Submitted:
03 April 2026
Posted:
06 April 2026
You are already at the latest version
Abstract
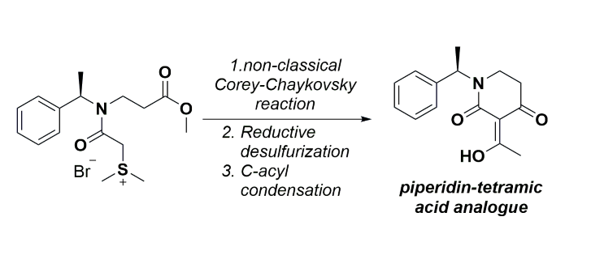
Herein, starting from (R)-(+)-α-methylbenzylamine, we report an efficient synthesis and full characterization of a new (R)-3-(1-hydroxyethylidene)-1-(1-phenylethyl)piperidine-2,4-dione, a new tetramic acid analogue. The key steps involved a non-classical Corey-Chaykovsky intramolecular cy-clization reaction to access the corresponding zwitterion, followed by a sequential desul-furization/reduction and condensation procedure. The key intermediate was obtained in 5 steps, and the desired product 7 with an overall 58% yield.
Keywords:
(R)-phenylethylamine
; non-classical Corey-Chaykovsky
; zwitterion
; 3-acyltetramic acid
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



