Submitted:
10 April 2026
Posted:
14 April 2026
You are already at the latest version
Abstract
Carbon Capture and Storage (CCS) and Direct Air Capture (DAC) technologies must improve quickly due to the escalating climate problem, which is caused by annual CO₂ emissions reaching 37 billion metric tons. Because of their remarkable surface areas and customizable pore topologies, Metal–Organic Frameworks (MOFs) have become very intriguing sorbents; yet, exploring their large chemical design space is still computationally prohibitive. The growing significance of machine learning (ML) in advancing CO₂ capture research within MOFs is methodically examined in this review. We evaluate state-of-the-art models in four important areas using a structured evaluation protocol: process-level application, mechanistic interpretability, descriptor physical relevance, and predictive performance. Recent developments in Machine Learning Interatomic Potentials (MLPs) challenge traditional rigid-lattice assumptions by showing that framework flexibility greatly affects diffusivity and adsorption thermodynamics. While physics-informed descriptor engineering achieves R² values ranging from 0.81 to 0.97 depending on gas species and pressure regime, generative techniques, such as Deep Reinforcement Learning and transformer-based topologies, enable the inverse construction of high-affinity frameworks. Crucially, the field is moving away from isolated property prediction and toward multiscale, process-integrated optimization, where machine learning models combine material characteristics with industrial performance indicators like recovery and CO₂ purity in pressure swing adsorption systems. All of these advancements point to the need for physics-informed, comprehensible designs that can connect molecular-scale discovery with experimentally reliable, water-stable materials appropriate for commercial use.
Keywords:
machine learning
; MOFs
; CO2
; green house gases
; adsorption
; artificial intelligence
Introduction
The average world temperature has been gradually increasing; in 2023, it was found to be about 1.2 °C higher than the preindustrial average. Greenhouse gas emissions are the primary cause of this warming, with carbon dioxide (CO₂) responsible for more than 60% of the impact. The urgent need to reach "net zero" targets by 2050 is highlighted by the fact that annual worldwide CO₂ emissions exceeded 37.15 billion tons in 2022. Carbon Capture and Storage (CCS) and Direct Air Capture (DAC) have shown promise in controlling these levels by reducing emissions from ambient air and industrial point sources. [1]
Finding materials with strong CO₂ affinity and good selectivity over other gases, such as ambient water vapor (H₂O) or nitrogen (N₂), is a major difficulty for these technologies.[2] Because of their ultrahigh surface areas, configurable pore diameters, and programmable functions, Metal-Organic Frameworks (MOFs), crystalline porous materials made of metal clusters connected by organic ligands have emerged as promising possibilities. MOFs can be specifically tailored with open metal sites (OMS) or particular chemical functional groups (like diamines) to improve CO₂ binding performance, in contrast to conventional adsorbents.[3]
But there are already more than 100,000 synthesized structures and trillions more proposed in silico, so the field of chemical design for MOFs is almost limitless. It is computationally and time-consuming to thoroughly evaluate these materials using conventional laboratory synthesis or high-fidelity molecular simulations like Density Functional Theory (DFT) or Grand Canonical Monte Carlo (GCMC).[4] To identify next-generation carbon capture materials more quickly, this bottleneck calls for a move toward data-driven computational screening and generative design. By providing a balance between the speed of classical force fields and the high precision of ab initio approaches, machine learning (ML) has emerged as a revolutionary tool for CO₂ remediation. ML applications have four main remediated purposes in the context of MOFs:
Tens of thousands of materials may be screened in a fraction of the time needed for conventional simulations thanks to machine learning (ML) models like Random Forests (RF) and Artificial Neural Networks (ANN), which are used to quickly forecast CO2 working capacity and selectivity.[5]
Researchers can describe framework flexibility using advanced Machine-Learning Interatomic Potentials (MLPs), which show that structural vibrations can increase CO₂ diffusivity by an order of magnitude when compared to stiff models. Additionally, the combination of high-throughput computational screening and artificial intelligence has improved predictive modelling, directing experimental efforts toward ideal materials and improving CO₂ adsorption efficiency by designing core-shell MOFs.[6]
For the inverse design of MOFs, methods such as Large Language Models and Deep Reinforcement Learning (DRL) are used to navigate deep subspaces and identify materials with severe CO2 affinity that are uncommon in current databases. By forecasting process-level parameters like CO2 purity, recovery, and energy productivity in pressure swing adsorption, machine learning (ML) helps close the gap between material attributes and industrial cycle performance.
Researchers can now quantify the impact of particular material characteristics, such as Lewis’s acidity, pore shape, and steric hindrance, by combining understandable machine learning models with SHAP or PDP analysis. This provides a theoretical foundation for the upcoming generation of CO₂ capture experiments.[7]
The following fundamental standards served as the basis for writing the reviews:Every evaluation lists the precise attributes that were anticipated (such as uptake, selectivity, or TOF) as well as the performance metrics (such as R2 or RMSE) attained in comparison to predetermined "ground truth" labels.Models were assessed according to whether their inputs (descriptors), such as pore limiting diameter, metal partial charges, or energy-based radial distribution functions, were from basic physics or chemical intuition. The reviews address the methods used to evaluate the models (such as k-fold cross-validation) and whether or not they showed transferability to new material classes or external experimental data. Each assessment emphasizes the novel scientific understanding offered by the model, such as the "bifurcation" of ideal pore sizes or the "coupling effect" between functional dopants and micropore volume, in addition to reporting accuracy.
Every analysis discusses the work's shortcomings, such as the failure to describe chemical reactions in humid streams, the dependence on rigid-framework assumptions, or the omission of open metal sites.The model's usefulness for industrial scalability and its contribution to the advancement of computational materials science are appropriately reflected in the review summary that results from this methodical foundation.
