Submitted:
02 April 2026
Posted:
03 April 2026
You are already at the latest version
Abstract
Human neutrophil elastase (HNE) is a central mediator of neutrophil-driven inflammation. Yet, despite decades of research and drug development, therapies targeting HNE have not consistently translated into clear clinical benefits. We suggest that this translational gap partly arises from how HNE has traditionally been conceptualized, as a single enzyme to inhibit. In biological systems, however, HNE operates within a complex and tightly regulated network of proteases and inflammatory mediators. This network is spatially compartmentalized and strongly in-fluenced by local redox conditions, making HNE activity highly context dependent. From a systems perspective, HNE acts as an amplifier of inflammation. Its extracellular activity connects several pathological processes, including activation of innate immunity, extracellular matrix degradation, disruption of epithelial and endothelial barriers, and the transition toward chronic inflammation. In this review, we integrate insights from enzymology, systems biology, and clinical research to reassess the development of HNE inhibitors, ranging from endogenous anti-proteases to more recent reversible synthetic compounds. Despite their chemical and pharma-cological diversity, many of these strategies have encountered similar limitations. We therefore argue that future therapeutic approaches should move beyond the inhibition of HNE as an iso-lated target and instead aim to modulate the broader protease network, with particular attention to drug–target kinetics and precise delivery to disease-relevant microenvironments.
Keywords:
human neutrophil elastase
; HNE inhibitors
; neutrophilic inflammation
; protease–antiprotease imbalance
; ARDS
; COPD
; drug discovery
1. Introduction
Inflammation represents an essential host defense program triggered by infection or tissue injury and orchestrated through coordinated interactions among innate and adaptive immune cells, soluble mediators such as cytokines and proteases, and local tissue responses. In a balanced setting, the inflammatory cascade subsides once the initiating stimulus has been neutralized, allowing restoration of tissue integrity and functional homeostasis. However, when self-amplifying pathways predominate, particularly those driven by activated neutrophils and their proteolytic machinery, the response can escalate into a dysregulated state described as a “protein storm” [1]. This phenomenon parallels the concept of a cytokine storm and involves uncontrolled protease activity, oxidative mediators, extracellular matrix degradation, and secondary waves of inflammatory signaling [1].
Among the molecules central to this process, human neutrophil elastase (HNE) occupies a pivotal position in both antimicrobial defense and tissue remodeling. During normal immune function, neutrophils confine HNE within phagolysosomal compartments to digest engulfed pathogens and modulate extracellular matrix components. In pathological contexts, however, excessive or misdirected extracellular release of the enzyme becomes harmful. Once outside the cell, HNE degrades structural proteins including elastin, collagen, and basement membrane constituents, contributing directly to tissue injury (2). In addition, it compromises epithelial and endothelial barrier integrity and activates pro-inflammatory signaling pathways, thereby reinforcing inflammatory amplification and promoting chronic damage [2]. These observations have positioned HNE as an attractive therapeutic target in diseases characterized by persistent neutrophilic inflammation, including chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory distress syndrome (ARDS), and certain autoimmune or neutrophilic dermatoses [3].The rationale for inhibiting HNE originated from early investigations into α1-antitrypsin (AAT) deficiency, in which unopposed elastase activity drives progressive pulmonary emphysema [4]. These findings established the protease–antiprotease equilibrium as a critical determinant of lung tissue preservation. Initial therapeutic strategies therefore focused on augmenting endogenous inhibitors, including AAT replacement and the administration of naturally occurring antiproteases such as secretory leukoprotease inhibitor (SLPI) and elafin [5]. More broadly, tissue stability depends on a tightly regulated balance between neutrophil-derived proteases and their endogenous antagonists, enabling effective pathogen clearance and controlled matrix remodeling without excessive collateral injury. Disruption of this equilibrium, whether through genetic deficiency, exaggerated neutrophil activation, or oxidative inactivation of antiproteases, permits unchecked HNE activity, which in turn sustains inflammation, accelerates tissue destruction, and drives disease progression [6]. Despite decades of investigation, the development of clinically successful natural HNE inhibitors has proven difficult. Achieving sufficient selectivity, metabolic stability, and an appropriate degree of inhibition remains challenging, as many compounds also affect related serine proteases, producing unintended immunological consequences [7,8]. Pharmacokinetic barriers further complicate translation, particularly the need to deliver adequate drug concentrations to inflamed pulmonary tissues while avoiding rapid degradation. Moreover, excessive suppression of elastase activity carries the risk of impairing host defense and tissue repair mechanisms. To overcome these limitations, efforts shifted toward synthetic inhibitors beginning in the late 1980s and early 1990s, resulting in several families of potent and selective molecules [9]. Early pharmacological studies in experimental lung injury models, reported by Diane Amy Trainor, suggested that such compounds possessed the characteristics necessary to rigorously evaluate the protease–antiprotease hypothesis in clinical settings [9]. Over the ensuing decades, global research initiatives have continued to pursue synthetic HNE inhibitors capable of preventing elastase-mediated tissue destruction while maintaining acceptable safety profiles, with the aim of improving outcomes in neutrophil-driven inflammatory diseases. Multiple authoritative reviews have catalogued inhibitor chemotypes, structural determinants, and mechanisms of enzyme interaction, providing important insights into structure–activity relationships and early discovery strategies [10,11,12]. Rather than reiterating these classifications, the present review adopts a complementary perspective centred on inflammatory system dysregulation, kinetic behavior, and clinical applicability. We focus specifically on reversible synthetic inhibitors, examining concepts such as enzyme residence time, spatially protected HNE pools, and the constraints of endogenous antiprotease control during intense neutrophilic responses. By integrating pathophysiological frameworks, including protease-dominated inflammatory amplification, compartmentalized enzyme activity, and disease-specific therapeutic demands, this review aims to clarify why many highly potent inhibitors have failed to translate clinically and to identify design principles that distinguish modern drug-like candidates from earlier generations. In this context, HNE inhibition is considered not merely as enzymatic blockade, but as a strategy to modulate protease-driven inflammatory escalation in both pulmonary and systemic disorders.
2. Description of the Work and Literature Search Strategy
This review is organized to provide a mechanistically oriented analysis of human neutrophil elastase (HNE) inhibition, beginning with the biological role of HNE in inflammatory diseases and the limitations of endogenous inhibitory mechanisms, followed by an overview of natural inhibitors and a comprehensive evaluation of synthetic compounds. Particular emphasis is placed on structure–activity relationships, pharmacological and kinetic properties, and clinical translatability. The final sections address current challenges, emerging strategies, and future therapeutic perspectives.
A literature search was conducted using PubMed/MEDLINE, ScienceDirect, Wiley Online Library and Nature Publishing Group to identify relevant publications from the 1990s to 2025. Search terms included combinations of “neutrophil elastase inhibitors,” “serine protease inhibitors,” “protease–antiprotease balance,” “sivelestat,” “alvelestat,” and some disease-related terms such as “ARDS” and “COPD”. Reference lists of selected articles were also screened to identify additional relevant studies. The search yielded approximately 600 records, of which 420 were screened by abstract after removal of duplicates. Full texts were evaluated for 200 articles, and approximately 120 publications were analyzed based on relevance and scientific quality. Inclusion criteria comprised original research, clinical studies, and authoritative reviews addressing HNE biology, inhibition, or therapeutic targeting. Studies not directly related to HNE, lacking sufficient methodological detail, duplicate reports, or non-English publications were excluded. As this is a narrative review, final study selection was guided by relevance to mechanistic and translational themes.
