Submitted:
27 February 2026
Posted:
28 February 2026
You are already at the latest version
Abstract
The effective treatment of neurodegenerative diseases (NDDs), such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis, remains a critical challenge in modern medicine. Given the limitations of current therapies, alternative strategies to slow neurodegeneration are urgently needed. This study presents a critical review of the current evidence regarding low-dose ionizing radiation (LDIR) as a promising modality for modulating neurodegenerative processes. This study examines current experimental data on the effects of LDIR on cellular protective and compensatory mechanisms, including evidence from in vivo models of NDDs. Our analysis demonstrates that LDIR enhances antioxidant activity and DNA repair, stimulates autophagy and neuroplasticity, and modulates neuroinflammatory signaling. Collectively, these findings support the hypothesis of the neuroprotective potential of LDIR, underscoring its translational viability provided that strict dosimetric guidelines are followed and individual biological responses are rigorously monitored.
Keywords:
neurodegeneration
; neuroprotection
; radiotherapy
; hormesis
; low-dose ionising radiation
; in vivo models
; dementia
; Alzheimer's disease
; Parkinson's disease
1. Introduction
Neurodegenerative diseases (NDDs) represent a global medical and socioeconomic challenge, driven by the worldwide aging population and a progressive burden on healthcare systems. The lack of effective disease-modifying therapies and the limitations of current protocols, which focus primarily on symptomatic relief, underscore the urgent need for fundamentally new therapeutic strategies. While conventional radiation therapy is traditionally employed for malignant neoplasms and is associated with destructive effects on tumor cells, recent experimental evidence suggests that low-dose ionizing radiation (LDIR) holds significant potential to counteract neurodegenerative disorders [1,2].
Over the past few decades, a substantial body of evidence has accumulated demonstrating the non-linear nature of the biological response to radiation exposure. Unlike high-dose ionizing radiation (HDIR, >10 Sv), which is typically deleterious to biological tissues, LDIR is capable of inducing proliferative activity, modulating immune responses, activating repair systems, and delaying the progression of neoplastic processes [3,4,5]. This suite of adaptive reactions is frequently characterized within the framework of the radiation hormesis phenomenon [6].
The thresholds defining LDIR remain somewhat arbitrary. Traditionally, the phenomenon of radiation hormesis is considered characteristic of low-linear energy transfer (low-LET) radiation (X-rays, γ-rays, β-particles) at absorbed doses up to 100 mGy. Numerous studies have confirmed that irradiation within this range can exert protective effects, enhancing the resistance of cells and the organism as a whole to subsequent exposure to damaging doses [7,8,9]. Furthermore, it has been shown that the upper threshold for the adaptive response can reach 0,5 Gy [8]. Exceeding this limit is thought to result in the exhaustion of compensatory reserves, primarily due to the critical accumulation of DNA double-strand breaks (DSBs) that outpaces the capacity of repair systems [8,10]. Nevertheless, recent findings are significantly reshaping these perspectives. It has been established that γ-radiation can induce an adaptive response even at relatively high doses (e.g., 3 Gy) [11]. Moreover, beneficial effects on the central nervous system (CNS) have been observed following exposure to high-LET radiation, such as heavy ions (56Fe, 147 keV/µm, 0,5 Gy) [11,12]. It is likely that at the systemic level, tissue-wide reparative and compensatory mechanisms play a decisive role in mediating the radiation effect, whereas DSB accumulation may not serve as a sufficiently reliable marker. The capacity of LDIR to initiate repair and compensatory processes in neural tissue is currently viewed as a promising foundation for developing neuroprotective methods and disease-modifying therapies for neurodegenerative disorders [13,14].
2. Molecular and Cellular Mechanisms of Radiation Hormesis in the Central Nervous System
The impact of low-dose ionizing radiation on the human body is multifaceted and necessitates a comprehensive analysis of its diverse effects. The biological response to LDIR is a complex, multi-level cascade governed not only by physical radiation parameters, such as the type of radiation, absorbed dose, dose rate, and exposure regimen (chronic, acute, fractionated, or combined), but also by the state of the cellular microenvironment. Together, these factors determine the integrated effect on physiological processes within target cells and tissues. In the context of neurodegeneration, a pivotal aspect of radiation hormesis is the modulation of fundamental physiological processes: i.e., a hierarchical system of interactions at the cellular, tissue, and organ levels that underpins CNS homeostasis and the organism's adaptive capacity (Figure 1).
Evidence from in vitro and in vivo studies demonstrates that exposure to low-dose radiation induces a complex adaptive response. This response not only mobilizes antioxidant defense systems and optimizes DNA repair processes, thereby fostering a radioresistant phenotype against subsequent high-dose irradiation [15,16,17,18], but also triggers a systemic anti-inflammatory response [5]. The latter is mediated through the repression of pro-inflammatory cytokines and the phenotypic polarization of microglia from a pro-inflammatory M1 state to a neuroprotective M2 state. Furthermore, a crucial aspect of LDIR action involves the activation of proteostasis and autophagy mechanisms, which facilitate the clearance of pathological protein aggregates, alongside the epigenetic modulation of gene expression responsible for synaptic plasticity and neuronal survival [15,16]. These mechanisms (Table 1) provide a theoretical foundation for developing therapeutic protocols aimed at slowing neuronal degradation.
2.1. Modulation of Redox Homeostasis and Antioxidant Defense
Reactive oxygen species (ROS), such as the superoxide anion (O₂⁻) and hydrogen peroxide (H₂O₂), are generated during redox reactions in living organisms and play a pivotal role in the development of oxidative stress associated with carcinogenesis, atherosclerosis, neurodegenerative diseases, and aging [19]. Under physiological conditions, protective cascades ensure the synthesis of key antioxidant enzymes: superoxide dismutase (SOD) catalyzes the conversion of O₂⁻ to H₂O₂, which is subsequently decomposed into water and oxygen by catalase (CAT) and glutathione peroxidase (GPx). The progression of NDDs, such as Alzheimer's and Parkinson's diseases, is fundamentally linked to the functional degradation of the Nrf2 signaling axis, which triggers a systemic failure of metabolic homeostasis [20]. In this context, the neuroprotective potential of low-dose radiation stems from LDIR's ability to act as a mild pro-oxidant stimulus that activates nuclear factor erythroid 2-related factor 2 (Nrf2) – the master regulator of the cellular antioxidant response, coordinating the expression of enzymes involved in antioxidant defense, detoxification, and repair [21] (Figure 2).
Under conditions of oxidative stress, Nrf2 is released from its cytoplasmic complex with the inhibitory Kelch-like ECH-associated protein 1 (KEAP1) [22] and translocates to the nucleus. There, it binds to antioxidant response elements (AREs) within the promoter regions of target genes, triggering the transcription of a broad spectrum of cytoprotective enzymes: heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST), as well as GPx, SOD, and CAT [23,24,25,26]. In the context of LDIR-induced radioresistance, particular importance is attributed to manganese superoxide dismutase (MnSOD) – the primary mitochondrial antioxidant that maintains bioenergetic homeostasis and establishes the basis for mitohormetic effects [27,28].
The crosstalk between the Nrf2 pathway and DNA repair mechanisms is further evidenced by findings that its activation (whether radiation-induced or chemically triggered by sulforaphane [29] or naphthoquinone [30]) promotes the attenuation of induced DSBs of DNA [30,31] and enhances radioresistance [32]. Within the central nervous system, the neuroprotective effect of Nrf2 is multifaceted, operating both at the neuronal level and through the modulation of the microenvironment. On one hand, Nrf2 provides direct neuronal protection by reducing radiation-induced apoptosis through the modulation of signaling pathways (specifically the Nrf2–Notch1 axis) and the mitigation of ROS levels [33,34]. On the other hand, Nrf2 acts as a systemic regulator of the neuroimmune response by inducing microglial polarization toward the anti-inflammatory M2 phenotype. This process is accompanied by the repression of pro-inflammatory cytokine production and the up-regulation of regenerative factors, such as arginase-1 (Arg-1) and CD206, which are critical for restoring CNS structural integrity in neurodegenerative conditions [35,36].
2.2. Neuroimmunomodulation and Systemic Anti-inflammatory Response
The biological effects of LDIR are orchestrated through the complex modulation of immune functions, evidenced by the ability to attenuate chronic inflammation and activate cellular and humoral defense mechanisms [37]. In contrast to the deleterious effects of high doses, LDIR initiates signaling cascades aimed at maintaining cellular homeostasis [38]. The acceleration of DNA repair following irradiation is supported by a specific reorganization of the cytokine profile: the activation of IL-1, IL-3, GM-CSF, and G-CSF promotes cell viability and optimizes hematopoiesis, while low concentrations of IL-6 may serve as a survival regulator, preventing the initiation of apoptotic programs [39,40,41].
A critical aspect of LDIR action in the pathogenesis of neurodegenerative diseases is its ability to significantly downregulate the production of key pro-inflammatory mediators, including IL-1β, IL-6, and TNF-α [42]. The reduction in these factors contributes to the dampening of the systemic inflammatory response and the minimization of CNS tissue damage. This effect is closely coupled with the aforementioned functional polarization of microglia, the transition from an aggressive pro-inflammatory state (M1 phenotype) to a neuroprotective and regenerative state (M2 phenotype) [35].
