Submitted:
27 March 2026
Posted:
31 March 2026
You are already at the latest version
Abstract
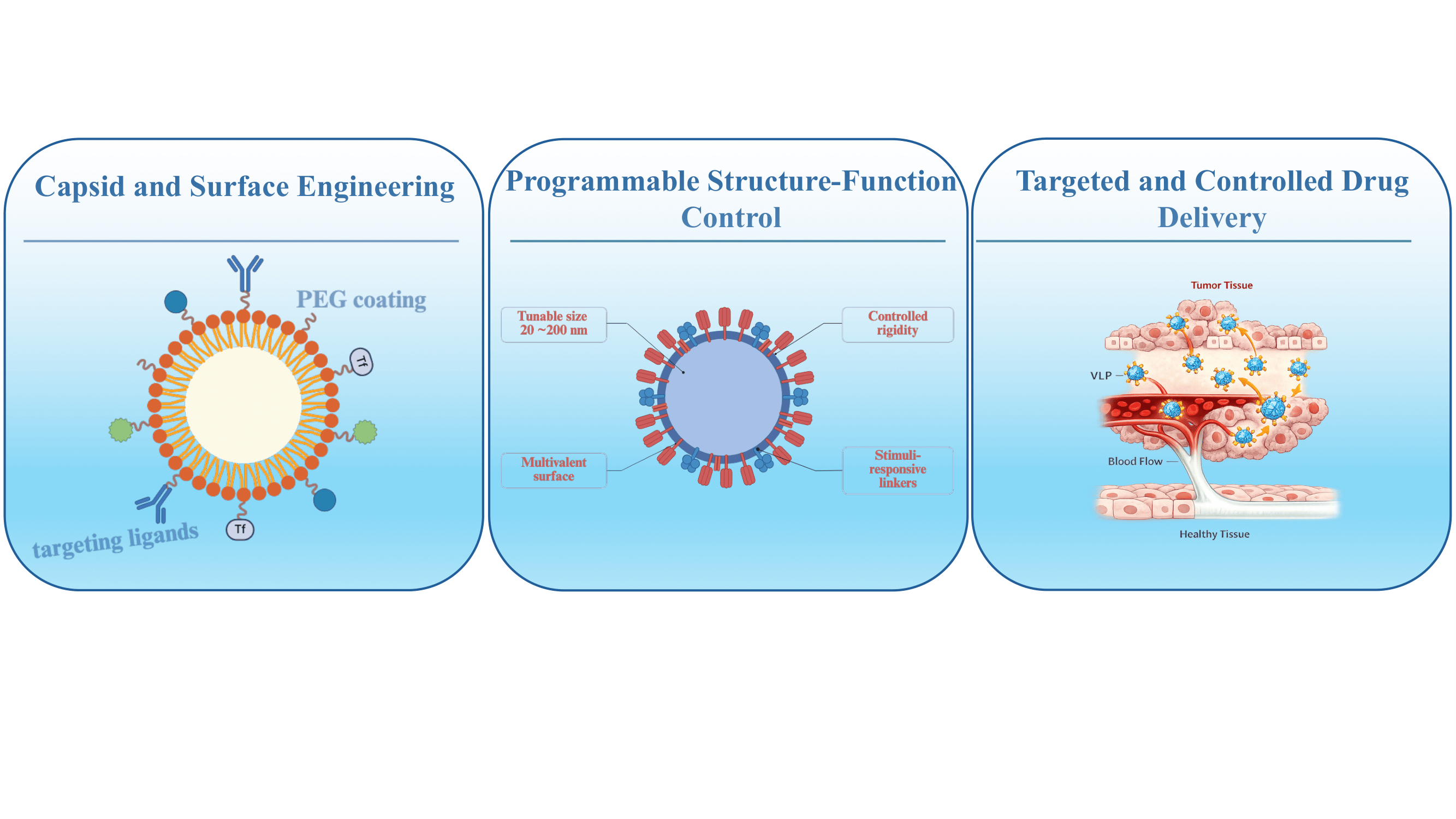
Virus-like particles (VLPs) are self-assembling protein nanostructures that replicate the structural precision of viral capsids while lacking genetic material, rendering them inherently safe and highly modular biomaterials. Their genetically encoded architecture enables precise control over size, symmetry, mechanical stability, surface topology, and internal cavity volume, positioning VLPs as programmable protein-based nanocarriers for chemotherapeutic delivery. Recent advances in capsid engineering, biorthogonal conjugation, and template-guided assembly have enabled fine tuning of cargo loading, targeting ligand display, and stimuli-responsive drug release. Unlike many synthetic nanocarriers, VLPs offer atomically defined structure–function relationships, allowing rational modulation of biodistribution, cellular uptake, immune recognition, and therapeutic performance. This review examines VLPs as engineered protein biomaterials for precision chemotherapy, highlighting strategies for internal cargo integration, interfacial surface modification, mechanical reinforcement, and microenvironment-triggered release. Here we discuss how physicochemical parameters govern biological interactions and translational feasibility. Clinical progress underscores both the promise and remaining challenges of scalable manufacturing and immune modulation. By integrating biomaterials design principles with translational constraints, this review outlines a framework for the rational development of clinically viable VLP-based chemotherapeutic systems.
Keywords:
virus-like particles (VLPs)
; targeted drug delivery
; chemotherapeutics
; nanomedicine
; precision oncology
1. Context and Unmet Needs in Cancer Treatment
Cancer remains a significant global health threat. In 2022, approximately 20 million new cases and 9.7 million cancer-related deaths were reported worldwide, underscoring the urgent need for more effective therapeutic strategies [1]. Although significant progress has been made in cancer prevention, early detection, and treatment, which has been contributing to a decline in mortality rates in many regions, the global burden of cancer is rapidly increasing [1]. This rise largely driven by population ageing and the growing prevalence of risk factors is linked to environmental and lifestyle changes [2,3].
Within this landscape, VLPs have emerged as a unique class of protein-based biomaterials for therapeutic delivery. VLPs are nanoscale assemblies formed by the self-organization of viral capsid proteins in the absence of genetic material, rendering them non-infectious while preserving the structural fidelity of native viruses offer several advantages as drug delivery systems (DDS). Their genetically encoded architecture enables precise control over particle size, symmetry, surface topology, and internal cavity volume, features that are difficult to replicate using fully synthetic nanocarriers. As a result, VLPs provide a modular scaffold in which structure–function relationships can be systematically tuned to influence biological performance.
VLPs traditionally known for their role vaccine development due to their intrinsic immunogenicity and ability to boost hormonal and cellular immune responses, are now gaining traction in the field of drug delivery [4]. Their biocompatibility, ability to penetrate biological barriers, innate cargo-carrying potential, and ease of surface modification make VLPs highly attractive as programmable platforms for the targeted delivery of therapeutic agents, including small molecules, chemotherapeutics, nucleic acids and proteins [5,6]. Although VLPs have been extensively reviewed as nanocarriers for a broad range of therapeutic and diagnostic cargos [4,5,6,7,8,9], their application as platforms for chemotherapeutic drug delivery has rarely been examined as a primary focus. Herein, we review recent advances in the design and functionalization of VLPs for chemotherapeutic drug delivery, highlighting their versatility, current challenges, and potential to enhance the specificity and efficacy of cancer treatments.
2. Architectural and Functional Basis
VLPs are nanoscale, protein-based assemblies that mimic the structural organization of native viruses. They are formed through the self-assembly of capsid proteins (CPs), but lack the viral genetic material, rendering them non-infectious. These nanostructures typically range from 20 to 200 nm in diameter, depending on their viral origin. Lacking genetic material and replication machinery, VLPs are non-infectious by design, yet retain intrinsic immunogenicity and strong safety profile, making them highly suitable for biomedical applications [9]. Viral structural proteins can be expressed in a variety of live or cell-free systems, after which they self-assemble into virus-like structures that can be further purified, engineered, or functionally reconstituted for specific applications. Cell-free systems are particularly attractive for VLP production as they allow tight control over reaction conditions and reduce the risk of host-cell contaminants, though they require virus-specific optimization and may not be compatible with all capsid types [10].
The structural diversity of VLPs reflects the wide range of architectures found among their viral sources. VLPs can adopt icosahedral, rod-shaped, or spherical geometries, and can be composed of one or multiple capsid proteins, forming either single- or multi-layered assemblies [11]. Some VLPs are non-enveloped, while others acquire a lipid envelope derived from the host cell membrane during the processes of assembly and budding [12]. In enveloped VLPs, such as those derived from HIV-1, Influenza and Ebola virus, viral glycoproteins embedded in the lipid membrane mediate cell entry via membrane fusion - a process governed by glycoprotein conformational changes and modulated by the lipid composition and protein–lipid interactions between the fusing membranes [12,13].