1. Physics-Informed Descriptor Engineering
1.1. Descriptor Engineering Strategies
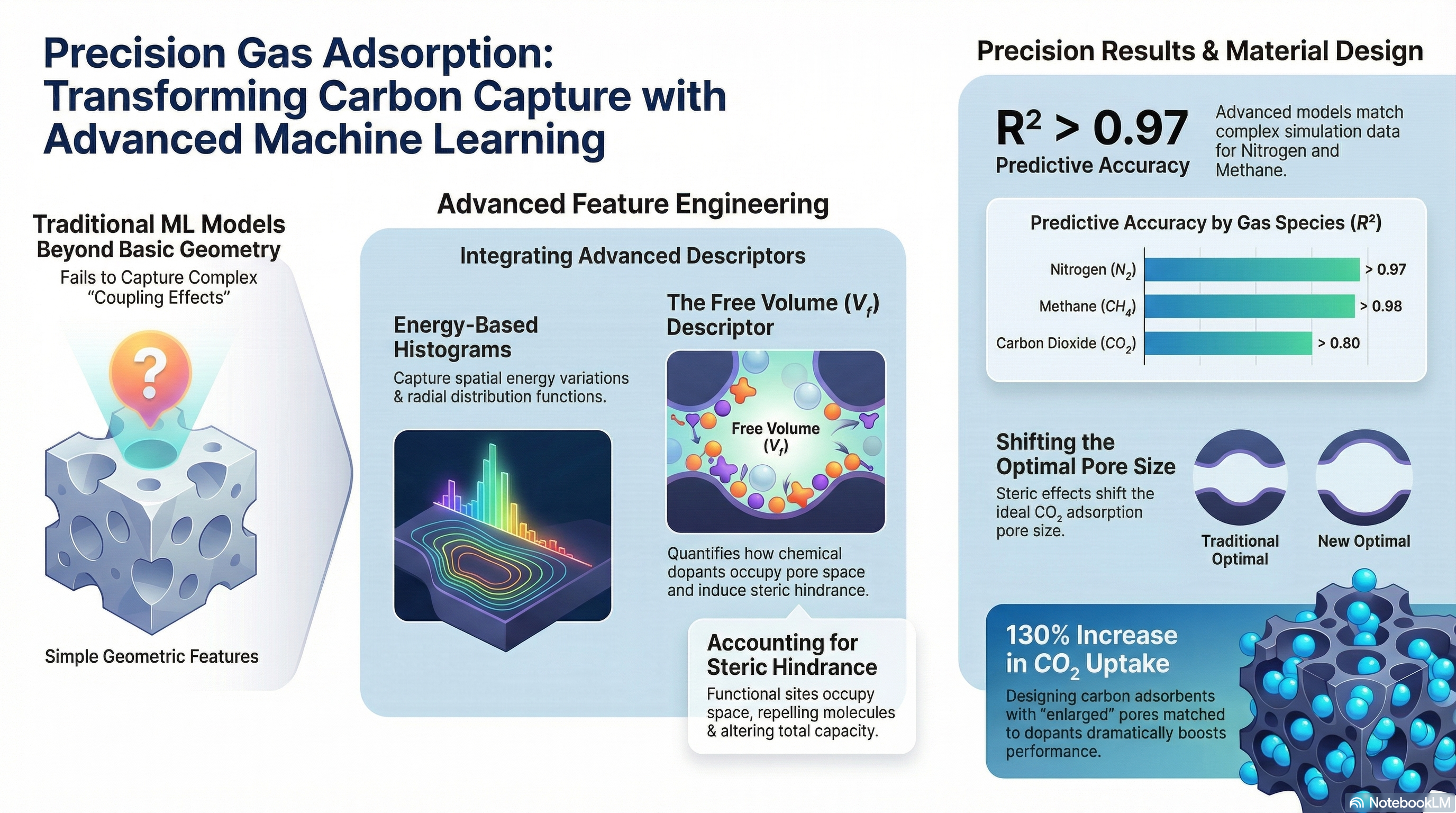
An important step toward understanding the intricate potential energy surfaces (PES) of porous materials is the incorporation of spatially aware energy descriptors. Researchers have obtained R²>0.97 for nitrogen and R²>0.87 for carbon dioxide isotherms by adding surface energy histograms and radial distribution functions (RDFs) to conventional geometric features and Henry's constants. By giving the ML model a physically valid representation of surface form, this method specifically addresses the "intermediate pressure bottleneck"—the region where both binding energy and spatial heterogeneity govern uptake.