3. Neutrophils, Elastase and the “Protein Storm”
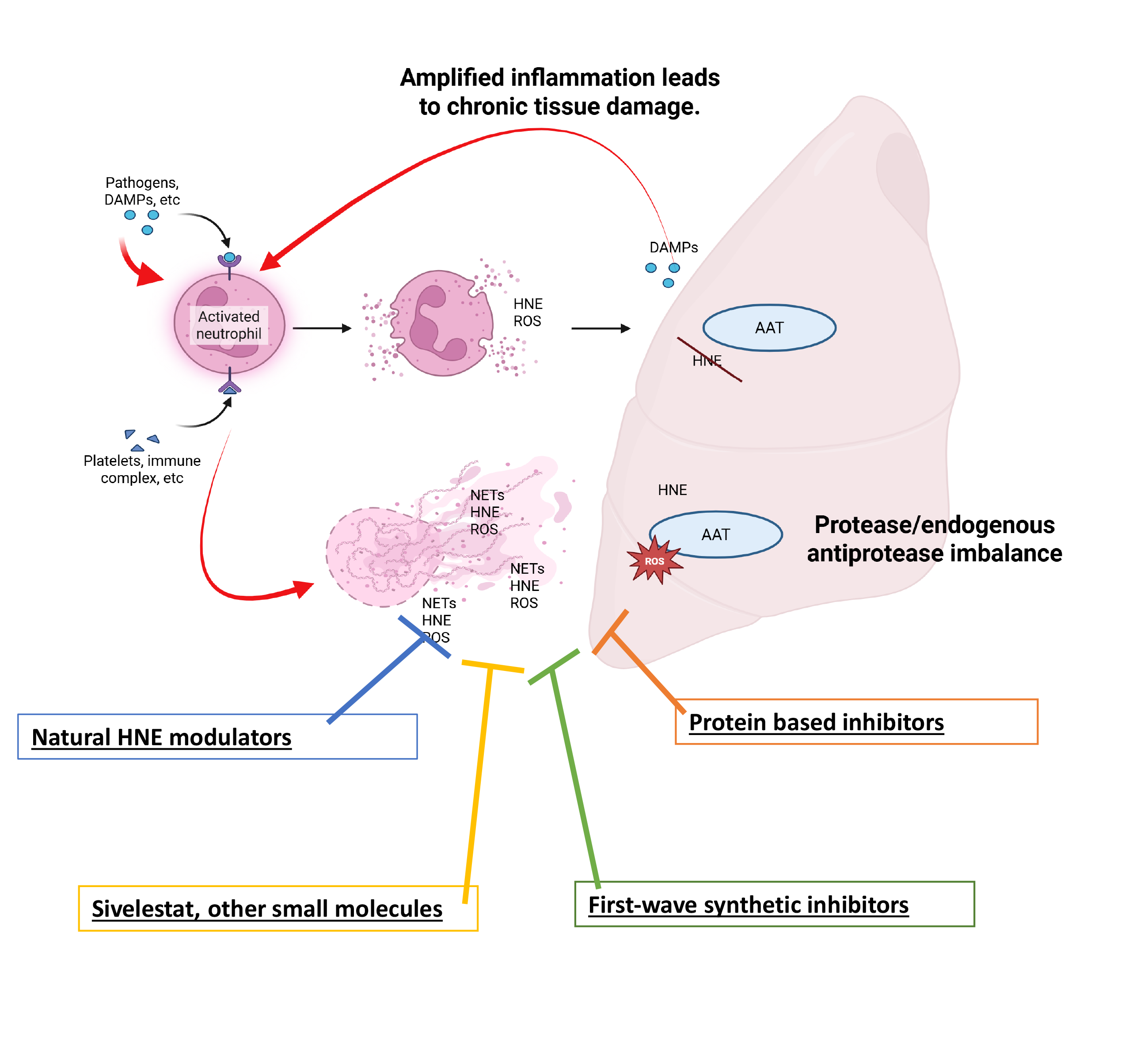
Building on the central role of neutrophil-derived proteases in inflammatory amplification, it is important to consider how neutrophil effector mechanisms contribute to both host protection and tissue injury. Neutrophils constitute a rapid-response component of innate immunity, migrating efficiently to sites of infection or tissue damage. Upon activation, neutrophils release granule-derived enzymes and generate reactive oxygen species (ROS). Under specific stimuli, they can also undergo NETosis, a distinct form of cell activation leading to the release of neutrophil extracellular traps (NETs), web-like structures composed of DNA, histones, and antimicrobial proteins that help immobilize and neutralize invading pathogens. While these responses are essential for host defense, their excessive or dysregulated activation contributes to a highly proteolytic and pro-inflammatory microenvironment. While these responses are essential for pathogen clearance, persistent or exaggerated neutrophil activation can promote collateral tissue injury and inflammatory pathology across a broad spectrum of disorders, including cardiovascular, autoimmune, and malignant diseases. In this context, sustained protease activity becomes a key driver of structural damage and inflammatory propagation. HNE exhibits broad substrate specificity, enabling degradation of extracellular matrix proteins such as elastin, collagen, fibronectin, and proteoglycans, as well as cleavage of cell-surface receptors and activation of latent cytokines or growth factors (2). When regulatory mechanisms fail, excessive elastase activity promotes progressive disruption of tissue architecture and host defence pathways, contributing to airway obstruction and structural destruction in chronic inflammatory lung diseases [13,14]. Importantly, the consequences of HNE activity extend beyond direct proteolysis. Matrix degradation products, including elastin fragments, collagen peptides, and basement membrane components, can function as damage-associated molecular patterns (DAMPs), stimulating leukocyte recruitment, macrophage activation, and downstream cytokine production. Concurrent oxidative stress further modifies proteins through lipid peroxidation–derived aldehydes such as 4-hydroxy-2-nonenal (4-HNE), which forms covalent adducts with nucleophilic amino acid residues and contributes to cellular dysfunction, metabolic disturbance, and chronic inflammation [15]. Although elastase can participate in the clearance of damaged proteins, this capacity is frequently insufficient during sustained inflammation, allowing accumulation of modified macromolecules and further tissue injury. Through these interconnected mechanisms, HNE links neutrophil activation to proteolytic damage, DAMP generation, and propagation of chronic inflammatory responses. Clinically, elevated extracellular elastase activity is a defining feature of several pulmonary disorders, including COPD, CF, and bronchiectasis, where matrix degradation promotes mucus hypersecretion and impairs airway clearance [16,17,18]. Increased HNE activity also correlates with worse outcomes in ARDS, contributing to barrier disruption, edema formation, and secondary injury [13]. Beyond the respiratory system, elastase released within inflamed joints degrades cartilage components and facilitates tissue invasion in rheumatoid arthritis [19], while in dermatological conditions such as psoriasis it promotes keratinocyte proliferation through epidermal growth factor receptor (EGFR) signaling and can delay wound repair by altering matrix remodeling processes [20]. Emerging evidence further suggests a role in tumor progression and metastasis, likely mediated by extracellular matrix degradation and generation of bioactive fragments [21]. The sequence of events underlying the protein storm is reported in Figure 1.
4. Factors Contributing to Inadequate Inhibition of HNE by Endogenous Inhibitors
There are several reasons why endogenous inhibitors of HNE may fail to adequately control its activity. Beyond the excessive local release of HNE during intense inflammatory responses, endogenous inhibition is often compromised by poor spatial colocalization with active enzyme [22,23]. HNE can bind to cell membranes, extracellular matrix components, and bacterial surfaces, and when it is membrane-associated or matrix-bound, it may be sterically shielded from soluble inhibitors such as AAT. Although SLPI and elafin are locally expressed, they may not access all microenvironments in which HNE is active. Consequently, elastolytic activity can persist despite apparently sufficient systemic levels of endogenous inhibitors. Furthermore, ROS generated by neutrophils and macrophages at inflammatory sites can chemically modify endogenous inhibitors. AAT in fact is particularly susceptible to oxidative damage, especially at methionine residues within its reactive centre loop (RCL), which markedly reduces its affinity for HNE [24]. Similarly, SLPI and elafin are vulnerable to both oxidative modification and proteolytic degradation. Thus, inflammation simultaneously promotes HNE release while impairing the function of its natural inhibitors. In addition, endogenous inhibitors are themselves substrates for proteolytic enzymes. HNE and other proteases, including cathepsins and metalloproteinases, can cleave SLPI and elafin, rendering them inactive. This loss of inhibitory capacity establishes a self-amplifying cycle in which elastase activity further diminishes its own regulation. The efficacy of endogenous inhibition is also limited by restricted tissue penetration. Being AAT a relatively large protein, it penetrates poorly into thick mucus, necrotic tissue, or biofilm-rich environments and is therefore inefficient at reaching intracellular or pericellular sites where HNE remains active [25,26]. This limitation is particularly evident in diseases such as cystic fibrosis and chronic bronchitis, where viscous secretions hinder inhibitor diffusion. Finally, in chronic inflammatory conditions, endogenous inhibitors are subjected to sustained demand. Persistent neutrophil recruitment maintains continuous elastase release, while inhibitor synthesis cannot indefinitely compensate for this burden. As a result, prolonged exposure to unregulated HNE activity leads to cumulative tissue damage despite the presence of regulatory mechanisms, explaining why endogenous inhibition is especially ineffective in chronic inflammatory disorders. In summary, insufficient control of HNE activity results in degradation of elastin and collagen, disruption of epithelial and endothelial barriers, amplification of inflammatory signaling, and impaired tissue repair and remodeling. These processes directly contribute to disease progression in pulmonary, cardiovascular, and systemic inflammatory conditions.