The variety of VLP shapes, sizes and surface chemistries allows for different cellular interactions, internalization pathways, biodistribution, clearance and transport within complex biological environments, such as the tumor microenvironment [14,15]. In all cases, their surface can be readily modified to incorporate targeting ligands, antigenic epitopes, or chemical linkers, enabling selective delivery to specific tissues or cells. Internally, VLPs can be loaded with a range of therapeutic payloads, including chemotherapeutic agents, nucleic acids, or imaging probes, either through covalent conjugation, encapsulation during assembly, or post-assembly diffusion [16]. Moreover, their nanoscale size allows their efficient tissue penetration and cellular uptake, while their virus-mimicking architecture can promote endosomal escape and intracellular delivery. In addition, certain VLPs naturally elude to the formation of a protein corona, a major challenge to nanoparticle delivery that can lead to nanoparticle aggregation, rapid clearance and masking of targeting ligands irrespective of administration route [17,18]. These properties make VLPs powerful candidates for programmable chemotherapeutic drug delivery systems, offering the potential to enhance treatment efficacy, reduce off-target toxicity, and overcome biological barriers that limit the performance of conventional nanocarriers.
In the following sections, we explore the strategies for engineering VLPs as chemotherapeutic carriers, including methods for cargo loading, surface functionalization, and design considerations that influence pharmacokinetics, biodistribution, and therapeutic outcomes.
3. Engineering VLPs for Drug Delivery
3.1. Assembly-Driven Modifications and Cargo Loading
A hallmark of the utility of VLPs as delivery vehicles lies in the intrinsic connection between their self-assembly processes and functional integration. Since protein cage shape influences particle-cell interactions, biodistribution, and transport in biological environments, structural manipulation allows direct tailoring of VLP properties to supply specific therapeutic requirements [19]. Consequently, assembly-driven engineering approaches have become central to the rational design of VLPs for drug delivery applications.
The structure of VLPs is genetically encoded and therefore highly tunable. CP subunits are evolutionarily optimized to self-assemble into symmetric, hierarchically ordered shells through specific inter-subunit interactions, which define particle geometry, size, and surface topology [20]. Modulating these intrinsic CP features through single-point mutations or domain insertions provides a direct means to guide self-assembly toward alternative morphologies or to enhance capsid stability [21,22]. For example, C-terminal histidine-peptide extension introduced into chimeric hepatitis B virus core antigen (HBcAg)-VLPs conferred enhanced resistance to chemical and physical stress [23]. In the same study, a naturally occurring disulfide bond at Cys61 was identified as an additional, independent stabilizing element, contributing to the overall structural integrity of the VLPs. In another example, the introduction of cysteines through double-site mutations (L102C/P300C) at the VP1 pentamer interface of SV40 VLPs favored the formation of a homogenous population of small, T=1-like VLPs with enhanced stability, attributed to engineered disulfide bonds [24].
VLPs can self-assemble either spontaneously from CPs alone or in the presence of templates such as nucleic acids, proteins, or synthetic polymers. Template-assisted assembly provides control over particle morphology based on the characteristics of the template and offers a streamlined approach for the co-packaging of cargo. For instance, cowpea chlorotic mottle virus (CCMV)-based virus-like particles can afford spherical, icosahedral or rod-shaped structures of various sizes when assembled around different lengths of single or double-stranded DNA [25]. Similarly, tobacco mosaic virus (TMV) VLPs can attain different aspect ratios dictated by the length of the RNA template used during assembly [26]. Access to a range of geometries and sizes from the same CPs has allowed the interrogation of the impact of these properties on biodistribution, cellular uptake, and drug delivery efficiency [26,27,28,29]. Importantly, the final structure and function of a VLP is also dictated by the assembly conditions, including pH, ionic strength, temperature, and presence of divalent cations or crowding agents [30,31]. Consequently, fine-tuning these parameters allows researchers to control VLP yield, cargo loading efficiency, and structural fidelity. pH-triggered disassembly and reassembly cycles are often employed to load small-molecule drugs post-assembly, while salt concentrations can modulate capsid rigidity and porosity [32,33].
A detailed understanding of capsid assembly is therefore critical for rational VLP engineering. Experimental techniques such as cryo-electron microscopy and X-ray crystallography enable high-resolution structural characterization of individual CPs and assembled particles [34,35], while atomic force microscopy allows topographical mapping of VLPs [36,37]. Complementary methods including dynamic light scattering and size-exclusion chromatography are commonly used to assess particle size distributions and assembly homogeneity [38,39], whereas isothermal titration calorimetry and surface plasmon resonance allow interrogation of protein-protein and protein-cargo interactions [40]. Computational modeling and molecular dynamics simulations have also proven valuable for predicting assembly pathways, evaluating how different templates influence particle morphology, and identifying energetically favorable configurations [41,42,43].
3.2. Surface Functionalization Strategies
VLPs offer multivalency for functionalization owing to the repetitive arrangement of their protein subunits, allowing high-density presentation of functional groups on both their external and internal surfaces. Table 1 summarizes the key functional groups commonly conjugated to VLP surfaces, including therapeutic agents, targeting ligands, cell-penetrating peptides and other supportive components, which can be installed through either genetic or chemical modifications, depending on their nature [44].
Genetic modification pertains to the addition of foreign peptide or protein sequences into the genes encoding the capsid-forming proteins, enabling their co-expression and incorporation into the VLP during self-assembly. The new sequence can be introduced in the form of an insertion or substitution at surface-exposed loops, as well as an extension of the N- or C-termini of capsid proteins, depending on the structural tolerance of the VLP [56,57]. This method typically yields highly homogeneous products with a defined stoichiometry and orientation of the inserted functional molecule but is limited by size constraints and the biochemical properties of the insert, such as charge, hydrophobicity, and the adopted secondary structure [58,59]. Fusion of large or structurally disruptive peptides may impair VLP assembly or reduce VLP stability [58,60]. Strategies to circumvent these limitations include, for instance, the tandem core approach in hepatitis B core antigen VLPs, which enables the display of large proteins or multiple functional motifs without compromising VLP assembly and stability [61], and the use of a nonsense suppression system in MS2 VLPs to control the density of scFv incorporation, ensuring proper folding of coat–scFv fusion proteins [62]. In most cases, structural modeling of capsid proteins can guide the rational placement of fusion sequences into permissive locations [63]. Finally, beyond the fusion of foreign proteins onto capsid proteins, single-point mutations can be introduced to reduce nonspecific interactions, by substituting binding residues with inert ones [64], or to incorporate addressable functional groups for site-specific chemical modification [65]. Genetic introduction of unnatural amino acid residues bearing bioorthogonal reaction handles, as well as the insertion of signal sequences for enzyme-mediated chemical conjugation, are highly sought-after strategies for VLP functionalization and are discussed below. The feasibility of such modifications is contingent on avoiding adverse effects on capsid assembly, stability, and any essential inbuilt functions, including receptor binding.
Chemical conjugation provides a versatile post-synthetic strategy for modifying assembled VLPs, enabling the attachment of therapeutic agents and other functional components beyond peptides and proteins. Conventional approaches exploit solvent-exposed natural amino acids on the VLP surface for selective covalent modification, most commonly lysine, cysteine, tyrosine, glutamic acid, and aspartic acid residues, which serve as reactive handles for conjugation [66]. Among these, lysine and carboxylate-containing residues are frequently targeted due to their abundance and high solvent accessibility, allowing high-density display of functional molecules. For example, Biabanikhankahdani et al. conjugated about 470 folic acid (FA) and 1600 DOX molecules to the external surface of truncated hepatitis B core antigen (tHBcAg) VLPs via primary amine and carboxylate groups, respectively, using EDC/Sulfo-NHS chemistry [67]. The resulting dual-conjugated VLPs enabled specific DOX delivery to FA receptor-overexpressing cancer cells. Alternatively, when a more precise attachment of functional groups is required, cysteines, which are rarer on VLP surfaces and often engaged in disulfide bonds, can be targeted through thiol-reactive chemistries [68,69]. In the absence of free cysteines, the genetic introduction of addressable cysteines at permissive sites is a common strategy for site-specific functionalization [68,70].
Chemical conjugation to natural amino acids is not always suitable, as nonspecific modification of abundant lysine or cysteine residues, particularly in antibodies, can lead to heterogeneous display and aggregation upon VLP functionalization [71]. To address this limitation, site-selective conjugation strategies based on genetically encoded unnatural amino acids (UAAs) have been developed, enabling the introduction of bioorthogonal reactive handles at defined positions. These engineered residues can be selectively modified using orthogonal “click” chemistries, such as azide-alkyne cycloaddition, which proceed with high specificity and efficiency under mild conditions [72,73]. For example, bacteriophage Qβ and MS2 VLPs have been functionalized via copper-catalyzed azide–alkyne cycloaddition (CuAAC) by globally incorporating azide- or alkyne-bearing methionine analogues during expression, allowing conjugation of diverse biomolecules without compromising capsid integrity [73]. While CuAAC affords stable triazole linkages, concerns over copper-induced cytotoxicity and protein oxidation have motivated the use of stabilizing ligands (e.g., TBTA or THPTA) or copper-free alternatives such as strain-promoted azide-alkyne cycloaddition [74,75].