Importantly, using XGBoost trained on more than 10,000 structures shows that the spatial decay of interaction sites (recorded via RDF) is the main differentiator for capacity in frameworks with comparable energy levels, whereas affinity distribution (f(E)) establishes the energetic baseline. The model's poor performance in the CO₂ chemisorption regime, however, emphasizes the single-charge probes' present limitations. Future physics-informed machine learning approaches must use dipole and quadrupole probes to account for the orientation-dependent packing and electrostatic multipoles of CO₂, bridging the gap between simplified physisorption models and practical selective separation, in order to achieve industrial-grade reliability.[8]
By moving beyond standalone pore or doping engineering, researchers have identified a critical micropore-dopant coupling mode that determines CO₂ transport in carbon-based adsorbents. Using a Random Forest architecture trained on multi-scale simulation data (R²=0.934), this study introduces Free volume (Vf) as a primary descriptor (accounting for 25% relative importance) to quantify the steric effects induced by surface functionalization. This approach reveals a significant thermodynamic shift: while basic dopants (e.g., NH₂) utilize Lewis’s acid-base interactions to optimize adsorption at 7 Å, physisorption-dominant dopants (e.g., oxygen groups) occupy available nano space, necessitating an enlarged optimal pore size of 8–10 Å to maximize capacity. These results were validated experimentally, as adsorbents designed with this particular coupling mode attained a leading-level capacity of 4 mmol/g, a 130% improvement over unoptimized frameworks. These findings highlight the need for physics-based descriptor engineering to settle long-standing disputes over the promoting vs. inhibitory effects of heteroatom doping in porous carbons. [9]
Compared to conventional structural models, the incorporation of advanced descriptor engineering—more precisely, the insertion of more than 700 "calculated" molecular features—has been demonstrated to increase CO₂ adsorption prediction accuracy by 15%–20%. The difficulties in capturing sparse gas-solid interactions are highlighted by the XGBoost framework's performance sensitivity at low pressures (0.01 bar), even though it achieves a high coefficient of determination (R²>0.94) at industrial pressures (2.5 bar). Importantly, the application of SHAP interpretability reveals a basic thermodynamic transition: as pressure rises, charge distribution and electronegativity features (representing Coulomb forces) gradually replace the predictive weight of atomic mass and number (representing van der Waals forces) at low pressures. However, there is a risk of overestimating real-world performance due to the use of a hypothetical dataset (hMOF) and simulated GCMC labels; in the absence of experimental validation, the model's capacity to account for structural defects or competitive adsorption in complex flue gases continues to be a crucial gap for industrial scalability.[10]
1.2. Hybrid Textural-Optimization and Outlier-Aware Predictive Modeling
One significant step toward overcoming the "black-box" constraints of conventional adsorption modeling is the transition from solo machine learning to hybridized optimization frameworks. Researchers successfully neutralized the hyperparameter tuning mistakes that cause underestimation in solo models at high uptake regimes by combining the Growth Optimization (GO) method with Least Square Support Vector Machines (LSSVM), achieving an astounding R² of 0.9798. SBET is the main structural driver of capture, outperforming pore volume and Langmuir surface area in prediction sensitivity, according to a database-driven analysis of 475 experimental points.
Crucially, Williams plot analysis provides a level of statistical robustness necessary for quick screening by confirming that 94.95% of the experimental data fits into the model's applicable domain. The current model's reliance on experimental textural measurements suggests a continuous barrier in purely computational discovery processes. Future physics-informed machine learning approaches must close the gap between these high-accuracy experimental models and high-throughput theoretical proxies to improve industrial scalability. This will ensure the utilization of the "metal-affinity" and "pore-filling" dynamics discovered here in the de novo design of next-generation carbon capture materials.[11]
1.3. Ensemble Interaction Mapping and Node-Affinity Hierarchies
A major advancement in closing the accuracy gap between individual machine learning models and intricate experimental adsorption data is the implementation of unique stacking ensemble structures. Researchers were able to attain a benchmark-surpassing R² of 0.9833 for CO₂ uptake across 1,212 experimental data points by combining the predictive powers of tree-based, kernel-based, and neural network learners. By using ablation studies and permutation importance to separate the impact of thermodynamics from framework architecture, this study goes beyond straightforward performance reporting to offer a multi-criteria interpretability framework.
Crucially, partial dependence plots that measure the stepwise shift in CO₂ binding efficacy support the analysis's identification of a "metal-affinity hierarchy," where copper and magnesium centres clearly outperform conventional zinc-based nodes in terms of productivity. The 70% prevalence of zinc-based items in existing experimental archives, however, draws attention to a recurring problem with data imbalances that puts well-studied materials in danger of model overfitting. To ensure that the discovery of high-performance MOFs extends into the underrepresented chemical subspaces of rare or complicated metal centres, future physics-informed ML strategies must include stratified sampling and synthetic oversampling (e.g., SMOTE) to improve commercial scalability.[12]
The fundamental language for quantitatively representing MOF structures is established via physics-informed descriptor engineering, but large-scale predictive procedures are where these capabilities truly shine. Supervised learning methods can quickly predict adsorption parameters over large chemical spaces once chemically meaningful descriptors are defined. The first significant acceleration point in ML-driven CO₂ remediation research is this transition from feature construction to high-throughput prediction.
2. High-Throughput Screening and Universal Property Prediction
2.1. High-Throughput Screening and Discovery
The move to hybrid physics-ML models makes it possible to screen composite materials more effectively than with conventional molecular simulations. Researchers demonstrated that transfer learning is a reliable method for predicting "unseen" materials like 6FDA-DAM based on known polymer features by training a stacked ensemble regression model on over 54,000 hybrid membranes and achieving an outstanding forecast accuracy of R² = 0.96. Importantly, big data mining of this dataset revealed that the MOF filler becomes the bottleneck for elite performance instead of the polymer matrix; in top-performing membranes, PLD and LCD importance spikes to over 30%, indicating that once a high-permeability baseline is established, pore-size engineering becomes more important than polymer selection.
Constructive evaluation, however, indicates a reliance on the ideal interface assumption of the Maxwell model. Interfacial gaps and polymer chain rigidity frequently undermine the theoretical "Robeson limit" performance in practical industrial scalability. To guarantee that high-throughput screening yields materials that maintain their separation efficiency under the mechanical stresses of flue gas streams, forthcoming physics-informed machine learning initiatives should integrate interfacial morphology descriptors to reflect the complex bonding between organic linkers and polyimide matrices.[13]
The shift from simulation-heavy datasets to experimental-based machine learning models provides a more rigorous benchmark for actual CO₂ capture. Researchers have shown that ensemble learning can traverse the diverse terrain of 236 distinct experimental MOFs with a 15% RMSE improvement over conventional gradient-boosting frameworks by employing the CATBoost architecture. The integration of SHAP interpretability reveals a deeper layer of chemical control: localized atomic environments and electronic state distributions (e.g., PEOE_VSA7) are the true differentiators for optimizing adsorption, even though the model confirms that pressure and surface area remain the dominant thermodynamic drivers.
