Submitted:
27 February 2026
Posted:
28 February 2026
You are already at the latest version
Abstract
The Neonatal Intensive Care Unit (NICU) represents a critical frontier in modern medicine, characterized by extreme physiological fragility and a profound reliance on off-label pharmacotherapy. This review examines the current state of neonatal pharmacology through the lens of developmental pharmacology, identifying systemic barriers including cytochrome P450 ontogeny (CYP3A7 dominance in early life), renal immaturity, and altered protein binding that create unpredictable drug responses. Through a forensic analysis of the cisapride arrhythmia case, we illustrate the lethal consequences of “ontogeny-blind” drug development, where negligible CYP3A7-mediated metabolism combined with hERG potassium channel blockade (IC50: 6.5–44.5 nM) led to fatal ventricular arrhythmias. We argue that the convergence of next-generation artificial intelligence technologies—specifically AlphaFold 3 for structural biology, Large Language Models for chemical space exploration, and emerging biological World Foundation Models—provides a promising, though still maturing, pathway for addressing the “therapeutic orphan” problem. We propose a conceptual framework for “generative redesign” of therapeutics that moves from reactive post-marketing withdrawal to proactive in silico safety optimization. Critical implementation barriers including neonatal-specific training data scarcity, regulatory pathway immaturity, and the need for international consortium building are addressed with a strategic roadmap spanning near-term (2–5 years) through long-term (10+ years) development priorities.
Keywords:
neonatal intensive care unit
; off-label prescribing
; developmental pharmacology
; CYP3A4/CYP3A7 ontogeny
; artificial intelligence
; AlphaFold 3
; hERG channel
; cardiotoxicity
; generative chemistry
; molecular docking
; precision medicine
1. Introduction
The term “therapeutic orphan,” coined by Harry Shirkey in 1968, remains an apt descriptor for the neonatal population [1]. The standard of care in the NICU is paradoxically defined by the widespread use of medications that have never been systematically tested or approved for use in newborns. A systematic review encompassing 78 studies and 23,999 neonates reveals the staggering scale of this phenomenon: in an analysis of 17,421 medication items, 96.4% of neonates were exposed to at least one off-label or unlicensed drug [2]. The burden falls disproportionately on the most vulnerable; 100% of extremely preterm infants (born before 28 weeks of gestation) receive off-label medicines during hospitalization [2,3].
This off-label reliance is a global reality with significant geographical variation. While Africa reports high prevalence rates (66%), developed nations are not immune; studies in Estonia and Germany report prevalence rates of 87% and 70%, respectively [2]. The clinical consequences are measurable: children prescribed medications off-label are more than three times more likely to experience adverse drug events compared to those receiving appropriately labeled therapies, a risk likely amplified in neonates given their developmental vulnerabilities [4]. Medication errors occur eight times more frequently in NICUs than in adult hospital settings [5]. In neonates, the risk is magnified by rapid physiologic change, small dosing margins, frequent polypharmacy, and limited availability of pediatric formulations. At the same time, the field has demonstrated that rigorous neonatal trials can meaningfully improve outcomes; caffeine therapy for apnea of prematurity is a prominent example of a neonatal intervention with durable clinical benefit [6].
Regulatory initiatives have evolved substantially over three decades. The Best Pharmaceuticals for Children Act (BPCA) provides six-month patent/exclusivity extensions for conducting FDA-requested pediatric studies, while the Pediatric Research Equity Act (PREA) authorizes the FDA to require pediatric studies for drugs expected to have substantial pediatric use. Together, these laws have generated more than 650 products with new pediatric labeling information [7]. However, neonates have benefited disproportionately less: from 1997 to 2010, of 406 pediatric labeling changes, only 24 (6%) included neonatal-specific information [8]. The roster of FDA-approved neonatal-specific drugs remains notably limited: caffeine citrate for apnea of prematurity, surfactant preparations, ibuprofen lysine and indomethacin for patent ductus arteriosus closure, and inhaled nitric oxide for hypoxic respiratory failure.
This review argues that neonatal pharmacotherapy must move beyond reactive safety management toward proactive “design for development.” We use cisapride—a drug widely used in infants before being withdrawn for torsadogenic risk—as a case study of developmental vulnerability. We then evaluate how modern structure-based computing and generative artificial intelligence (AI) can support safer drug redesign, provided these methods are constrained by developmental biology, validated prospectively, and aligned with emerging regulatory expectations.
Scope and approach: This is a narrative review. Evidence was identified through targeted searches of peer-reviewed literature and regulatory sources with an emphasis on systematic reviews, pivotal pharmacology studies, and drug-safety analyses relevant to neonates. Because the neonatal evidence base is sparse for many drugs, we explicitly highlight areas where conclusions remain uncertain or depend on extrapolation.
2. Developmental Pharmacology: The Biological Barrier
To understand pharmacological failure in the NICU, one must appreciate the ontogeny of drug metabolism. The neonate is not a “small adult” but a distinct biological entity with fundamentally different pharmacokinetic parameters that change dramatically over days to weeks of postnatal development.
2.1. The Cytochrome P450 “Isoform Switch”
The metabolism of xenobiotics is primarily handled by the cytochrome P450 (CYP) enzymes. In adults, CYP3A4 metabolizes nearly 50% of marketed drugs. However, in the fetus and early neonate, CYP3A7 is the dominant isoform [9]. While sharing 87% sequence identity with CYP3A4, CYP3A7 has distinct substrate specificity. Crucially, CYP3A7 is generally a poor metabolizer of standard CYP3A4 substrates; the rate of biotransformation for certain drugs by CYP3A4 can be orders of magnitude higher than by CYP3A7 [9,10].
CYP3A4 is negligible or absent at birth and only reaches adult levels by 1–2 years of age [11]. CYP1A2 is barely detectable at birth and increases over 1–3 months. CYP2C19 immaturity explains why diazepam half-life extends to 50–90 hours in neonates versus 20–50 hours in adults. Phase II conjugation enzymes follow similarly protracted maturation: UDP-glucuronosyltransferase protein abundances increase 8- to 55-fold from neonates to adults across different isoforms, with the estimated age for reaching 50% of adult protein abundance ranging from 2.6 to 10.3 years [12]. UGT2B7, responsible for morphine glucuronidation, reaches adult activity only by 2–6 months—explaining why neonates younger than 10 days require 50% dose reductions for morphine [13].
2.2. Renal Immaturity and Pharmacokinetic Variability
Glomerular filtration rate (GFR) at birth measures only 2–4 mL/min (even lower in preterm neonates), increasing 2- to 4-fold in the first four weeks and reaching adult levels of approximately 120 mL/min by 8–12 months [14]. This dramatically affects aminoglycoside, vancomycin, and other renally cleared drug disposition. Population pharmacokinetic modeling of amikacin in 205 extremely preterm neonates (24–30 weeks postconceptional age) found clearance increasing from 0.486 L/h per 70 kg at 24 weeks to 0.940 L/h per 70 kg by 30 weeks—nearly doubling over just six weeks of maturation [15].
Body composition differences further complicate predictions. Total body water comprises 85% of preterm neonate mass versus 60% in adults, while body fat constitutes only 1% in preterm infants versus 18–25% in adults. Plasma albumin concentrations measure 75–80% of adult levels with reduced binding affinity [14]. The clinical consequence: standard vancomycin dosing achieves target trough concentrations in less than 30% of neonates, while model-informed precision dosing can increase target attainment to 62–94% [16].