5. Why Nature-Inspired Inhibition Has Not Solved the HNE Problem?
Although a broad array of naturally occurring substances has been reported to attenuate HNE activity, their development trajectory reveals a recurring pattern: biochemical promise rarely translates into durable clinical benefit. Rather than a lack of potency, the central issue appears to be contextual performance, how these molecules behave within the dynamic, protease-rich, and oxidatively stressed environments in which HNE drives pathology. Natural HNE modulators span chemically and biologically diverse space, from small secondary metabolites (terpenoids, tannins, flavonoids, and other polyphenols) to macromolecular inhibitors such as proteins and peptides isolated from plants, invertebrates, and microorganisms [12]. Many demonstrate measurable in vitro inhibition and are often characterized by low intrinsic cytotoxicity and acceptable aqueous solubility. Yet these favorable attributes do not necessarily confer therapeutic viability. Rapid metabolic turnover limited epithelial permeability, instability in inflamed tissues, and inefficient accumulation at sites of neutrophil degranulation collectively restrict effective target engagement in vivo. In this regard, the disconnect between enzyme inhibition under controlled assay conditions and functional suppression of tissue-destructive elastolysis in patients remains substantial. Plant-derived phenolics illustrate this dichotomy particularly well. Their anti-elastase activity frequently coexists with antioxidant and immunomodulatory effects, complicating mechanistic attribution and dose optimization [27,28,29,30,31,32]. Rather than acting as dedicated elastase antagonists, many function as multi-target redox-active agents whose indirect modulation of inflammatory signaling may overshadow direct catalytic inhibition. Consequently, therapeutic interpretation becomes confounded: reductions in inflammatory markers do not necessarily equate to sustained control of elastase-mediated matrix degradation. More structurally complex biological matrices, such as insect-derived extracts or venom peptides, highlight the evolutionary ingenuity of elastase regulation in nature [33,34,35]. However, their structural complexity, immunogenic potential, manufacturing challenges, and pharmacokinetic unpredictability limit scalability and regulatory feasibility. These agents often serve better as mechanistic templates than as drug candidates in their native form. Protein-based inhibitors represent a more clinically established paradigm, exemplified by augmentation strategies using AAT. Inhaled formulations of recombinant or plasma-derived AAT aim to restore the protease–antiprotease equilibrium within the lung [36,37]. Nevertheless, practical constraints, including high dosing requirements, prolonged inhalation times, variable pulmonary deposition, and susceptibility to oxidative inactivation, restrict their efficiency [38]. Moreover, the broader clinical impact of systemic AAT supplementation remains debated, as improvements in biochemical endpoints do not always translate into consistent long-term disease modification [39,40,41]. Beyond molecule-specific challenges, translational barriers are amplified by trial design limitations [42]. Reliable surrogate biomarkers for elastase activity in vivo are lacking, necessitating prolonged and resource-intensive clinical studies to detect structural or functional endpoints. This slows iterative optimization and obscures early signals of therapeutic efficacy. In addition, HNE operates within microenvironments such as neutrophil extracellular traps, membrane-bound compartments, and oxidatively modified matrices, contexts that many natural inhibitors were never evolved to access or withstand. Taken together, the experience with naturally derived HNE inhibitors suggests that future progress will depend less on identifying new sources of biochemical inhibition and more on engineering context-resilient modulators. Ideal next-generation agents must retain activity under oxidative stress, penetrate relevant tissue compartments, selectively target active enzyme pools, and achieve controllable pharmacokinetics. A critical reassessment of the limitations inherent to natural scaffolds therefore provides not merely a historical overview, but a strategic blueprint for rational design of more durable elastase-directed therapies. Key distinctions among endogenous, natural and synthetic HNE inhibitors are summarized in Table 1.
6. Synthetic Inhibitors of HNE as Drug Candidates: Definition and General Concepts
In contrast to naturally derived peptides and proteins, synthetic inhibitors of HNE are typically rationally designed small molecules encompassing a wide range of chemical scaffolds [43,44]. Their reduced molecular size and tunable structural features enable efficient access to the catalytic pocket of HNE and direct suppression of its proteolytic activity. Compared with large protein-based inhibitors, small-molecule HNE inhibitors generally exhibit more favorable pharmacokinetic and pharmaceutical properties, including improved bioavailability, lower immunogenicity, and reduced risk of systemic toxicity. However, in the context of elastase-targeted drug discovery, maximal inhibitory potency alone is insufficient to ensure therapeutic success. The defining requirement for synthetic HNE inhibitors is a high degree of selectivity for HNE over other members of the serine protease family [43,44,45]. This challenge is particularly acute given the close structural and functional similarity between HNE and related proteases such as trypsin, chymotrypsin, proteinase 3, cathepsin G, and several key enzymes of the coagulation cascade [45,46]. Even modest cross-inhibition can disrupt essential physiological processes and severely limit clinical utility [47]. Importantly, HNE displays a relatively restricted expression pattern, being predominantly localized to neutrophils and released at sites of inflammation. Selective inhibition therefore offers the opportunity to attenuate pathological proteolysis while preserving host defense mechanisms and normal tissue homeostasis. Conversely, insufficient selectivity can lead to profound off-target effects, including bleeding complications, immune dysregulation, and impaired wound healing, outcomes that have contributed to the clinical failure of multiple elastase inhibitors.
7. First-Wave Synthetic Strategies Targeting HNE
The earliest attempts to attenuate excessive activity of HNE centred on small, synthetically tractable molecules engineered to interact directly with the catalytic machinery of the enzyme. An influential survey by Edwards and Bernstein over thirty years ago [48], catalogued the diverse chemotypes pursued during this formative period, reflecting the exploratory nature of inhibitor design at the time. Both peptide-derived and non-peptidic scaffolds were investigated, including chloromethyl ketones, peptidyl aldehydes and ketones, as well as a range of heterocyclic and non-heterocyclic small molecules. Among these, peptide-based transition-state mimics and short oligopeptides demonstrated particularly high potency, frequently achieving nanomolar inhibition and validating structure-activity relationship-driven optimization as a viable strategy. Building on these findings, subsequent medicinal chemistry efforts focused on enhancing selectivity and improving drug-like characteristics. For example, incorporation of α-ketobenzoxazole motifs into peptidyl backbones was explored as a means to increase metabolic resilience and oral exposure [49], while Cregge and colleagues introduced pentafluoroethyl ketone functionalities that conferred measurable oral efficacy [50]. Crystallographic analyses of optimized leads confirmed productive orientation within the HNE active site and stable enzyme–inhibitor complex formation. Additional peptide-oriented designs included carboxylate-containing transition-state analogues described by Sato and co-workers [51], where the strategic inclusion of a valyl residue was instrumental in balancing inhibitory potency with reduced toxicity. Concurrently, several non-peptidic series were advanced. Imaki and collaborators reported pivaloyloxybenzene-based compounds with notable selectivity, and certain sulfonanilide derivatives demonstrated intravenous activity in preclinical models [52].
Separately, Ohmoto’s group developed desamino 5-amino-2-phenylpyrimidin-6-one derivatives equipped with an α-keto-1,3,4-oxadiazole electrophile capable of covalently engaging the catalytic Ser195 residue [53]. Despite encouraging biochemical potency and mechanistic validation, most early synthetic inhibitors did not advance beyond preclinical stages. Limitations commonly arose from inadequate discrimination against related serine proteases, unfavorable pharmacokinetic behavior, including limited oral bioavailability and rapid systemic clearance, and safety concerns linked to highly reactive warheads. Collectively, these obstacles curtailed clinical translation and highlighted the necessity for more refined design principles in subsequent generations of HNE inhibitors.
8. Emergence of Modern Synthetic HNE Inhibitors
Recognition of the limitations inherent to early HNE inhibitors catalyzed a shift toward more sophisticated design strategies, prioritizing clinical translatability alongside enzymatic potency. Rather than focusing exclusively on maximal inhibition, modern approaches emphasized selectivity, improved pharmacokinetic behavior, and reduced toxicity. This transition was enabled by advances in high-resolution HNE crystal structures, which facilitated structure-based drug design and more precise optimization of active-site interactions [54,55,56]. Within this evolving landscape, sivelestat represents a pivotal example of a rationally designed HNE inhibitor [55,56]. Although developed earlier than many contemporary agents, it embodies key principles, such as balanced potency and improved drug-like properties, that continue to inform current small-molecule inhibitor development. As such, sivelestat occupies an intermediate position between early empirical inhibitors and later generations of elastase-targeted therapeutics, serving as a conceptual bridge toward modern HNE inhibitor design.
A scheme showing the evolution of HNE inhibitors is shown in Figure 2.
9. Sivelestat and the Protease Inhibition Paradox: Success in Chronic Disease, Struggle in Acute Critical Care
Sivelestat is the first clinically approved inhibitor of HNE [55,56] and represents a pivotal case study in protease-targeted therapy. Its development highlights a broader paradox: while protease inhibition has transformed outcomes in chronic viral diseases such as HIV and hepatitis C, translation in acute critical care syndromes like ARDS has been inconsistent. Despite strong mechanistic rationale and encouraging early results, sivelestat achieved only regional approval and limited global adoption. This divergence reflects a fundamental challenge in ARDS therapeutics, the disconnect between molecular target engagement and meaningful clinical benefit in biologically heterogeneous syndromes [57,58,59,60,61].