Nevertheless, this study also functions as a critical assessment of the limits of generalization. The discrepancy between the model's validation score (R2 = 0.84) and training performance (R2 = 0.99) highlights a recurring problem with overfitting when training sophisticated models on sparse experimental data. In order to guarantee that high-throughput screening of hypothetical frameworks can consistently transfer into industrially scalable, high-stability sorbents, future efforts in physics-informed machine learning must shift toward dataset diversification, embracing a larger spectrum of chemical functions.[14]
2.2. Universal Property Prediction and Isotherm Generalization
An important development in zeolite-based carbon capture is the shift away from material-specific empirical models and toward unified experimental datasets. Researchers have developed a "universal" predicting capability that greatly surpasses conventional Langmuir isotherm fittings in both accuracy and generalization by training a Gradient Boosted Trees (GBT) model using more than 5,700 experimental data sets. By identifying the Si/Al ratio and cation composition as crucial chemical factors, this method enables the model to generalize over a variety of frameworks (such as FAU, ZSM-5, and 13X) and high operating environments up to 45 bar.
Crucially, the model's resilience is confirmed by external validation on unseen datasets, which successfully resolves the overfitting issues typical of datasets derived from literature. A constructive criticism, however, shows that the dataset is still biased toward low-uptake regimes (<2 mmol/g) and that 32% of the training data did not report surface area. Future physics-informed machine learning approaches should prioritize the inclusion of high-uptake experimental benchmarks and uniform reporting of structural parameters to further optimize model sensitivity in peak performance regimes in order to improve industrial scalability.[15]
2.3. Property-Driven ML Applications
In this study, two particular properties—CO₂ working capacity and CO₂/N₂ selectivity—are predicted using machine learning. In order to quickly screen Metal-Organic Frameworks and achieve high predictive accuracy (R²) for carbon capture metrics, Artificial Neural Networks (ANNs) offer a computationally efficient substitute for Graph Neural Networks. This model quantifies a crucial design insight by combining industrial, field, and simulation data: CO₂ working capacity is primarily determined by pore size and surface area, exhibiting a weak negative association with greater chemical complexity. The CO₂ capacity predictions are reliable (MAE = 0.8 mmol/g), but the CO₂/N₂ selectivity predictions show significant dispersion (MAE = 25), indicating that intrinsic properties might not adequately capture the competitive adsorption physics needed for high-selectivity forecasting. In addition, the model's ability to generalize to new, uncharacterized structural fragments is limited in the absence of specific thermodynamic benchmarks (fixed T, P) or external experimental validation. Future physics-informed directions must address the high uncertainty in selectivity modeling to guarantee that quick AI-based screening translates into reliable industrial carbon reduction technologies.[16]
Predictive accuracy by itself does not ensure scientific comprehension, even when high-throughput screening greatly reduces computing costs. The physicochemical principles regulating adsorption behaviour may be obscured by black-box models. In order to derive mechanistic insights from trained models and guarantee that predictions are rooted in adsorption physics, interpretability frameworks and thermodynamic mapping techniques are crucial.
3. Model Interpretability and Thermodynamic Mapping
3.1. Model Interpretability and Physical Insight
Machine-learning potentials (MLPs) based on quantum chemistry data offer a high-fidelity substitute for the rigid-lattice assumptions commonly used in molecular simulations of gas transport. Researchers were able to achieve remarkable forecast accuracy (R² = 0.9916) for system energies by using the DeepPot-SE model to reflect the dynamic flexibility of MgMOF-74. This method shows that structural flexibility plays a major role in CO₂ diffusivity prediction; rigid models greatly overestimate adsorption free-energy barriers, leading to diffusion coefficients that are over ten times slower than those found in flexible frameworks.