Bacterial-derived ligation systems, such as sortase-mediated conjugation and the SpyTag/SpyCatcher system, provide comparable site-selectivity and bioorthogonality. Sortase-mediated conjugation enables site-specific attachment of functional proteins or peptides through the catalysis of a transpeptidation reaction between a C-terminal LPXTG motif engineered into the VLP and an N-terminal oligoglycine on the cargo [76]. The SpyTag/SpyCatcher system, derived from a split protein domain of Streptococcus pyogenes, enables spontaneous and irreversible covalent bond formation between a short peptide tag (SpyTag) and its protein partner (SpyCatcher). Unlike strategies based on unnatural amino acid incorporation, this genetically encoded system enables efficient, site-specific VLP functionalization under mild conditions without the need for specialized reagents or complex expression protocols, enhancing simplicity and scalability [77].
Lastly, non-covalent modification strategies, such as affinity tag interactions, provide a modular and reversible alternative to covalent conjugation while preserving the bioactivity of the cargo [78]. The trimeric decoration protein (Dec) from bacteriophage L binds P22 VLPs with high affinity and can serve as a scaffold for surface display without CP modification. Fusion of Dec to either the soluble region of murine CD40L or a CD47-derived “self-peptide” enabled their individual presentation on the capsid exterior, with CD47-Dec VLPs showing reduced phagocytic uptake, highlighting a strategy to prolong P22 VLP circulation time [79]. Similarly, engineered affinity tags, such as His-tag/trisNTA, have been used to decorate norovirus VLPs with peptides or fluorescent probes for targeted cellular uptake [80].
4. Current Progress in Chemotherapy Delivery
Chemotherapy remains a cornerstone of cancer treatment, yet its clinical effectiveness is often compromised by dose-limiting toxicity, inadequate specificity, and poor water solubility [81,82]. Non-selective biodistribution leads to off-target effects against healthy cells and insufficient drug accumulation at tumor sites, resulting in partial remission and the emergence of drug-resistant cell populations [81,83]. To address these limitations, recent efforts in cancer nanomedicine have focused on developing multicomponent carriers with tumor-targeting capabilities, controlled drug release, immune evasion features and other supportive components [84]. Among these, VLPs stand out as versatile nanocarriers capable of meeting the complex requirements of chemotherapeutic delivery. Herein, we highlight recent advances in the design of VLP-based carriers for chemotherapy delivery, with a focus on rational design strategies, chemically switchable systems and progress toward clinical translation.
A drug delivery system based on Flock House Virus (FHV) VLPs has been developed for targeted delivery of hydrophobic chemotherapeutic drugs [85]. FHV VLPs are icosahedral particles of about 30 nm with T=3 symmetry, assembled from 180 identical protein subunits [86]. Capsid proteins were engineered to contain a serine to lysine mutation at position 268 (S268K), located on an outer, solvent-exposed loop, for efficient attachment of the tumor-homing peptide tLyP-1 (CGNKRTR). This peptide is a linear, C-terminally truncated version of Lyp-1, a circular peptide known for its ability to specifically recognize tumor cells [87]. Peptide conjugation was carried out in two steps, using a heterobifunctional PEGylated SMCC crosslinker as the anchoring moiety, to avoid nonspecific uptake and improve biocompatibility. FHV particles exhibit dynamic behavior, a preference for hydrophobic molecules, and are stabilized by Ca2+ ions. These properties were exploited to encapsulate two hydrophobic drugs, DOX and ellipticine (EPT), via partial capsid destabilization induced by Ca2+ removal. The resulting Dox_tLyP-1_S268K and EPT_tLyP-1_S268K VLPs were stable and capable of selectively targeting MDA-MB-231 breast tumor cells in vitro, inducing apoptosis [85]. Owing to the pH-sensitive profile of FHV particles and PEGylated surface, drug release occurred in a controlled, endosome-specific manner, triggered by the acidic endosomal environment.
Another drug delivery system enabling pH-responsive cargo release was constructed from Physalis mottle virus (PhMV) VLPs [88]. These VLPs are composed of 180 identical capsid protein subunits that self-assemble into ~30 nm icosahedral particles. The uniform size, small dimensions, biocompatibility and long circulation times in vivo make these particles well-suited for drug delivery applications [89]. Furthermore, the capsid crystal structure reveals four solvent-exposed lysines on the exterior and one internal solvent-exposed cysteine, allowing site-specific modification. PhMV VLPs were loaded with the prodrug 6-maleimidocaproyl-hydrazone doxorubicin (DOX-EMCH) via thiol-maleimide conjugation to the interior addressable cysteine and PEGylated through NHS esterification of exterior lysins using methoxy-PEG N-hydroxysuccinimide (mPEG-NHS), to prevent non-specific uptake and improve biocompatibility. The inclusion of the acid-sensitive hydrazine linker EMCH allowed controlled release of DOX under acidic conditions, mimicking those of the tumor microenvironment or acidic endosomal/lysosomal compartments. DOX-PhMV-PEG particles induced cytotoxicity and DNA damage across a panel of cancer cell lines and exhibited significantly greater efficacy in vivo compared to free DOX in a breast tumor mouse model, achieving a 3.4-fold higher therapeutic efficacy.
Certain VLPs possess inherent tumor-homing capabilities that can be directly exploited for targeted drug delivery without the need for additional modifications. One such example is the Potato Virus X (PVX) VLP, which has been harnessed for targeted delivery to Non-Hodgkin’s B cell lymphomas [45]. PVX is a filamentous plant virus with demonstrated tumor-homing properties in tumor-bearing mice and preferential accumulation within B cell follicles [90]. This property has been explored for targeted delivery of the cytotoxic drug MMAE to malignant B cells, without introducing external targeting motifs. Specifically, vcMMAE was conjugated to surface-exposed cysteine residues on Potato virus X (PVX) VLPs using thiol-maleimide chemistry. The resulting PVX-vcMMAE conjugates demonstrated selective binding to CD45+ Raji B lymphoma cells and exhibited significantly enhanced cytotoxicity compared to free vcMMAE. In vivo studies in a Raji B cell lymphoma mouse model revealed that PVX-vcMMAE treatment led to tumor growth inhibition and improved survival, with no observed toxicity to normal B cells [35]. These findings underscore the potential of PVX VLPs as a platform for targeted delivery of cytotoxic agents to B cell malignancies.
A VLP-based delivery system has also been harnessed for image-guided chemotherapy, combining PET–fluorescence dual imaging and convection-enhanced delivery (CED) of epirubicin (EPI) in brain tumor therapy [91]. The base vehicle for this system is the Bacteriophage Qβ-derived VLP, whose capsid proteins were co-expressed with green fluorescent protein (GFP) to generate GFP-encapsulated Qβ virus-like particles (gVLPs). These gVLPs were surface modified with a cell penetrating peptide (CPP) to enhance cell uptake efficiency, and passively loaded with EPI under physiological conditions. DOTA moieties were conjugated for radiolabeling with 68Ga, enabling in vivo tracking via PET imaging alongside intrinsic fluorescence. CED delivery of EPI@CPP-gVLPs enabled broader intratumoral distribution and significantly reduced neurotoxicity compared to free EPI in mice brain, attributed to the stable encapsulation and CPP-controlled release of the drug. Repeated intracranial administration of EPI@CPP-gVLPs through CED infusion in tumor-bearing mice eradicated tumors without recurrence or serious brain damage, demonstrating the potential of this platform as a CED infusate for safe and effective local chemotherapy.
In another example, a papillomavirus-like particle-drug conjugate, named AU-011, has advanced to clinical evaluation for multiple tumor types. AU-011 is a drug-VLP system consisting of recombinantly expressed human papillomavirus (HPV) capsid proteins L1 and L2, conjugated to a phthalocyanine photosensitizer, IRDye 700DX, which exerts its cytotoxic effect upon photoactivation with near-infrared light (NIR) [92]. HPV capsids naturally interact with modified heparan sulfate proteoglycans (HSPGs), which have been shown to be a primary factor required for HPV entry into cells [93]. These modifications are typically restricted to basement membranes in normal tissues but become aberrantly exposed on the surface of many tumors. This tumor-tropic feature enables HPV VLPs to target a broad spectrum of tumor types without requiring additional modifications [93]. Effectively, AU-011 is currently in clinical trials for multiple types of cancer, including small choroidal melanoma (ClinicalTrials.gov: NCT06007690), bladder cancer (ClinicalTrials.gov: NCT05483868) and choroidal metastasis from breast or lung primary tumors (ClinicalTrials.gov: NCT06643884). Further, AU-011 itself may serve as an additional immunogenic catalyst within the tumor; In uveal melanoma cell lines, NIR-activated AU-011 induces calreticulin and HSP90 membrane exposure, indicative of its ability to trigger immunogenic cell death (ICD) in addition to direct cytotoxicity [94]. In another study, a single in vivo dose followed by NIR further promoted ICD hallmarks and immune activation in the tumor microenvironment, effects that were amplified by co-administration of checkpoint inhibitors, highlighting AU-011’s potential in combination immunotherapy [95].