9.1. Protease Inhibition in Chronic Diseases vs. Acute Critical Care
Chronic viral infections rely on essential, non-redundant proteases with stable expression and clearly defined causal roles [62,63]. Sustained inhibition directly disrupts replication, and therapeutic effects are predictable. Similarly, in some chronic inflammatory disorders, protease activity acts as a persistent and central disease driver, allowing prolonged dosing to maintain effective suppression. In contrast, ARDS is a rapidly evolving syndrome characterized by network-level inflammation, interpatient heterogeneity, and functional redundancy among proteases such as cathepsins, matrix metalloproteinases, and proteinase 3 [64,65,66]. Within this adaptive system, elastase contributes to tissue injury but is not universally dominant. Compensatory pathways may blunt the impact of selective inhibition, and the therapeutic window is narrow and highly time-dependent. Consequently, although sivelestat effectively inhibited HNE, modulation of a single enzymatic node was often insufficient to alter the broader inflammatory trajectory.
9.2. The Temporal and Stratification Mismatch
In chronic disease, protease inhibitors are typically introduced before irreversible tissue damage occurs. In ARDS, treatment often begins after substantial alveolar injury is established [67,68]. Elastase inhibition may therefore limit further damage without reversing structural destruction. Sivelestat’s short half-life and need for continuous infusion further illustrate the challenge of aligning pharmacokinetics with the brief period of protease-driven injury. Patient selection represents an additional constraint. Chronic therapies target individuals with confirmed molecular pathology, whereas ARDS trials have relied primarily on clinical criteria rather than biomarkers of neutrophil-predominant inflammation [68,69,70,71]. The absence of stratification likely diluted potential benefits in elastase-dominant subgroups.
9.3. Reinterpreting the Sivelestat Experience
Rather than reflecting simple inefficacy, sivelestat illustrates structural barriers to protease targeting in acute critical illness: biological heterogeneity, protease redundancy, narrow therapeutic windows, and limited biomarker guidance. Its trajectory underscores the need for early intervention, endotype stratification, and potentially combination approaches addressing parallel inflammatory pathways. Sivelestat therefore represents a proof of pharmacological feasibility coupled with a translational lesson: selective HNE inhibition is achievable, but clinical success depends on precise alignment between drug properties, disease phase, and patient biology.
10. From First-Generation Lessons to Next-Generation Design
The experience with sivelestat reframed elastase inhibitor development. The challenge was not insufficient potency, but insufficient contextual fit between inhibitor design and disease biology. Early compounds exposed key constraints, including pharmacokinetic limitations, incomplete tissue exposure, protease-network redundancy, and lack of biomarker-guided deployment. These insights shifted the field from a potency-driven model toward an integrated strategy incorporating drug kinetics, delivery, and disease stratification. Importantly, they did not invalidate the therapeutic rationale for HNE targeting; rather, they clarified the requirements for achieving clinical impact. Subsequent medicinal chemistry efforts therefore focused on improving stability, reversibility, selectivity, and suitability for chronic administration. The emergence of second-generation small molecules reflects this conceptual evolution, from proof-of-concept inhibitors to structurally refined agents designed for sustained and context-appropriate modulation of HNE activity.
11. Overcoming First-Generation Limitations: Design Drivers for Next-Generation HNE Inhibitors
Development of next-generation HNE inhibitors was guided by limitations identified with early compounds. First-generation agents demonstrated in vivo feasibility but revealed shortcomings in metabolic stability, selectivity over related serine proteases, route of administration, and suitability for prolonged therapy. Second-generation strategies prioritized:(i) replacement of reactive or hydrolytically labile groups with stable scaffolds;(ii) optimization of reversible, high-affinity non-covalent interactions;(iii) pharmacokinetic profiles compatible with sustained exposure; and (iv) improved selectivity to reduce off-target protease inhibition. This transition marked a shift from empirically derived inhibitors toward rationally engineered molecules balancing potency with drug-like properties. In this continuum, compounds such as sivelestat and alvelestat represent developmental milestones that informed later optimization. Their clinical and pharmacological experience established design principles enabling the emergence of highly refined reversible inhibitors with improved selectivity and translational potential.
12. Small-Molecule Optimization Trajectories: From Transitional Scaffolds to Clinically Advanced Candidate
12.1. Transitional Chemical Space and Early Rational Optimization
Sivelestat and alvelestat represent a pivotal transitional phase in the evolution of synthetic HNE inhibitors, bridging early reactive inhibitors and later structure-guided small molecules. Their design marked a movement away from broadly reactive serine-targeting warheads toward inhibitors engineered to exploit the physicochemical architecture of the HNE catalytic pocket through carefully tuned non-covalent interactions. Both compounds incorporate sulfonyl-containing motifs and polar substituents capable of stabilizing interactions within the S1 pocket and adjacent subsites, enabling reversible yet high-affinity binding. This strategy improved selectivity compared with earlier peptidic or covalent inhibitors while maintaining sufficient potency for in vivo evaluation. Importantly, these molecules demonstrated that effective elastase inhibition could be achieved without permanent enzyme inactivation, establishing reversibility as a viable therapeutic paradigm. From a medicinal chemistry perspective, these transitional scaffolds also highlighted the importance of balancing enzymatic potency with pharmacokinetic and safety considerations. Although their inhibitory strength remained modest compared with later agents, their chemical frameworks provided a foundation for subsequent optimization by identifying key interaction motifs and structural features necessary for selective HNE engagement. Clinically, sivelestat achieved regulatory approval in limited indications, validating HNE as a tractable therapeutic target, while alvelestat advanced as an orally available alternative with improved pharmacokinetic characteristics suitable for chronic respiratory diseases. However, both compounds also underscored the complexities of translating biochemical inhibition into clinical benefit, emphasizing the need for further refinement.
12.2. Second-Generation Refinement: The Case of Alvelestat
The development of alvelestat (AZD9668) reflects a deliberate evolution in elastase inhibitor design based on lessons learned from earlier agents [72]. Rather than refining the same reactive pharmacology, alvelestat represents a strategic pivot toward chemical stability, reversibility, and suitability for chronic administration. Conceptually, it can be viewed as an attempt to reconcile potent neutrophil elastase inhibition with the pharmacological demands of long-term inflammatory disease management. Its scaffold departs from ester-based, hydrolytically labile inhibitors and adopts a chemically robust, non-peptidic architecture that enhances metabolic stability while preserving high affinity for the elastase active site. Multiple heteroaromatic elements create a rigid molecular framework supporting defined interactions within the catalytic pocket. Structural features such as the trifluoromethyl substituent contribute to pharmacokinetic stability, while the methylsulfonyl group promotes precise engagement with catalytic and adjacent residues. Collectively, these modifications represent a move toward structural durability and optimized drug-like properties. Mechanistically, alvelestat acts as a fully reversible competitive inhibitor that binds non-covalently within the catalytic pocket containing Ser195, preventing substrate access without forming a covalent intermediate. Kinetic analyses indicate a slow-binding mechanism characterized by rapid formation of an initial enzyme–inhibitor complex followed by conformational stabilization into a more tightly bound state, resulting in prolonged residence time while maintaining reversibility [73]. Enzymatic activity is restored upon dilution or inhibitor removal, confirming the absence of permanent inactivation. Alvelestat advanced into phase II clinical trials for chronic respiratory conditions, including COPD, bronchiectasis, and AATD [74,75]. Although improvements in lung function endpoints were not consistently demonstrated, the compound exemplifies modern HNE inhibitor design, combining selectivity, oral bioavailability, and suitability for prolonged administration.
12.3. High-Affinity Optimization and Clinical Advancement: BAY 85-8501
Further refinement of non-covalent inhibitor design led to the development of BAY 85-8501, a highly potent and selective reversible inhibitor intended for inflammatory lung diseases [76]. Preclinical studies demonstrated picomolar inhibitory potency together with strong selectivity over related serine proteases, indicating highly efficient target engagement compared with earlier molecules that typically exhibited nanomolar activity. The compound also displayed pharmacokinetic properties compatible with oral administration, reinforcing the feasibility of systemic delivery for elastase inhibition. In clinical evaluation involving patients with non-cystic fibrosis bronchiectasis, oral administration over several weeks was generally well tolerated, with mostly mild to moderate adverse events [77]. Despite its favorable biochemical profile, however, clinical studies did not demonstrate significant improvements in key functional outcomes over short treatment durations [77]. These findings highlighted an important translational challenge: achieving high enzymatic inhibition does not necessarily translate into measurable clinical benefit, particularly in complex inflammatory diseases where drug exposure at the site of pathology, disease heterogeneity, and compensatory biological mechanisms may influence therapeutic response.