2.3. Pharmacogenomic Considerations
The pharmacogenomic landscape in neonates is underexplored despite high exposure to relevant drugs. Analysis of 19,195 NICU patients from 2005–2018 found that 75% of NICU admissions had at least one exposure to a drug class with Clinical Pharmacogenetics Implementation Consortium guidelines during early childhood [17]. Relevant gene–drug pairs include CYP2D6 affecting codeine conversion to morphine (creating toxicity risk in ultra-rapid metabolizers among breastfed infants), UGT1A1 variants associated with hyperbilirubinemia, and MT-RNR1 variants (m.1555A>G) conferring aminoglycoside-induced ototoxicity risk—the latter now detectable via a 26-minute point-of-care test [18]. Yet applying adult pharmacogenomic data to neonates is complicated by ontogeny–genotype interactions where polymorphism effects only manifest after enzyme maturation.

Table 1.
Developmental determinants that modulate neonatal drug exposure and safety, with implications for design and evaluation.
Table 1.
Developmental determinants that modulate neonatal drug exposure and safety, with implications for design and evaluation.
| Domain | Neonatal feature | Design / evaluation implication | Example |
|---|---|---|---|
| Absorption | Variable gastric pH, motility, and feeding patterns | Oral bioavailability is unpredictable; prefer exposure-guided strategies | Feeding intolerance drugs |
| Distribution | High total body water; low fat stores; lower albumin in preterm infants | Larger Vd for hydrophilic drugs; higher free fraction for highly bound drugs | Aminoglycosides; cisapride protein binding |
| Metabolism | CYP3A7 predominance early; low CYP3A4/UGT activity at birth | High vulnerability to DDIs; metabolite profiles differ from adults | CYP3A inhibitors increasing cisapride exposure |
| Excretion | Rapid maturation of GFR and tubular transport | Clearance changes over days–weeks; dosing must be dynamic | Vancomycin MIPD improves target attainment |
| Electrophysiology | Limited repolarization reserve and co-morbidity burden | Safety margins for hERG blockade may be smaller; require developmental safety assays | Torsadogenic risk signals |
3. The Cisapride Paradigm: A Forensic Case Study
Cisapride, a 5-hydroxytryptamine (5-HT4) receptor agonist, was widely used for gastroesophageal reflux disease and feeding intolerance in infants prior to its withdrawal from the U.S. market. Its clinical story illustrates how developmental pharmacology and polypharmacy can transform a seemingly useful therapy into a high-risk intervention. The withdrawal of the promotility agent cisapride serves as a proof-of-concept for why “ontogeny-aware” safety testing is essential and illustrates how AI-driven approaches could prevent similar complications [19].
3.1. Mechanism of Action and Cardiotoxicity
Cisapride, a substituted piperidinyl benzamide discovered by Janssen Pharmaceuticals in 1980, acts as a selective 5-HT4 receptor agonist (EC50 = 140 nM) that stimulates acetylcholine release from the enteric nervous system, increasing gastric motility [19]. The FDA approved it in July 1993 for nocturnal heartburn associated with gastroesophageal reflux disease, and it rapidly gained widespread use including off-label application in neonates for feeding intolerance.
The mechanism of cardiotoxicity involves blockade of the hERG (KCNH2) potassium channel responsible for the rapid delayed rectifier potassium current (IKr), with an IC50 of 6.5–44.5 nM (most studies: 15–20 nM) [20]. This channel blockade delays cardiac repolarization, prolongs the QT interval on electrocardiography, and creates an arrhythmogenic substrate capable of triggering torsades de pointes—a polymorphic ventricular tachycardia that can degenerate into ventricular fibrillation. The critical pharmacological problem is selectivity: with hERG IC50 approximately 10-fold lower than the 5-HT4 EC50, the therapeutic window depended entirely on the 98% protein binding that limits free drug concentrations [21].
From July 1993 through December 1999, the FDA received reports of 341 cardiac arrhythmias including 80 deaths [22]. In approximately 85% of serious adverse events, patients had identifiable risk factors: concurrent CYP3A4 inhibitors (clarithromycin, ketoconazole, ritonavir), other QT-prolonging medications, electrolyte disturbances, or underlying cardiac conditions. Janssen announced the voluntary withdrawal of cisapride on March 23, 2000, with an effective date of July 14, 2000; 23 additional deaths were reported between the announcement and the completion of withdrawal [22].
3.2. Developmental Vulnerability: CYP3A-Mediated Exposure and the Neonatal “Double-Hit”
The problem in the NICU was driven by a metabolic “double-hit.” Cisapride is a substrate for CYP3A4, which clears it efficiently in adults. However, neonates rely on CYP3A7, which cannot metabolize cisapride. Studies using recombinant enzymes showed that the rate of cisapride biotransformation by CYP3A7 was negligible (0.01 nmol/24 h) compared to CYP3A4 (0.77 nmol/24 h)—a 77-fold difference [23,24]. In vivo studies detected no cisapride metabolism in neonates younger than 7 days [25]. Clearance increases linearly with postconceptional age, meaning the youngest patients accumulate drug even without classic CYP3A4 inhibitor interactions. When combined with the frequent NICU need for antifungals and antibiotics (and thus DDIs), this creates a “double-hit” exposure–toxicity scenario: higher parent-drug exposure plus potent hERG blockade.
Cardiac electrophysiology also differs developmentally. The rapid delayed rectifier potassium current (IKr) plays a proportionally more prominent role in cardiac repolarization during the neonatal period compared to adulthood, as other repolarizing currents (particularly IKs) are less mature [26,27]. QTc shows maturational changes: decreasing in the first week, increasing until 2 months, then decreasing again. Preterm infants demonstrate longer baseline QTc values than term infants, creating less reserve before reaching arrhythmogenic thresholds. Pharmacokinetic data grouped by gestational age confirmed significantly lower clearance in the youngest gestational age group (CL/F: 0.45 ± 0.26 L/h/kg versus 0.85 ± 0.69 in older neonates) [28]. Crucially, randomized controlled trials ultimately showed that cisapride does not reduce feed intolerance or improve gastric emptying in normal preterm neonates, suggesting the risk–benefit calculation was unfavorable even before withdrawal [29]. (Figure 1)
4. The Artificial Intelligence Landscape: Tools for Discovery and Safety
The field of drug discovery is transitioning from empirical approaches to computational design. Three AI technologies—AlphaFold 3, Large Language Models, and World Foundation Models—provide the strategic toolkit for this transformation, though implementation readiness varies significantly across applications.
4.1. AlphaFold 3: Protein-Ligand Structure Prediction and Molecular Docking
DeepMind’s AlphaFold 3 (AF3), released in May 2024, represents a significant advance in modeling biological structures [30]. Unlike its predecessor, AF3 utilizes a diffusion-based architecture—analogous to image generation models—to denoise atomic coordinates and predict the structure of multimolecular complexes including proteins, DNA, RNA, ligands, ions, and modified residues in a unified framework [31]. For protein–ligand interactions, AF3 achieves 50% greater accuracy than traditional physics-based docking methods (such as AutoDock Vina) on the PoseBusters benchmark, with a 58% success rate for ligand binding prediction versus 24% for AutoDock Vina [30].
For neonatal drug safety applications, AF3 can model the hERG channel in its various conformational states (open, closed, inactivated) and predict the precise binding pose of drug candidates, identifying specific pharmacophores responsible for channel blockade [32]. Recent publications (2024–2025) demonstrate that AlphaFold-guided structural modeling can predict multiple hERG conformational states and that incorporating these predictions substantially improves correlation with experimental drug binding affinities [33]. The finding that “most drugs bind more effectively in the inactivated state and are trapped in the closed state” enables more accurate state-dependent docking predictions.
However, critical limitations constrain direct drug discovery applications. AlphaFold 3 predicts static structures and cannot model protein dynamics or conformational changes—struggling with complexes involving greater than 5 Å RMSD conformational changes [34]. It cannot predict quantitative binding affinities, shows 4.4% chirality violation rates on benchmarks, and demonstrates reduced accuracy for ligands with low similarity to training data or for “orphan” proteins lacking sequence homologs [35]. Environmental factors such as pH, temperature, and solvent effects remain unmodeled. Commercial partnerships with Eli Lilly and Novartis (reportedly totaling approximately $6 billion in deals) underscore pharmaceutical industry confidence, yet AF3 remains a hypothesis-generation tool requiring experimental validation [36].