Recent studies in breast cancer models, where metastasis is associated with the epithelial-mesenchymal transition (EMT) and the acquisition of a cancer stem cell (CSC) phenotype, demonstrate that the M1-to-M2 transition is largely mediated by the inhibition of the JAK1/STAT3 signaling pathway. The activation of the JAK1/STAT3 pathway serves as a key trigger for both neuroinflammation and carcinogenesis; its suppression by LDIR (regulated by SOCS and PIAS proteins) leads to a downregulation of damage markers, including a reduction in the CD44+/CD24- cell population, while simultaneously enhancing the synthesis of regenerative factors that facilitate structural tissue repair and the clearance of metabolic waste products [43]. Furthermore, additional modulation of the innate immune system under LDIR is achieved through the activation of the cGAS-STING cascade, initiated by the release of mitochondrial DNA (mtDNA) fragments into the cytosol. The mtDNA-mediated activation of the cGAS-STING pathway under LDIR conditions represents a pivotal link in non-targeted effects (NTE), ensuring 'controlled' immunomodulation and the induction of an adaptive response [44]. Thus, LDIR-mediated immunomodulation is a coordinated process that shifts the biological system from destructive inflammation toward a pathway of effective neuroprotection.
2.3. Regulation of Cellular Signaling, Proliferation, and Synaptic Plasticity
Experimental evidence indicates that LDIR acts as a precision modulator of intracellular signaling, activating cascades associated with ataxia-telangiectasia mutated (ATM) serine/threonine kinase, ERK, MAPK, c-Jun N-terminal kinase (JNK), and p53 [45,46,47]. Within the framework of radiation hormesis, these pathways mediate an adaptive response aimed at maintaining genomic stability and establishing a radioresistant phenotype. A fundamental distinction in the biological action of LDIR lies in its selectivity: low doses stimulate the proliferation of normal cells (e.g., the 2BS cell line) via the activation of the MAPK/ERK and PI3K/AKT axes, whereas such effects are not observed in tumor cells (e.g., the NCI-H446 cell line) [46]. The capacity of LDIR to optimize specific cellular functions without inducing genotoxic stress is further supported by other models; for instance, a dose of 25 mGy transiently enhances the secretory activity of pancreatic β-cells through the p38 MAPK pathway and the PDX-1 transcription factor without inducing DSB of DNA, in stark contrast to the effects of HDIR at 2.5 Gy [45,48].
In the context of neuroprotection, the most significant effect of modulating these signaling cascades is the stimulation of neural stem cell proliferation. LDIR has been shown to enhance hippocampal neurogenesis, which correlates with improved cognitive function and learning abilities in experimental models [49,50]. Transcriptomic analysis has revealed fundamental differences in cellular responses to low versus high radiation doses. While HDIR induces DNA damage response programs, cell cycle arrest, and apoptosis, LDIR predominantly activates genes responsible for antioxidant defense, MAPK/ERK signaling, glycolysis, and mitochondrial function [51]. This further confirms the role of ROS as signaling molecules that initiate metabolic adaptation.
Nevertheless, despite this pronounced neuroprotective potential, the impact of LDIR on cell cycle regulation remains an open question. In particular, the dual role of p21, a key cell cycle regulator that may determine the balance between neuronal survival and the activation of apoptotic programs, requires further investigation [47]. Given the dose-dependent nature of chromosomal damage within the 10–200 mGy range, precise control of dosimetric parameters remains a critical prerequisite for the safe translation of these effects into the therapy of NDDs.
2.4. Induction of Proteostasis and DNA Repair Mechanisms
A critical component of the adaptive response to LDIR is the activation of systems dedicated to maintaining proteostasis. A central role in this process is played by heat shock proteins (HSPs), a family of molecular chaperones whose expression is induced in response to thermal, oxidative, and radiation stress. The primary function of HSPs is to ensure correct protein folding, prevent aggregation, and protect cells from apoptosis, which is of paramount importance for neuronal survival [52].
Research confirms that high basal expression of HSP70 and HSP27 directly correlates with the development of a stable radioresistant phenotype. Specifically, in head and neck squamous cell carcinoma (HNSCC) lines and breast cancer side population (SP) cells (MCF-7), the levels of these chaperones increase significantly following irradiation, ensuring cell survival under radiation exposure [53,54]. For instance, it has been demonstrated in HNSCC lines that cells with elevated HSP70 content maintain higher proliferative potential when exposed to doses ranging from 2 to 12 Gy compared to normal human dermal fibroblasts (NHFs) and human dermal microvascular endothelial cells (HDMECs) [53]. A similar pattern is observed in the MCF-7 line, where the increased radioresistance of the SP subpopulation is attributed to the marked overexpression of HSP27 and HSP70 in response to a 5 Gy radiation exposure [54]. Notably, the realization of protective effects via the HSP system follows the general laws of hormesis and is characterized by a narrow 'therapeutic window'. Analogous to thermal stress, where beneficial effects on viability are observed only within a strictly limited range of conditions [55], the efficacy of radiation-induced proteostasis in the CNS is critically dependent on the precision of dosimetric parameters.
In parallel with chaperone systems, LDIR activates high-precision DNA repair mechanisms, specifically the DNA double-strand break repair (DSBR) system [56]. The central regulator of this process is the ATM kinase, which recognizes breaks and initiates a phosphorylation cascade of effector proteins. This enzyme regulates mitochondrial function, induces cell cycle arrest via p53 activation, and coordinates the choice between DNA repair and the induction of apoptosis [57]. Another critical element is poly(ADP-ribose) polymerase 1 (PARP1), which detects DNA breaks, stabilizes damaged sites, and recruits additional repair factors [58]. According to current understanding, LDIR-induced radioresistance is achieved through the synergistic activation of the aforementioned ATM kinase, PARP1, the transcription factor STAT1 [59], and the ATM/ERK/NF-κB signaling pathway [48]. Together, these components form the 'first line of defense' for DNA, facilitating the rapid initiation of signaling cascades that prevent lesions from progressing into lethal chromosomal aberrations. Notably, LDIR activates not only DSBR but also a broad spectrum of genome surveillance systems, including mismatch repair (MMR). For instance, a significant increase (up to five-fold) in the expression of DNA repair genes (hMSH2 and hMSH6) has been observed in medical personnel with prolonged occupational exposure to LDIR [60]. Thus, adaptation to radiation exposure is characterized by a dose-dependent and cumulative nature, enhancing the efficiency of quality control systems for both protein integrity and genetic stability.
2.5. Mitohormesis and Metabolic Remodeling
The phenomenon of mitohormesis, triggered by exposure to LDIR, represents an adaptive restructuring of the mitochondrial network aimed at restoring the bioenergetic capacity of neurons and maintaining a dynamic equilibrium between biogenesis and mitophagy. Mitochondrial dysfunction, manifested by impaired energy metabolism and exacerbated oxidative stress, is a cornerstone of the pathogenesis of major neurodegenerative diseases [61]. Paradoxically, the moderate oxidative stress induced by LDIR acts as a signaling stimulus that activates compensatory systems through mitochondrial adaptation mechanisms, whereas HDIR causes direct cellular damage [62,63,64,65].
A central role in mediating this response is played by the activation of the SIRT1/PGC-1α axis, which coordinates the expression of nuclear and mitochondrial genes responsible for organelle biogenesis and the efficiency of oxidative phosphorylation [66,67]. LDIR stimulates the transcription of respiratory chain components and modulates the activity of the integral outer mitochondrial membrane mitochondrial fission protein 1 (FIS1) and Mitofusin-1 (MFN1), which are essential for maintaining organelle integrity [62].
A crucial aspect of metabolic remodeling is the enhancement of the protein and organelle quality control systems. Hormetic radiation doses likely facilitate the timely clearance of damaged mitochondria via Pink1/PARK2/mROS-dependent mitophagy, thereby preventing excessive ROS generation and the initiation of apoptotic cascades [68,69]. Thus, the dual role of ROS under LDIR exposure lies, on one hand, in triggering cytoprotective pathways (such as the aforementioned Nrf2/ARE) that elevate cellular antioxidant status, and on the other, in maintaining signaling oxidation levels required for the selective removal of irreversibly damaged organelles, thereby ensuring neuronal metabolic plasticity. The stimulation of mitohormesis represents a promising neuroprotective strategy that promotes the long-term reinforcement of bioenergetic homeostasis in the CNS and delays neurodegeneration [70].
3. Neurobiological Effects of Low-Dose Ionizing Radiation in Experimental In Vivo Models of Neurodegenerative Diseases
The application of the radiation hormesis concept within the context of neurodegenerative diseases offers unique opportunities to modulate pathological processes by stimulating endogenous defense systems. Unlike high doses that induce overt tissue damage, LDIR acts as a moderate stressor, triggering a cascade of adaptive neurobiological reactions, as evidenced by numerous experimental studies [73,74]. A pivotal mechanism in this process is the activation of programs responsible for maintaining cellular homeostasis [65]. Particularly within the doses around 100 mGy, experimental data have further confirmed that LDIR can induce epigenetic modifications that regulate the expression of antioxidant defense and repair genes [75], as well as influence neuronal differentiation [70], thereby contributing to an increase in overall lifespan [76].
The capacity of LDIR to modulate the structural and functional plasticity of the brain has become a focal point of in vivo research. It has been established that low radiation doses can stimulate the proliferation of neural stem cells and hippocampal neurogenesis [50], as well as normalize synaptic transmission through the modulation of neurotrophic factor levels, such as BDNF and NGF [77]. Collectively, these effects, particularly those framed within the context of radiation hormesis, establish a biological foundation for the neuroprotective potential of LDIR. This potential is further substantiated by observed improvements in cognitive function and the attenuation of neurodegeneration across various experimental models of NDDs discussed hereafter (Table 2).
3.1. Effects of LDIR in Experimental Models of Alzheimer’s Disease
The application of the radiation hormesis concept in the therapy of Alzheimer’s disease (AD) is based on the ability of low-dose ionizing radiation to modulate key pathogenic pathways: the accumulation of amyloid-beta (Aβ) aggregates, the formation of microtubule-associated protein tau neurofibrillary tangles, and the development of chronic neuroinflammation [78].