13. Translational Lessons and Pharmacological Differentiation Across Small-Molecule HNE Inhibitors
Comparative evaluation of these small-molecule inhibitors reveals several recurring themes that extend beyond individual compounds. First, the relationship between systemic pharmacokinetics and local airway exposure appears critical. Oral administration may not consistently achieve sufficient concentrations within airway secretions to neutralize extracellular elastase activity, particularly in diseases characterized by thick mucus and impaired drug penetration. Second, selective targeting of HNE alone may not fully address the broader protease-antiprotease imbalance present in chronic inflammatory lung disorders. Endogenous antiproteases and multiple proteolytic enzymes contribute to tissue injury, suggesting that narrow inhibition of a single protease may yield limited clinical impact unless combined with additional therapeutic strategies. Third, clinical endpoints such as lung function or symptom scores may require longer treatment durations or earlier intervention stages to demonstrate benefit. These considerations collectively emphasize that biochemical potency, while essential, is insufficient on its own to ensure clinical success. The experience with transitional and advanced small-molecule inhibitors therefore provided critical insights into pharmacodynamic requirements, delivery challenges, and disease-specific factors that must be addressed in future therapeutic development.
14. Scaffold Diversification and Medicinal Chemistry Expansion in HNE Inhibitor Development
Recognition of the limitations associated with early inhibitors stimulated extensive medicinal chemistry efforts aimed at scaffold diversification and optimization. Derivative programs originating from sivelestat-related chemistry explored modifications intended to improve metabolic stability, pharmacokinetic behavior, and suitability for chronic administration. One representative outcome of these efforts was the identification of freselestat (ONO-6818), which, although emerging from the same developmental lineage, incorporated a structurally distinct core architecture. This compound demonstrated oral activity together with high affinity and selectivity for HNE in preclinical studies, illustrating the potential of scaffold redesign to enhance pharmacological properties [78,79]. However, clinical development was discontinued because of safety and tolerability concerns observed during longer-term administration, including elevations in liver function parameters. These findings underscored the importance of balancing potency with systemic safety, particularly for chronic indications requiring sustained exposure. Additional analogues derived from modifications of related scaffolds, including peptidic variants designed to improve metabolic stability and potency, have also been investigated. Although none has yet achieved clinical success beyond earlier inhibitors, these programs contributed substantially to understanding structure–activity relationships and identified key determinants governing selectivity, stability, and pharmacokinetic behavior [80,81].
15. Beyond Small Molecules: Peptide-Based and Biologically Inspired Inhibition Strategies
While small-molecule inhibitors dominate the therapeutic landscape, alternative approaches have explored biologically inspired strategies aimed at achieving higher specificity and more physiologically relevant inhibition. Depelestat (DX-890, EPI-hNE4) represents a peptide-based inhibitor engineered to target HNE with high selectivity [82]. This recombinant protein, composed of 56 amino acids, is structurally modelled on natural protease inhibitors and functions as a reversible inhibitor by forming stable enzyme–inhibitor complexes. Unlike low-molecular-weight compounds, its peptide framework mimics endogenous regulatory mechanisms, offering the potential for highly specific elastase neutralization. Preclinical studies and ex vivo analyses demonstrated effective inhibition of both membrane-associated and soluble elastase activity, supporting its therapeutic rationale in inflammatory lung disorders [83]. Clinical investigations progressed into early-phase trials in conditions such as acute respiratory distress syndrome and cystic fibrosis, where excessive neutrophil elastase activity contributes to tissue damage and inflammatory amplification [83]. However, peptide-based inhibitors present distinct pharmacokinetic and formulation challenges, including parenteral administration requirements, susceptibility to proteolytic degradation, and limited tissue penetration. These constraints have complicated clinical translation despite promising biological activity. Nevertheless, depelestat has provided important proof-of-concept for biologic targeting of HNE and has contributed to validating elastase inhibition as a therapeutic strategy [84]. Its development also highlights the potential value of hybrid approaches, such as peptidomimetics or engineered biologics, aimed at combining high specificity with improved stability and drug-like properties. The principal biologic and synthetic HNE inhibitors discussed in this review are summarized in Table 2.
16. Emerging Combination or Multi-Mechanism Strategies
Nanoparticle-based and multifunctional therapeutic approaches are emerging as promising strategies to modulate neutrophil-driven pathology by simultaneously targeting neutrophil activation, oxidative stress, and NET formation, three interconnected processes that contribute to tissue injury in inflammatory lung diseases. Nanocarriers such as liposomes, polymeric nanoparticles, dendrimers, and biomimetic particles (e.g., cell membrane–coated nanoparticles) can be engineered to deliver drugs selectively to inflamed tissues or activated neutrophils, thereby increasing local drug concentration while minimizing systemic toxicity [85,86,87,88,89]. For example, nanoparticles loaded with elastase inhibitors, antioxidants (such as N-acetylcysteine or catalytic ROS scavengers), or anti-inflammatory agents have demonstrated the ability to reduce reactive oxygen species production, inhibit NET release, and attenuate protease-mediated tissue damage in preclinical models of acute lung injury and sepsis [85,88]. Multifunctional platforms are particularly attractive because they can combine several therapeutic mechanisms within a single construct, for instance, ROS-responsive nanoparticles that release inhibitors in oxidative environments, or particles functionalized with targeting ligands that recognize neutrophil surface markers or inflamed endothelium. In addition, enzyme-mimetic nanomaterials (“nanozymes”) with superoxide dismutase or catalase-like activity are being explored to directly neutralize oxidative stress while indirectly limiting NETosis and inflammatory signalling.
The conceptual framework for next-generation strategies targeting human neutrophil elastase is reported in Figure 3.
Although these approaches remain largely preclinical, they offer a conceptual advantage over single-target drugs by addressing the complex, self-amplifying nature of neutrophil-mediated inflammation and may represent an important future direction for treating conditions such as acute respiratory distress syndrome, chronic obstructive pulmonary disease, cystic fibrosis, and bronchiectasis. It is our opinion that the future of elastase inhibition lies not in stronger inhibitors, but in smarter alignment between drug, biology, and patient. The recurring determinants of translational failure across HNE inhibitor development programs are summarized in Table 3.
5. Conclusions
Despite decades of mechanistic investigation and extensive medicinal chemistry efforts, the central paradox of HNE-directed therapy remains unresolved: why has robust biochemical inhibition so rarely translated into durable clinical benefit? We contend that this disconnect reflects not simply technical shortcomings in drug design, but a conceptual misalignment in how HNE has been framed, not as a dynamically regulated node within a spatially compartmentalized, redox-sensitive, and redundant protease-inflammatory network, but as a solitary enzymatic target. Accordingly, this review does not catalogue inhibitor classes or reiterate structure-activity relationships. Instead, it proposes a systems-oriented translational framework integrating enzyme kinetics, inflammatory amplification circuits, endogenous antiprotease constraints, and disease-specific pathophysiology. By reframing HNE inhibition as context-dependent modulation of a protease-driven network, rather than straightforward enzymatic blockade, we seek to clarify the mechanistic roots of prior translational failures and to outline guiding principles for the development of next-generation, clinically resilient inhibitors capable of achieving sustained therapeutic benefit.
Author Contributions
Conceptualization, S.V. and P.I.; writing—original draft preparation, P.I., S.V., M.A.G.; writing—review and editing, P.I., S.V., M.A.G. M.G. T.R. and G.P.; visualization, S.V. and M.A.G.; supervision, P.I. and S.V.; funding acquisition, S.V. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was funded by the Italian Ministry of Research and University in the framework of the project PRIN 2022F5N25M.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors are deeply grateful to Dr. Monica Campagnoli (Department of Molecular Medicine, University of Pavia, Italy) for her tireless input in the paper drafting. During the preparation of this manuscript the authors used ChatGPT for the purposes of improving language and readability. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of this review or in the writing of the manuscript.