The "hopping" mechanism between open metal sites is highlighted by this integration of thermodynamics and machine learning, although the model's capacity for generalization still has to be critically assessed. The MLP's success is inextricably linked to the accuracy of the underlying semi-empirical lot since it was trained solely on simulated DFT-MD trajectories from a single 30 ps frame. Additionally, the absence of direct experimental validation for Mg-MOF-74 diffusivity highlights a more general requirement for standardized experimental benchmarks to confirm the industrial scalability of such physics-informed machine learning models. Although studies that depend on small simulated datasets are advantageous in identifying physical patterns such as flexibility-enhanced diffusion, they should be used with caution in real-world applications where complicated gas mixtures and structural flaws may change the kinetics of transport.[17]
3.2. Ensemble-Averaged Thermodynamics and Potential Energy Surface (PES) Mapping
The creation of transferable machine learning force fields (MLFFs) represents a paradigm shift in the high-throughput screening of porous materials for direct air capture. Researchers have achieved thermodynamic ab initio quality at computing rates equivalent to classical approaches by fine-tuning a foundation model (MACE-MP) using a specific GoldDAC dataset. This method shows that in chemically complex hybrid settings, typical UFF+DDEC models have systemic errors, especially in lanthanide-based frameworks where H2O adsorption energies are overstated by up to 17.8%. The DAC-SIM package has identified 161 promising candidates by transitioning from single-point interaction energies to ensemble-averaged properties. This study demonstrates that sustaining a high CO₂/H₂O selectivity (KH ratio > 100) necessitates the "selective pocket" mechanism facilitated by parallel benzene rings (PAR) and uncoordinated nitrogen. The existing reliance on rigid-framework assumptions continues to be a barrier to adequately capturing the regeneration costs and chemisorption dynamics essential for large-scale industrial deployment, even if these models successfully root screening in real-world physics.[18]
The "cooperative insertion mechanism" is less about static binding and more about a dynamic redistribution that favours faster kinetics, according to this blend of thermodynamics and machine learning. The study's dependence on a solely computational MLP framework underscores the difficulties in simulating chemical transitions to carbamates or carbonates in humid streams, even though it uses actual NMR data for confirmation. Future physics-informed machine learning approaches must close the gap between these transport models and the intricate stability and regeneration cycles needed in high-humidity flue gas conditions to guarantee industrial scalability
4. Molecular Transport and Multicomponent Separation Mechanisms
4.1. Molecular-Level Transport and Adsorption Mechanisms
The long-standing computational bottleneck of modeling flexible frameworks with open metal sites (OMS) at ab initio accuracy is addressed by the application of fragment-based Neural Network Potentials (NNPs). In comparison to SCXRD experimental standards, this study obtained a remarkable force RMSE of 0.039 eV/Å while preserving great transferability with only 0.54% variance in lattice parameters by using an E (3)-equivariant architecture (NequIP) trained on a small collection of ~2,000 DFT conformers. Importantly, a hybrid MD/GCMC workflow that integrates thermodynamics and machine learning shows that structural dynamics are crucial for realistic adsorption modeling: framework flexibility promotes structural relaxation, which delocalizes CO₂ molecules and optimizes the energy landscape at low pressures (0.1–1.0 bar).
At 298 K, conventional rigid-lattice GCMC models offer a respectable approximation, but, at higher temperatures, they greatly underestimate uptake due to their inability to account for the required structural modifications between the host and guest. Even though this active-learning-driven screening was successful, a critical assessment of the fragment-based approach indicates that, although transferable, these models need to be thoroughly validated against various chemical environments to make sure they don't "forget" complex interactions outside of their original training fragment. These high-fidelity potentials should be used in future physics-informed machine learning approaches to investigate the cooperative insertion processes and regeneration cycles necessary for large-scale commercial carbon capture.[19]
4.2. Multicomponent Gas Separation and Structural Design Strategies
An important step towards the practical implementation of MOF-based carbon capture is the shift from binary/ternary gas models to real 6-component natural gas mixtures (N₂, CO₂, CH₄, C₂H₆, C₃H₈, H₂S). This work achieved a predictive accuracy of R²=0.922 for material renderability by effectively identifying 10 elite MOF candidates from the 12,020-structure CoRE-MOF database by combining GCMC simulations with a Random Forest architecture. A "volcanic" structure-property relationship is revealed by a database-driven analysis of the data, where the best separation performance is limited to a density window of 0.5–1.7 g/cm³; frameworks outside of this range experience either kinetic exclusion or a loss of selectivity because of oversized pores.
The study outlines a clear tripartite design strategy for next-generation adsorbents: (i) replacing metal nodes, such as substituting Cd with Mn to quadruple working capacity; (ii) incorporating nitrogen-rich organic linkers, such as pyridine or azoles, to take advantage of electrostatic interactions. While the rigid-framework assumption successfully identified top-tier materials like XIGWUF and ETECOX, future physics-informed ML directions must go beyond static lattice models to incorporate the thermodynamic and kinetic impacts of framework flexibility in real-world, high-pressure natural gas streams.[20]
Local chemical conditions inside the framework ultimately determine adsorption selectivity and catalytic activation, even though transport modeling clarifies how CO2 moves through porous designs. In addition to merely structural optimization, engineering Lewis acid–base interactions, electronic structure modification, and synergistic site arrangements connects chemical reactivity and adsorption thermodynamics.
5. Active-Site and Electronic Structure Engineering
5.1. Lewis Acid-Base Site Engineering and Catalytic Kinetics
The merging of explainable machine learning with low-cost descriptors makes rapid screening of catalytic frameworks possible without the prohibitive expense of density functional theory (DFT). Researchers were able to predict CO₂ cycloaddition activity with an astounding 97% accuracy by using 372 high-yield experimental data points to train a Random Forest architecture. By measuring the "optimal Lewis acidity" of metal nodes using SHAP and PDP analysis, this study goes beyond "black-box" predictions. The findings indicate that a metal partial charge between 1.2 and 2.0 maximizes epoxide substrate activation while preventing active site deactivation through saturated coordination.
The effective experimental validation of MOF-76(Y), which attained a top-tier TOF of 64.72 h⁻¹, demonstrates that machine learning (ML) may bridge the gap between CO₂ use in the real world and hypothetical structure formation. However, a rigorous analysis indicates that to assure comparability across non-linear reaction profiles, the dependence on datasets collected from the literature requires extensive data cleaning (e.g., filtering for high yields). Future physics-informed machine learning directions should concentrate on standardizing experimental benchmarks to further enhance the commercial scalability of screened catalysts for a variety of catalytic reactions.[21]
5.2. Electronic Property Modulation and Electrocatalytic Selectivity
A superior class of 2D conjugated MOFs (2D c-MOFs) that outperform conventional Cu(211) benchmarks in CO₂ electroreduction performance has been found by combining DFT-driven discovery with Gradient Boosting Regression (GBR) designs. This study establishes a fundamental stability hierarchy (TMN4>TMN2O2>TMO4) and finds catalysts with very low limiting potentials, like NiN4−HDQ (UL=−0.04V for CO), by methodically altering both the metal active sites and organic ligands (HDQ series).