5. Challenges and Future Directions
Extensive efforts have been directed toward engineering VLPs with tunable drug release profiles, enhanced targeting, and VLP transfer across various physiological barriers. Intrinsically, VLPs possess several features that may help address key obstacles in the clinical translation of cancer nanomedicine, particularly those encountered by synthetic nanocarriers. Their diversity in structure, surface properties, and tropism provides a versatile toolbox for precision chemotherapeutic delivery. Beyond single-agent delivery, recent studies have highlighted the capacity of VLPs to co-deliver chemotherapeutics with synergistic payloads, such as siRNA or immunomodulatory proteins, to enhance antitumor efficacy [96,97].
Building on VLPs intrinsic advantages, incorporating material science strategies can further enhance VLP capabilities. Reinforcement of VLP architecture using synthetic polymers has emerged as a promising route to fine-tune physicochemical properties, where strategies such as surface cross-linking can increase VLP resilience under harsh conditions, including acidic pH, enzymatic degradation, and mechanical stress [98]. PEGylation remains the most widely used ‘stealth’ modification, reducing immunogenicity and prolonging circulation time by shielding the particle surface from opsonization [99]. Some alternative hydrophilic polymers, such as poly(2-oxazoline)s and zwitterionic coatings, are being explored to achieve comparable stealth performance while avoiding PEG-associated drawbacks [100]. Internally, polymeric cross-linkers and controlled polymerization reactions have been employed to stabilize the capsid scaffold and support higher-density small molecule cargo loading without compromising structural integrity [101,102].
Complementary to these advances, in silico tools may gain a prominent role in VLP design and translational efforts. Molecular modeling and molecular dynamics simulations enable assessment of nanoparticle structural stability under diverse conditions while providing atomistic insight into nano-bio and nano-drug interactions that are difficult to capture experimentally [103,104]. Computational approaches have already been applied to guide VLP design: for example, iterative histidine-scanning and folding free-energy calculations have been used to design pH-responsive P22 capsids that assemble and disassemble in a controlled, pH-dependent manner [104]. More broadly, AI-driven tools hold promise for advancing VLP research and development, supporting capsid structure prediction, analysis of assembly pathways, image-based particle characterization, and optimization of production and purification processes [105].
Although VLPs offer high modularity and apparent biocompatibility for drug delivery, their innate biological properties can pose significant challenges. Native tropism may drive unplanned interactions with host cell receptors, while recognition by the immune system can elicit immunogenic responses that compromise targeting specificity, circulation time and therapeutic efficacy.
A compelling case is that of cowpea mosaic virus (CPMV), a plant virus frequently used in nanomedical applications due to its stability and ease of functionalization. Despite its botanical origin, CPMV demonstrates a notable capacity to associate with mammalian cells [106]. This unexpected interaction has been linked to the presence of vimentin on the surface of certain human cells, including cancer cells and activated endothelial cells [107,108]. Vimentin, typically a cytoskeletal protein, can under some physiological and pathological conditions translocate to the cell surface, where it becomes accessible to circulating particles. The interaction between CPMV and surface-expressed vimentin facilitates uptake into non-target mammalian cells, raising concerns about off-target biodistribution. While this property can be harnessed for applications that aim to exploit surface vimentin as a biomarker, such as tumor targeting, it may compromise specificity in therapies that require strict tissue selectivity or immune stealth. To address this, researchers have explored surface engineering strategies such as PEGylation, which effectively reduces nonspecific interactions of CPMV with mammalian cells [109]. While not directly evaluated, this likely includes vimentin-mediated interactions. The extent of suppression depends on PEG density, with coverage above 80 PEG500 or 60 PEG2000 chains per particle shown to significantly minimize off-target binding [109]. Understanding and managing native tropism is thus critical to maximizing the therapeutic potential of VLPs while mitigating risks related to off-target effects.
Despite the strong preclinical performance and growing sophistication of VLP-based chemotherapeutic delivery systems, only one VLP platform has advanced into clinical trials, revealing challenges in the translation of this technology. This gap is not unique to VLPs as antitumor nanotherapeutics but reflects broader obstacles across cancer nanomedicine. Effective clinical translation requires an integrated approach that accounts for manufacturability, scalability, and regulatory constraints from the earliest stages of nanoparticle design [110,111].
A major bottleneck is the ability to scale production in a cost-effective and reproducible manner, both for nanomedicines in general and for VLPs in particular [112]. While the biomimicry of native viral architectures confers advantages such as biocompatibility and biodegradability, these biological origins also introduce challenges related to their structural heterogeneity and complex purification from host-derived contaminants. The production of VLPs relies on complex cell culture and protein expression systems leading to comparatively high manufacturing costs [112]. Enhancing production efficiency and reducing expenses are pivotal for clinical application. As VLPs evolve toward increasingly complex, multicomponent architectures, design choices must therefore anticipate downstream constraints. The synthetic and regulatory burdens associated with nanoparticle multifunctionality have been widely discussed, suggesting that inherently versatile, minimalist designs may offer a more direct path to clinical translation [113,114,115]. Notably, the investigational VLP-drug conjugate AU-011 exemplifies this principle, employing a base viral vehicle with inherent tumor-tropic properties and a single conjugated chemotherapeutic agent. These considerations highlight key design parameters governing VLP biomaterial performance (Table 2).
6. Conclusions
Programmable virus-like particles represent a powerful and adaptable platform for precision chemotherapy, offering fine control over drug loading, targeting, and release while retaining the advantageous biological features of viral architectures. Advances in capsid engineering and site-specific functionalization have enabled VLPs to overcome key limitations of conventional chemotherapeutic delivery, including poor selectivity and systemic toxicity. Nonetheless, limitations related to native tropism, immune recognition, and scalable manufacturing reinforce the need for mechanism-driven design frameworks that anticipate translational constraints. Clinical progress exemplified by AU-011 suggests that exploiting intrinsic viral properties through minimalist engineering may provide a more viable route to translation than over more complex nanocarrier designs.
Future success in VLP-based chemotherapy will depend on integrating molecular programmability with manufacturability and regulatory feasibility, positioning VLPs as a unifying platform at the interface of nanomedicine, synthetic biology, and precision oncology.
Funding
This work was financed by national funds from FCT - Fundação para a Ciência e a Tecnologia, I.P., under the scope of the project UID/50006/2023 of the Associate Laboratory for Green Chemistry - LAQV REQUIMTE and UID/Multi/04349/2020 (DOI: 10.54499/UIDB/04349/2020) of the Center of Nuclear Sciences and Technology (C2TN) from University of Lisbon.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this review.
Acknowledgments
This work was financed by national funds from FCT - Fundação para a Ciência e a Tecnologia, I.P., under the scope of the project UID/50006/2023 of the Associate Laboratory for Green Chemis-try - LAQV REQUIMTE and UID/Multi/04349/2020 (DOI: 10.54499/UIDB/04349/2020) of the Center of Nuclear Sciences and Technology (C2TN) from University of Lisbon.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CCMV | Cowpea chlorotic mottle virus |
| CED | Convection-enhanced delivery |
| CP | Capsid protein |
| CPMV | Cowpea mosaic virus |
| CPP | Cell penetrating peptide |
| CuAAC | Copper-catalyzed azide–alkyne cycloaddition |
| DDS | Drug delivery system |
| Dec | Decoration protein |
| DOX | Doxorubicin |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EGFR | Epithelial growth factor receptor |
| EMCH | 6-maleimidocaproyl-hydrazone |
| EPI | Epirubicin |
| EPT | Ellipticin |
| FA | Folic acid |
| FHV | Flock house virus |
| GFP | Green fluorescent protein |
| HBcAg | Hepatitis B virus core antigen |
| HER2 | Human epidermal growth factor 2 |
| HIV-1 | Human immunodeficiency virus 1 |
| HPV | Human papillomavirus |
| HSPG | Heparan sulfate proteoglycan |
| ICD | Immunogenic cell death |
| MMAE | Monomethyl auristatin |
| NHS | N-hydroxysulfosuccinimide |
| NIR | Near-infrared light |
| PEG | Polyethylene glycol |
| PhMV | Physalis mottle virus |
| PVX | Potato virus X |
| RGD | Arginylglycylaspartic acid |
| scFv | Single-chain variable fragment |
| siRNA | Small interfering RNA |
| SV40 | Simian virus 40 |
| TMV | Tobacco mosaic virus |
| UAA | Unnatural amino acid |
| VLP | Virus-like particle |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN.
- Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263.
- OECD and European Commission EU country cancer profiles synthesis report 2025. OECD Publishing; Paris, France, 2025. [Google Scholar]
- Global cancer burden growing, amidst mounting need for services. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 3rd of March 2026).