In vivo experimental studies demonstrate the significant therapeutic potential of LDIR in reducing amyloid burden. A seminal study by Marples et al. first demonstrated that localized cranial irradiation promotes a reduction in amyloid plaque count in transgenic B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/J mice [79]. Mechanistically, this effect may be attributed to both direct physicochemical impact, such as the disruption of hydrogen bonds within the β-sheet structure of amyloid fibrils and the depolymerization of glycosaminoglycans [14,80], and the activation of biological clearance systems. Specifically, in the 5xFAD mouse model, it was established that fractionated irradiation (total dose 10 Gy, 2 Gy/fraction) induces the expression of the triggering receptor expressed on myeloid cells 2 (TREM2) on microglia, facilitating a phenotypic shift from a pro-inflammatory M1-like state to a phagocytic M2-like phenotype [36]. This transition is accompanied by a decrease in pro-inflammatory cytokine levels (e.g., TNF-α) and an increase in anti-inflammatory factors (e.g., TGF-β), correlating with improved spatial memory in the subjects [36,81]. Further evidence of the multi-target nature of such exposure (within the 0,5–2 Gy dose range) is provided by data showing the concomitant reduction of both amyloid burden and phosphorylated tau levels, which collectively ensures robust neuroprotection [82].
Current evidence suggests that even complex combined irradiation (γ-rays and 12C carbon ions) exerts a regulatory influence on the immune system. In 5xFAD transgenic mice, such exposure led to a "rebalancing" of the cytokine profile in the prefrontal cortex and hippocampus, alongside improved odor recognition memory. Furthermore, 3,5 months post-irradiation, these mice exhibited increased levels of macrophage inflammatory protein-1α (MIP-1α) in the prefrontal cortex and a reduction in IL-2β levels in the hippocampus [81].
The neuroprotective effects of radiation also effectively attenuate tau pathology. Experimental studies using the 3xTg-AD line (B6;129-Tg(APPSwe,tauP301L) 1LfaPsen1tm1Mpm) demonstrated that a course of right-hemisphere brain irradiation (2 Gy for 5 days) results in a concurrent reduction of Aβ-fractions and phosphorylated tau protein levels [83]. Similarly, in the Tau P301S homozygous model, combined irradiation (0,24 Gy γ-rays and 0,18 Gy 12C) significantly improved locomotor function and endurance while exerting an anxiolytic effect [84]. This progression is associated with pronounced modulation of microglial activity and the upregulation of cytokines in the hippocampus and cerebellum, indicating the activation of adaptive CNS resources in response to combined radiation stress in irradiated animals [84]. Additionally, LDIR stimulates regeneration via the induction of vascular endothelial growth factor (VEGF) and growth-associated protein 43 (GAP-43), supporting axonal growth and myelination [85].
However, there are grounds to believe that the efficacy of LDIR may be limited by sexual dimorphism. In the TgF344-AD rat model, it was established that females are less sensitive to the anti-amyloid effects of ionizing radiation (2 Gy for 5 days), despite a decrease in microglial inflammatory markers (CD68) [80]. This underscores the critical importance of personalizing radiation protocols to achieve the desired biological response.
The translational potential of these findings is supported by clinical observations. Pilot trials conducted at the University of Virginia in 2023 confirmed the safety of fractionated irradiation (2 Gy for 5 days) in patients with AD and its ability to slow cognitive decline [86]. Cases of low-dose CT (40–80 mGy) application also indicate the possibility of restoring basic functions in patients with severe dementia [86,87]. Despite these promising results, these data require verification in large-scale randomized controlled trials.
3.2. Impact of LDIR on Pathogenetic Targets in in vivo Models of Parkinson’s Disease
Recent experimental studies highlight the multi-level neuroprotective effects of LDIR in Parkinson’s disease (PD) models. LDIR exposure emerges as a promising strategy for PD, aimed at activating preconditioning stimuli and enhancing the endogenous defense mechanisms of dopaminergic neurons.
A key mechanism underlying the hormetic response in PD is the effective suppression of oxidative stress and neuroinflammation. In studies utilizing classical MPTP-induced models, LDIR exposure (specifically, 50 cGy total-body irradiation) was found to induce a rapid surge in antioxidant enzyme activity (CAT and GSH) as early as 3 hours post-exposure [74]. Furthermore, it has been demonstrated that LDIR significantly reduces the intensity of oxidative stress and the severity of apoptosis in neurons of the substantia nigra pars compacta and striatum. These findings are complemented by contemporary research confirming that low doses (approximately 60 cGy) exert anti-inflammatory effects by suppressing the expression of pro-inflammatory and glial activation markers in the striatum (e.g., GFAP, CD54) [88].
Of particular interest is the selective impact of LDIR on key molecular targets associated with the disease. Studies in large mammals (minipigs) revealed a specific reduction in LRRK2 protein levels in the striatum 28 days post-irradiation, a critical genetic risk factor for PD [89]. Notably, under these irradiation protocols (cumulative dose < 2 Gy), the levels of other proteins, including α-synuclein, PARK2, and tyrosine hydroxylase, remained stable, with no detectable pathological changes, vascular lesions, or reactive gliosis in the brain tissue [90]. Such selectivity suggests the potential for targeted intervention on specific molecular pathways without systemic disruption of neuronal metabolism, further confirming the existence of a safe "therapeutic window" for LDIR application.
Beyond targeted monotherapy, combination therapy represents a promising avenue for PD treatment, demonstrating pronounced synergistic effects. For instance, the combination of Ginkgo biloba extract (EGb761) and fractionated low-dose γ-irradiation (0,25 Gy weekly for 6 weeks; total cumulative dose of 1,5 Gy) in a reserpine-induced parkinsonism model not only restored striatal dopamine levels but also normalized antioxidant system markers, including GSH, MDA, and iron ion concentrations [91]. These integrated approaches provide neuroprotection by addressing mitochondrial dysfunction and stabilizing cellular homeostasis. Consequently, LDIR emerges as a promising tool for developing innovative PD therapeutic strategies, capable of slowing degenerative processes through the stimulation of adaptive CNS resources and the selective modulation of target proteins.
3.3. Experimental Evidence and Prospects for Modulating Amyotrophic Lateral Sclerosis Pathogenesis via LDIR
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by the selective loss of motor neurons, leading to paresis, progressive muscular atrophy, and terminal respiratory failure [92]. Unlike Alzheimer’s and Parkinson’s diseases, research into the effects of LDIR on experimental ALS models is in its infancy; however, preliminary data suggest a complex and non-linear nature of this interaction.
In a recent fundamental in vivo study using a minipig model, it was demonstrated that a single dose of total-body irradiation (1,79 Gy) can modulate the expression and intracellular distribution of key ALS-associated proteins, including FUS/TLS, C9orf72, STMN2, and pTDP-43 [93]. Notably, the observed changes were strictly tissue- and cell-specific, varying across different brain regions and cellular compartments (nucleus vs. cytoplasm). The authors emphasize that these molecular shifts cannot be unequivocally interpreted as the activation of a pathological cascade; rather, they may represent an adaptive response or a hormetic effect that could exert neuroprotective actions in certain contexts. These findings open a new avenue in ALS etiology research, suggesting that low-intensity external radiation factors may potentially influence the molecular triggers of the disease.
In contrast to fundamental research on pathogenesis, clinical application of radiotherapy in ALS is currently limited to palliative care and does not aim to modify the neurodegenerative process. Low-dose irradiation of the salivary glands is a recognized clinical practice to manage sialorrhea in ALS patients, providing a temporary improvement in quality of life [94]. This therapeutic approach remains purely symptomatic and does not target the underlying pathogenetic mechanisms. Consequently, further investigation into dose-dependent effects and radiation hormesis mechanisms in animal models of ALS represents a promising research direction. Understanding the conditions under which LDIR triggers adaptive protective responses (aimed at preserving respiratory and locomotor functions) versus those that may promote the aggregation of pathological proteins (e.g., pTDP-43, FUS/TLS) could be key to developing novel preventive or therapeutic strategies to maintain neuromuscular functionality.
3.4. Potential Applications of LDIR in Traumatic Injuries of the Nervous System
Experimental studies across various models demonstrate a broad spectrum of neuroprotective effects of LDIR that extend beyond classical neurodegenerative models. For instance, in a model of retinitis pigmentosa (rd10 mice; B6.CXB1-Pde6brd10/J), LDIR at a dose of 650 mGy exhibited a pronounced protective effect, significantly delaying photoreceptor cell death and restoring functional activity. The molecular mechanism underlying this protection is associated with the activation of the antioxidant enzyme gene peroxiredoxin-2 (Prdx2), with a prolonged therapeutic effect achieved through repeated irradiation protocols [95]. Of particular significance are data obtained from models of peripheral nervous system injuries. It has been established that low-intensity irradiation at a dose of 700 cGy following sciatic nerve injury in rats stimulates regenerative processes, suppresses fibrous scar formation, and significantly improves electrophysiological parameters of conduction recovery [96].
A critical aspect of these effects is systemic immunomodulation. LDIR has been shown to optimize neural tissue repair by activating T-cell-mediated immunity and modulating associated cytokine profiles; notably, the absence of a regenerative response in immunodeficient animals confirms the systemic nature of this adaptation [71]. Furthermore, the modulation of neuroprotective factors, such as neuropeptide Y (NPY) and Purkinje cell protein 4 (PCP4), identified in intact swine, suggests that LDIR can induce a state of preconditioning, potentially increasing CNS resistance to subsequent neurodegenerative and behavioral impairments [72].
In conclusion, the gathered evidence confirms the universal nature of the radiation hormesis phenomenon within the nervous system. The ability of LDIR to activate endogenous repair mechanisms and proteostasis, among other effects, opens new perspectives for developing comprehensive rehabilitation strategies for both traumatic and degenerative conditions of the CNS and peripheral nerves.
