Submitted:
02 April 2026
Posted:
03 April 2026
You are already at the latest version
Abstract
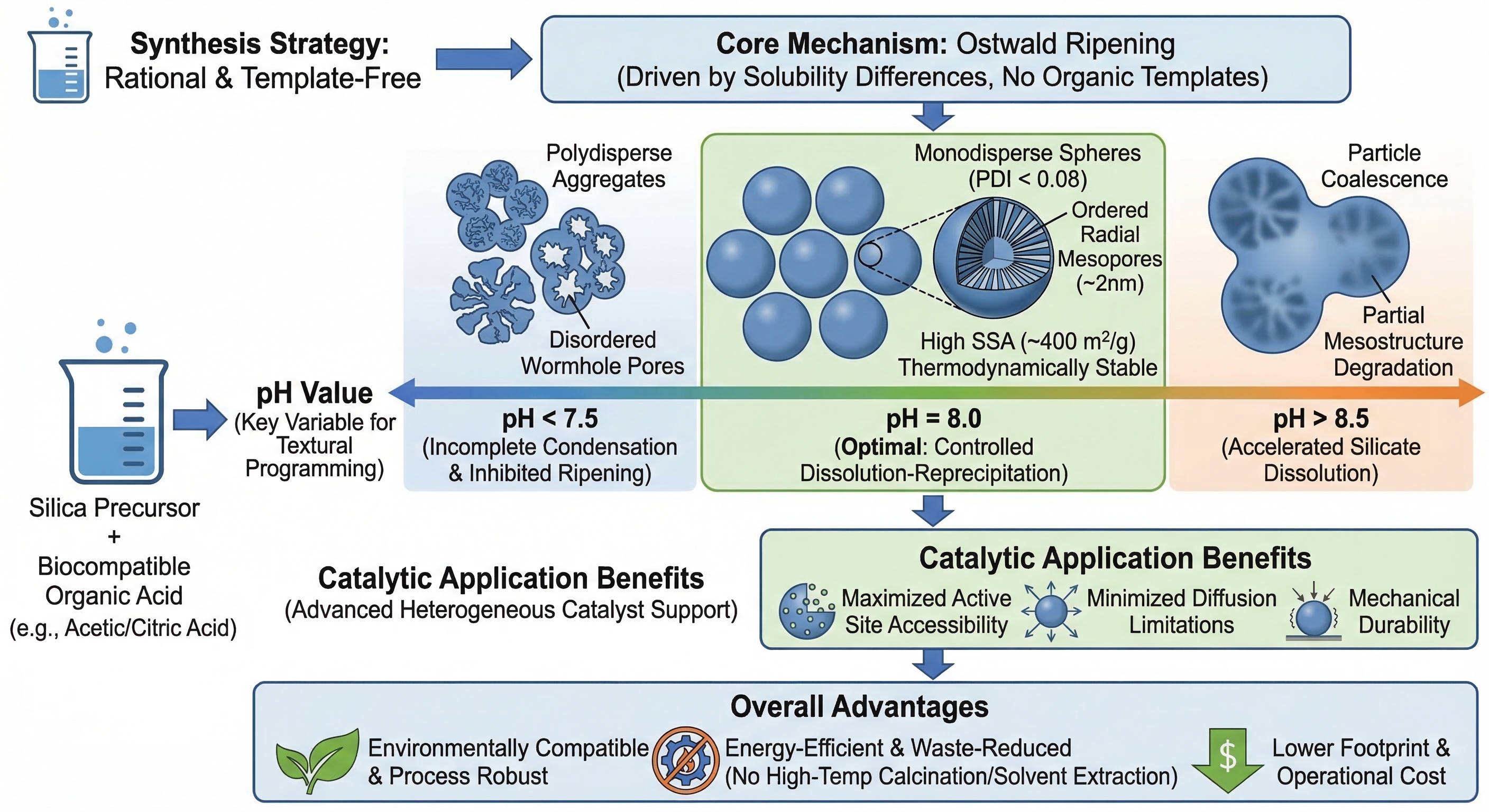
This study introduces a rational, template-free synthetic strategy for the scalable preparation of high-performance monodisperse spherical mesoporous silica particles (MSPs), engineered specifically as advanced heterogeneous catalytic supports. Leveraging Ostwald ripening as the core morphogenetic driver—rather than conventional organic structure-directing agents—the approach achieves both environmental compatibility and process robustness. Precise pH modulation to 8.0 using biocompatible organic acids (e.g., acetic or citric acid) enables controlled silica dissolution–reprecipitation kinetics, yielding MSPs with exceptional sphericity (PDI < 0.08), narrow size distribution, a specific surface area of up to 484 m²/g, uniform pore diameters centered at ~2 nm, and radially aligned, thermodynamically stabilized mesochannels—structural attributes that collectively satisfy stringent design criteria for high-efficiency catalytic carriers, including maximized active-site accessibility, minimized diffusion limitations, and mechanical resilience under reaction conditions. A systematic pH-screening study reveals a distinct structural transition: at pH < 7.5, incomplete condensation and suppressed ripening yield polydisperse aggregates with disordered worm-like porosity; at pH > 8.5, accelerated silicate dissolution induces particle coalescence and partial mesostructural degradation. Critically, pore ordering, channel dimensionality, surface area, and particle morphology are all quantitatively modulated by pH—establishing it as a master variable for hierarchical textural programming. This study compares the methoxychlor (MXC) degradation efficiency of polyhedral Bi2WO6 and MSP/Bi2WO6 under identical irradiation conditions to assess MSP’s catalytic impact. Mechanistic analysis of charge dynamics, interfacial electron transfer, and active species reveals how MSP enhances photocatalytic activity.
Keywords:
surfactant-free synthesis
; mesoporous silica
; sodium silicate
; pH-regulated Ostwald ripening
; green synthesis
; catalysis applications
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