- Shahrivarkevishahi, A.; Hagge, L.M.; Brohlin, O.R.; Kumari, S.; Ehrman, R.; Benjamin, C.; Gassensmith, J.J. Virus-like particles: a self-assembled toolbox for cancer therapy. Mater. Today Chem. 2022, 24, 100808. [Google Scholar]
- Chung, Y.H.; Cai, H.; Steinmetz, N. F. Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv. Drug Deliv. Rev. 2020, 156, 214–235. [Google Scholar] [CrossRef]
- Kim, K.R.; Lee, A.S.; Kim, S.M.; Heo, H.R.; Kim, C.S. Virus-like nanoparticles as a theranostic platform for cancer. Front. Bioeng. Biotechnol. 2023, 10, 1106767. [Google Scholar] [CrossRef]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnology 2021, 19, 59. [Google Scholar] [CrossRef]
- Kim, K.R.; Lee, A.S.; Kim, S.M.; Heo, H.R.; Kim, C.S. Virus-like nanoparticles as a theranostic platform for cancer. Front. Bioeng. Biotechnol. 2023, 10, 1106767. [Google Scholar] [CrossRef]
- Yuan, B.; Liu, Y.; Lv, M.; Sui, Y.; Hou, S.; Yang, T.; Belhadj, Z.; Zhou, Y.; Chang, N.; Ren, Y.; Sun, C. Virus-like particle-based nanocarriers as an emerging platform for drug delivery. J. Drug Target 2023, 31, 433–455. [Google Scholar] [PubMed]
- Le, D.T.; Müller, K.M. In Vitro Assembly of Virus-Like Particles and Their Applications. Life (Basel) 2021, 11 11, 334. [Google Scholar]
- Alvandi, N.; Rajabnejad, M.; Taghvaei, Z.; Esfandiari, N. New generation of viral nanoparticles for targeted drug delivery in cancer therapy. J. Drug Target 2022, 30, 151–165. [Google Scholar] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 13 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kreutzberger, A. J. B.; Odongo, L.; Nelson, E. A.; Nyenhuis, D. A.; Kiessling, V.; Liang, B.; Cafiso, D. S.; White, J. M.; Tamm, L. K. Ebola virus glycoprotein interacts with cholesterol to enhance membrane fusion and cell entry. Nat. Struct. Mol. Biol. 2021, 28, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Nkanga, C.I.; Steinmetz, N.F. The pharmacology of plant virus nanoparticles. Virology 2021, 15 556, 39–61. [Google Scholar] [CrossRef]
- Chariou, P. L.; Lee, K. L.; Pokorski, J. K.; Saidel, G. M.; Steinmetz, N. F. Diffusion and Uptake of Tobacco Mosaic Virus as Therapeutic Carrier in Tumor Tissue: Effect of Nanoparticle Aspect Ratio. J. Phys. Chem. B. 2016, 120, 6120–6129. [Google Scholar]
- Ikwuagwu, B.; Tullman-Ercek, D. Virus-like particles for drug delivery: a review of methods and applications. Curr. Opin. Biotechnol. 2022, 78, 102785. [Google Scholar] [CrossRef]
- Berardi, A.; Bombelli, F.B.; Thuenemann, E.C.; Lomonossoff, G.P. Viral nanoparticles can elude protein barriers: exploiting rather than imitating nature. Nanoscale 2019, 11, 2306–2316. [Google Scholar] [CrossRef]
- Zackova Suchanova, J.; Hejtmankova, A.; Neburkova, J.; Cigler, P.; Forstova, J.; Spanielova, H. The Protein Corona Does Not Influence Receptor-Mediated Targeting of Virus-like Particles. Bioconjug. Chem. 2020, 31, 1575–1585. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small 2013, 9, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- McNeale, D.; Dashti, N.; Cheah, L. C.; Sainsbury, F. Protein cargo encapsulation by virus-like particles: Strategies and applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2023, 15, e1869. [Google Scholar] [CrossRef] [PubMed]
- Pistono, P.E.; Xu, J.; Huang, P.; Fetzer, J.L.; Francis, M.B. Exploring the Effects of Intersubunit Interface Mutations on Virus-Like Particle Structure and Stability. Biochemistry 2024, 63, 1913–1924. [Google Scholar] [CrossRef]
- Biela, A. P.; Naskalska, A.; Fatehi, F.; Twarock, R.; Heddle, J. G. Programmable polymorphism of a virus-like particle. Commun. Mater. 2022, 3, 7. [Google Scholar] [CrossRef]
- Schumacher, J.; Bacic, T.; Staritzbichler, R.; Daneschdar, M.; Klamp, T.; Arnold, P.; Jägle, S.; Türeci, Ö.; Markl, J.; Sahin, U. Enhanced stability of a chimeric hepatitis B core antigen virus-like-particle (HBcAg-VLP) by a C-terminal linker-hexahistidine-peptide. J. Nanobiotechnology 2018, 16, 39. [Google Scholar] [CrossRef]
- Xu, C.; Zhu, W.; Mao, H.; Zhang, W.; Yin, G. Q.; Zhang, X. E.; Li, F. Switch from Polymorphic to Homogenous Self-Assembly of Virus-Like Particles of Simian Virus 40 through Double-Cysteine Substitution. Small 2020, 16, e2004484. [Google Scholar] [CrossRef]
- de Ruiter, M. V.; van der Hee, R. M.; Driessen, A. J. M.; Keurhorst, E. D.; Hamid, M.; Cornelissen, J. J. L. M. Polymorphic assembly of virus-capsid proteins around DNA and the cellular uptake of the resulting particles. J. Control. Release 2019, 307, 342–354. [Google Scholar] [CrossRef]
- Shukla, S.; Eber, F. J.; Nagarajan, A. S.; DiFranco, N. A.; Schmidt, N.; Wen, A. M.; Eiben, S.; Twyman, R. M.; Wege, C.; Steinmetz, N. F. The Impact of Aspect Ratio on the Biodistribution and Tumor Homing of Rigid Soft-Matter Nanorods. Adv. Healthc. Mater. 2015, 4, 874–882. [Google Scholar] [CrossRef]
- Bruckman, M. A.; Randolph, L. N.; VanMeter, A.; Hern, S.; Shoffstall, A. J.; Taurog, R. E.; Steinmetz, N. F. Biodistribution, pharmacokinetics, and blood compatibility of native and PEGylated tobacco mosaic virus nano-rods and -spheres in mice. Virology 2014, 449, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Ablack, A. L.; Wen, A. M.; Lee, K. L.; Lewis, J. D.; Steinmetz, N. F. Increased tumor homing and tissue penetration of the filamentous plant viral nanoparticle Potato virus X. Mol. Pharm. 2013, 10, 33–42. [Google Scholar] [CrossRef]
- Finbloom, J. A.; Aanei, I. L.; Bernard, J. M.; Klass, S. H.; Elledge, S. K.; Han, K.; Ozawa, T.; Nicolaides, T. P.; Berger, M. S.; Francis, M. B. Evaluation of Three Morphologically Distinct Virus-Like Particles as Nanocarriers for Convection-Enhanced Drug Delivery to Glioblastoma. Nanomaterials (Basel) 2018, 8, 1007. [Google Scholar] [CrossRef] [PubMed]
- Raguram, A.; An, M.; Chen, P. Z.; Liu, D. R. Directed evolution of engineered virus-like particles with improved production and transduction efficiencies. Nat. Biotechnol. 2025, 43, 1635–1647. [Google Scholar] [CrossRef]
- Strugała, A.; Jagielski, J.; Kamel, K.; Nowaczyk, G.; Radom, M.; Figlerowicz, M.; Urbanowicz, A. Virus-Like Particles Produced Using the Brome Mosaic Virus Recombinant Capsid Protein Expressed in a Bacterial System. Int. J. Mol. Sci. 2021, 22, 3098. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Guenther, R. H.; Sit, T. L.; Opperman, C. H.; Lommel, S. A.; Willoughby, J. A. Loading and release mechanism of red clover necrotic mosaic virus derived plant viral nanoparticles for drug delivery of doxorubicin. Small 2014, 10, 5126–5136. [Google Scholar] [CrossRef]
- Wong, S. M.; Ren, Y. In Vitro-Reassembled Plant Virus-Like Particles of Hibiscus Chlorotic Ringspot Virus (HCRSV) as Nano-Protein Cages for Drugs. Methods Mol. Bio 2018, 1776, 229–236. [Google Scholar]
- Hu, L.; Salmen, W.; Chen, R.; Zhou, Y.; Neill, F.; Crowe, J. E., Jr.; Atmar, R. L.; Estes, M. K.; Prasad, B. V. V. Atomic structure of the predominant GII.4 human norovirus capsid reveals novel stability and plasticity. Nat. Commun. 2022, 13, 1241. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N. T.; Hesketh, E. L.; Saxena, P.; Meshcheriakova, Y.; Ku, Y. C.; Hoang, L. T.; Johnson, J. E.; Ranson, N. A.; Lomonossoff, G. P.; Reddy, V. S. Crystal Structure and Proteomics Analysis of Empty Virus-like Particles of Cowpea Mosaic Virus. Structure 2016, 24, 567–575. [Google Scholar] [CrossRef]