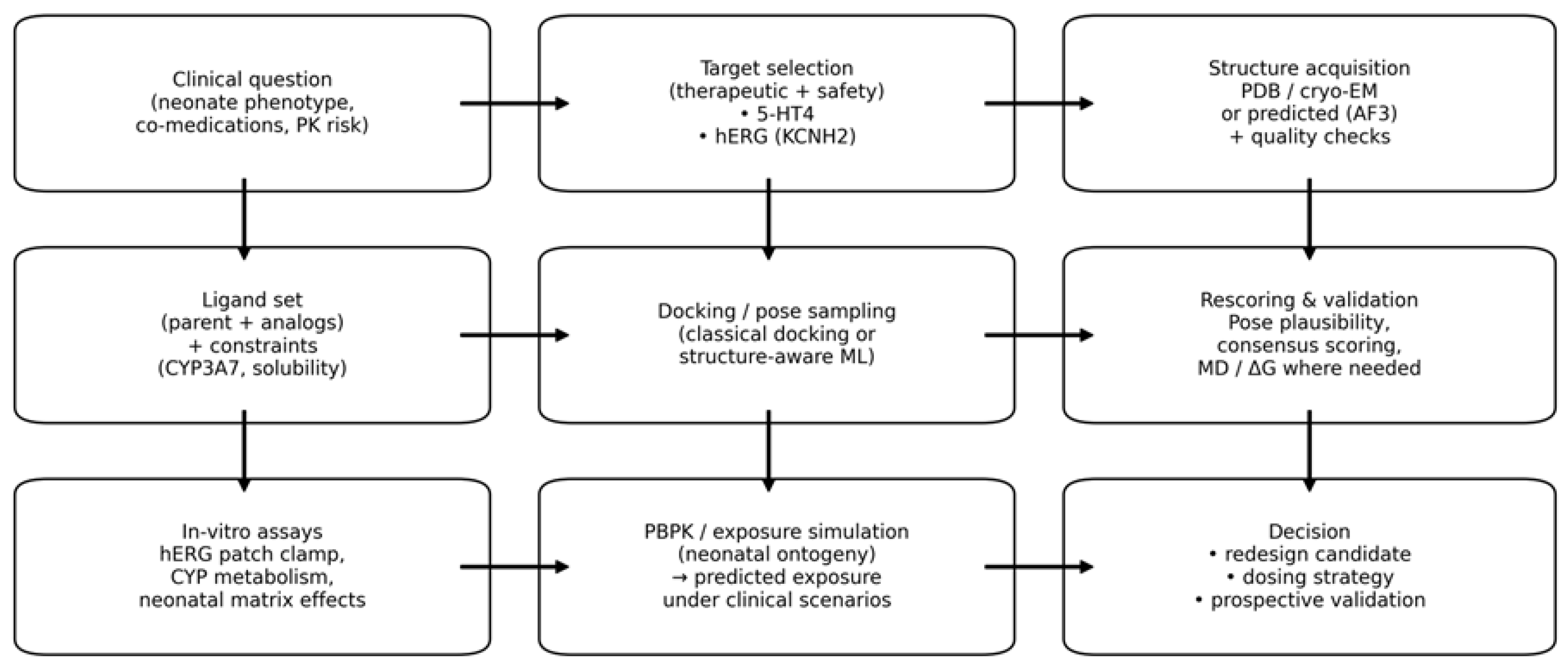
Figure 2.
Suggested structure-based workflow for integrating molecular docking with neonatal clinical constraints (PK risk, DDIs) and developmental validation assays. Practical implication: In neonatal use cases, docking should be treated as a decision-support layer within a broader validation stack (Figure 1). When a safety liability is well-defined (e.g., hERG blockade), docking and machine-learning predictors can be used to triage and redesign molecules, but only if predictions are repeatedly cross-checked against experimental electrophysiology and neonatal exposure models.
Figure 2.
Suggested structure-based workflow for integrating molecular docking with neonatal clinical constraints (PK risk, DDIs) and developmental validation assays. Practical implication: In neonatal use cases, docking should be treated as a decision-support layer within a broader validation stack (Figure 1). When a safety liability is well-defined (e.g., hERG blockade), docking and machine-learning predictors can be used to triage and redesign molecules, but only if predictions are repeatedly cross-checked against experimental electrophysiology and neonatal exposure models.
4.2. Large Language Models and Neuro-Symbolic Constraints in Chemistry
Large Language Models (LLMs) operating on chemical text strings (SMILES notation) allow for systematic exploration of chemical space. Models such as ChemBERTa create dense vector representations of molecules, capturing complex features like valency and aromaticity that enable similarity searching and property prediction [37]. For toxicity prediction, LLM-based predictors have achieved significant advances: Logic Tensor Networks (LTN) have demonstrated accuracies as high as 93% in classifying hERG blockers, significantly outperforming traditional Random Forest models [38].
Generative models like MolGPT can produce novel molecular structures token-by-token [39]. By conditioning the generation process, researchers can steer models to produce molecules with desired property profiles—for instance, high 5-HT4 affinity but low hERG affinity. ADMET-AI achieves the highest average rank on the Therapeutics Data Commons ADMET Leaderboard and can screen 1 million molecules in 3.1 hours [40]. For hERG cardiotoxicity specifically, specialized tools such as DeepHIT and CardioTox net achieve external validation accuracy exceeding 0.77–0.81 with area under the ROC curve metrics of approximately 0.83–0.94 [41].
The most compelling drug repurposing success involves baricitinib for COVID-19: BenevolentAI’s knowledge graph identified this JAK inhibitor within 48 hours, predicting dual antiviral and anti-inflammatory mechanisms subsequently validated in Phase 3 trials, leading to FDA Emergency Use Authorization and WHO-recommended treatment status with demonstrated mortality reduction [42]. Yet no AI-discovered drug has achieved full clinical approval as of late 2025. The first AI-designed molecule to reach Phase 2 with positive results is Insilico Medicine’s ISM001-055 (rentosertib) for idiopathic pulmonary fibrosis, with Phase 2a topline results announced in November 2024 and full results published in Nature Medicine in June 2025 [43]. Timeline compression is notable: Insilico Medicine achieved target-to-preclinical candidate in 18 months versus typical 4–6 year timelines, and Exscientia reports design cycles 70% faster with 10× fewer synthesized compounds required [44].
Despite these advances, important limitations warrant emphasis. LLMs trained on SMILES strings can generate chemically invalid or synthetically intractable molecules, a manifestation of “hallucination” in molecular generation that necessitates robust post-generation filtering [37,39]. The “activity cliff” problem—wherein structurally similar molecules exhibit dramatically different biological activities—remains a fundamental challenge for property prediction models that rely on molecular similarity assumptions [41]. Benchmark inflation through data leakage, where test-set molecules share high structural similarity with training data, can produce overly optimistic performance estimates that do not generalize to novel chemical scaffolds [40]. Critically, for neonatal applications, ADMET prediction models are almost exclusively trained on adult pharmacokinetic endpoints, and no validated neonatal-specific absorption, distribution, metabolism, and excretion datasets exist to calibrate these predictions for the unique developmental physiology of newborns.
Neuro-symbolic approaches, including logic tensor networks, have been proposed as a way to embed mechanistic constraints and improve generalization in safety prediction; however, many such models remain pre-publication or lack prospective validation, and they should be viewed as hypothesis-generating rather than regulatory-grade evidence [38].