4. Clinical Translation Prospects of LDIR as a Systemic Modulator of Neurodegeneration
The collective evidence, including findings from in vivo models, demonstrates the complex and multifactorial neuroprotective potential of LDIR, which targets key pathogenetic nodes of NDDs. While monoclonal Aβ-antibody therapies are characterized by limited efficacy and a high risk of amyloid-related imaging abnormalities (ARIA) [97,98,99], and current treatments for PD, ALS, and other NDDs remain largely symptomatic, LDIR exposure is capable of triggering a comprehensive physiological response. These observations underscore the need to re-evaluate current therapeutic paradigms and justify the search for novel, fundamental approaches to NDD treatment.
Notably, the clinical translation of this approach has already commenced, including prospective randomized trials (e.g., NCT05635968). Importantly, these LDIR protocols were not associated with the serious adverse events typical of Aβ-immunotherapy and resulted in documented improvements in neurocognitive function tests [100]. Consequently, in vivo experimental data position LDIR as a foundation for a pathogenetically grounded three-stage therapeutic approach to NDDs:
First stage: induction of apoptosis in senescent cells and dysfunctional neurons;
Second stage: activation of proteome clearance systems (e.g., targeting Aβ-aggregates, tau protein, and FUS/TLS protein);
Third stage: stimulation of endogenous neurorepair and neurogenesis.
5. Concluding Remarks and Future Directions
The synthesized in vivo data confirm that low-dose ionizing radiation initiates a systemic adaptive response mediated through the mechanisms of radiation hormesis, including antioxidant system activation, neuroimmunomodulation, and the stimulation of neuroplasticity and proteostasis maintenance systems. The capacity of LDIR to target the fundamental pathogenetic nodes of neurodegenerative diseases positions this method not merely as a tool for symptomatic relief but as a promising instrument for the disease-modifying modulation of the neurodegenerative process.
Despite its pronounced therapeutic potential, the transition to widespread clinical practice necessitates the precision standardization of dosimetric protocols, accounting for individual biological variability and sexual dimorphism. Future research should focus on elucidating the underlying nature of radiation hormesis, exploring the synergy between LDIR and pharmacological agents, and verifying neuroprotective effects through large-scale randomized controlled trials. Systematizing these approaches will establish the foundation for personalized pathogenetic strategies in nuclear medicine, aimed at the effective management of neurodegenerative disorder progression.
Author Contributions
V.V.G. and Ya.S.K. drafted the work; V.G.K. and V.S.K. helped in writing the manuscript and approved the submitted version; Ya.S.K. reviewed the literature, V.V.G. and V.S.K. contributed to the conception of the work, and S.G.M. substantively revised it and approved the submitted version. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by state contract (unique project ID: FGFU-2025-0004) and by RSF grant (agreement No.22-75-10036-P).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Karina Ruslanova, Nadia El-Maula, Anastasia Chernomordova and Vsevolod Derbenev for managing ongoing tasks and workflows, thereby allowing the authors to devote the necessary time to finalize this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 2BS | Human embryonic lung diploid fibroblast cell line |
| A549 | Adenocarcinomic human alveolar basal epithelial cell line |
| AD | Alzheimer's disease |
| Akt (PKB) | Protein kinase B |
| ALS | Amyotrophic lateral sclerosis |
| ARE | Antioxidant response element |
| ATM | Ataxia-telangiectasia mutated (serine/threonine kinase) |
| Aβ | Amyloid-beta peptide |
| BDNF | Brain-derived neurotrophic factor |
| C9orf72 | Chromosome 9 open reading frame 72 protein |
| CAT | Catalase |
| CCR2 | C-C chemokine receptor type 2 |
| CCS | Copper chaperone for superoxide dismutase |
| cGAS | Сyclic GMP-AMP synthase |
| CNS | Central nervous system |
| CSCs | Cancer stem cells |
| CT | Computed tomography |
| DNA | Deoxyribonucleic acid |
| DSB | DNA double-strand break |
| DSBR | Double-strand break repair |
| EGb761 | Standardized extract of Ginkgo biloba |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| FIS1 | Mitochondrial fission protein 1 |
| FUS/TLS | Fused in Sarcoma/ Translocated in Liposarcoma protein |
| GAP-43 | Growth-associated protein 43 |
| G-CSF | Granulocyte colony-stimulating factor |
| GFAP | Glial fibrillary acidic protein |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| GST | Glutathione S-transferase |
| Gy | Gray |
| HDIR | High-dose ionising radiation |
| HDMEC | Human dermal microvascular endothelial cells |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| HO-1 | Heme oxygenase-1 |
| HSP27/HSP70 | Heat shock protein 27 / Heat shock protein 70 |
| IL-1β, IL-6, IL-10, IL-2β | Interleukin-1 beta / 6 / 10 / 2 beta |
| JAK1 | Janus kinase 1 |
| JNK | c-Jun N-terminal kinases |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LDIR | Low-dose ionising radiation |
| LET | Linear energy transfer |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPK | Mitogen-activated protein kinase |
| MCF-7 | Breast cancer side population cells |
| MCP-1 | Monocyte chemoattractant protein-1 (also known as CCL2) |
| MDA | Malondialdehyde |
| MFN1 | Mitofusin-1 |
| MIP-1α | Macrophage inflammatory protein-1α |
| MMR | Mismatch repair |
| MnSOD | Manganese superoxide dismutase (also known as SOD2) |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mROS | Mitochondrial reactive oxygen species |
| mtDNA | Mitochondrial DNA |
| NDDs | Neurodegenerative diseases |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| NHF | Normal human fibroblasts |
| Notch1 | Neurogenic locus notch homolog protein 1 |
| NPY | Neuropeptide Y |
| NQO1 – NAD(P)H | Quinone oxidoreductase 1 – NAD(P)H |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLC | Non-small cell lung cancer |
| p53 | Tumor protein p53 |
| PARK2 | Parkin RBR E3 ubiquitin protein ligase (PRKN, PARKIN) |
| PARP1 | Poly [ADP-ribose] polymerase 1 |
| PCP4 | Purkinje cell protein 4 (also known as PEP-19) |
| PD | Parkinson's disease |
| PET | Positron emission tomography |
| PI3K | Phosphoinositide 3-kinase |
| PIAS | Protein inhibitor of activated STAT |
| Prdx2 | Peroxiredoxin-2 |
| pTDP43 | Phosphorylated TAR DNA-binding protein 43 |
| ROS | Reactive oxygen species |
| SOCS | Suppressor of cytokine signaling |
| SOD | Superoxide dismutase |
| SP cells | Side population cells |
| STAT1/STAT3 | Signal transducer and activator of transcription 1 / 3 |
| STING | Stimulator of interferon genes |
| STMN2 | Stathmin-2 (also known as SCG10) |
| Sv | Sievert |
| TGF- β | Transforming growth factor-beta |
| TNF-α | Tumor necrosis factor-alpha |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VEGF | Vascular endothelial growth factor |
References
- Vaiserman, A.; Cuttler, J.M.; Socol, Y. Low-dose ionizing radiation as a hormetin: experimental observations and therapeutic perspective for age-related disorders. Biogerontology 2021, 22, 145–164. [Google Scholar] [CrossRef]
- Paithankar, J.G.; Gupta, S.C.; Sharma, A. Therapeutic potential of low dose ionizing radiation against cancer, dementia, and diabetes: evidences from epidemiological, clinical, and preclinical studies. Mol Biol Rep 2023, 50, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Luckey, T.D. Physiological benefits from low levels of ionizing radiation. Health Phys 1982, 43, 771–789. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Li, W.; Jiang, H.; Liang, X.; Zhao, Y.; Yu, D.; Zhou, L.; Wang, G.; Tian, H.; Han, F.; et al. Low-dose radiation may be a novel approach to enhance the effectiveness of cancer therapeutics. Int J Cancer 2016, 139, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Au, N.P.B.; Wu, T.; Kumar, G.; Jin, Y.; Li, Y.Y.T.; Chan, S.L.; Lai, J.H.C.; Chan, K.W.Y.; Yu, K.N.; Wang, X.; et al. Low-dose ionizing radiation promotes motor recovery and brain rewiring by resolving inflammatory response after brain injury and stroke. Brain Behav Immun 2024, 115, 43–63. [Google Scholar] [CrossRef]
- Macklis, R.M. Radithor and the era of mild radium therapy. JAMA 1990, 264, 614–618. [Google Scholar] [CrossRef]
- Wolff, S. Is Radiation All Bad? The Search for Adaptation. Radiation research 1992, 131. [Google Scholar] [CrossRef]
- Desouky, O.; Ding, N.; Zhou, G. Targeted and non-targeted effects of ionizing radiation. Journal of Radiation Research and Applied Sciences 2015, 8, 247–254. [Google Scholar] [CrossRef]
- Tang, F.R.; Loke, W.K.; Khoo, B.C. Low-dose or low-dose-rate ionizing radiation–induced bioeffects in animal models. Journal of radiation research 2017, 58, 165–182. [Google Scholar] [CrossRef]
- Tubiana, M.; Arengo, A.; Averbeck, D.; Masse, R. Low-Dose Risk Assessment. Radiation research 2007, 167, 742–744. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Dobynde, M.I. The Effects of Galactic Cosmic Rays on the Central Nervous System: From Negative to Unexpectedly Positive Effects That Astronauts May Encounter. Biology (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Liu, B.; Hinshaw, R.G.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Shi, Q.; Holton, P.; et al. Space-like (56)Fe irradiation manifests mild, early sex-specific behavioral and neuropathological changes in wildtype and Alzheimer's-like transgenic mice. Scientific reports 2019, 9, 12118. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, N.; Gokul, M.; Narayanam, H.; Ananya, A.K. Low-dose radiation research insights in experimental animals: A gateway to therapeutic implications. Vet World 2024, 17, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.M.; Pereira, L.; Teubig, P.; Santos, P.; Mendes, F.; Vinals, S.; Galaviz, D.; Herrera, F. Radiation as a Tool against Neurodegeneration-A Potential Treatment for Amyloidosis in the Central Nervous System. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liu, J.; Mai, Y.; Hong, Y.; Jia, Z.; Tian, G.; Liu, Y.; Liang, H.; Liu, J. Current advances and future trends of hormesis in disease. NPJ Aging 2024, 10, 26. [Google Scholar] [CrossRef]
- Le, D.D.; Jang, Y.S.; Truong, V.; Yu, S.; Dinh, T.; Lee, M. Bioactivities of Quinic Acids from Vitex rotundifolia Obtained by Supercritical Fluid Extraction. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Hosztafi, S.; Galambos, A.R.; Koteles, I.; Karadi, D.A.; Furst, S.; Al-Khrasani, M. Opioid-Based Haptens: Development of Immunotherapy. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Haiying Wang, W.Z.; Cao, Yi. Radiation-induced cellular senescence and adaptive response: mechanistic interplay and implications 2025, Volume 6.