The primary electronic "handles" for tuning activity are electron affinity (EA) and electronegativity (χ), which together account for more than 34% of feature importance, according to a critical ML-based sensitivity analysis. The analysis indicates that ligand-induced orbital overlap is the main cause of intermediate adsorption strength, even though these 2D c-MOFs maintain pristine surfaces in aqueous conditions and show great thermal stability at 400 K. Future physics-informed machine learning approaches must connect these high-fidelity C1 models with multi-carbon (C2+) coupling kinetics in order to guarantee industrial scalability and fully realize the potential of these highly conductive, adjustable frameworks for artificial carbon cycle applications.[22]
5.3. Synergistic Site Engineering and Composite Pore Modulation
The computational "time trap" of simulating complicated composite adsorbents is addressed by integrating Convolutional Neural Networks (CNNs) with inception modules, offering a reliable framework for multi-criteria screening. Through training on 700 GCMC-validated structures, our model successfully navigated the inherent trade-offs between CO₂ working capacity and selectivity, achieving a high predictive fidelity (R² ≈ 0.90). Importantly, big data analysis of the 1,631-composite pool shows that unusual "synergistic" frameworks like IL@MARJAQ use the IL to create completely new potential energy minima for CO₂, whereas most ionic liquids improve selectivity by physically limiting N₂ transport.
Additionally, this study quantifies the non-linear impact of IL loading, proving that "more is not always better"; for instance, by adjusting the amount of injected molecules, the selectivity of IL@GUBKUL may be modified from 614 to over 7,000. Precision loading solutions replace trial-and-error post-synthetic alteration in this field. However, the rigid-framework assumption of the current model and its propensity to underestimate uptake in frameworks with open metal sites (OMS) indicate that lattice dynamics must be included in future physics-informed ML directions to fully capture the regeneration efficiencies and process economics needed for industrial flue gas separation.[23]
Insights gained from electrical modulation and active-site engineering naturally inform the next evolutionary stage, inverse design. Generative machine learning techniques use learned structure-property connections to suggest completely new MOF designs optimized for many aims instead of screening existing materials. This transition from predictive modeling to autonomous material generation essentially redefines the discovery paradigm.
6. Multi-Objective Inverse Design and Generative MOF Discovery
6.1. Multi-Objective Inverse Design and Chemical Subspace Exploration
The difficulty of navigating the almost infinite chemical space of porous crystals is addressed by the switch from brute-force screening to Deep Reinforcement Learning (DRL) for inverse design. Researchers have successfully produced physisorbents for Direct Air Capture with Qst values more than 40 kJ/mol and CO₂/H₂O selectivities greater than 1 by incorporating transformer-based predictors into a reward-driven environment. A database-driven analysis of these findings reveals a crucial trade-off in material "genes": high selectivity is controlled by particular Cu and Zn clusters that occupy a distinct subspace in the chemical design landscape, while high affinity for CO₂ is frequently driven by open metal sites (such as Mn-based N₁₃₁ nodes) that concurrently attract water.
The DRL approach's current reliance on classical force fields restricts its capacity to predict chemisorptive interactions involving charge transfer, despite its strong extrapolation capability—producing structures that compete with top-performing experimental MOFs like KAUST-7. Future physics-informed machine learning approaches must use DFT-derived charges and active learning loops to refine the predictors to improve industrial scalability. This will guarantee that inverse-designed candidates can continue to be effective in the humid, high-dilution environments typical of real-world atmospheric capture.[24]
The discovery of materials suited for particular carbon capture regimes has advanced significantly with the transition from manual high-throughput screening to reinforcement learning-enhanced generative design. Using the MOFGPT framework, researchers have shown that a reward-guided RL strategy can achieve 100% validity even when aiming for extreme CO₂ adsorption performance (mean + 2σ), but standard supervised fine-tuning is unable to produce a single chemically valid structure (0% validity). By optimizing MOFid string sequences, which encode both SMILES-based chemistry and RCSR-based topologies, this method enables the exploration of an almost unlimited chemical space without requiring the manual assembly of building blocks.
The RL model incorporates underlying structure-property correlations, as demonstrated by a database-driven examination of the generated candidates. For instance, open Cu²⁺ paddle wheel units and nitrogen-rich linkers that improve electrostatic interactions are consistently associated with strong CO₂ uptake. Additionally, the framework shows a capacity to investigate underrepresented areas of the chemical landscape that conventional screening could miss, with novelty rates consistently exceeding 63%. However, because it ignores the dynamic flexibility that is frequently essential for CO2 transport kinetics, the existing reliance on rigid-lattice assumptions continues to be a significant drawback. To close the gap between artificial intelligence-designed strings and artificially robust industrial sorbents, future physics-informed machine learning approaches should incorporate 3D structural building and DFT-based stability filters.[25]
Material-level optimization by itself does not ensure commercial viability, notwithstanding impressive gains in generative discovery. In the end, adsorption performance needs to be assessed under actual process settings, such as cycles of temperature and pressure swings. Therefore, integrating machine learning with process simulation closes the gap between system-scale performance and molecular design, allowing for the conversion of computer prediction into deployable carbon capture solutions.
7. Multiscale Generative Design and Process-Oriented Optimization
7.1. Process-Integrated Generative Design and Material Optimization
A major step forward from basic property screening to process-level material design is the creation of a multiscale generative workflow. Researchers have successfully navigated a terrain of trillions of potential structures using MOF-NET, an architecture based on NLP-style word embeddings, to identify candidates that "substantially outperform" benchmarks like CALF-20 and 13X zeolite. Strict size exclusion (3–5 Å pores) or the creation of high-density binding pockets (6–30 Å pores) where CO₂ molecules are stabilized by numerous oxygen-rich nodes are the two ways to achieve optimal performance, according to a database-driven analysis of the best-performing materials.