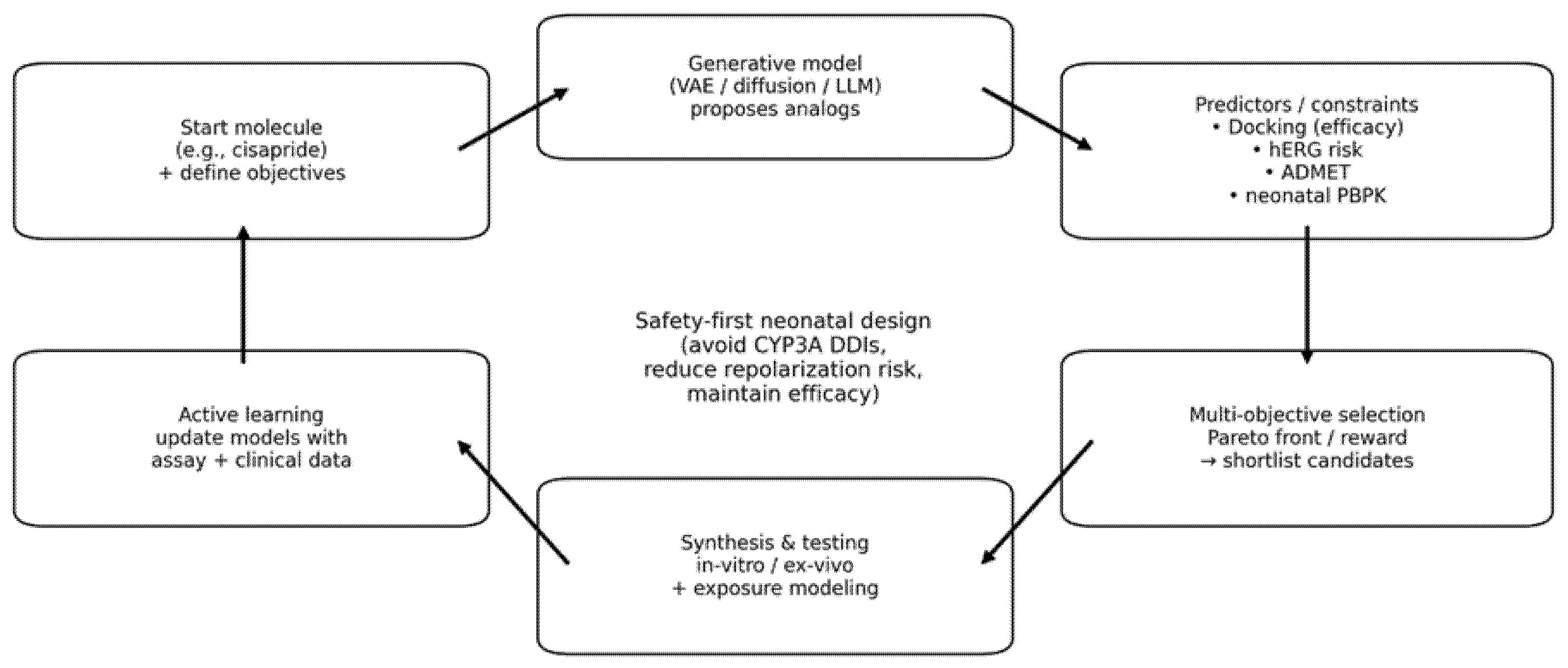
Figure 3.
Conceptual generative redesign loop for safety-first neonatal drug development. Candidate molecules are proposed, filtered by efficacy and safety predictors, evaluated under neonatal PBPK constraints, and iteratively improved through active learning with experimental data.
Figure 3.
Conceptual generative redesign loop for safety-first neonatal drug development. Candidate molecules are proposed, filtered by efficacy and safety predictors, evaluated under neonatal PBPK constraints, and iteratively improved through active learning with experimental data.
4.3. World Foundation Models: Simulating the Biological System
The concept of a “World Model” is evolving from robotics to the life sciences. A World Foundation Model (WFM) is a generative model trained on massive datasets to maintain an internal representation of a system’s physics and dynamics [45]. In physical AI (e.g., NVIDIA Cosmos), WFMs simulate friction and motion [46]. In biology, companies such as Isomorphic Labs are building WFMs to simulate the “laws of biology”—predicting how molecules will behave in biological contexts [47].
For neonatal applications, a biological WFM could theoretically simulate the “virtual neonate” by incorporating ontogeny data. Such a model would input a drug structure and a “neonate state vector” (high CYP3A7, low CYP3A4, low GFR, altered protein binding) to simulate the metabolic fate of the drug over time, predicting metabolic stagnation and accumulation before clinical trials. Single-cell foundation models such as scGPT (trained on 33 million cells) enable cell type annotation, gene network inference, and drug response prediction [48]. The Therapeutics Data Commons (TDC-2), released in 2024, now encompasses over 1,000 multimodal datasets across 85 million cells integrated with the PrimeKG knowledge graph [49].
However, the TDC authors acknowledge that “even the strongest algorithms fall short of solving key therapeutics challenges, including distributional shifts, multi-scale modeling, and robust generalization to novel data points” [49]. For neonatal applications specifically, foundation model readiness remains at Technology Readiness Level 2–3. No large-scale neonatal single-cell RNA-seq databases exist for training. Most toxicity databases focus on adult endpoints without developmental toxicity data. Current models trained on adult data cannot capture immature hepatic and renal function, fluctuating fluid compartments, rapid developmental changes, or organ maturation variations essential to neonatal physiology.
5. Generative Redesign: A Cisapride 2.0 Conceptual Framework
The existence of marketed safer 5-HT4 agonists validates the premise that cisapride’s toxicity is chemically correctable. Generative AI now provides systematic tools to solve the multi-objective optimization problem required to create safer alternatives. It is important to note that the framework proposed below is conceptual; the validation examples (prucalopride, mosapride, ATI-7505) were historically developed through traditional medicinal chemistry rather than AI-guided approaches, but they demonstrate that the chemical space for safer alternatives exists and is amenable to the computational strategies described herein.
5.1. Structure–Activity Relationships and the Optimization Objective
The critical structural feature distinguishing cisapride from safer alternatives is the 4-atom linking group between the piperidine nitrogen and fluorophenyl ring—nearly identical to haloperidol and terfenadine, both potent hERG blockers [50]. Mosapride, with only a 1-carbon linker, shows virtually no hERG blocking. Mutagenesis studies identified Y652 (tyrosine) and F656 (phenylalanine) as critical hERG residues mediating cisapride binding through cation–π interactions and π-stacking [51].
To create “Cisapride 2.0” for neonatal use, multi-objective optimization must target: (1) Efficacy: maintaining high-affinity 5-HT4 agonism (EC50 < 200 nM); (2) Safety: eliminating hERG binding (IC50 > 10 μM, ideally > 100 μM) to achieve a safety margin exceeding 1000-fold; (3) Metabolism: achieving clearance via pathways active in neonates (e.g., ester hydrolysis, sulfation) rather than CYP3A4-dependent hepatic metabolism; and (4) Lipophilicity reduction: lower logP reduces hERG binding propensity.
5.2. The Generative Workflow
Current workflows utilize reinforcement learning (RL). A generator (e.g., MolGPT) proposes molecular structures, which are evaluated by a “critic” array: AlphaFold 3 for binding predictions, hERG-specific neural networks for cardiotoxicity screening, and ADMET-AI for broader pharmacokinetic assessment [52]. The RL agent updates the generator to maximize a reward function defined by safety and efficacy constraints.
DeepHIT, a deep learning framework combining dense neural networks with graph convolutional networks, achieves external test accuracy of 0.773 with sensitivity of 0.833 [53]. Its most innovative feature is an in silico chemical transformation module generating virtual compounds from seed structures based on 78,471 known chemical transformation patterns—providing proof-of-concept by generating urotensin II receptor antagonists without hERG activity from a hERG-blocking parent compound. Studies using ScafVAE (Scaffold-constrained Variational Autoencoder) have successfully optimized molecules to minimize hERG inhibition while maximizing solubility, explicitly demonstrating the capability to “design out” cardiotoxicity [54].
