX: Eudragit coated PND-PEG-TPP/DOX; ND: nanodiamond;PEG: Polyethylene glycol; PND: ND coated with polydopamine; PND-PEG: PEG-Functionalized PND; PND-PEG-TPP: TPP-functionalized PND-PEG; PND-PEG-TPP/DOX: Doxorrubicin-loaded- PND-PEG-TPP. Developed using PowerPoint, adapted from [72].

Figure 5.
A – Zeta potential variation of the developed ES@PND-PEG-TPP/DOX through time; B – In vitro drug release profiles of the developed nanosystems; C – Animal tumor weight variation after treatment administration ; D – Animal body weight variation after treatment administration ; E – Tumor photographs 14 days after treatment; F –TUNEL and caspase-3 tumor analysis after treatment (a – saline; b – saline + laser; c – free DOX; d – PND-PEG-TPP/DOX; e – ES@PND-PEG-TPP/DOX; f – PND-PEG-TPP/DOX + NIR; g – ES@PND-PEG-TPP/DOX + NIR); DOX: Doxorrubicin hydrochloride; ES@PND-PEG-TPP/DOX: Eudragit coated PND-PEG-TPP/DOX; ND: nanodiamond; NIR – Near Infrared; PND-PEG; PND-PEG-TPP/DOX – Doxorrubicin loaded- PND-PEG-TPP adapted from Su et al. [72].
Figure 5.
A – Zeta potential variation of the developed ES@PND-PEG-TPP/DOX through time; B – In vitro drug release profiles of the developed nanosystems; C – Animal tumor weight variation after treatment administration ; D – Animal body weight variation after treatment administration ; E – Tumor photographs 14 days after treatment; F –TUNEL and caspase-3 tumor analysis after treatment (a – saline; b – saline + laser; c – free DOX; d – PND-PEG-TPP/DOX; e – ES@PND-PEG-TPP/DOX; f – PND-PEG-TPP/DOX + NIR; g – ES@PND-PEG-TPP/DOX + NIR); DOX: Doxorrubicin hydrochloride; ES@PND-PEG-TPP/DOX: Eudragit coated PND-PEG-TPP/DOX; ND: nanodiamond; NIR – Near Infrared; PND-PEG; PND-PEG-TPP/DOX – Doxorrubicin loaded- PND-PEG-TPP adapted from Su et al. [72].

Figure 6.
A – Scanning electron microscopy image of BIBF coarse crystals; B – Scanning electron microscopy image of BIBF-NC; C – Scanning electron microscopy image BIBF-NCs@L100; D – Particle size stability studies at pH 6.8; E – Particle size stability studies at pH 7.4; F – In vitro dissolution profiles of BIBF soft capsules, uncoated BIBF-NCs, BIBF-NCs@L100, and their physical mixture. BIBF: nintedanib; BIBF-NCs: BIBF nanocrystals; BIBF-NCs@L100: Eudragit-coated BIBF-NCs; PM: physical mixture. Adapted from Che et al. [74].
Figure 6.
A – Scanning electron microscopy image of BIBF coarse crystals; B – Scanning electron microscopy image of BIBF-NC; C – Scanning electron microscopy image BIBF-NCs@L100; D – Particle size stability studies at pH 6.8; E – Particle size stability studies at pH 7.4; F – In vitro dissolution profiles of BIBF soft capsules, uncoated BIBF-NCs, BIBF-NCs@L100, and their physical mixture. BIBF: nintedanib; BIBF-NCs: BIBF nanocrystals; BIBF-NCs@L100: Eudragit-coated BIBF-NCs; PM: physical mixture. Adapted from Che et al. [74].

Figure 7.
A – In vitro drug release profiles of the developed liposomal formulations (LP – Liposomes without bile salts; SG/LP-bile salt-containing LPs; ES-SG/LP- Eudragit-coated bile salt-containing LPs); B – Caco-2 cell uptake of the developed formulations, recorded by confocal laser scanning microscopy; C – Transmission electron microscopy images of the developed nanosystems; adapted from Alghurabi et al. [75].
Figure 7.
A – In vitro drug release profiles of the developed liposomal formulations (LP – Liposomes without bile salts; SG/LP-bile salt-containing LPs; ES-SG/LP- Eudragit-coated bile salt-containing LPs); B – Caco-2 cell uptake of the developed formulations, recorded by confocal laser scanning microscopy; C – Transmission electron microscopy images of the developed nanosystems; adapted from Alghurabi et al. [75].