Abbreviations
The following abbreviations are used in this manuscript:
| AAT | α1-antitrypsin |
| AATD | α1-antitrypsin deficiency |
| ARDS | Acute respiratory distress syndrome |
| CF | Cystic fibrosis |
| COPD | Chronic obstructive pulmonary diseases |
| EGFR | Epidermal growth factor receptor |
| DAMPs | Damage associated molecular patterns |
| HNE | Human neutrophil elastase |
| NETs | Neutrophil extracellular trap |
| ROS | Reactive oxygen species |
| SLPI | Secretory leukoprotease inhibitor |
| 4-HNE | 4-hydroxy-2-nonenal |
References
- Nie, J.; Zhou, L.; Tian, W.; Liu, X.; Yang, L.; Yang, X.; Zhang, Y.; Wei, S.; Wang, D.W.; Wei, J. Deep insight into cytokine storm: from pathogenesis to treatment. Signal Transduct. Target. Ther. 2025, 10, 112. [CrossRef]
- Voynow, J.A.; Shinbashi, M. Neutrophil elastase and chronic lung disease. Biomolecules 2021, 11, 1065. [CrossRef]
- Kelly-Robinson, G.A.; Reihill, J.A.; Lundy, F.T.; McGarvey, L.P.; Lockhart, J.C.; Litherland, G.J.; Thornbury, K.D.; Martin, S.L. The serpin superfamily and their role in the regulation and dysfunction of serine protease activity in COPD and other chronic lung diseases. Int. J. Mol. Sci. 2021, 22, 6351. [CrossRef]
- Viglio, S.; Bak, E.G.; Schouten, I.G.M.; Iadarola, P.; Stolk, J. Protease-specific biomarkers to analyse protease inhibitors for emphysema associated with alpha-1 antitrypsin deficiency: an overview of current approaches. Int. J. Mol. Sci. 2021, 22, 1065. [CrossRef]
- Liu, X.; Ucakar, B.; Vanvarenberg, K.; Marbaix, E.; Vanbever, R. Impact of administration route and PEGylation on alpha-1 antitrypsin augmentation therapy. J. Control. Release 2025, 382, 113643. [CrossRef]
- Oriano, M.; Amati, F.; Gramegna, A.; De Soyza, A.; Mantero, M.; Sibila, O.; Chotirmall, S.H.; Voza, A.; Marchisio, P.; Blasi, F.; et al. Protease–antiprotease imbalance in bronchiectasis. Int. J. Mol. Sci. 2021, 22, 5996. [CrossRef]
- Lucas, S.D.; Costa, E.; Guedes, R.C.; Moreira, R. Targeting COPD: advances on low-molecular-weight inhibitors of human neutrophil elastase. Med. Res. Rev. 2013, 33, E73–E101. [CrossRef]
- Tousif, M.I.; Nazir, M.; Riaz, N.; Saleem, M.; Tauseef, S.; Azam, S.M.; Arfan Yawer, M.; Zengin, G. Terpenoids as human neutrophil elastase inhibitors: a comprehensive review. ChemBioChem 2023, 24, e202300346. [CrossRef]
- Trainor, D.A. Synthetic inhibitors of human neutrophil elastase. Trends Pharmacol. Sci. 1987, 8, 303–307. [CrossRef]
- Ohbayashi, H. Current synthetic inhibitors of human neutrophil elastase in 2005. Expert Opin. Ther. Pat. 2005, 15, 759–771. [CrossRef]
- Crocetti, L.; Quinn, M.T.; Schepetkin, I.A.; Giovannoni, M.P. A patenting perspective on human neutrophil elastase inhibitors (2014–2018) and their therapeutic applications. Expert Opin. Ther. Pat. 2019, 29, 555–578. [CrossRef]
- Ocampo-Gallego, J.S.; Pedroza-Escobar, D.; Caicedo-Ortega, R.A.; Berumen-Murra, M.T.; Novelo-Aguirre, A.L.; de Sotelo-León, R.D.; Delgadillo-Guzmán, D. Human neutrophil elastase inhibitors: classification, biological-synthetic sources and their relevance in related diseases. Fundam. Clin. Pharmacol. 2024, 38, 13–32. [CrossRef]
- Wang, Z.; Chen, F.; Zhai, R.; Zhang, L.; Su, L.; Lin, X.; Thompson, T.; Christiani, D.C. Plasma neutrophil elastase and elafin imbalance is associated with acute respiratory distress syndrome development. PLoS ONE 2009, 4, e4380. [CrossRef]
- Cagnone, M.; Piloni, D.; Ferrarotti, I.; Di Venere, M.; Viglio, S.; Magni, S.; Bardoni, A.; Salvini, R.; Fumagalli, M.; Iadarola, P.; et al. Balance between proteases and α1-antitrypsin in bronchoalveolar lavage fluid of lung transplant recipients. High-Throughput 2019, 8, 5. [CrossRef]
- Barrera, G.; Pizzimenti, S.; Ciamporcero, E.S.; Daga, M.; Ullio, C.; Arcaro, A.; Cetrangolo, G.P.; Ferretti, C.; Dianzani, C.; Lepore, A.; et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid. Redox Signal. 2015, 22, 1681–1702. [CrossRef]
- Prasad, B. Chronic obstructive pulmonary disease (COPD). Int. J. Pharm. Res. Technol. 2020, 10, 67–71.
- Galiniak, S.; Mołoń, M.; Rachel, M. Links between disease severity, bacterial infections and oxidative stress in cystic fibrosis. Antioxidants 2022, 11, 887. [CrossRef]
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637. [CrossRef]
- Balogh, E.; Veale, D.J.; McGarry, T.; Orr, C.; Szekanecz, Z.; Ng, C.T.; Fearon, U.; Biniecka, M. Oxidative stress impairs energy metabolism in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 95. [CrossRef]
- Ferreira, A.V.; Perelshtein, I.; Perkas, N.; Gedanken, A.; Cunha, J.; Cavaco-Paulo, A. Detection of human neutrophil elastase on wound dressings as marker of inflammation. Appl. Microbiol. Biotechnol. 2017, 101, 1443–1454. [CrossRef]
- Cipak Gasparovic, A.; Milkovic, L.; Borovic Sunjic, S.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [CrossRef]
- Korkmaz, B.; Attucci, S.; Jourdan, M.L.; Juliano, L.; Gauthier, F. Inhibition of neutrophil elastase by α1-protease inhibitor at neutrophil surface. J. Immunol. 2005, 175, 3329–3339. [CrossRef]
- Janciauskiene, S.; Wrenger, S.; Immenschuh, S.; Olejnicka, B.; Greulich, T.; Welte, T.; Chorostowska-Wynimko, J. The Multifaceted Effects of Alpha1-Antitrypsin on Neutrophil Functions. Front. Pharmacol. 2018, 9, 341. [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of α1-antitrypsin causes loss of activity. J. Biol. Chem. 2000, 275, 27258–27265. [CrossRef]
- Li, Z.; Alam, S.; Wang, J.; Sandstrom, C.S.; Janciauskiene, S.J.; Mahadeva, R. Oxidized α1-antitrypsin stimulates MCP-1 release. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L388–L400. [CrossRef]
- Brand, P.; Schulte, M.; Wencker, M.; Herpich, C.H.; Klein, G.; Hanna, K.; Meyer, T. Lung deposition of inhaled alpha1-proteinase inhibitor. Eur. Respir. J. 2009, 34, 354–360. [CrossRef]
- Casaloti, L.G.; Santos, J.A.N. Inhibition of elastase by Sedum dendroideum extracts. Multidiscip. Sci. J. 2019, 7, 140–147. [CrossRef]
- Kacem, R. Phenolic compounds from medicinal plants as Natural anti-elastase products for the therapy of pulmonary emphysema. J. Med. Plants Res. 2013, 7, 3499–3507.