5.3. Validation: Prucalopride and Mosapride as Proof-of-Concept
History confirms that safer alternatives are pharmacologically achievable. Prucalopride, FDA-approved in 2018 (Motegrity), employs a benzofurancarboxamide scaffold completely different from the benzamide class. Its hERG IC50 of approximately 5.7 μM is 20-fold weaker than cisapride, with a 5-HT4 selectivity margin exceeding 150-fold over other receptors and a 300-fold difference between 5-HT4 and hERG affinity [55]. Mosapride replaces the piperidine core with morpholine and uses a single-carbon linker, resulting in no significant hERG effect [56,57]. ATI-7505 was designed to be metabolically stable and cleared by non-CYP pathways [57,58]. An AI-driven workflow trained on the cisapride failure data could have accelerated the discovery of these safer alternatives and, prospectively, could identify novel scaffolds optimized for neonatal metabolic pathways.
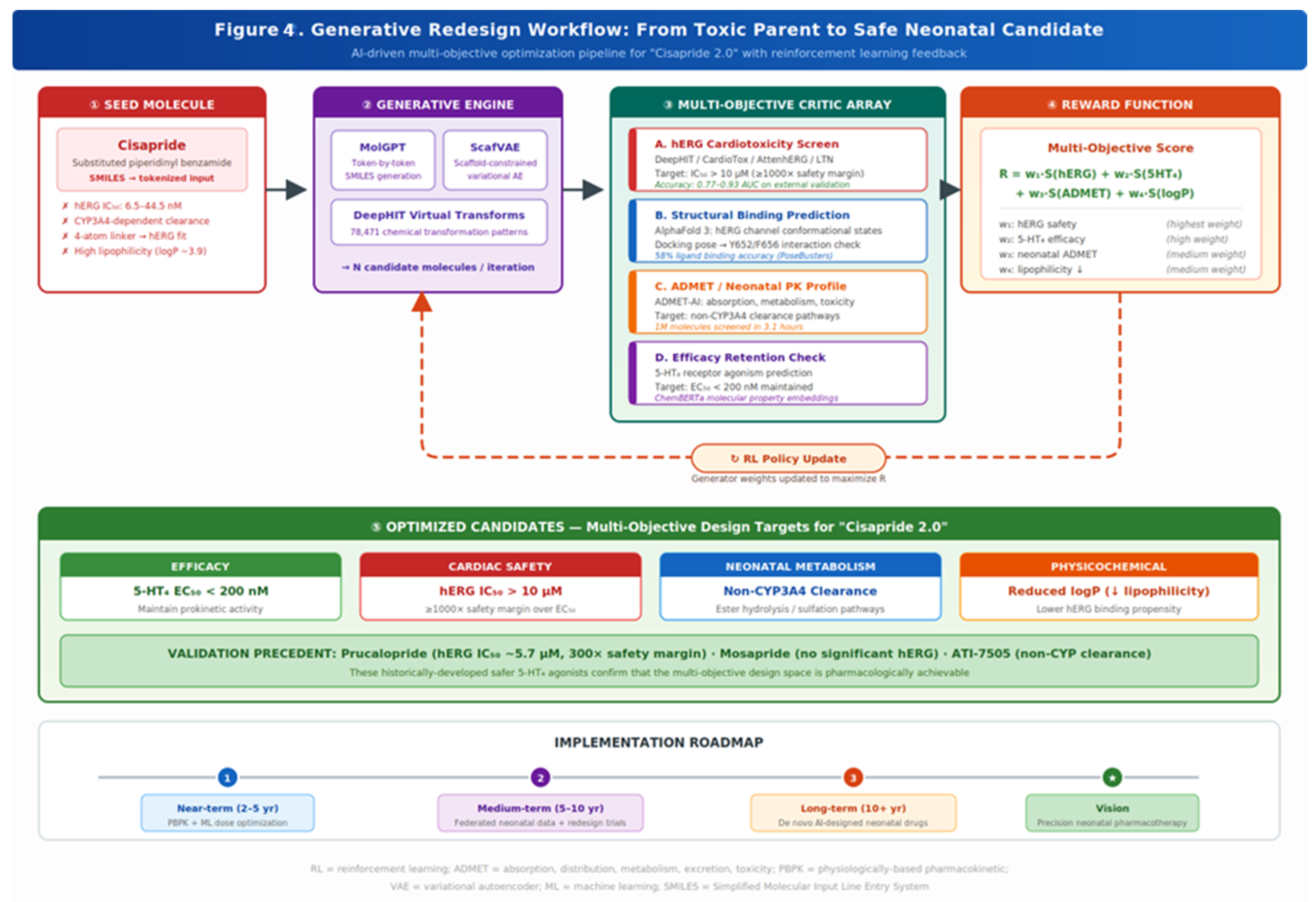
Figure 4.
Conceptual generative redesign workflow for “Cisapride 2.0.” Stage 1: The seed molecule (cisapride) is analyzed for toxicity-associated features (hERG IC50 6.5–44.5 nM, CYP3A4 dependence, 4-atom linker, high logP). Stage 2: Generative engines (MolGPT, ScafVAE, DeepHIT) propose virtual chemical transformations from 78,471 known patterns. Stage 3: A multi-objective critic array evaluates candidates for (A) hERG cardiotoxicity (target IC50 > 10 μM), (B) structural binding via AlphaFold 3 conformational modeling, (C) neonatal-appropriate ADMET profiles (non-CYP3A4 clearance), and (D) 5-HT4 efficacy retention (EC50 < 200 nM). Stage 4: A reward function R = w1·S(hERG) + w2·S(5-HT4) + w3·S(ADMET) + w4·S(logP) ranks candidates. Stage 5: Optimized leads meeting all four design criteria. Historical validation precedents (prucalopride, mosapride, ATI-7505) confirm the pharmacological feasibility of this multi-objective optimization.
Figure 4.
Conceptual generative redesign workflow for “Cisapride 2.0.” Stage 1: The seed molecule (cisapride) is analyzed for toxicity-associated features (hERG IC50 6.5–44.5 nM, CYP3A4 dependence, 4-atom linker, high logP). Stage 2: Generative engines (MolGPT, ScafVAE, DeepHIT) propose virtual chemical transformations from 78,471 known patterns. Stage 3: A multi-objective critic array evaluates candidates for (A) hERG cardiotoxicity (target IC50 > 10 μM), (B) structural binding via AlphaFold 3 conformational modeling, (C) neonatal-appropriate ADMET profiles (non-CYP3A4 clearance), and (D) 5-HT4 efficacy retention (EC50 < 200 nM). Stage 4: A reward function R = w1·S(hERG) + w2·S(5-HT4) + w3·S(ADMET) + w4·S(logP) ranks candidates. Stage 5: Optimized leads meeting all four design criteria. Historical validation precedents (prucalopride, mosapride, ATI-7505) confirm the pharmacological feasibility of this multi-objective optimization.
6. Implementation Barriers and Strategic Roadmap
6.1. Data Scarcity and the Neonatal Training Gap
The data scarcity problem is paramount. With 90% off-label use indicating minimal clinical data generation, foundation models lack the training substrate needed for neonatal-specific predictions. Neonatal disease mechanisms for conditions such as necrotizing enterocolitis, bronchopulmonary dysplasia, and retinopathy of prematurity remain incompletely characterized, creating target identification gaps [59]. Ethical constraints on prospective data generation in critically ill neonates fundamentally limit the ability to generate training data. International consortium building using federated learning approaches—aggregating data across NICUs while protecting patient privacy—represents a potentially critical enabling factor, though the infrastructure and governance frameworks required for such initiatives remain in early stages of development [60].