Figure 8.
A – Schematic representation of the developed NLCs (TAC-NLCs - Tacrolimus-loaded nanostructured lipid carriers; TAC-NLCs/E FS100 - Tacrolimus-loaded, Eudragit-coated), including the performed assays; B – 3D response surface plots of particle size (top row), zeta potential (middle row), and entrapment efficiency (bottom row); C – In vitro cytotoxicity results of the developed NLCs in macrophages and colon cells; adapted from Altaf et al. [79].
Figure 8.
A – Schematic representation of the developed NLCs (TAC-NLCs - Tacrolimus-loaded nanostructured lipid carriers; TAC-NLCs/E FS100 - Tacrolimus-loaded, Eudragit-coated), including the performed assays; B – 3D response surface plots of particle size (top row), zeta potential (middle row), and entrapment efficiency (bottom row); C – In vitro cytotoxicity results of the developed NLCs in macrophages and colon cells; adapted from Altaf et al. [79].


Table 1.
Description of several commercially available Eudragit grades.
| Grade | Chemical name | pH-dependent solubility | Glass Transition Temperature (ºC) | Physical appearance | Applications | Reference |
| Eudragit E100 | Poly (butyl methacrylate, (2-dimethylaminoethyl) methacrylate, methyl methacrylate) 1:2:1 | Soluble in gastric fluid below pH 5 | 48 |
Granules | Increased bioavailability and dissolution profile; high oral bioavailability. | [10,38,39,60,61,62,63]. |
| Eudragit E PO | Powder | |||||
|
Eudragit L 100 |
Poly (methacrylic acid, methyl methacrylate) 1:1 | Soluble in intestinal fluid from pH 6 | 150 | Powder | Increased oral absorption, increased taste masking, controlled release, enteric targeted drug delivery, delayed release profile, high oral bioavailability. | |
| Eudragit L30 D-55 | Poly (methacrylic acid, ethyl acrylate) 1:1 | Soluble in intestinal fluid from pH 5.5 (resistant to gastric juice but readily dissolves at pH above 5.5) | 110 | Aqueous dispersion | Enteric coating | |
| Eudragit S100 | Poly (methacrylic acid, methyl methacrylate) 1:2 | Soluble in intestinal fluid from pH 7 | 150 | Powder | Increased oral absorption, increased taste masking, controlled release, colonic-specific drug delivery, delayed release profile, high oral bioavailability. | |
| Eudragit FS30 D | Poly (methyl acrylate, methyl methacrylate, methacrylic acid) 7:3:1 | Soluble in intestinal fluid from pH 7 | 48 | Aqueous dispersion | Increased oral absorption, increased taste masking, controlled release, colonic-specific drug delivery, delayed release profile, high oral bioavailability. | |
| Eudragit FS100 | Soluble in intestinal fluid from pH 7 | 55.6 | Granules | |||
| Eudragit RL100 | Poly (ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride) 1:2:0.2 | High permeability, insoluble (pH-independent) | 70 | Granules | Sustained release, improved permeation and increased bioavailability and shelf life. | |
| Eudragit RS100 | Poly (ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride) 1:2:0.1 | Low permeability, insoluble (pH-independent) | 64 |
Granules | Sustained release, improved permeation and increased bioavailability and shelf life. | |
| Eudragit RS PO | Powder |
Table 2.
Outline of several Eudragit-containing nanoparticulate systems.
| Nanoparticle Type | Drug | Eudragit Grade | Production Method | Physicochemical Properties | Target Site | Reference |
| Mesoporous Silica Nanoparticles | Catechin |
Eudragit S100 | Rotary evaporation method followed by polymeric coating | Size:150 nm ZP: - 40.25 mV PDI: 0.23±0.09 AE: 82% |
Colon | [68] |