- Wittenauer, J.; Mäckle, S.; Sußmann, D.; Schweiggert-Weisz, U.; Carle, R. Polyphenols from grape pomace inhibit elastase. Fitoterapia 2015, 101, 179–187. [CrossRef]
- Tan, X.F.; Kim, D.W.; Song, Y.H.; Kim, J.Y.; Yuk, H.J.; Wang, Y.; Curtis-Long, M.J.; Park, K.H. Human neutrophil elastase inhibitory potential of flavonoids from Campylotropis hirtella and their kinetics. J. Enzyme Inhib. Med. Chem. 2016, 31, 16–22. [CrossRef]
- Uddin, Z.; Li, Z.; Song, Y.H.; Kim, J.Y.; Park, K.H. Visconata inhibits human neutrophil elastase. Tetrahedron Lett. 2017, 58, 2507–2511. [CrossRef]
- Saleem, M.; Nazir, M.; Hussain, H.; Tousif, M.I.; Elsebai, M.F.; Riaz, N.; Akhtar, N. Natural Phenolics as Inhibitors of the Human Neutrophil Elastase (HNE) Release: An Overview of Natural Anti-inflammatory Discoveries during Recent Years. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2018, 17, 70–94. [CrossRef]
- Tang, Y.; Wang, Y.; Pei, Z.; Li, W.; Zhang, D.; Liu, L.; Kong, L.; Liu, S.; Jiang, X.; Ma, H. A serine protease inhibitor from Musca domestica larva exhibits inhibitory activity against elastase and chymotrypsin. Biotechnol. Lett. 2016, 38, 1147–1153. [CrossRef]
- Luan, N.; Zhao, Q.; Duan, Z.; Ji, M.; Xing, M.; Zhu, T.; Mwangi, J.; Rong, M.; Liu, J.; Lai, R. Identification and Characterization of ShSPI, a Kazal-Type Elastase Inhibitor from the Venom of Scolopendra hainanum. Toxins 2019, 11, 708. [CrossRef]
- Wan, H.; Lee, K.S.; Kim, B.Y.; Yuan, M.; Zhan, S.; You, H.; Li, J.; Jin, B.R. A spider (Araneus ventricosus) chymotrypsin inhibitor that acts as an elastase inhibitor and a microbial serine protease inhibitor. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2013, 165, 36–41. [CrossRef]
- Ahmad, S.; Saleem, M.; Riaz, N.; Lee, Y.S.; Diri, R.; Noor, A.; Almasri, D.; Bagalagel, A.; Elsebai, M.F. The Natural Polypeptides as Significant Elastase Inhibitors. Front. Pharmacol. 2020, 11, 688. [CrossRef]
- Marinaccio, L.; Stefanucci, A.; Scioli, G.; Della Valle, A.; Zengin, G.; Cichelli, A.; Mollica, A. Peptide Human Neutrophil Elastase Inhibitors from Natural Sources: An Overview. Int. J. Mol. Sci. 2022, 23, 2924. [CrossRef]
- Cantin, A.M.; Woods, D.E. Aerosolized prolastin suppresses bacterial proliferation. Am. J. Respir. Crit. Care Med. 1999, 160, 1130–1135. [CrossRef]
- Gottlieb, D.J.; Luisetti, M.; Stone, P.J.; Allegra, L.; Cantey-Kiser, J.M.; Grassi, C.; Snider, G.L. Short-term supplementation therapy in AAT deficiency. Am. J. Respir. Crit. Care Med. 2000, 162, 2069–2072. [CrossRef]
- McCarthy, C.; Dimitrov, B.D. Augmentation therapy for alpha-1 antitrypsin deficiency. COPD 2010, 7, 234–236. [CrossRef]
- Strange, C. Anti-proteases and alpha-1 antitrypsin augmentation therapy. Respir. Care 2018, 63, 690–698. [CrossRef]
- Griese, M.; Scheuch, G. Delivery of alpha-1 antitrypsin to airways. Ann. Am. Thorac. Soc. 2016, 13, S346–S351. [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [CrossRef]
- Bode, W.; Huber, R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta 2000, 1477, 241–252. [CrossRef]
- Turk, B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [CrossRef]
- Edwards, P.D.; Bernstein, P.R. Synthetic inhibitors of elastase. Med. Res. Rev. 1994, 14, 127–194. [CrossRef]
- Edwards, P.D.; Zottola, M.A.; Davis, M.; Williams, J.; Tuthill, P.A. Peptidyl inhibitors of human neutrophil elastase. J. Med. Chem. 1995, 38, 3972–3982. [CrossRef]
- Cregge, R.J.; Durham, S.L.; Farr, R.A.; Gallion, S.L.; Hare, C.M.; Hoffman, R.V.; Janusz, M.J.; Kim, H.O.; Koehl, J.R.; Mehdi, S.; et al. Inhibition of human neutrophil elastase. 4. Design, synthesis, X-ray crystallographic analysis, and structure-activity relationships for a series of P2-modified, orally active peptidyl pentafluoroethyl ketones. J. Med. Chem. 1998, 41, 2461–2480. [CrossRef]
- Sato, F.; Inoue, Y.; Omodani, T.; Imano, K.; Okazaki, H.; Takemura, T.; Komiya, M. Design and synthesis of peptide-based carboxylic acid-containing transition-state inhibitors of human neutrophil elastase. Bioorg. Med. Chem. Lett. 2002, 12, 551–555. [CrossRef]
- Imaki, K.; Okada, T.; Nakayama, Y.; Nagao, Y.; Kobayashi, K.; Sakai, Y.; Mohri, T.; Amino, T.; Nakai, H.; Kawamura, M. Non-peptidic inhibitors of human neutrophil elastase: the design and synthesis of sulfonanilide-containing inhibitors. Bioorg. Med. Chem. 1996, 4, 2115–2134. [CrossRef]
- Ohmoto, K.; Yamamoto, T.; Okuma, M.; Horiuchi, T.; Imanishi, H.; Odagaki, Y.; Kawabata, K.; Sekioka, T.; Hirota, Y.; Matsuoka, S.; et al. Development of orally active nonpeptidic inhibitors of human neutrophil elastase. J. Med. Chem. 2001, 44, 1268–1285. [CrossRef]
- Kawabata, K.; Suzuki, M.; Sugitani, M.; Imaki, K.; Toda, M.; Miyamoto, T. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem. Biophys. Res. Commun. 1991, 177*, 814–820. [CrossRef]
- Kawabata, K.; Moore, A.R.; Willoughby, D.A. Impaired activity of protease inhibitors towards neutrophil elastase bound to human articular cartilage. Ann. Rheum. Dis. 1996, 55, 248–252. [CrossRef]
- Navia, M.A.; McKeever, B.M.; Springer, J.P.; Lin, T.Y.; Williams, H.R.; Fluder, E.M.; Dorn, C.P.; Hoogsteen, K. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84-A resolution. Proc. Natl. Acad. Sci. USA 1989, 86, 7–11. [CrossRef]
- Aikawa, N.; Kawasaki, Y. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther. Clin. Risk Manag. 2014, 10, 621–629. [CrossRef]
- Kido, T.; Muramatsu, K.; Yatera, K.; Asakawa, T.; Otsubo, H.; Kubo, T.; Fujino, Y.; Matsuda, S.; Mayumi, T.; Mukae, H. Efficacy of early sivelestat administration on acute lung injury and acute respiratory distress syndrome. Respirology 2017, 22, 708–713. [CrossRef]
- Iwata, K.; Doi, A.; Ohji, G.; Oka, H.; Oba, Y.; Takimoto, K.; Igarashi, W.; Gremillion, D.H.; Shimada, T. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern. Med. 2010, 49, 2423–2432. [CrossRef]
- Sahebnasagh, A.; Saghafi, F.; Safdari, M.; Khataminia, M.; Sadremomtaz, A.; Talaei, Z.; Rezai Ghaleno, H.; Bagheri, M.; Habtemariam, S.; Avan, R. Neutrophil elastase inhibitor (sivelestat) may be a promising therapeutic option for management of acute lung injury/acute respiratory distress syndrome or disseminated intravascular coagulation in COVID-19. J. Clin. Pharm. Ther. 2020, 45, 1515–1519. [CrossRef]
- Ding, Q.; Wang, Y.; Yang, C.; Tuerxun, D.; Yu, X. Effect of Sivelestat in the Treatment of Acute Lung Injury and Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Intensive Care Res. 2023, 1–10. [CrossRef]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [CrossRef]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [CrossRef]
- Shapiro, S.D. Proteolysis in the lung. Eur. Respir. J. Suppl. 2003, 44, 30s–32s. [CrossRef]
- Calfee, C.S.; Delucchi, K.; Parsons, P.E.; Thompson, B.T.; Ware, L.B.; Matthay, M.A.; NHLBI ARDS Network. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir. Med. 2014, 2, 611–620. [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [CrossRef]
- Bos, L.D.J.; Artigas, A.; Constantin, J.M.; Hagens, L.A.; Heijnen, N.; Laffey, J.G.; Meyer, N.; Papazian, L.; Pisani, L.; Schultz, M.J.; et al. Precision medicine in acute respiratory distress syndrome: workshop report and recommendations for future research. Eur. Respir. Rev. 2021, 30, 200317. [CrossRef]
- Matera, M.G.; Rogliani, P.; Ora, J.; Calzetta, L.; Cazzola, M. Investigational elastase inhibitors for ARDS. Expert Opin. Investig. Drugs 2023, 32, 793–802. [CrossRef]
- Li, Y.; Zhao, J.; Wei, J.; Zhang, Y.; Zhang, H.; Li, Y.; Liao, T.; Hu, Y.; Yuan, B.; Zhang, X.; et al. Neutrophil elastase inhibitor (Sivelestat) in the treatment of acute respiratory distress syndrome induced by COVID-19: a multicenter retrospective cohort study. Respir. Res. 2025, 26, 28. [CrossRef]
- Tremblay, G.M.; Janelle, M.F.; Bourbonnais, Y. Anti-inflammatory activity of neutrophil elastase inhibitors. Curr. Opin. Investig. Drugs 2003, 4, 556–565.