- Tapio, S.; Jacob, V. Radioadaptive response revisited. Radiat Environ Biophys 2007, 46, 1–12. [Google Scholar] [CrossRef]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer's Disease. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Kaur, K.; Narang, R.K.; Singh, S. Role of Nrf2 in Oxidative Stress, Neuroinflammation and Autophagy in Alzheimer's Disease: Regulation of Nrf2 by Different Signaling Pathways. Curr Mol Med 2025, 25, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Song, S.; Wang, Y.; Bian, Y.; Wu, M.; Wu, H.; Shi, Y.; Duan, H. NAD(P)H: quinone oxidoreductase 1 attenuates oxidative stress and apoptosis by regulating Sirt1 in diabetic nephropathy. J Transl Med 2022, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, C.; Li, Z.; Huang, W.; Zheng, S.; Liu, C.; Shi, X.; Ma, Y.; Ni, Q.; Tan, W.; et al. Astaxanthin activates the Nrf2/Keap1/HO-1 pathway to inhibit oxidative stress and ferroptosis, reducing triphenyl phosphate (TPhP)-induced neurodevelopmental toxicity. Ecotoxicol Environ Saf 2024, 271, 115960. [Google Scholar] [CrossRef]
- Jiang, X.; Yu, M.; Wang, W.K.; Zhu, L.Y.; Wang, X.; Jin, H.C.; Feng, L.F. The regulation and function of Nrf2 signaling in ferroptosis-activated cancer therapy. Acta Pharmacol Sin 2024, 45, 2229–2240. [Google Scholar] [CrossRef]
- Shan, C.; Wang, Y.; Wang, Y. The Crosstalk between Autophagy and Nrf2 Signaling in Cancer: from Biology to Clinical Applications. Int J Biol Sci 2024, 20, 6181–6206. [Google Scholar] [CrossRef]
- Eldridge, A.; Fan, M.; Woloschak, G.; Grdina, D.J.; Chromy, B.A.; Li, J.J. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radic Biol Med 2012, 53, 1838–1847. [Google Scholar] [CrossRef]
- Yamaoka, K.; Edamatsu, R.; Itoh, T.; Mori, A. Effects of low-dose X-ray irradiation on biomembrane in brain cortex of aged rats. Free Radic Biol Med 1994, 16, 529–534. [Google Scholar] [CrossRef]
- Lu, W. Sulforaphane regulates AngII-induced podocyte oxidative stress injury through the Nrf2-Keap1/ho-1/ROS pathway. Ren Fail 2024, 46, 2416937. [Google Scholar] [CrossRef]
- Khan, N.M.; Sandur, S.K.; Checker, R.; Sharma, D.; Poduval, T.B.; Sainis, K.B. Pro-oxidants ameliorate radiation-induced apoptosis through activation of the calcium-ERK1/2-Nrf2 pathway. Free Radic Biol Med 2011, 51, 115–128. [Google Scholar] [CrossRef]
- Mathew, S.T.; Bergstrom, P.; Hammarsten, O. Repeated Nrf2 stimulation using sulforaphane protects fibroblasts from ionizing radiation. Toxicol Appl Pharmacol 2014, 276, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int J Biol Sci 2015, 11, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Mao, A.; Yan, J.; Sun, C.; Di, C.; Zhou, X.; Li, H.; Guo, R.; Zhang, H. Downregulation of Nrf2 promotes radiation-induced apoptosis through Nrf2 mediated Notch signaling in non-small cell lung cancer cells. Int J Oncol 2016, 48, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int J Biol Sci 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Wu, H.; Wu, J.; Jiang, J.; Qian, Z.; Yang, S.; Sun, Y.; Cui, H.; Li, S.; Zhang, P.; Zhou, Z. Compound 7 regulates microglia polarization and attenuates radiation-induced myelopathy via the Nrf2 signaling pathway in vivo and in vitro studies. Mol Med 2024, 30, 198. [Google Scholar] [CrossRef]
- Kim, S.; Chung, H.; Ngoc Mai, H.; Nam, Y.; Shin, S.J.; Park, Y.H.; Chung, M.J.; Lee, J.K.; Rhee, H.Y.; Jahng, G.H.; et al. Low-Dose Ionizing Radiation Modulates Microglia Phenotypes in the Models of Alzheimer's Disease. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Seong, K.M.; Kim, C.S.; Lee, B.S.; Nam, S.Y.; Yang, K.H.; Kim, J.Y.; Park, J.J.; Min, K.J.; Jin, Y.W. Low-dose radiation induces Drosophila innate immunity through Toll pathway activation. J Radiat Res 2012, 53, 242–249. [Google Scholar] [CrossRef]
- Feinendegen, L.E.; Pollycove, M.; Neumann, R.D. Feinendegen. Low-dose cancer risk modeling must recognize up-regulation of protection. Dose Response 2009, 8, 227–252. [Google Scholar] [CrossRef]
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in radiobiological responses: a review. Radiat Res 2012, 178, 505–523. [Google Scholar] [CrossRef]
- Kiang, J.G.; Smith, J.T.; Hegge, S.R.; Ossetrova, N.I. Circulating Cytokine/Chemokine Concentrations Respond to Ionizing Radiation Doses but not Radiation Dose Rates: Granulocyte-Colony Stimulating Factor and Interleukin-18. Radiat Res 2018, 189, 634–643. [Google Scholar] [CrossRef]
- Chen, H.; Liao, S.B.; Cheung, M.P.; Chow, P.H.; Cheung, A.L.; W.S., O. Effects of sperm DNA damage on the levels of RAD51 and p53 proteins in zygotes and 2-cell embryos sired by golden hamsters without the major accessory sex glands. Free Radic Biol Med 2012, 53, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.K.; Wang, J.Y.; Chen, C.F.; Chao, K.Y.; Chang, M.C.; Chen, W.M.; Hung, S.C. Early Passage Mesenchymal Stem Cells Display Decreased Radiosensitivity and Increased DNA Repair Activity. Stem Cells Transl Med 2017, 6, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.; Kim, M.J.; Kim, R.K.; Kumar Kaushik, N.; Seong, K.M.; Nam, S.Y.; Lee, S.J. Low-dose radiation decreases tumor progression via the inhibition of the JAK1/STAT3 signaling axis in breast cancer cell lines. Sci Rep 2017, 7, 43361. [Google Scholar] [CrossRef] [PubMed]
- Averbeck, D. Low-Dose Non-Targeted Effects and Mitochondrial Control. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int J Radiat Biol 2015, 91, 13–27. [Google Scholar] [CrossRef]
- Liang, X.; Gu, J.; Yu, D.; Wang, G.; Zhou, L.; Zhang, X.; Zhao, Y.; Chen, X.; Zheng, S.; Liu, Q.; et al. Low-Dose Radiation Induces Cell Proliferation in Human Embryonic Lung Fibroblasts but not in Lung Cancer Cells: Importance of ERK1/2 and AKT Signaling Pathways. Dose Response 2016, 14, 1559325815622174. [Google Scholar] [CrossRef]
- Khan, M.G.M.; Wang, Y. Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle. Cells 2022, 11. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, K.; An, R.; Wang, C.; Zhou, X.; Tian, Z.; Liao, Z. Single Low-Dose Ionizing Radiation Transiently Enhances Rat RIN-m5F Cell Function via the ROS/p38 MAPK Pathway Without Inducing Cell Damage. Antioxidants (Basel) 2025, 14. [Google Scholar] [CrossRef]
- Wei, L.C.; Ding, Y.X.; Liu, Y.H.; Duan, L.; Bai, Y.; Shi, M.; Chen, L.W. Low-dose radiation stimulates Wnt/beta-catenin signaling, neural stem cell proliferation and neurogenesis of the mouse hippocampus in vitro and in vivo. Curr Alzheimer Res 2012, 9, 278–289. [Google Scholar] [CrossRef]
- Betlazar, C.; Middleton, R.J.; Banati, R.B.; Liu, G.J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol 2016, 9, 144–156. [Google Scholar] [CrossRef]