6.2. Regulatory Framework Immaturity
The FDA’s January 2025 draft guidance on “Considerations for the Use of AI to Support Regulatory Decision Making for Drug and Biological Products” provides a credibility assessment framework but explicitly excludes drug discovery applications that do not directly impact patient safety, drug quality, or study reliability [61]. Over 500 AI-related submissions have reached the Center for Drug Evaluation and Research (CDER) since 2016, but regulatory pathways for AI-derived predictions in the drug approval process remain immature [62]. The validation requirements for AI-designed molecules in vulnerable populations such as neonates have not been established.
6.3. A Phased Development Roadmap
Near-term opportunities (2–5 years) include AI-assisted clinical trial optimization for existing neonatal studies, integration of physiologically-based pharmacokinetic (PBPK) modeling with machine learning for dose optimization, deployment of model-informed precision dosing for drugs with established population PK models (vancomycin, aminoglycosides, morphine), and AI-powered adverse event signal detection from NICU electronic health records [63].
Medium-term goals (5–10 years) should focus on building the data infrastructure necessary for neonatal-specific foundation models through international consortia, developing federated learning approaches, and focusing drug redesign efforts on compounds such as cisapride where mechanisms of toxicity are understood and safer chemical space is mapped.
The longest-term ambition—de novo AI-designed drugs specifically for neonatal diseases—will require fundamental advances in understanding developmental biology at molecular resolution and will likely remain beyond the 10-year horizon without substantial investment in basic neonatal science.
6.4. Ethical and Equity Considerations
The application of AI-driven drug development in neonatal populations raises important ethical considerations that warrant deliberate attention. First, the use of retrospective clinical data and electronic health records to train AI models in neonatal medicine must navigate complex consent frameworks, particularly given the vulnerability of the patient population and the inability of neonates to provide assent. Institutional review boards and regulatory bodies have not yet established standardized guidelines for the secondary use of neonatal clinical data in AI model training, and practices vary widely across jurisdictions [59,60].
Second, equity concerns are paramount. As noted, Africa reports 66% off-label drug use prevalence in neonates [2], yet the AI tools described in this review—AlphaFold 3, LLM-based molecular generators, and WFMs—require substantial computational infrastructure, specialized expertise, and ongoing maintenance costs. Without deliberate mechanisms for technology transfer and capacity building, AI-driven drug development risks exacerbating the existing disparity between high-resource and low-resource neonatal care settings. International consortia must explicitly incorporate equity provisions, ensuring that training datasets reflect global populations and that resulting therapeutic innovations are accessible to the settings with the greatest unmet need.
Third, algorithmic transparency and interpretability remain critical for clinical adoption. Clinicians responsible for prescribing decisions in vulnerable neonatal populations will require explainable outputs from AI-driven safety predictions—not merely confidence scores, but mechanistic rationale for why a given molecular modification reduces toxicity risk. The development of interpretable AI frameworks tailored to pharmacological decision-making represents both a technical and a regulatory imperative.
7. Conclusions
The 90% off-label burden in neonatal pharmacotherapy represents both a profound unmet need and an indictment of historical pharmaceutical development priorities. The history of neonatal pharmacotherapy is marked by the struggle to adapt adult tools to a unique physiological reality. The tragedy of cisapride—where negligible CYP3A7-mediated metabolism combined with potent hERG blockade created fatal arrhythmias in the most vulnerable patients—underscores the lethal consequences of ignoring ontogeny.
Today, the convergence of AlphaFold 3 for structural prediction, Large Language Models for molecular design and toxicity screening, and emerging World Foundation Models for biological simulation offers a promising pathway forward. AI tools do not eliminate the fundamental challenges of developmental pharmacology, but they offer the potential to systematically redesign legacy drugs for neonatal safety, optimize dosing in the face of maturational variability, and accelerate the generation of evidence where none exists. Important caveats remain: LLM-based molecular generation is susceptible to hallucination and activity cliff artifacts, ADMET models lack neonatal-specific training data, and WFMs remain at an early stage of technological readiness for developmental biology applications.
Realizing this potential will require sustained investment in neonatal-specific data generation, regulatory framework evolution, international consortium building, deliberate attention to equity and access, and a fundamental commitment to studying the smallest and most vulnerable patients whose therapeutic needs have been orphaned for too long. By simulating the interaction between drug molecules and the immature neonatal system, we can move from the era of the “therapeutic orphan” to an era of precision, AI-designed neonatal pharmacotherapy.
Abbreviations.
The following abbreviations are used in this manuscript:
| 5-HT4 | 5-Hydroxytryptamine Receptor Type 4 |
| ADMET | Absorption, Distribution, Metabolism, Excretion, and Toxicity |
| AF3 | AlphaFold 3 |
| BPCA | Best Pharmaceuticals for Children Act |
| CDER | Center for Drug Evaluation and Research |
| CYP | Cytochrome P450 |
| EC50 | Half-Maximal Effective Concentration |
| GFR | Glomerular Filtration Rate |
| hERG | Human Ether-à-go-go-Related Gene |
| IC50 | Half-Maximal Inhibitory Concentration |
| IKr | Rapid Delayed Rectifier Potassium Current |
| LLM | Large Language Model |
| NICU | Neonatal Intensive Care Unit |
| PBPK | Physiologically-Based Pharmacokinetic |
| PK | Pharmacokinetic |
| PREA | Pediatric Research Equity Act |
| QTc | Corrected QT Interval |
| RL | Reinforcement Learning |
| RMSD | Root Mean Square Deviation |
| SMILES | Simplified Molecular Input Line Entry System |
| TDC | Therapeutics Data Commons |
| TRL | Technology Readiness Level |
| UGT | UDP-Glucuronosyltransferase |
| WFM | World Foundation Model |
Author Contributions
Conceptualization, writing—original draft preparation, writing—review and editing, supervision, George T. El-Ferzli. The author has read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Generative AI tools (ChatGPT 5.2 Pro, OpenAI) were used exclusively for language refinement, formatting assistance, and improving clarity of expression. All scientific concepts, interpretations, conclusions, analyses, and the full intellectual content of the manuscript were conceived, generated, and verified by the authors. No AI tools were used to generate data, perform analysis, create scientific claims, or determine results. The authors thoroughly reviewed, validated, and edited all AI-assisted text and take full responsibility for the final manuscript.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Shirkey, H. Therapeutic orphans. J. Pediatr. 1968, 72, 119–120. [Google Scholar] [CrossRef]
- Costa, H.T.M.L.; Costa, T.X.; Martins, R.R.; Oliveira, A.G. Use of off-label and unlicensed medicines in neonatal intensive care. PLoS ONE 2018, 13, e0204427. [Google Scholar] [CrossRef]
- Nir-Neuman, H.; Abu-Kishk, I.; Toledano, M.; et al. Unlicensed and Off-Label Medication Use in Pediatric and Neonatal Intensive Care Units: No Change Over a Decade. Adv. Ther. 2018, 35, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Horen, B.; Montastruc, J.L.; Lapeyre-Mestre, M. Adverse drug reactions and off-label drug use in paediatric outpatients. Br. J. Clin. Pharmacol. 2002, 54, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, R.; Bates, D.W.; Landrigan, C.; et al. Medication errors and adverse drug events in pediatric inpatients. JAMA 2001, 285, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.H.S.; Lipshultz, S.E. Caffeine and clinical outcomes in premature neonates. Children 2019, 6, 118. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Pediatric Labeling Changes. Available online: https://www.fda.gov/science-research/pediatrics/pediatric-labeling-changes (accessed on 13 February 2026).
