Submitted:
07 April 2026
Posted:
08 April 2026
You are already at the latest version
Abstract
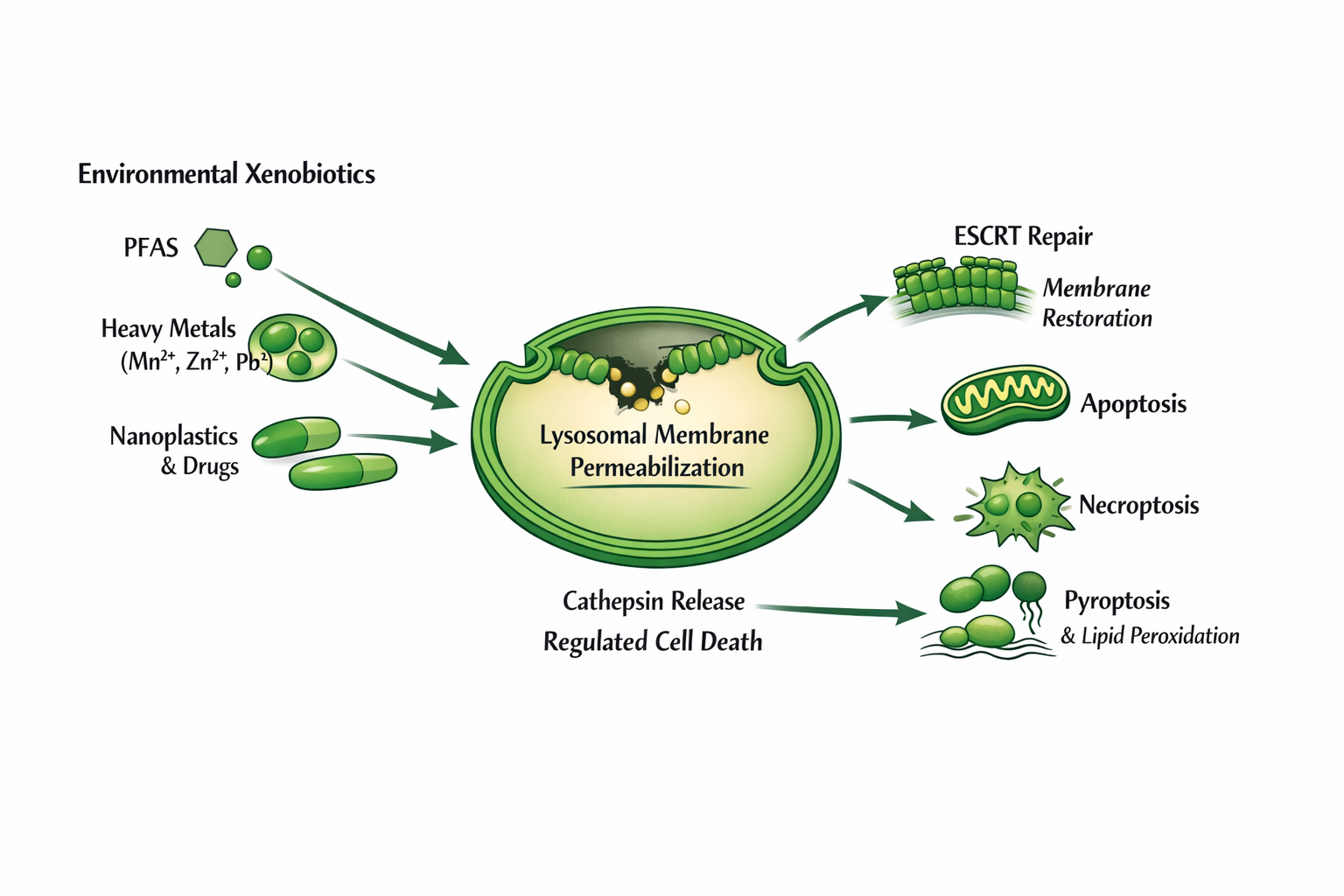
Lysosomes are acidic organelles central to cellular degradation, nutrient sensing, and regulated cell death. The stability of the lysosomal membrane is essential for cell viability, yet it is highly vulnerable to disruption by diverse environmental and endogenous stressors. This review synthesizes current understanding of the molecular mechanisms driving lysosomal membrane permeabilization (LMP) and lysosomal membrane rupture (LMR) in response to xenobiotics, including cationic amphiphilic drugs, perfluorinated alkyl substances (PFAS), and redox‑active heavy metals. We review lysosomal injury, namely intralysosomal ion trapping, metal‑catalyzed Fenton chemistry, and lipid peroxidation, which converges to generate oxidative membrane pores. We also examine cellular membrane‑repair systems, particularly the Endosomal Sorting Complex Required for Transport (ESCRT)-III machinery and ER–lysosome lipid transfer pathways, that act to restore lysosomal integrity. Failure of these protective mechanisms initiates distinct regulated cell death programs, including apoptosis, ferroptosis, and pyroptosis. Finally, we discuss the dual role of LMP in human health: both as a mediator of environmental toxicant‑induced injury and as a promising therapeutic target for overcoming multidrug resistance in cancer. By integrating findings from emerging non‑mammalian model systems and advanced imaging modalities, this review provides a unified framework for understanding lysosomal membrane dynamics under chemical stress.
Keywords:
lysosome
; lysosomal membrane permeabilization (LMP)
; xenobiotics
; Fenton chemistry
; ESCRT repair
; regulated cell death
; PFAS
; heavy metals
; cancer resistance
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