- Collett, S.; Torresi, J.; Earnest-Silveira, L.; Christiansen, D.; Elbourne, A.; Ramsland, P. A. Probing and pressing surfaces of hepatitis C virus-like particles. J. Colloid Interface Sci. 2019, 545, 259–268. [Google Scholar] [CrossRef]
- McCormick, R. A.; Ralbovsky, N. M.; Gilbraith, W.; Smith, J. P.; Booksh, K. S. Analyzing atomic force microscopy images of virus-like particles by expectation-maximization. NPJ Vaccines 2014, 9, 112. [Google Scholar]
- Wu, C. Y.; Yeh, Y. C.; Yang, Y. C.; Chou, C.; Liu, M. T.; Wu, H. S.; Chan, J. T.; Hsiao, P. W. Mammalian Expression of Virus-Like Particles for Advanced Mimicry of Authentic Influenza Virus. PLoS One 2010, 5, e9784. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Tover, A.; Berger, E.; Jungbauer, A. Quantification and characterization of virus-like particles by size-exclusion chromatography and nanoparticle tracking analysis. J. Chromatogr. A. 2017, 1487, 89–99. [Google Scholar] [PubMed]
- Murali, S.; Rustandi, R. R.; Zheng, X.; Payne, A.; Shang, L. Applications of Surface Plasmon Resonance and Biolayer Interferometry for Virus–Ligand Binding. Viruses 2022, 14, 717. [Google Scholar] [CrossRef]
- Zhang, L.; Lua, L.H.L.; Middelberg, A.P.J.; Sun, Y.; Connors, N.K. Biomolecular engineering of virus-like particles aided by computational chemistry methods. Chem. Soc. Rev. 2015, 44, 8608–8618. [Google Scholar] [CrossRef]
- Elrad, O. M.; Hagan, M. F. Mechanisms of Size Control and Polymorphism in Viral Capsid Assembly. Nano Lett. 2008, 8, 3850–3857. [Google Scholar] [CrossRef]
- Luo, H.; Ma, Y.; Ren, Y.; Li, Z.; Sheng, Y.; Wang, Y.; Su, Z.; Bi, J.; Zhang, S. Study of self-assembling properties of HBc-VLP derivatives aided by molecular dynamic simulations from a thermodynamic perspective. J. Biomol. Struct. Dyn. 2024, 42, 12822–12835. [Google Scholar] [CrossRef] [PubMed]
- Wijesundara, Y. H.; Herbert, F. C.; Kumari, S.; Howlett, T.; Koirala, S.; Trashi, O.; Trashi, I.; Al-Kharji, N. M.; Gassensmith, J. J. Rip it, stitch it, click it: A chemist’s guide to VLP manipulation. Virology 2022, 577, 105–123. [Google Scholar] [CrossRef]
- Shukla, S.; Roe, A. J.; Liu, R.; Veliz, F. A.; Commandeur, U.; Wald, D. N.; Steinmetz, N. F. Affinity of plant viral nanoparticle potato virus x (PVX) towards malignant b cells enables cancer drug delivery. Biomater. Sci. 2020, 8, 3935–3943. [Google Scholar] [CrossRef]
- Li, M.; Liu, Z.; Wang, D.; Ye, J.; Shi, Z.; Pan, C.; Zhang, Q.; Ju, R.; Zheng, Y.; Liu, Y. Intraocular mRNA delivery with endogenous MmPEG10-based virus-like particles. Exp. Eye Res. 2024, 243, 109899. [Google Scholar] [CrossRef]
- Gan, B. K.; Rullah, K.; Yong, C. Y.; Ho, K. L.; Omar, A. R.; Alitheen, N. B.; Tan, W. S. Targeted delivery of 5-fluorouracil-1-acetic acid (5-FA) to cancer cells overexpressing epithelial growth factor receptor (EGFR) using virus-like nanoparticles. Sci. Rep. 2020, 10, 16867. [Google Scholar] [CrossRef]
- Thong, Q. X.; Biabanikhankahdani, R.; Ho, K. L.; Alitheen, N. B.; Tan, W. S. Thermally-responsive virus-like particle for targeted delivery of cancer drug. Sci. Rep. 2019, 9, 3945. [Google Scholar] [CrossRef] [PubMed]
- Hovlid, M. L.; Steinmetz, N. F.; Laufer, B.; Lau, J. L.; Kuzelka, J.; Wang, Q.; Hyypiä, T.; Nemerow, G. R.; Kessler, H.; Manchester, M.; Finn, M. G. Guiding Plant Virus Particles to Integrin-Displaying Cells. Nanoscale 2012, 4, 3698–3705. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Liu, A. P.; Voss, N. R.; Schmid, S. L.; Finn, M. G. Multivalent Display and Receptor-Mediated Endocytosis of Transferrin on Virus-Like Particles. Chembiochem 2010, 11, 1273–1279. [Google Scholar] [CrossRef]
- Charoenphol, P.; Bermudez, H. Aptamer-targeted DNA nanostructures for therapeutic delivery. Mol. Pharm. 2014, 11, 1721–1725. [Google Scholar] [CrossRef]
- Martins, S. A.; Santos, J.; Cabo Verde, S.; Correia, J. D. G.; Melo, R. Construction of HER2-specific HIV-1-based VLPs. Bioengineering (Basel) 2022, 9, 713. [Google Scholar] [CrossRef]
- Wang, G.; Jia, T.; Xu, X.; Chang, L.; Zhang, R.; Fu, Y.; Li, Y.; Yang, X.; Zhang, K.; Lin, G.; Han, Y.; Li, J. Novel miR-122 delivery system based on MS2 virus like particle surface displaying cell-penetrating peptide TAT for hepatocellular carcinoma. Oncotarget 2016, 7, 59402–59416. [Google Scholar] [CrossRef]
- Muñoz-Morris, M. A.; Heitz, F.; Divita, G.; Morris, M. C. The peptide carrier Pep-1 forms biologically efficient nanoparticle complexes. Biochem. Biophys. Res. Commun. 2007, 355, 877–882. [Google Scholar] [CrossRef]
- Liu, B. R.; Lo, S. Y.; Liu, C. C.; Chyan, C. L.; Huang, Y. W.; Aronstam, R. S.; Lee, H. J. Endocytic Trafficking of Nanoparticles Delivered by Cell-penetrating Peptides Comprised of Nona-arginine and a Penetration Accelerating Sequence. PLoS One 2013, 8, e67100. [Google Scholar] [CrossRef]
- Zhao, L.; Kopylov, M.; Potter, C.S.; Carragher, B.; Finn, M.G. Engineering the pp7 virus capsid as a peptide display platform. ACS Nano 2019, 13, 4443–4454. [Google Scholar] [CrossRef]
- Dang, M.; Wu, L. J.; Zhang, S. R.; Zhu, J. R.; Hu, Y. Z.; Yang, C. X.; Zhang, X. Y. Ms2 virus-like particles as a versatile peptide presentation platform: Insights into the deterministic abilities for accommodating heterologous peptide lengths. ACS Synth. Biol. 2023, 12, 3704–3715. [Google Scholar] [CrossRef]
- Essus, V. A.; Souza Júnior, G. S. E.; Nunes, G. H. P.; Oliveira, J. D. S.; de Faria, B. M.; Romão, L. F.; Cortines, J. R. Bacteriophage P22 capsid as a pluripotent nanotechnology tool. Viruses 2023, 15, 516. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J. O.; Vaca, L. Interaction between virus-like particles (VLPs) and pattern recognition receptors (PRRs) from dendritic cells (DCs): Toward better engineering of VLPs. Front Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Smith, M. L.; Lindbo, J. A.; Dillard-Telm, S.; Brosio, P. M.; Lasnik, A. B.; McCormick, A. A.; Nguyen, L. V.; Palmer, K. E. Modified tobacco mosaic virus particles as scaffolds for display of protein antigens for vaccine applications. Virology 2006, 348, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Peyret, H.; Gehin, A.; Thuenemann, E. C.; Blond, D.; El Turabi, A.; Beales, L.; Clarke, D.; Gilbert, R. J.; Fry, E. E.; Stuart, D. I.; Holmes, K.; Stonehouse, N. J.; Whelan, M.; Rosenberg, W.; Lomonossoff, G. P.; Rowlands, D. J. Tandem fusion of hepatitis B core antigen allows assembly of virus-like particles in bacteria and plants with enhanced capacity to accommodate foreign proteins. PLoS One 2015, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lino, C. A.; Caldeira, J. C.; Peabody, D. S. Display of single-chain variable fragments on bacteriophage MS2 virus-like particles. J. Nanobiotechnology 2017, 15, 13. [Google Scholar] [CrossRef]
- Grataitong, K.; Huault, S.; Chotwiwatthanakun, C.; Jariyapong, P.; Thongsum, O.; Chawiwithaya, C.; Chakrabandhu, K.; Hueber, A. O.; Weerachatyanukul, W. Chimeric virus-like particles (VLPs) designed from shrimp nodavirus (MrNV) capsid protein specifically target EGFR-positive human colorectal cancer cells. Sci. Rep. 2021, 11, 16579. [Google Scholar]
- Martino, M. L.; Crooke, S. N.; Manchester, M.; Finn, M. G. Single-point mutations in Qβ virus-like particles change binding to cells. Biomacromolecules 2021, 22, 3332–3341. [Google Scholar] [CrossRef]