Crucially, this study provides experimentalists with a clear design guideline by identifying Cu-based nodes and fluorinated short-linkers as the "genetic markers" of elite adsorbents. The study identifies a continuous innovation gap even while the best-in-class small-pore material (HJM + N387 + E44) shows a notable productivity advantage over CALF-20 because of its improved N₂ rejection. The next frontier for physics-informed machine learning (ML) should be the integration of synthetic accessibility (SA) scores and lattice dynamics, ensuring that these "theoretical possibilities" can withstand the transition from computer to laboratory, as many computationally designed materials still encounter challenges in synthesizability and water stability.[4]
Conclusions
The paradigm of carbon capture research has changed as a result of the incorporation of Machine Learning (ML) into the identification and optimization of Metal-Organic Frameworks (MOFs). Large-scale brute-force screening has given way to data-driven approaches that combine generative design, prediction, and interpretation into cohesive computational workflows. The realization that framework flexibility is essential to adsorption thermodynamics and transport kinetics has been a key development. Rigid-lattice approximations can underestimate diffusivity and misinterpret adsorption near open metal sites, as shown by machine-learning interatomic potentials and hybrid simulation frameworks. These results emphasize how crucial dynamic structural influences are to effectively simulating CO₂ behaviour in porous structures. Concurrently, material discovery has moved from passive screening to proactive exploration of chemical subspaces thanks to inverse design approaches made possible by reinforcement learning and transformer-based generative models. These models shed light on how pore geometry, functional group distribution, and confinement effects interact to affect adsorption performance when combined with physically significant descriptors. There is still a gap between computational discovery and industrial implementation despite quick methodological advancements. Many models continue to favour well-characterized chemistries and frequently overlook deployment-critical elements like water stability, multicomponent gas competition, and synthetic viability. To close this gap, generative and predictive frameworks must incorporate process-level limitations, synthetic accessibility measurements, and stability-aware targets. In the future, physics-informed, interpretable machine learning architectures that function across molecule, material, and process scales appear to be the most viable avenue. Future research can expedite the creation of scalable and experimentally feasible adsorbent platforms in line with global net-zero goals by combining dynamic adsorption modelling with industrial performance assessment.
Comparative Overview of ML Models for CO2 and Gas Remediation:
| Study Focus | Primary ML Algorithm(s) | Key Descriptor(s) | Predictive Performance (R2) | Core Scientific Insight |
| Pore Energy Mapping | XGBoost | Energy-based RDFs & Surface Histograms | > 0.81 CO2 > 0.97 N2 | Spatially aware energy RDFs resolve the "intermediate pressure bottleneck" in isotherms. |
| Composite Modulation | CNN (Inception) | Geometric + Chemical (Ionic Liquids) | ≈ 0.90 | Ionic liquids can act as synergistic sites, creating new potential energy minima for CO2. |
| Kinetic Transport | DeepPot-SE (MLP) | Atomic coordinates (Flexible) | 0.9916 (Energy) | Framework flexibility accelerates CO2 diffusivity by 10x compared to rigid models. |
| Generative Design | MOFGPT (Transformer) | MOFid (NLP-based strings) | 35–100% Validity | Reinforcement learning effectively navigates the "extreme tail" of property distributions. |
| Process-Level Design | MOF-NET (ANN) | Word Embeddings of Building Blocks | Elite purity/recovery | Optimal design bifurcates into small-pore exclusion vs. large-pore binding. |
| Mixed Matrix Membranes | Stacking Ensemble | Polymer FFV + MOF PLD/LCD | 0.96 | A "10x permeability rule" exists where filler must exceed polymer permeability for gain. |
| Experimental Benchmarking | Stacking (RF/XGB/MLP) | Textural (BET) + Operational (P, T) | 0.9833 | Identified a metal-affinity hierarchy where Mg and Cu centers provide superior binding sites. |
| Hybrid Optimization | LSSVM-GO | Textural + Operational | 0.9798 | Growth Optimization (GO) significantly reduces prediction errors in high-uptake regimes. |
| Multicomponent Separation | Random Forest | Structural + Chemical Descriptors | 0.922 (R%) | MOF renderability is optimized within a specific density window of 0.5–1.7 g/cm³. |
| Electrocatalytic Selectivity | Gradient Boosting (GBR) | Electronic (EA, chi, d-band) | 0.9998 | Catalytic activity is primarily governed by electron affinity and electronegativity. |
| Universal Zeolite Prediction | GBT / RF / DL | Si/Al Ratio + Cation type | 0.936 | Provides a universal framework without case-specific parameter fitting required by Langmuir models. |
References
- Ozkan, M.A., Amir-Ali & Coley, William & Shang, Ruoxu & Ma, Yi, Progress in carbon dioxide capture materials for deep decarbonization. Chem. 8. 141-173. 2022. [CrossRef]