- Laughon, M.M.; Avant, D.; Tripathi, N.; et al. Drug labeling and exposure in neonates. JAMA Pediatr. 2014, 168, 130–136. [Google Scholar] [CrossRef]
- Stevens, J.C. New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov. Today 2006, 11, 440–445. [Google Scholar] [CrossRef]
- Hines, R.N. Ontogeny of human hepatic cytochromes P450. J. Biochem. Mol. Toxicol. 2007, 21, 169–175. [Google Scholar] [CrossRef]
- de Wildt, S.N.; Kearns, G.L.; Leeder, J.S.; et al. Cytochrome P450 3A: Ontogeny and drug disposition. Clin. Pharmacokinet. 1999, 37, 485–505. [Google Scholar] [CrossRef]
- Bhatt, D.K.; Mehrotra, A.; Gaedigk, A.; et al. Age- and Genotype-Dependent Variability in the Protein Abundance and Activity of Six Major Uridine Diphosphate-Glucuronosyltransferases in Human Liver. Clin. Pharmacol. Ther. 2019, 105, 131–141. [Google Scholar] [CrossRef]
- Knibbe, C.A.J.; Krekels, E.H.J.; van den Anker, J.N.; et al. Morphine Glucuronidation in Preterm Neonates, Infants and Children Younger than 3 Years. Clin. Pharmacokinet. 2009, 48, 371–385. [Google Scholar] [CrossRef]
- van den Anker, J.; Reed, M.D.; Allegaert, K.; et al. Developmental changes in pharmacokinetics and pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, C.M.; Svahn, S.; Van der Linden, A.; et al. Individualised dosing of amikacin in neonates: a pharmacokinetic/pharmacodynamic analysis. Eur. J. Clin. Pharmacol. 2009, 65, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Kalamees, R.; Soeorg, H.; Ilmoja, M.; et al. Prospective validation of a model-informed precision dosing tool for vancomycin treatment in neonates. Antimicrob. Agents Chemother. 2024, 68, e01591-23. [Google Scholar] [CrossRef] [PubMed]
- Gallaway, K.A.; Cann, K.; Oetting, K.; et al. The potential impact of preemptive pharmacogenetic genotyping in the neonatal intensive care unit. J. Pediatr. 2023, 259, 113489. [Google Scholar] [CrossRef]
- McDermott, J.H.; Mahaveer, A.; James, R.A.; et al. Rapid point-of-care genotyping to avoid aminoglycoside-induced ototoxicity in neonatal intensive care. JAMA Pediatr. 2022, 176, 486–492. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Cisapride: What can we learn from the rise and fall of a prokinetic? J. Dig. Dis. 2011, 12, 147–156. [Google Scholar] [CrossRef]
- Mohammad, S.; Zhou, Z.; Gong, Q.; et al. Blockage of the HERG human cardiac K+ channel by the gastrointestinal prokinetic agent cisapride. Am. J. Physiol. 1997, 273, H2534–H2538. [Google Scholar] [CrossRef]
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Wysowski, D.K.; Corken, A.; Gallo-Torres, H.; et al. Postmarketing reports of QT prolongation and ventricular arrhythmia in association with cisapride and Food and Drug Administration regulatory actions. Am. J. Gastroenterol. 2001, 96, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Treluyer, J.M.; Rey, E.; Sonnier, M.; et al. Evidence of impaired cisapride metabolism in neonates. Br. J. Clin. Pharmacol. 2001, 52, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.E.; Gotschall, R.R.; Kearns, G.L.; Leeder, J.S. Cytochrome P450 involvement in the biotransformation of cisapride and racemic norcisapride in vitro: Differential activity of individual human CYP3A isoforms. Drug Metab. Dispos. 2001, 29, 1548–1554. [Google Scholar] [PubMed]
- Kearns, G.L.; Robinson, P.K.; Wilson, J.T.; et al. Cisapride disposition in neonates and infants: In vivo reflection of cytochrome P450 3A4 ontogeny. Clin. Pharmacol. Ther. 2003, 74, 312–325. [Google Scholar] [CrossRef]
- Cordeiro, J.M.; Panama, B.K.; Goodrow, R.; et al. Developmental changes in expression and biophysics of ion channels in the canine ventricle. J. Mol. Cell. Cardiol. 2013, 65, 34–45. [Google Scholar] [CrossRef]
- Vandenplas, Y.; Benatar, A.; Cools, F.; et al. Efficacy and tolerability of cisapride in children. Paediatr. Drugs 2001, 3, 559–573. [Google Scholar] [CrossRef]
- Roden, D.M. Drug-induced prolongation of the QT interval. N. Engl. J. Med. 2004, 350, 1013–1022. [Google Scholar] [CrossRef]
- Barnett, C.P.; Omari, T.; Davidson, G.P.; et al. Effect of cisapride on gastric emptying in premature infants with feed intolerance. J. Paediatr. Child Health 2001, 37, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Google DeepMind. AlphaFold 3 predicts the structure and interactions of all of life’s molecules. Available online: https://blog.google/technology/ai/google-deepmind-isomorphic-alphafold-3-ai-model/ (accessed on 13 February 2026).
- Kryshtafovych, A.; Schwede, T.; Topf, M.; et al. Critical assessment of methods of protein structure prediction (CASP)—Round XV. Proteins 2023, 91, 1539–1549. [Google Scholar] [CrossRef]
- Ngo, K.; Yang, P.C.; Yarov-Yarovoy, V.; Clancy, C.E.; Vorobyov, I. Harnessing AlphaFold to Reveal hERG Channel Conformational State Secrets. eLife 2025, 13, RP104901. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Lin, H.; Alade, A.A.; et al. AlphaFold3 in Drug Discovery: A Comprehensive Assessment of Strengths and Limitations. bioRxiv 2025. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Koumadorakis, D.E.; Lazaros, K.; et al. AlphaFold 3: An overview of applications and performance insights. Int. J. Mol. Sci. 2025, 26, 3671. [Google Scholar] [CrossRef] [PubMed]
- Isomorphic Labs. Partnerships with Eli Lilly and Novartis. Available online: https://www.isomorphiclabs.com/ (accessed on 13 February 2026).
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Hossain, D.; Al Abir, F.; Chen, J.Y. hERG-LTN: A New Paradigm in hERG Cardiotoxicity Assessment Using Neuro-Symbolic and Generative AI Embedding Approach. bioRxiv 2025. [Google Scholar] [CrossRef]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; et al. MolGPT: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2022, 62, 2064–2076. [Google Scholar] [CrossRef]
- Swanson, K.; Walters, P.; Sellers, B. ADMET-AI: A machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics 2024, 40, btae416. [Google Scholar] [CrossRef]
- Yang, T.; Ding, X.; McMichael, E.; et al. AttenhERG: A reliable and interpretable graph neural network framework for predicting hERG channel blockers. J. Cheminform. 2024, 16, 95. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef]
- Xu, Z.; Ren, F.; Wang, P.; et al. A generative AI-discovered TNIK inhibitor for idiopathic pulmonary fibrosis: A randomized phase 2a trial. Nat. Med. 2025, 31, 2602–2610. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- NVIDIA. World Models Definition. NVIDIA Glossary. Available online: https://www.nvidia.com/en-us/glossary/world-models/ (accessed on 13 February 2026).
- NVIDIA. Cosmos: Physical AI World Foundation Models. Available online: https://www.nvidia.com/en-us/ai/cosmos/ (accessed on 13 February 2026).
- Noetik, AI. OCTO—World Models as Simulators of Patient Biology. Available online: https://www.noetik.ai/octo (accessed on 13 February 2026).