- Servid, A.; Jordan, P.; O’Neil, A.; Prevelige, P.; Douglas, T. Location of the bacteriophage P22 coat protein C-terminus provides opportunities for the design of capsid-based materials. Biomacromolecules 2013, 14, 2989–2995. [Google Scholar] [CrossRef]
- Pokorski, J. K.; Steinmetz, N. F. The art of engineering viral nanoparticles. Mol. Pharm. 2011, 8, 29–43. [Google Scholar]
- Biabanikhankahdani, R.; Ho, K. L.; Alitheen, N. B.; Tan, W. S. A dual bioconjugated virus-like nanoparticle as a drug delivery system and comparison with a pH-responsive delivery system. Nanomaterials (Basel) 2018, 8, 236. [Google Scholar] [CrossRef]
- Edwardson, T. G. W.; Levasseur, M. D.; Tetter, S.; Steinauer, A.; Hori, M.; Hilvert, D. Protein cages: From fundamentals to advanced applications. Chem. Rev. 2022, 122, 9145–9197. [Google Scholar] [CrossRef] [PubMed]
- Chen, P. L.; Wang, M.; Ou, W. C.; Lii, C. K.; Chen, L. S.; Chang, D. Disulfide bonds stabilize JC virus capsid-like structure by protecting calcium ions from chelation. FEBS Lett. 2001, 500, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Finbloom, J. A.; Han, K.; Aanei, I. L.; Hartman, E. C.; Finley, D. T.; Dedeo, M. T.; Fishman, M.; Downing, K. H.; Francis, M. B. Stable disk assemblies of a tobacco mosaic virus mutant as nanoscale scaffolds for applications in drug delivery. Bioconjug. Chem. 2016, 27, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chariou, P. L.; Steinmetz, N. F. Site-specific antibody conjugation strategy to functionalize virus-based nanoparticles. Bioconjug. Chem. 2020, 31, 1408–1416. [Google Scholar] [CrossRef]
- Strable, E.; Prasuhn, D. E., Jr.; Udit, A. K.; Brown, S.; Link, A. J.; Ngo, J. T.; Lander, G.; Quispe, J.; Potter, C. S.; Carragher, B.; Tirrell, D. A.; Finn, M. G. Unnatural amino acid incorporation into virus-like particles. Bioconjug. Chem. 2009, 19, 866–875. [Google Scholar] [CrossRef]
- Patel, K. G.; Swartz, J. R. Surface functionalization of virus-like particles by direct conjugation using azide–alkyne click chemistry. Bioconjug. Chem. 2011, 22, 376–387. [Google Scholar]
- Chen, Z.; Li, N.; Li, S.; Dharmarwardana, M.; Schlimme, A.; Gassensmith, J. J. Viral chemistry: the chemical functionalization of viral architectures to create new technology. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 512–534. [Google Scholar] [CrossRef]
- Jewett, J. C.; Bertozzi, C. R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef]
- Patterson, D.; Schwarz, B.; Avera, J.; Western, B.; Hicks, M.; Krugler, P.; Terra, M.; Uchida, M.; McCoy, K.; Douglas, T. Sortase-Mediated Ligation as a Modular Approach for the Covalent Attachment of Proteins to the Exterior of the Bacteriophage P22 Virus-like Particle. Bioconjug. Chem. 2017, 28, 2114–2124. [Google Scholar] [CrossRef]
- Brune, K. D.; Leneghan, D. B.; Brian, I. J.; Ishizuka, A. S.; Bachmann, M. F.; Draper, S. J.; Biswas, S.; Howarth, M. Plug-and-Display: decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef]
- Rohovie, M. J.; Nagasawa, M.; Swartz, J. R. Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioeng. Transl. Med. 2017, 2, 43–57. [Google Scholar] [PubMed]
- Schwarz, B.; Madden, P.; Avera, J.; Gordon, B.; Larson, K.; Miettinen, H. M.; Uchida, M.; LaFrance, B.; Basu, G.; Rynda-Apple, A.; Douglas, T. Symmetry Controlled, Genetic Presentation of Bioactive Proteins on the P22 Virus-like Particle Using an External Decoration Protein. ACS Nano 2015, 9, 9134–9147. [Google Scholar] [CrossRef]
- Koho, T.; Ihalainen, T. O.; Stark, M.; Uusi-Kerttula, H.; Wieneke, R.; Rahikainen, R.; Blazevic, V.; Marjomäki, V.; Tampé, R.; Kulomaa, M. S.; Hytönen, V. P. His-tagged norovirus-like particles: A versatile platform for cellular delivery and surface display. Eur. J. Pharm. Biopharm. 2015, 96, 22–31. [Google Scholar]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery (Oxford) 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Gala, U. H.; Miller, D. A.; Williams, R. O., 3rd. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188319. [Google Scholar] [PubMed]
- Khan, S. U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Comun. Signal 2024, 22, 109. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical cancer nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, M. A smart viral vector for targeted delivery of hydrophobic drugs. Sci. Rep. 2021, 11, 7030. [Google Scholar] [CrossRef] [PubMed]
- Odegard, A.; Banerjee, M.; Johnson, J. E. Flock house virus: A model system for understanding nonenveloped virus entry and membrane penetration. Curr. Top. Microbiol. Immunol. 2010, 343, 1–22. [Google Scholar] [PubMed]
- Song, N.; Zhao, L.; Zhu, M.; Zhao, J. Recent progress in LyP-1-based strategies for targeted imaging and therapy. Drug Deliv. 2019, 26, 363–375. [Google Scholar] [CrossRef]
- Hu, H.; Steinmetz, N. F. Doxorubicin-loaded physalis mottle virus particles function as a pH-responsive prodrug enabling cancer therapy. Biotechnol. J. 2020, 15, e2000077. [Google Scholar] [CrossRef]
- Hu, H.; Masarapu, H.; Gu, Y.; Zhang, Y.; Yu, X.; Steinmetz, N. F. Physalis mottle virus-like nanoparticles for targeted cancer imaging. ACS Appl Mater Interfaces 2019, 11, 18213–18223. [Google Scholar] [CrossRef]
- Shukla, S.; Wen, A. M.; Ayat, N. R.; Commandeur, U.; Gopalkrishnan, R.; Broome, A. M.; Lozada, K. W.; Keri, R. A.; Steinmetz, N. F. Biodistribution and clearance of a filamentous plant virus in healthy and tumor-bearing mice. Nanomedicine (Lond.) 2019, 9, 221–235. [Google Scholar] [CrossRef]
- Pang, H. H.; Chen, P. Y.; Wei, K. C.; Huang, C. W.; Shiue, Y. L.; Huang, C. Y.; Yang, H. W. Convection-enhanced delivery of a virus-like nanotherapeutic agent with dual-modal imaging for besiegement and eradication of brain tumors. Theranostics 2019, 9, 752–1763. [Google Scholar] [CrossRef]
- Kines, R. C.; Varsavsky, I.; Choudhary, S.; Bhattacharya, D.; Spring, S.; McLaughlin, R.; Kang, S. J.; Grossniklaus, H. E.; Vavvas, D.; Monks, S.; MacDougall, J. R.; de Los Pinos, E.; Schiller, J. T. An infrared dye–conjugated virus-like particle for the treatment of primary uveal melanoma. Mol. Cancer Ther. 2018, 17, 565–574. [Google Scholar] [CrossRef]
- Kines, R. C.; Cerio, R. J.; Roberts, J. N.; Thompson, C. D.; de Los Pinos, E.; Lowy, D. R.; Schiller, J. T. Human papillomavirus capsids preferentially bind and infect tumor cells. Int. J. Cancer 2016, 138, 901–911. [Google Scholar] [CrossRef]
- Ma, S.; Huis In’t Veld, R. V.; Houy, A.; Stern, M. H.; Rich, C.; Ossendorp, F. A.; Jager, M. J. In vitro testing of the virus-like drug conjugate belzupacap sarotalocan (AU-011) on uveal melanoma suggests BAP1-related immunostimulatory capacity. Invest. Ophthalmol. Vis. Sci. 2023, 64, 10. [Google Scholar] [CrossRef]
- Kines, R. C.; Thompson, C. D.; Spring, S.; Li, Z.; de Los Pinos, E.; Monks, S.; Schiller, J. T. Virus-like particle-drug conjugates induce protective, long-lasting adaptive anti-tumor immunity in the absence of specifically targeted tumor antigens. Cancer Immunol. Res. 2021, 9, 693–706. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Liu, Y.; Zhang, X.; Shan, W.; Ye, S.; Zhou, X.; Ge, Y.; Wang, X.; Ren, L. Nanoparticle-based co-delivery of siRNA and paclitaxel for dual-targeting of glioblastoma. Nanomedicine (Lond.) 2020, 15, 1391–1409. [Google Scholar] [CrossRef]