- Stevens, T.; Ekholm, K.; Gränse, M.; Lindahl, M.; Kozma, V.; Jungar, C.; Ottosson, T.; Falk-Håkansson, H.; Churg, A.; Wright, J.L.; et al. AZD9668: pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J. Pharmacol. Exp. Ther. 2011, 339, 313–320. [CrossRef]
- Gunawardena, K.A.; Gullstrand, H.; Perrett, J. Pharmacokinetics and safety of AZD9668, an oral neutrophil elastase inhibitor, in healthy volunteers and patients with COPD. Int. J. Clin. Pharmacol. Ther. 2013, 51, 288–304. [CrossRef]
- Stockley, R.; De Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [CrossRef]
- Wells, J.M.; Titlestad, I.L.; Tanash, H.; Turner, A.M.; Chapman, K.R.; Hatipoğlu, U.Ş.; Goldklang, M.P.; D’’Armiento, J.M.; Pirozzi, C.S.; Drummond, M.B.; et al. Two randomised controlled phase 2 studies of the oral neutrophil elastase inhibitor alvelestat in alpha-1 antitrypsin deficiency. Eur. Respir. J. 2025, 66, 2501019. [CrossRef]
- von Nussbaum, F.; Li, V.M.; Allerheiligen, S.; Anlauf, S.; Bärfacker, L.; Bechem, M.; Delbeck, M.; Fitzgerald, M.F.; Gerisch, M.; Gielen-Haertwig, H.; et al. Freezing the Bioactive Conformation to Boost Potency: The Identification of BAY 85-8501, a Selective and Potent Inhibitor of Human Neutrophil Elastase for Pulmonary Diseases. ChemMedChem 2015, 10, 1163–1173. [CrossRef]
- Watz, H.; Nagelschmitz, J.; Kirsten, A.; Pedersen, F.; van der Mey, D.; Schwers, S.; Bandel, T.J.; Rabe, K.F. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85-8501 for the treatment of non-cystic fibrosis bronchiectasis: A randomized controlled trial. Pulm. Pharmacol. Ther. 2019, 56, 86–93. [CrossRef]
- Kuraki, T.; Ishibashi, M.; Takayama, M.; Shiraishi, M.; Yoshida, M. A novel oral neutrophil elastase inhibitor (ONO-6818) inhibits human neutrophil elastase-induced emphysema in rats. Am. J. Respir. Crit. Care Med. 2002, 166, 496–500. [CrossRef]
- Yoshimura, Y.; Hiramatsu, Y.; Sato, Y.; Homma, S.; Enomoto, Y.; Jikuya, T.; Sakakibara, Y. ONO-6818, a novel, potent neutrophil elastase inhibitor, reduces inflammatory mediators during simulated extracorporeal circulation. Ann. Thorac. Surg. 2003, 76, 1234–1239. [CrossRef]
- Ohmoto, K.; Okuma, M.; Yamamoto, T.; Kijima, H.; Sekioka, T.; Kitagawa, K.; Yamamoto, S.; Tanaka, K.; Kawabata, K.; Sakata, A.; et al. Design and synthesis of new orally active inhibitors of human neutrophil elastase. Bioorg. Med. Chem. 2001, 9, 1307–1323. [CrossRef]
- Stockley, R.A. Neutrophils and the pathogenesis of COPD. Chest 2002, 121, 151S–155S. [CrossRef]
- Attucci, S.; Gauthier, A.; Korkmaz, B.; Delépine, P.; Martino, M.F.; Saudubray, F.; Diot, P.; Gauthier, F. EPI-hNE4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J. Pharmacol. Exp. Ther. 2006, 318, 803–809. [CrossRef]
- Mac Sweeney, R.; McAuley, D.F. Do Nonventilatory Strategies for Acute Lung Injury and ARDS Work? In Evidence-Based Practice of Critical Care; Saunders: Philadelphia, PA, USA, 2010; pp. 73–81. [CrossRef]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [CrossRef]
- Kang, H.; Seo, J.; Yang, E.J.; Choi, I.H. Silver Nanoparticles Induce Neutrophil Extracellular Traps Via Activation of PAD and Neutrophil Elastase. Biomolecules 2021, 11, 317. [CrossRef]
- Kim, J.; Sahay, G. Nanomedicine hitchhikes on neutrophils to the inflamed lung. Nat. Nanotechnol. 2022, 17, 1–2. [CrossRef]
- Rezaei, N.; Zadory, M.; Babity, S.; Marleau, S.; Brambilla, D. Therapeutic applications of nanoparticles targeting neutrophil and extracellular traps. J. Control. Release 2023, 358, 636–653. [CrossRef]
- Li, H.; Li, C.; Fu, C.; Wang, Y.; Liang, T.; Wu, H.; Wu, C.; Wang, C.; Sun, T.; Liu, S. Innovative nanoparticle-based approaches for modulating neutrophil extracellular traps in diseases: from mechanisms to therapeutics. J. Nanobiotechnology 2025, 23, 88. [CrossRef]
- Lee, H.J.; Lee, N.K.; Kim, J.; Kim, J.; Seo, D.; Shin, H.E.; Kim, J.; Ahn, J.H.; Kim, S.N.; Kim, H.S.; et al. Sequential nanoparticle therapy targeting neutrophil hyperactivation to prevent neutrophil-induced pulmonary fibrosis. J. Nanobiotechnology 2025, 23, 381. [CrossRef]
Figure 1.
Schematic view of the sequence of events that characterize the protein storm.

Figure 2.
Evolution of HNE inhibitor development.

Figure 3.
Next-generation therapeutic strategies targeting the HNE-driven proteolytic network.


Table 1.
Comparison between endogenous, natural, and synthetic HNE inhibitors.
| Property | Endogenous Inhibitors (AAT, SLPI, Elafin) | Natural Small-Molecule Inhibitors | Synthetic Small-Molecule Inhibitors |
Peptide or Biologic Inhibitors |
| Molecular size | Large proteins | Small–medium | Small | Medium-sized |
| Selectivity for HNE | High | Variable | Tunable, often high | Very high |
| Oxidative stability | Poor (susceptible to ROS) | Variable | Optimizable | Moderate |
| Tissue penetration | Limited | Often limited | Generally good tissue penetration | Limited |
| Pharmacokinetics | Short half-life | Rapid metabolism | Optimizable | Limited stability |
| Immunogenicity risk | Low–moderate | Low | Low | Potential risk |
| Manufacturing complexity | High | Moderate | Moderate | High |
| Clinical success | Partial (AAT therapy) | Minimal | Moderate (regional approvals, clinical trials) | Limited |
| Main limitation | Oxidative inactivation and limited compartment access | Low potency and metabolic instability | Context-dependent efficacy and translational challenges | Delivery constraints and metabolic instability |
Table 2.
Major synthetic and biologic inhibitors of HNE.
| Compound | Type | Clinical Development/ Indication | Key Advantages | Main Limitations | Reference |
| Early peptidyl ketones/aldehydes | Peptide-derived inhibitors | Preclinical | High potency; early mechanistic validation | Poor pharmacokinetics; toxicity | [49,50,51] |
| α-ketoheterocycles/oxadiazoles | Synthetic small molecules | Preclinical | Improved selectivity compared with early inhibitors | Limited clinical translation | [52,53] |
| Sivelestat (ONO-5046) | Small-molecule inhibitor | Approved in Japan/Korea for ARDS; trials elsewhere | First clinically approved HNE inhibitor; proof of target validity | Short half-life; continuous infusion; limited global efficacy | [54,57,58,59,60,61,70] |
| Alvelestat (AZD9668) | Oral small-molecule inhibitor | Phase II trials in COPD, bronchiectasis, AAT deficiency | Oral bioavailability; high selectivity; suitable for chronic therapy | Limited improvement in lung function endpoints | [72,73,74,75] |
| BAY 85-8501 | Potent small-molecule inhibitor | Phase IIa bronchiectasis | Very high potency and selectivity; good tolerability | Limited clinical efficacy demonstrated | [76,77] |
| Freselestat (ONO-6818) | Small-molecule inhibitor (sivelestat lineage) | Phase II (discontinued) in COPD, AAT deficiency | Oral activity; improved scaffold stability | Liver toxicity signals; program discontinued | [78,79] |
| Depelestat (DX-890 / EPI-hNE4) | Recombinant peptide biologic | Phase I–II ARDS, cystic fibrosis | High specificity; mimics endogenous inhibition | Poor tissue penetration; proteolytic instability | [82,83] |
Table 3.
Key limiting factors contributing to the translational failure of HNE inhibitors.
| Limiting Factor | Mechanism | Consequence | Clinical Implication |
| Biological redundancy | Compensatory activity of other proteases | Partial pathway suppression | Limited efficacy in complex syndromes (e.g., ARDS) |
| Enzyme compartmentalization | Membrane-bound, NET-associated, or matrix-bound HNE becomes inaccessible | Incomplete target engagement | Persistent elastase activity despite therapy |
| Oxidative microenvironment | ROS modify inhibitors and endogenous antiproteases | Reduced inhibitory capacity | Reduced effectiveness in inflamed tissues |
| Pharmacokinetic–pharmacodynamic mismatch | Insufficient drug concentration at disease site | Suboptimal inhibition | Failure despite potent in vitro activity |
| Timing of intervention | Intervention after irreversible tissue damage | Limited reversibility | Poor outcomes in acute disease |
| Patient heterogeneity | Variable neutrophil burden and disease endotypes | Diluted treatment effect | Negative or inconclusive trials |
| Biomarker limitations | Lack of validated biomarkers of elastase activity | Poor patient selection | Inefficient clinical trial design |
| Delivery constraints | Poor penetration into mucus or inflamed tissue | Reduced local exposure | Particularly relevant in lung diseases |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