- Sampadi, B.; Vermeulen, S.; Misovic, B.; Boei, J.J.; Batth, T.S.; Chang, J.G.; Paulsen, M.T.; Magnuson, B.; Schimmel, J.; Kool, H.; et al. Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm (2020) 2022, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Muschter, D.; Geyer, F.; Bauer, R.; Ettl, T.; Schreml, S.; Haubner, F. A comparison of cell survival and heat shock protein expression after radiation in normal dermal fibroblasts, microvascular endothelial cells, and different head and neck squamous carcinoma cell lines. Clin Oral Investig 2018, 22, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Matchuk, O.N.; Zamulaeva, I.A. High Level of Radiation-Induced Heat Shock Protein with a Molecular Weight of 27 and 70 kDa is the Hallmark of Radioresistant SP Cells of MCF-7 Breast Cancer Culture. Radiats Biol Radioecol 2016, 56, 382–388. [Google Scholar]
- Lagisz, M.; Hector, K.L.; Nakagawa, S. Life extension after heat shock exposure: assessing meta-analytic evidence for hormesis. Ageing Res Rev 2013, 12, 653–660. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G(2)-phase. Sci Rep 2019, 9, 8255. [Google Scholar] [CrossRef]
- Laconi, E.; Cheri, S.; Fanti, M.; Marongiu, F. Aging and Cancer: The Waning of Community Bonds. Cells 2021, 10. [Google Scholar] [CrossRef]
- Li, W.H.; Wang, F.; Song, G.Y.; Yu, Q.H.; Du, R.P.; Xu, P. PARP-1: a critical regulator in radioprotection and radiotherapy-mechanisms, challenges, and therapeutic opportunities. Front Pharmacol 2023, 14, 1198948. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A 2004, 101, 1714–1719. [Google Scholar] [CrossRef]
- Machi, A., Jr.; Moreira Perez, M.; Luciano da Veiga, G.; Cristiano Pereira, E.; Adami, F.; Alves, B.; Fonseca, F. Expression of DNA repair genes in association with ionizing radiation. Acta Biomed 2022, 93. [Google Scholar] [CrossRef]
- Yang, H.M. Mitochondrial Dysfunction in Neurodegenerative Diseases. Cells 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Kostyuk, S.V.; Proskurnina, E.V.; Konkova, M.S.; Abramova, M.S.; Kalianov, A.A.; Ershova, E.S.; Izhevskaya, V.L.; Kutsev, S.I.; Veiko, N.N. Effect of Low-Dose Ionizing Radiation on the Expression of Mitochondria-Related Genes in Human Mesenchymal Stem Cells. Int J Mol Sci 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Ristow. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Yuyun, X.; Jinjun, Q.; Minfang, X.; Jing, Q.; Juan, X.; Rui, M.; Li, Z.; Jing, G. Gao. Effects of Low Concentrations of Rotenone upon Mitohormesis in SH-SY5Y Cells. Dose Response 2013, 11, 270–280. [Google Scholar] [CrossRef]
- Sosin, D.V.; Baranovskii, D.S.; Nechaev, D.N.; Sosina, M.A.; Shaposhnikov, A.V.; Trusov, G.A.; Titova, A.G.; Krasnikov, B.F.; Lomov, A.N.; Makarov, V.V.; et al. Population Studies and Molecular Mechanisms of Human Radioadaptive Capabilities: Is It Time to Rethink Radiation Safety Standards? Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, Y.; Yang, Y.; Huang, X.; Sun, C. SIRT1 Protects Dopaminergic Neurons in Parkinson's Disease Models via PGC-1alpha-Mediated Mitochondrial Biogenesis. Neurotox Res 2021, 39, 1393–1404. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1alpha Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front Mol Neurosci 2017, 10, 443. [Google Scholar] [CrossRef]
- Yu, L.; Yang, X.; Li, X.; Qin, L.; Xu, W.; Cui, H.; Jia, Z.; He, Q.; Wang, Z. Pink1/PARK2/mROS-Dependent Mitophagy Initiates the Sensitization of Cancer Cells to Radiation. Oxid Med Cell Longev 2021, 2021, 5595652. [Google Scholar] [CrossRef]
- Kurtman, C; Üçöz, Ö.M.; Karakoyun Çelik, M; Sokur, Ö; Kemal Özbilgin, I.M. Mitophagy in the A549 lung cancer cell line, radiation-induced damage, and the effect of ATM and PARKIN on the mitochondria. Int J Radiat Res 2022, 20, 9–13. [Google Scholar] [CrossRef]
- Bajinskis, A.; Lindegren, H.; Johansson, L.; Harms-Ringdahl, M.; Forsby, A. Low-dose/dose-rate gamma radiation depresses neural differentiation and alters protein expression profiles in neuroblastoma SH-SY5Y cells and C17.2 neural stem cells. Radiat Res 2011, 175, 185–192. [Google Scholar] [CrossRef]
- Kipnis, J.; Avidan, H.; Markovich, Y.; Mizrahi, T.; Hauben, E.; Prigozhina, T.B.; Slavin, S.; Schwartz, M. Low-dose gamma-irradiation promotes survival of injured neurons in the central nervous system via homeostasis-driven proliferation of T cells. Eur J Neurosci 2004, 19, 1191–1198. [Google Scholar] [CrossRef]
- Iacono, D.; Hatch, K.; Murphy, E.K.; Post, J.; Cole, R.N.; Perl, D.P.; Day, R.M. Proteomic changes in the hippocampus of large mammals after total-body low dose radiation. PLoS One 2024, 19, e0296903. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singla, N.; Chadha, V.D.; Dhawan, D.K. A concept of radiation hormesis: stimulation of antioxidant machinery in rats by low dose ionizing radiation. Hell J Nucl Med 2019, 22, 43–48. [Google Scholar] [PubMed]
- Kojima, S.; Matsuki, O.; Nomura, T.; Yamaoka, K.; Takahashi, M.; Niki, E. Elevation of antioxidant potency in the brain of mice by low-dose gamma-ray irradiation and its effect on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced brain damage. Free Radic Biol Med 1999, 26, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, S.; Sreetharan, S.; Kulesza, A.V.; Boreham, D.R.; Tai, T.C. Low-Dose Ionizing Radiation Exposure, Oxidative Stress and Epigenetic Programing of Health and Disease. Radiat Res 2017, 188, 525–538. [Google Scholar] [CrossRef]
- Caratero, A.; Courtade, M.; Bonnet, L.; Planel, H.; Caratero, C. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology 1998, 44, 272–276. [Google Scholar] [CrossRef]
- Dimberg, Y.; Vazquez, M.; Soderstrom, S.; Ebendal, T. Effects of X-irradiation on nerve growth factor in the developing mouse brain. Toxicol Lett 1997, 90, 35–43. [Google Scholar] [CrossRef]
- Lin, S.; Zhan, Y.; Wang, R.; Pei, J. Decoding neuroinflammation in Alzheimer's disease: a multi-omics and AI-driven perspective for precision medicine. Front Immunol 2025, 16, 1616899. [Google Scholar] [CrossRef]
- Marples, B.; McGee, M.; Callan, S.; Bowen, S.E.; Thibodeau, B.J.; Michael, D.B.; Wilson, G.D.; Maddens, M.E.; Fontanesi, J.; Martinez, A.A. Cranial irradiation significantly reduces beta amyloid plaques in the brain and improves cognition in a murine model of Alzheimer's Disease (AD). Radiother Oncol 2016, 118, 579–580. [Google Scholar] [CrossRef]
- Ceyzeriat, K.; Tournier, B.B.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Zilli, T. Low-Dose Radiation Therapy: A New Treatment Strategy for Alzheimer's Disease? J Alzheimers Dis 2020, 74, 411–419. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Levashova, A.I.; Nesterov, M.S.; Pikalov, V.A.; Chicheva, M.M. Combined Ionizing Radiation Caused Cognition and Non-Cognition Behavior Benefits and Modulated Microglial Activity in Wild-Type and Alzheimer's-like Transgenic Mice. Biology (Basel) 2025, 14. [Google Scholar] [CrossRef]
