
Submitted:
19 February 2026
Posted:
26 February 2026
You are already at the latest version
Abstract
“Targeted cancer therapy” is commonly operationalized as mutation-matched pharmacologic therapy. These therapies represent genuine scientific advances. However, population-level analyses indicate that eligibility for genome-targeted drugs remains in the low-teens percentage range, with estimated durable benefit affecting only a small single-digit fraction of metastatic patients once intratumoral heterogeneity (ITH) and evolutionary resistance are considered[1–4].Cancer is not solely a genomic disease; genomic alterations arise and operate within a broader systems-level biological context. Tumor survival and evolutionary persistence depend on distributed metabolic and redox programs—including redox fragility[5] and metabolic inflexibility[6]—that transcend individual mutations and span diverse tumor types and tumor cell populations. Because these programs reflect functional survival infrastructure rather than rare mutational events, they represent broader and potentially more universally exploitable therapeutic targets.This paper argues that oncology has, at times, conflated molecular precision with systemic comprehensiveness. We introduce a dual-axis evaluative framework distinguishing Specificity of Target from Distribution of Vulnerability, and propose a hierarchical model of cancer targeting encompassing:1. Genomic targeting (mutation-specific drugs)2. Redox targeting (pharmacologic ascorbate / high-dose intravenous vitamin C; HDIVC)[7–10]3. Metabolic/systemic targeting (ketogenic metabolic therapy; insulin–IGF modulation)[11–14]Using published eligibility data, ITH metrics, peroxide detoxification studies, and pan-cancer metabolic stratification analyses, we construct a semi-quantitative comparison of “Targetable Fraction” and “Effective Target Coverage” across these layers. We demonstrate that genomic targeting is highly specific but structurally narrow, whereas redox and metabolic targeting operate at broader biological levels with wider theoretical distribution. Importantly, these layers are complementary rather than mutually exclusive, and a systems-oncology doctrine should evaluate their combined role in expanding effective target coverage.This manuscript expands the conceptual taxonomy of targeted cancer therapy and establishes a formal record that redox and metabolic interventions constitute biologically coherent targeting modalities within a layered oncology framework.
Keywords:
targeted cancer therapy
; precision oncology
; intratumoral heterogeneity
; pharmacologic ascorbate
; high-dose intravenous vitamin C
; redox targeting
; cancer metabolism
; ketogenic metabolic therapy
; insulin–IGF signaling
; systems oncology
; distribution of vulnerability
; tumor evolution
Figure 1. Global Vulnerability Architecture for Cancer Targeting (conceptual).
Three biological targeting layers sit on top of a broader vulnerability landscape.
- Layer A: Genomic nodes (mutation-defined, narrow breadth, high precision).
- Layer B: Redox nodes (H2O2 detoxification / catalase-peroxidase capacity; phenotype-distributed).
- Layer C: Metabolic-systemic nodes (glucose–insulin/IGF axis, substrate restriction, metabolic inflexibility).
These layers connect to shared pan-cancer vulnerabilities: replication stress, proteotoxic stress, hypoxia adaptation, acidic microenvironment, amino-acid dependence, ferroptosis susceptibility, immune evasion.
Clinically, the ‘targetable fraction’ expands as one moves from Layer A to Layers B/C, at the cost of lower node-specific precision.
[Genomic (A): EGFR/ALK/HER2/BRAF/KRAS…] → narrow subset
│
▼
[Redox (B): low H2O2 clearance / catalase vulnerability] → phenotype-broad
│
▼
[Metabolic (C): insulin–IGF dependence / metabolic inflexibility] → phenotype-broad
│
▼
[Global landscape: replication stress · proteotoxic stress · hypoxia · acidity · amino-acid addiction · ferroptosis · immune evasion]
1. The Targeted Therapy Paradigm Reconsidered
The phrase “targeted therapy” carries three implicit promises:
- Precision
- Broad applicability
- Comprehensive tumor control
The data robustly support the first promise. They do not consistently support the latter two.
In this context, ‘targeted’ should be understood as referring to molecular specificity, not population-wide effectiveness.
1.1. Population-Level Impact
Marquart et al. (2018) estimated that approximately 15.4% of metastatic cancer patients were eligible for genome-driven therapy, with an estimated 4.9% deriving clinical benefit [1].