- Cui, H.; Wang, C.; Maan, H.; et al. scGPT: Toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 2024, 21, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Velez-Arce, A.; Huang, K.; Li, X.L.; et al. TDC-2: Multimodal Foundation for Therapeutic Science. bioRxiv 2024. [Google Scholar] [CrossRef]
- Fermini, B.; Fossa, A.A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; et al. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 12329–12333. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; et al. Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Lee, M.Y.; Lee, J.H.; et al. DeepHIT: a deep learning framework for prediction of hERG-induced cardiotoxicity. Bioinformatics 2020, 36, 3049–3055. [Google Scholar] [CrossRef]
- Dong, T.; You, L.; Chen, CY-C..; et al. Multi-objective drug design with a scaffold-aware variational autoencoder. Chem. Sci. 2025, 16, 13352–13367. [Google Scholar] [CrossRef]
- Camilleri, M.; Kerstens, R.; Rykx, A.; et al. A placebo-controlled trial of prucalopride for severe chronic constipation. N. Engl. J. Med. 2008, 358, 2344–2354. [Google Scholar] [CrossRef]
- Carlsson, L.; Amos, G.J.; Andersson, B.; et al. Electrophysiological Characterization of the Prokinetic Agents Cisapride and Mosapride in Vivo and in Vitro: Implications for Proarrhythmic Potential? J. Pharmacol. Exp. Ther. 1997, 282, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Tack, J.; Camilleri, M.; Chang, L.; et al. Systematic Review: Cardiovascular Safety Profile of 5-HT4 Agonists Developed for Gastrointestinal Disorders. Aliment. Pharmacol. Ther. 2012, 35, 745–767. [Google Scholar] [CrossRef]
- Camilleri, M.; Vazquez-Roque, M.I.; Burton, D.; et al. Pharmacodynamic effects of a novel prokinetic 5-HT receptor agonist, ATI-7505, in humans. Neurogastroenterol. Motil. 2007, 19, 30–38. [Google Scholar] [CrossRef]
- Allegaert, K.; van de Velde, M.; van den Anker, J. Neonatal clinical pharmacology. Paediatr. Anaesth. 2014, 24, 30–38. [Google Scholar] [CrossRef]
- Allegaert, K.; Yalcin, N.; Flint, R.B.; et al. Neonatal Clinical Pharmacology. Children 2024, 11, 1102. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products. Draft Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents (accessed on 13 February 2026).
- U.S. Food and Drug Administration. Artificial Intelligence for Drug Development. Available online: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/artificial-intelligence-drug-development (accessed on 13 February 2026).
- Conte, L.; Decembrino, N.; Arribas, C.; et al. Leveraging Artificial Intelligence for Decision Support in Neonatal and Pediatric Pharmacotherapy: A Scoping Review. Semin. Fetal Neonatal Med. 2025, 19, 101691. [Google Scholar] [CrossRef]
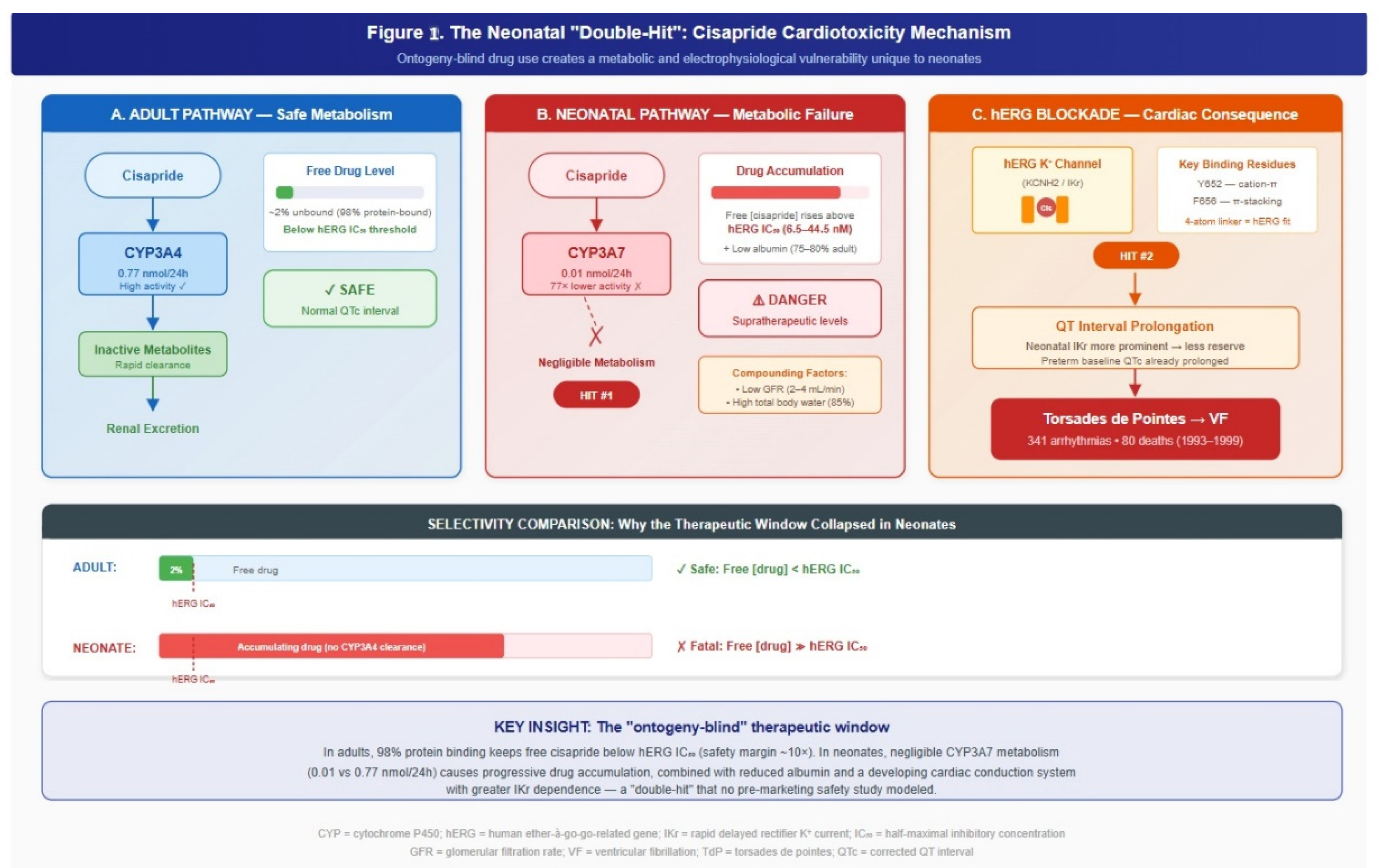
Figure 1.
The neonatal “double-hit” mechanism of cisapride cardiotoxicity. Panel A: In adults, CYP3A4 efficiently metabolizes cisapride (0.77 nmol/24 h), maintaining free drug concentrations below the hERG IC50 threshold. Panel B: In neonates, CYP3A7 dominance results in negligible metabolism (0.01 nmol/24 h; 77-fold reduction), causing drug accumulation compounded by low GFR (2–4 mL/min) and high total body water (85%). Panel C: Supratherapeutic concentrations block the hERG potassium channel at Y652 and F656 binding residues, prolonging the QT interval and precipitating torsades de pointes. The bottom panel compares adult versus neonatal free drug concentrations relative to the hERG IC50 safety threshold.
Figure 1.
The neonatal “double-hit” mechanism of cisapride cardiotoxicity. Panel A: In adults, CYP3A4 efficiently metabolizes cisapride (0.77 nmol/24 h), maintaining free drug concentrations below the hERG IC50 threshold. Panel B: In neonates, CYP3A7 dominance results in negligible metabolism (0.01 nmol/24 h; 77-fold reduction), causing drug accumulation compounded by low GFR (2–4 mL/min) and high total body water (85%). Panel C: Supratherapeutic concentrations block the hERG potassium channel at Y652 and F656 binding residues, prolonging the QT interval and precipitating torsades de pointes. The bottom panel compares adult versus neonatal free drug concentrations relative to the hERG IC50 safety threshold.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