- Ricciardi, N.R.; Modarresi, F.; Lohse, I.; Andrade, N.S.; Newman, I.R.; Brown, J.M.; Borja, C.; Marples, B.; Wahlestedt, C.R.; Volmar, C.H. Investigating the Synergistic Potential of Low-Dose HDAC3 Inhibition and Radiotherapy in Alzheimer's Disease Models. Mol Neurobiol 2023, 60, 4811–4827. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.D.; Wilson, T.G.; Hanna, A.; Fontanesi, G.; Kulchycki, J.; Buelow, K.; Pruetz, B.L.; Michael, D.B.; Chinnaiyan, P.; Maddens, M.E.; et al. Low Dose Brain Irradiation Reduces Amyloid-beta and Tau in 3xTg-AD Mice. J Alzheimers Dis 2020, 75, 15–21. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Ageldinov, R.A.; Anokhin, P.K.; Shamakina, I.Y. Combined Ionizing Radiation Exposure Improves Behavioral Symptoms and Modulates Brain Innate Immune System Activity in the Tau P301S Mice Line. Biochemistry (Mosc) 2025, 90, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, Y.; Zhao, J.; She, C.; Zhou, X.; Dong, Q.; Wang, P. Effects of Localized X-Ray Irradiation on Peripheral Nerve Regeneration in Transected Sciatic Nerve in Rats. Radiat Res 2017, 188, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Cuttler, J.M.; Moore, E.R.; Hosfeld, V.D.; Nadolski, D.L. Treatment of Alzheimer Disease With CT Scans: A Case Report. Dose Response 2016, 14, 1559325816640073. [Google Scholar] [CrossRef]
- Cuttler, J.M.; Moore, E.R.; Hosfeld, V.D.; Nadolski, D.L. Update on a Patient With Alzheimer Disease Treated With CT Scans. Dose Response 2017, 15, 1559325817693167. [Google Scholar] [CrossRef]
- Park, M.; Ha, J.; Lee, Y.; Choi, H.S.; Kim, B.S.; Jeong, Y.K. Low-moderate dose whole-brain gamma-ray irradiation modulates the expressions of glial fibrillary acidic protein and intercellular adhesion molecule-1 in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease mouse model. Neurobiol Aging 2023, 132, 175–184. [Google Scholar] [CrossRef]
- Iacono, D.; Murphy, E.K.; Stimpson, C.D.; Perl, D.P.; Day, R.M. Low-dose radiation decreases Lrrk2 levels in the striatum of large mammalian brains: New venues to treat Parkinson's disease? Parkinsonism Relat Disord 2024, 124, 107024. [Google Scholar] [CrossRef]
- Murphy, E.K.; Perl, D.P.; Day, R.M.; Iacono, D. Evaluating Parkinson's disease biomarkers in substantia nigra following sublethal gamma-radiation exposure in a large animal model. NPJ Parkinsons Dis 2025, 11, 286. [Google Scholar] [CrossRef]
- El-Ghazaly, M.A.; Sadik, N.A.; Rashed, E.R.; Abd-El-Fattah, A.A. Neuroprotective effect of EGb761(R) and low-dose whole-body gamma-irradiation in a rat model of Parkinson's disease. Toxicol Ind Health 2015, 31, 1128–1143. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Iacono, D.; Murphy, E.K.; Perl, D.P.; Day, R.M. gamma-Radiation induces region-specific subcellular alterations of amyotrophic lateral sclerosis and frontotemporal dementia markers in swine brain. Sci Rep 2026, 16, 5627. [Google Scholar] [CrossRef] [PubMed]
- Harriman, M.; Morrison, M.; Hay, J.; Revonta, M.; Eisen, A.; Lentle, B. Use of radiotherapy for control of sialorrhea in patients with amyotrophic lateral sclerosis. J Otolaryngol 2001, 30, 242–245. [Google Scholar] [CrossRef]
- Otani, A.; Kojima, H.; Guo, C.; Oishi, A.; Yoshimura, N. Low-dose-rate, low-dose irradiation delays neurodegeneration in a model of retinitis pigmentosa. Am J Pathol 2012, 180, 328–336. [Google Scholar] [CrossRef]
- Gocmen, S.; Sirin, S.; Oysul, K.; Ulas, U.H.; Oztas, E. The effects of low-dose radiation in the treatment of sciatic nerve injury in rats. Turk Neurosurg 2012, 22, 167–173. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Manolopoulos, A.; Ferrucci, L.; Kapogiannis, D. Critical assessment of anti-amyloid-beta monoclonal antibodies effects in Alzheimer's disease: a systematic review and meta-analysis highlighting target engagement and clinical meaningfulness. Scientific reports 2024, 14, 25741. [Google Scholar] [CrossRef]
- Honig, L.S.; Sabbagh, M.N.; van Dyck, C.H.; Sperling, R.A.; Hersch, S.; Matta, A.; Giorgi, L.; Gee, M.; Kanekiyo, M.; Li, D.; et al. Updated safety results from phase 3 lecanemab study in early Alzheimer's disease. Alzheimers Res Ther 2024, 16, 105. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer's Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11. [Google Scholar] [CrossRef]
- Kim, BH; Park, K.K.; Kim, WY; Hong, A; Kim, EH; Cho, JY; Rhee, SJ; Chung, HY. WK. Low-Dose Radiation Therapy in Alzheimer’s disease: An Interim Neurocognitive Analysis from a Multicenter Phase II Clinical Trial. Alzheimers Dement. 2025, 20, e084076. [Google Scholar] [CrossRef]
- Teipel, S.J.; Tang, Y.; Khachaturian, A. Sex differences in treatment effects of lecanemab and donanemab: A Bayesian reanalysis of CLARITY-AD and TRAILBLAZER-ALZ2. Alzheimers Dement (N Y) 2025, 11, e70155. [Google Scholar] [CrossRef]
- Andrews, D.; Ducharme, S.; Chertkow, H.; Sormani, M.P.; Collins, D.L.; Alzheimer's Disease Neuroimaging, I. The higher benefit of lecanemab in males compared to females in CLARITY AD is probably due to a real sex effect. Alzheimers Dement 2025, 21, e14467. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the molecular and cellular mechanisms of radiation hormesis in the central nervous system. LDIR initiates a cascade of adaptive responses aimed at maintaining neuronal homeostasis. Key regulatory pathways include: (1) Antioxidant response: activation of Nrf2 and subsequent induction of enzymatic defenses (SOD, CAT, GPx); (2) Mitohormesis: modulation of mitochondrial function via FIS1/MFN1 proteins; (3) Proliferation and survival: activation of the MAPK/ERK and PI3K/Akt pathways, alongside p53-mediated cell cycle regulation; (4) Neuroimmunomodulation: microglial polarization toward the anti-inflammatory M2 phenotype with a concomitant shift in the cytokine profile (increased IL-10/IL-4; decreased IL-1β, IL-6, and TNF-α); (5) Proteostasis and repair: induction of chaperones (HSP70, HSP27) and activation of the JAK1/STAT3 signaling pathway, promoting DNA repair and neurogenesis. Collectively, these processes establish a state of enhanced radioresistance and neuroprotection. Created with BioRender.com (Agreement number: ID29DHHNXG).
Figure 1.
Schematic representation of the molecular and cellular mechanisms of radiation hormesis in the central nervous system. LDIR initiates a cascade of adaptive responses aimed at maintaining neuronal homeostasis. Key regulatory pathways include: (1) Antioxidant response: activation of Nrf2 and subsequent induction of enzymatic defenses (SOD, CAT, GPx); (2) Mitohormesis: modulation of mitochondrial function via FIS1/MFN1 proteins; (3) Proliferation and survival: activation of the MAPK/ERK and PI3K/Akt pathways, alongside p53-mediated cell cycle regulation; (4) Neuroimmunomodulation: microglial polarization toward the anti-inflammatory M2 phenotype with a concomitant shift in the cytokine profile (increased IL-10/IL-4; decreased IL-1β, IL-6, and TNF-α); (5) Proteostasis and repair: induction of chaperones (HSP70, HSP27) and activation of the JAK1/STAT3 signaling pathway, promoting DNA repair and neurogenesis. Collectively, these processes establish a state of enhanced radioresistance and neuroprotection. Created with BioRender.com (Agreement number: ID29DHHNXG).
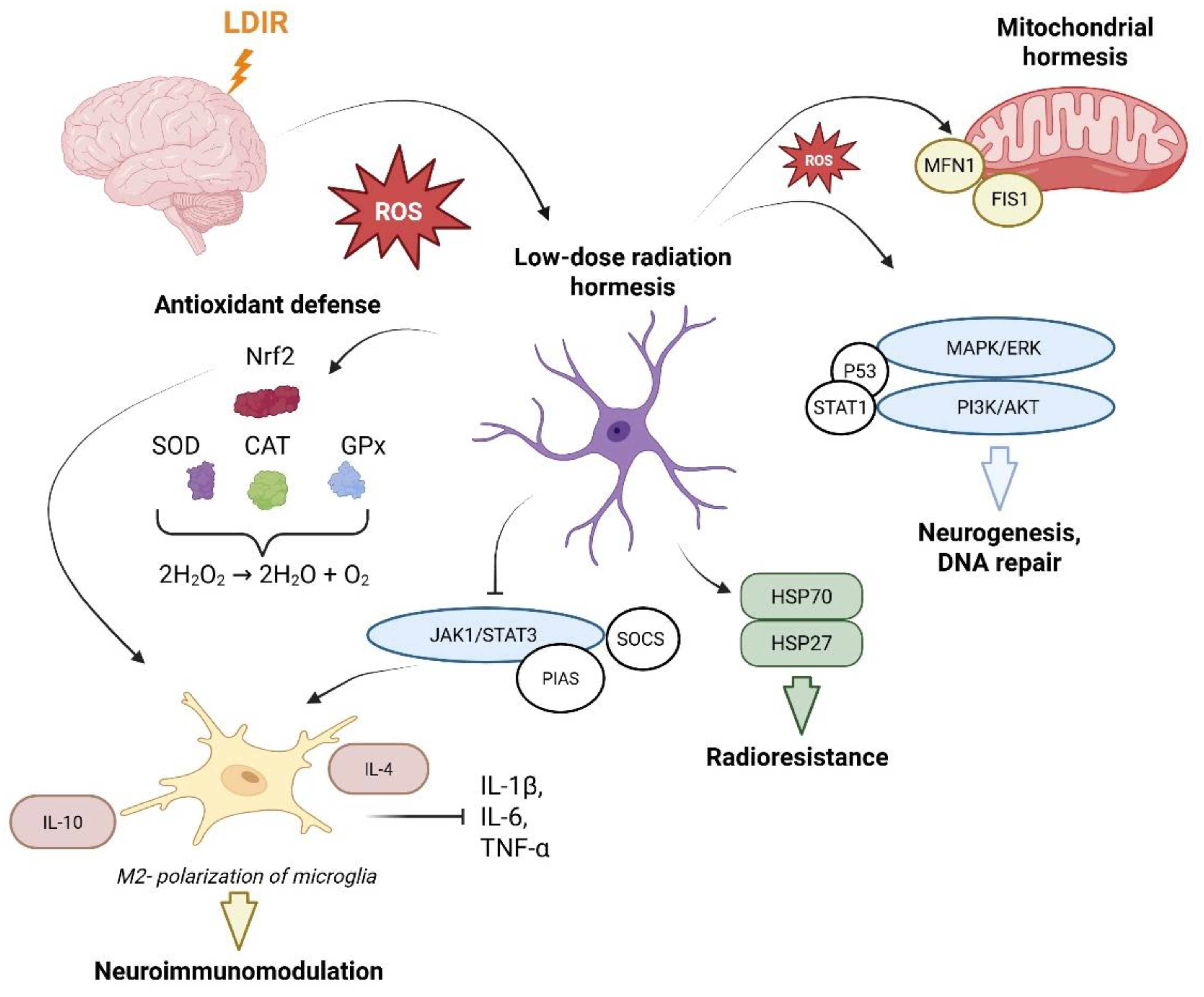
Figure 2.