- Carrascal-Hernández, D.C.G.-T., C.D.; Mendez-Lopez, M.; Insuasty, D.; García-Freites, S.; Sanjuan, M.; Márquez, E, CO₂ Capture: A Comprehensive Review and Bibliometric Analysis of Scalable Materials and Sustainable Solutions. . Molecules 2025, 30, 563, 2025. [CrossRef]
- Shreya Mahajan, M.L., Recent progress in metal-organic frameworks (MOFs) for CO2 capture at different pressures. Journal of Environmental Chemical Engineering,, 2022. [CrossRef]
- Sarkisov, Z.D.a.L., Multi-Scale Computational Design of Metal–Organic Frameworks for Carbon Capture Using Machine Learning and Multi-Objective Optimization. Chemistry of Materials 2024 36 (19), 9806-9821, 2024. [CrossRef]
- Hussin, F.N., Siti Aqilah & Mohamed Hatta, Nur Syahirah & Aroua, Mohamed & Mazari, Shaukat Ali, A systematic review of machine learning approaches in carbon capture applications. . Journal of CO2 Utilization, 2023. [CrossRef]
- François-Xavier Coudert, Recent advances in stimuli-responsive framework materials: Understanding their response and searching for materials with targeted behavior. Coordination Chemistry Reviews,, 2025. Volume 539. [CrossRef]
- Fathalian, F.A., Sepehr & Ghaemi, Ahad & Hemmati, Alireza, Intelligent prediction models based on machine learning for CO2 capture performance by graphene oxide-based adsorbents. . Scientific Reports. 12. 21507., 2022. [CrossRef]
- Z. Deng, L.S., Engineering machine learning features to predict adsorption of carbon dioxide and nitrogen in metal–organic frameworks. J. Phys. Chem. C (2024), 2024. [CrossRef]
- J. Zuo, F.S., Z. Qu, C. Yang, L. Xie, Y. Zhang, X. Li, J. Li, Unraveling the coupling effect of micropore confinement and functional sites of carbon-based adsorbents on flue gas CO₂ adsorption: A machine learning study based on multi-scale simulations. Carbon Capture Sci. Technol. (2025). , 2025. [CrossRef]
- Y. Teng, G.S., Interpretable machine learning for materials discovery: Predicting CO₂ adsorption properties of metal–organic frameworks. 2024. [CrossRef]
- P.O. Longe, S.D., M. Mehrad, D.A. Wood, , Robust machine-learning model for prediction of carbon dioxide adsorption on metal–organic frameworks. J. Alloys Compd. (2024), 2024. [CrossRef]
- Z. Iyiola, E.T.B., N.J. Okeke, K. Sanni, P. Longe, , Carbon capture using metal-organic frameworks (MOFs): Novel custom ensemble learning models for prediction of CO₂ adsorption. Processes, 2025. [CrossRef]
- H. Wan, Y.F., M. Hu, S. Guo, Z. Sui, X. Huang, Z. Liu, Y. Zhao, H. Liang, Y. Wu, H. Gao, and Z. Qiao, Interpretable machine learning and big data mining to predict the CO₂ separation in polymer–MOF mixed matrix membranes. Adv. Sci. (2024), 2024. [CrossRef]
- S. Achour, Z.H., ML-driven models for predicting CO₂ uptake in metal–organic frameworks (MOFs), . Can. J. Chem. Eng. (2024). 2024. [CrossRef]
- Kirtil, E., Universal prediction of CO₂ adsorption on zeolites using machine learning: A comparative analysis with Langmuir isotherm models. ChemEngineering 2025. [CrossRef]
- E.V. Kotov, J.S., G. Logabiraman, H. Dhall, M. Chandna, P. Madan, V. Sharma, Carbon capture and storage optimization with machine learning using an ANN model. E3S Web Conf. 588 (2024) 01003, 2024. [CrossRef]
- B. Zheng, G.X.G., C. dos Santos, R.N.B. Ferreira, M. Steiner, B. Luan, Simulating CO₂ diffusivity in rigid and flexible Mg-MOF-74 with machine-learning force fields. APL Mach. Learn. 2 (2024) 026115, 2024. [CrossRef]
- Yunsung Lim, H.P., Aron Walsh, Jihan Kim,, Accelerating CO2 direct air capture screening for metal-organic frameworks with a transferable machine learning force field,. Matter, 2025. [CrossRef]
- Tayfuroglu O, K.S., Modeling CO2 adsorption in flexible MOFs with open metal sites via fragment-based neural network potentials. . . J Chem Phys. 2025 Aug 7;163(5):054704., 2025. [CrossRef]
- Y. Zhou, S.J., S. He, W. Fan, L. Zan, L. Zhou, X. Ji, G. He, Machine-learning-assisted high-throughput screening of metal–organic frameworks for CO₂ separation from CO₂-rich natural gas. Ind. Eng. Chem. Res. (2024). , 2024.
- X. Bai, Y.L., Y. Xie, Q. Chen, X. Zhang, J.-R. Li, High-throughput screening of CO₂ cycloaddition MOF catalyst with an explainable machine learning model. Green Energy Environ. (2024), 2024. [CrossRef]
- G. Xing, S.L., G. Sun, J.-Y. Liu, , Modification of metals and ligands in two-dimensional conjugated metal–organic frameworks for CO₂ electroreduction: A combined DFT and machine learning study. SSRN Electron. J. (2024). , 2024. [CrossRef]
- Mengjia Sheng, X.Z., Hongye Cheng, Zhen Song, Zhiwen Qi,, Multi-criteria computational screening of [BMIM][DCA]@MOF composites for CO2 capture,. Green Chemical Engineering,, 2025. [CrossRef]
- Park, H., et al., Inverse design of metal–organic frameworks for direct air capture of CO2via deep reinforcement learning. Digital Discovery, 2024. 3(4): p. 728-741. [CrossRef]
- Srivathsan Badrinarayanan, R.M., Akshay Antony, Radheesh Sharma Meda, and Amir Barati Farimani, MOFGPT: Generative Design of Metal–Organic Frameworks using Language Models. Journal of Chemical Information and Modeling 2025 65 (17), 9049-9060, 2025. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