- Deo, V. K.; Kato, T.; Park, E. Y. Virus-like particles displaying recombinant short-chain fragment region and interleukin 2 for targeting colon cancer tumors and attracting macrophages. J. Pharm. Sci. 2016, 105, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ganguillet, S.; Turgay, Y.; Keys, T. G.; Causa, E.; Fradique, R.; Lutz-Bueno, V.; Chesnov, S.; Tan-Lin, C. W.; Lentsch, V.; Kotar, J.; Cicuta, P.; Mezzenga, R.; Slack, E.; Radiom, M. Surface cross-linking by macromolecular tethers enhances virus-like particles’ resilience to mucosal stress factors. ACS Nano 2024, 18, 3382–3396. [Google Scholar] [CrossRef] [PubMed]
- Owens, D. E., 3rd; Peppas, N. A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Beasock, D.; Fessler, A.; Szebeni, J.; Ljubimova, J.Y.; Afonin, K.A.; Dobrovolskaia, M.A. To PEGylate or not to PEGylate: immunological properties of nanomedicine’s most popular component, poly(ethylene) glycol and its alternatives. Adv Drug Deliv. Rev. 2021, 180, 114079. [Google Scholar] [CrossRef]
- Wu, Z.; Bayón, J. L.; Kouznetsova, T. B.; Ouchi, T.; Barkovich, K. J.; Hsu, S. K.; Craig, S. L.; Steinmetz, N. F. Virus-like particles armored by an endoskeleton. Nano Lett. 2024, 24, 2989–2997. [Google Scholar] [CrossRef]
- Lucon, J.; Qazi, S.; Uchida, M.; Bedwell, G. J.; LaFrance, B.; Prevelige, P. E., Jr.; Douglas, T. Use of the interior cavity of the p22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat. Chem. 2012, 4, 781–788. [Google Scholar] [CrossRef]
- Kim, K. J.; Kim, G.; Bae, J. H.; Song, J. J.; Kim, H. S. A pH-responsive virus-like particle as a protein cage for a targeted delivery. Adv. Healthc. Mater. 2024, 13, e2302656. [Google Scholar] [CrossRef]
- Luo, H.; Ma, Y.; Su, Z.; Gu, Y.; Zhang, S.; Gerstweiler, L. Investigating the stability of chimeric murine polyomavirus VP1 Capsomeres via molecular dynamics simulations and experimental analysis. Int. J. Biol. Macromol. 2025, 286, 138372. [Google Scholar] [CrossRef]
- Laxmi, B.; Devi, P. U. M.; Thanjavur, N.; Buddolla, V. The applications of artificial intelligence (AI)-driven tools in virus-like particles (VLPs) research. Curr. Microbiol. 2024, 81, 234. [Google Scholar] [CrossRef]
- Beatty, P. H.; Lewis, J. D. Cowpea mosaic virus nanoparticles for cancer imaging and therapy. Adv. Drug Deliv. Rev. 2019, 145, 130–144. [Google Scholar] [CrossRef]
- Koudelka, K. J.; Destito, G.; Plummer, E. M.; Trauger, S. A.; Siuzdak, G.; Manchester, M. Endothelial targeting of cowpea mosaic virus (CPMV) via surface vimentin. PLoS Pathog. 2009, 5, e1000417. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N. F.; Cho, C. F.; Ablack, A.; Lewis, J. D.; Manchester, M. Cowpea Mosaic Virus Nanoparticles Target Surface Vimentin on Cancer Cells. Nanomedicine (Lond.) 2011, 6, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N. F.; Ablack, A. L.; Hickey, J. L.; Ablack, J.; Manocha, B.; Mymryk, J. S.; Luyt, L. G.; Lewis, J. D. Intravital Imaging of Human Prostate Cancer Using Viral Nanoparticles Targeted to Gastrin-Releasing Peptide Receptors. Small 2011, 7, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Herdiana, Y. Bridging the Gap: The Role of Advanced Formulation Strategies in the Clinical Translation of Nanoparticle-Based Drug Delivery Systems. Int. J. Nanomedicine 2025, 20, 13039–13053. [Google Scholar] [CrossRef] [PubMed]
- Metselaar, J. M.; Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef]
- Bachmann, M. F.; van Damme, P.; Lienert, F.; Schwarz, T. F. Virus-like particles: a versatile and effective vaccine platform. Expert Rev. Vaccines 2025, 24, 444–456. [Google Scholar] [CrossRef]
- Huynh, E.; Zheng, G. Engineering multifunctional nanoparticles: all-in-one versus one-for-all. WIREs Nanomed. Nanobiotechnol. 2013, 5, 250–265. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, K.; Zheng, G. Nanomedicines Lost in Translation. ACS Nano 2019, 13, 13620–13626. [Google Scholar] [CrossRef] [PubMed]
- Gessner, I. Optimizing nanoparticle design and surface modification toward clinical translation. MRS Bull. 2021, 46, 643–649. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Functional components commonly integrated into virus-like particle–based protein biomaterials for chemotherapeutic drug delivery and targeting.
Table 1.
Functional components commonly integrated into virus-like particle–based protein biomaterials for chemotherapeutic drug delivery and targeting.
| Functional Group Type | Compounds | Function/Target | Application | Reference |
|---|---|---|---|---|
| Therapeutic Agents | Doxorubicin (DOX) | DNA intercalation; inhibits topoisomerase II | Chemotherapy | 29 |
| Monomethyl auristatin (MMAE) | Human B lymphoma cells targeting | Chemotherapy | 45 | |
| mRNA | Protein expression | Immunotherapy, gene therapy | 46 | |
| 5-fluorouracil-1-acetic acid | Binds epithelial growth factor receptor (EGFR) |
Chemotherapy | 47 | |
| Targeting Ligands | Folic Acid | Binds folate receptor | Targeting folate receptor–positive tumors | 48 |
| RGD peptides | Binds integrin αvβ3 | Anti-angiogenesis, tumor targeting | 49 | |
| Transferrin | Binds transferrin receptor | Targeting rapidly dividing cells | 50 | |
| Aptamer (e.g., AS1411) | Targets nucleolin | Selective tumor targeting | 51 | |
| Anti-HER2 antibody | Binds HER2 receptor | Targeting HER2-positive breast cancer | 52 | |
| Cell-Penetrating Peptides | TAT (HIV-derived) | Facilitates nuclear and cytosolic delivery | Intracellular delivery of drugs or genes | 53 |
| Pep-1 | Non-covalent cargo delivery | Protein or peptide delivery | 54 | |
| R9 (nona-arginine) | Promotes uptake | Enhanced internalization | 55 |
Table 2.
Design Parameters Governing Virus-Like Particle Biomaterial Performance in Chemotherapeutic Delivery.
Table 2.
Design Parameters Governing Virus-Like Particle Biomaterial Performance in Chemotherapeutic Delivery.
| Design Parameter | Biomaterial Feature | Controls | Design Trade-Off | References |
|---|---|---|---|---|
| Capsid geometry | Size and aspect ratio | Tumor penetration, circulation | Small particles enhance diffusion; elongated forms alter clearance |
[15,26,27,28,29] |
| Capsid stability | Inter-subunit cohesion, crosslinking | Circulatory integrity, endosomal resistance | Excess rigidity may hinder intracellular release | [23,24,98,101] |
| Surface charge | Zeta potential | Cellular uptake, serum interaction |
Positive charge increases uptake but accelerates clearance | [19,66] |
| Surface shielding | P[49,50,63EG or hydrophilic coatings | Immune evasion, reduced opsonization |
High density may mask targeting ligands | [99,100,109] |
| Ligand density | Multivalent display | Receptor engagement, selectivity |
Overcrowding may cause steric hindrance | [49,50,63] |
| Cargo loading strategy |
Encapsulation or conjugation |
Drug ratio, release kinetics | High loading can destabilize capsids |
[32,33,67,102] |
| Stimuli-responsive elements | pH-, redox-, or enzyme-sensitive linkers |
Site-specific drug release |
Premature activation reduces efficacy |
[32,88,103] |
| Native tropism | Intrinsic receptor affinity |
Target specificity | May cause off-target accumulation |
[106,107,108,109] |
| Manufacturing platform |
Expression and purification system |
Scalability, reproducibility |
Complex designs increase cost | [110,111,112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