| Budesonide | Eudragit S100 | CTAB-templated method followed by polymeric coating |
Size: 110 nm (spherical); 115 x 45 nm (rod-like); 100 nm (dendritic) (these values correspond to uncoated particles) ZP: -28±0.38 mV (spherical); -21.61±1.35 mV (rod-like); -22.23±1.21 mV (dendritic) EE: 44% (spherical, rod-like, dendritic) |
Colon | [69] | |
| Prednisolone and Budesonide | Eudragit S100 | Rotary evaporation method followed by polymeric coating | Size: ≈238 nm (Pred); ≈242 nm (BUD) ZP: -19.2±2.8 mV (Pred); -19.5±3.5 mV (BUD) PDI: 0.42 ± 0.11 (Pred); 0.45 ± 0.16 (BUD) LE: 95.2% (Pred); 82.0% (BUD) |
Colon | [70] | |
| Meropenem | Eudragit S100 | CTAB-templated method followed by polymeric coating | Size: 645 nm ZP: -9 mV LC: 24.1% |
Small intestine | [52] | |
| Ofloxacin and Doxorubicin | Eudragit S100 Eudragit L100 | Modified co-precipitation, silica coating and rhodamine labelling, followed by polymeric coating | HD: ≈450 nm ZP: -24.0±1.7 mV LC: 287.5 ugDox/mgNP; 274.0 ugDox/mgNP + 140 ugOflo/mgNP; 182.8 ugOflo/mgNP |
Colon | [71] | |
| Nanodiamond-Based Nanoparticles | Doxorubicin | Eudragit S100 | Polydopamine coating followed by solvent evaporation | Size: 221.4 ± 5.86 nm ZP: -28.1 ± 0.7 PDI: 0.207 LC: 12.86 ± 0.61% |
Colon | [72] |
| Nanocrystals | Ivermectin | Eudragit L100-55 | Wet milling, followed by spray drying | Size: 294 ± 4 nm PDI: 0.278 EE: 92.3 ± 1.23% (polymer concentration of 25%) |
Small intestine | [73] |
| Nintedanib | Eudragit L100 | Wet milling followed by spray drying | Size: 295.72 ± 3.04 nm ZP: -34.33 ± 0.68 mV PDI: 0.186 ± 0.054 |
Small intestine | [74] | |
| Liposomes | 5-aminosalicylic acid | Eudragit S100 | Thin-film hydration followed by probe sonication and layer-by-layer electrostatic deposition | Size: ≈ 240 nm ZP: ≈ - 26 mV PDI: ≈ 0.25 EE: ≈ 50-55% (values of ES100 coated LPs with 1mg/mL of sodium glycocholate) |
Colon | [75] |
| Budesonide | Eudragit S100 | Thin-film hydration followed by probe sonication and layer-by-layer electrostatic deposition | Size: 275 nm ZP: -38 mV PDI: 0.128 |
Colon | [76] | |
| Solid Lipid Nanoparticles | Saxagliptin | Eudragit RS100 | Modified solvent injection | Size: 212 to 442 nm ZP: - 41.09 ± 0.11 to 30.86 ± 0.63 mV PDI: 0.26 ± 0.051 to 0.45 ± 0.017 |
Small intestine | [77] |
| Oxaliplatin | Eudragit S100 | Solvent emulsification followed by pelletisation | Size: 116.81 ± 1.37 nm EE: 81.12 ± 0.26 % |
Colon | [78] | |
| Nanostructured Lipid Carriers | Tacrolimus | Eudragit FS100 | Modified microemulsion method followed by polymeric coating | Size: 198.7 nm ZP: -47.6 mV PDI: 0.176 EE: 78% |
Colon | [79] |
| 5-Fluorouracil | Eudragit S100 | High pressure homogenisation followed by polymeric coating | Size: 154 ± 3.17 nm ZP: -21.7 ± 2.02 mV PDI: 0.29 ± 0.07 EE: 89.81 ± 2.6% |
Colon | [80] | |
| Polymeric Nanoparticles | Iridoid glycoside | Eudragit S100 Eudragit L30-D 55 |
Single emulsion solvent evaporation followed by polymeric coating | Size: 247 ± 26 nm ZP: -22.4 ± 1.76 mV PDI: 0.21± 0.05 EE: 39.47 ± 2.69% |
Colon | [81] |
| Dasatinib | Eudragit L100 | Single emulsion solvent evaporation followed by polymeric coating | Size: 202.1 ± 5.7 nm ZP: -18 ± 1.01 mV EE: 93.11 ± 0.2% |
Small intestine | [82] |
AE: adsorption efficiency; BUD: Budenoside; CTAB: cetyl trimethyl ammonium bromide; EE: entrapment efficiency; HD: hydrodynamic diameter; LC: loading capacity; LE: loading efficiency; Pred: prednisolone; PDI: polydispersity index; ZP: zeta potential.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