Molecular mechanisms coupling antioxidant and immunomodulatory responses under LDIR exposure. Low-dose irradiation induces the generation of moderate levels of ROS, which act as signaling molecules to trigger the dissociation of the Nrf2/Keap1 complex. Free Nrf2 then translocates to the nucleus, where it binds to antioxidant response elements (AREs), initiating the expression of detoxification and antioxidant defense genes (SOD, CAT, GPx, HO-1, NQO1, GST). Concurrently, LDIR modulates the CNS microenvironment, promoting the functional polarization of microglia from a pro-inflammatory M1 phenotype to a neuroprotective M2 phenotype. This process is accompanied by a reorganization of the cytokine profile, characterized by decreased levels of pro-inflammatory factors (IL-1β, IL-6, TNF-α) and increased synthesis of anti-inflammatory mediators (IL-10, TGF-β). The integration of these cascades provides a comprehensive neuroprotective effect. Created with BioRender.com (Agreement number: LC29DWV7IV).
Figure 2.
Molecular mechanisms coupling antioxidant and immunomodulatory responses under LDIR exposure. Low-dose irradiation induces the generation of moderate levels of ROS, which act as signaling molecules to trigger the dissociation of the Nrf2/Keap1 complex. Free Nrf2 then translocates to the nucleus, where it binds to antioxidant response elements (AREs), initiating the expression of detoxification and antioxidant defense genes (SOD, CAT, GPx, HO-1, NQO1, GST). Concurrently, LDIR modulates the CNS microenvironment, promoting the functional polarization of microglia from a pro-inflammatory M1 phenotype to a neuroprotective M2 phenotype. This process is accompanied by a reorganization of the cytokine profile, characterized by decreased levels of pro-inflammatory factors (IL-1β, IL-6, TNF-α) and increased synthesis of anti-inflammatory mediators (IL-10, TGF-β). The integration of these cascades provides a comprehensive neuroprotective effect. Created with BioRender.com (Agreement number: LC29DWV7IV).
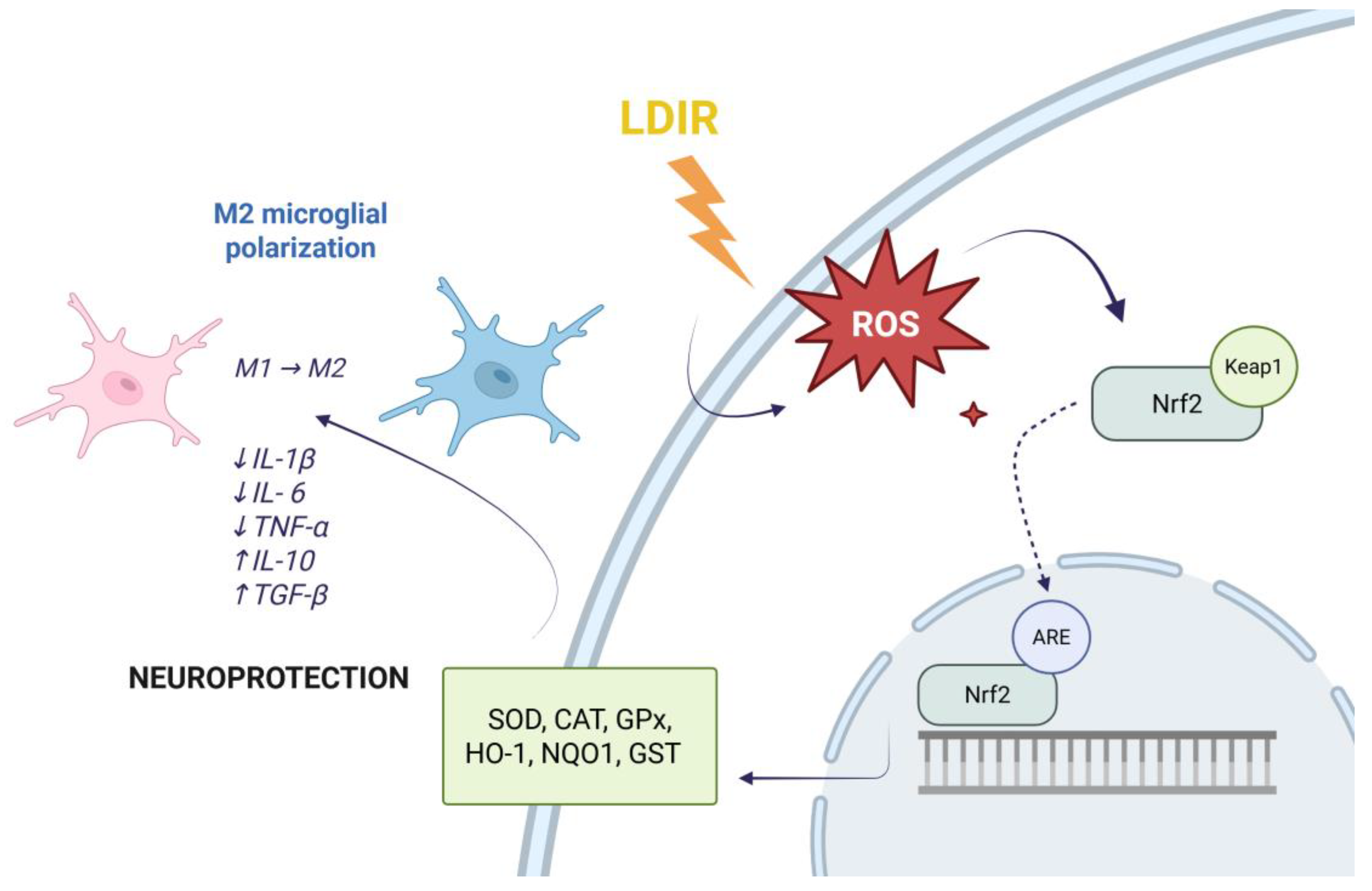

Table 1.
Interplay between intracellular adaptive mechanisms and systemic biological effects induced by low-dose ionizing radiation.
Table 1.
Interplay between intracellular adaptive mechanisms and systemic biological effects induced by low-dose ionizing radiation.
| Protective and Adaptive Mechanisms | Key Markers and Signaling Pathways | Biological Effects | References | |||
|---|---|---|---|---|---|---|
| Antioxidant Defense | Nrf2/ARE pathway, SOD, MnSOD, CAT, GPx, HO-1, NQO1, GST |
ROS neutralization, reduction of oxidative stress, neuronal protection against apoptosis |
[27,28,30,32,33] | |||
| Neuroimmunomodulation | Microglial polarization (M1 → M2), ↓IL-1β, ↓IL-6, ↓TNF-α, ↑IL-10, ↑TGF-β, ↑T-cell response |
Mitigation of chronic neuroinflammation, establishment of a regenerative CNS microenvironment | [35,36,39,40,41,42,71] | |||
| Proliferation and Neuroplasticity | MAPK/ERK pathway, PI3K/Akt, p53, p38/MAPK/PDX-1 pathway | Stimulation of hippocampal neurogenesis, improvement of cognitive functions and synaptic transmission | [45,46,47,48,49,50,51] | |||
| Proteostasis and DNA Repair | HSP70, HSP27, ATM kinase, PARP1, STAT1, ATM/ERK/NF-κB, ↑hMSH2 and ↑hMSH6, NPY, PCP4 | Correct protein folding, prevention of protein aggregation and proteopathy, restoration of genomic integrity | [53,54,56,59,60,72] | |||
| Mitohormesis | SIRT1/PGC-1α pathway, FIS1/MFN1, Pink1/PARK2 pathway* | Restoration of neuronal bioenergetics, clearance of damaged mitochondria (mitophagy) | [62,66,67,68,69] |
* Hypothetically
Table 2.
Neuroprotective effects of LDIR in experimental models of NDDs and traumatic CNS injuries in vivo.
Table 2.
Neuroprotective effects of LDIR in experimental models of NDDs and traumatic CNS injuries in vivo.
| Pathology | Subject (model) | Irradiation regimen | Key biological effects | Refs. |
|---|---|---|---|---|
| Alzheimer’s disease | Mice (5XFAD, 3xTg-AD, APPswePSEN1) | γ-rays 0,5–2 Gy (fractionated); γ-rays 0,24 Gy + 12C nuclei 0,18 Gy (combined) | Reduction of amyloid burden (↓Aβ); M1→M2 microglia polarization (TREM2); increased neuronal viability; ↑VEGF, ↑GAP-43; improvement of cognitive functions | [81,82,84,85] |
| Parkinson’s disease | Mice (MPTP); rats (reserpine); minipigs |
γ-rays 0.5–0.6 Gy (single dose); γ-rays 0.25 Gy x 6 weeks (cumulative dose 1,5 Gy) | Activation of CAT and GSH; ↓LRRK2 in the striatum; suppression of neuroinflammation (↓GFAP, ↓CD54); synergy with Ginkgo biloba – restoration of dopamine, GSH, MDA levels, and iron ions in the striatum | [74,88,89,91] |
| Amyotrophic lateral sclerosis | Minipigs | γ-rays 1,79 Gy (single dose) | Modulation of expression and distribution of FUS/TLS, C9orf72, STMN2, and pTDP43 proteins | [93] |
| Retinitis pigmentosa | rd10 mice | γ-rays 0,65 Gy (repeated) | Delayed photoreceptor cell death; activation of the antioxidant enzyme Prdx2 | [95] |
| Sciatic nerve injury | Female Wistar rats | γ-rays 7 Gy (single dose) | Stimulation of regeneration; suppression of scarring | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