Haslam et al. (2021) reported updated estimates of approximately 13.6% eligibility and ~7.0% response in 2020[2].
Even under modern assumptions, mutation-matched targeted therapy remains a minority-impact modality at the population level.
1.2. Tumor-Cell Coverage: Intratumoral Heterogeneity
Multi-region sequencing studies demonstrate that many tumors are not genomically uniform.
Gerlinger et al. reported that 63–69% of somatic mutations were not ubiquitous across sampled tumor regions [3].
In TRACERx NSCLC, median mutation heterogeneity was approximately 30%, with ~48% heterogeneous copy-number alterations [4].
Thus, even within eligible patients, mutation-layer targeting may not encompass all malignant clones present within a tumor ecosystem.
1.3. Durability Constraints
Durability of response further narrows effective impact. Examples include:
Interim Conclusion
Genomic targeted therapy is highly precise. However, it is simultaneously:
- Population-restricted
- Clone-restricted
- Evolutionarily constrained
Molecular specificity at a single genomic locus does not equate to comprehensive tumor-system control. A therapy may be exquisitely specific yet structurally narrow in biological reach. Conflating precision with breadth has shaped oncology doctrine more than the distributed architecture of cancer biology itself.
Cancer biology is organized around shared functional hallmarks rather than isolated mutational events [10,11]. Non-oncogene dependencies and stress-adaptation programs—including oxidative buffering, replication stress tolerance, and proteostasis maintenance—represent distributed liabilities that transcend individual driver mutations [12,13,14,15].
2. Layer 1—Genomic Targeted Therapy: Precision With Structural Breadth Limits
2.1. Population-Level Eligibility
Marquart et al. (2018) estimated [1]:
- 15.4% eligibility for genome-informed therapy
- 4.9% estimated response benefit
Haslam et al. (2021) updated these figures [2]:
- ~13.6% eligibility
- ~7.0% response estimate
Thus, population-level effective reach remains in low single-digit to low-teen ranges.
2.2. Tumor-Cell Coverage: Intratumoral Heterogeneity
Gerlinger et al.:
- 63–69% of somatic mutations not ubiquitous across tumor regions [3]
TRACERx NSCLC:
Implication: mutation-layer targeting frequently does not cover all malignant clones within an individual tumor.
2.3. Durability Constraints
Examples:
2.4. Semi-Quantitative Model: Genomic Effective Target Coverage (GETC)
Define: GETC ≈ Eligibility × Response Rate × Tumor-Cell Coverage × Durability Factor
Illustrative sensitivity framing (conceptual): Each term in GETC is constrained by real-world structural factors—population eligibility, partial tumor-clone coverage due to ITH, and time-limited durability due to resistance. When these constraints are compounded multiplicatively, the resulting effective coverage (i.e., the fraction of the total metastatic population experiencing meaningful, durable, multi-clonal control) can shrink substantially even when each individual factor appears clinically meaningful in isolation.
Illustrative scenario (conceptual): Using published estimates of eligibility (~14%[1,2]), response among eligible (often ~40–60% depending on tumor type [5,6,7]), partial clonal coverage [3,4], and time-limited durability (based on reported median PFS durations [5,6,7]), the resulting effective coverage would approximate 2–3% of the overall metastatic population under these assumptions.
These parameters vary substantially across tumor types, biomarkers, and lines of therapy; this calculation is intended to illustrate multiplicative constraint rather than provide a definitive epidemiologic estimate.
This formulation is not intended as a definitive measurement. Rather, it formalizes a systems principle: single-node genomic precision does not automatically translate into system-wide tumor control, because eligibility, clonal coverage, and durability are structurally limiting multipliers.
The practical implication is that high molecular precision does not necessarily translate into broad tumor-system impact.
3. A Dual-Axis Framework: Specificity vs Distribution
Targeted therapies should be evaluated along two axes:
- Axis A — Specificity of Target: Molecular precision.
- Axis B — Distribution of Vulnerability: Prevalence of the exploited vulnerability across tumor types and tumor cells.
Genomic targeting: High specificity / Low distribution.
Redox targeting: Moderate specificity / Moderate-to-broad distribution.
Metabolic targeting: Moderate specificity / Broad distribution.
4. Layer 2 — Redox Targeting and the Catalase Axis
4.1. Redox Fragility in Cancer
Cancer cells often operate under elevated reactive oxygen species (ROS) levels and exist near oxidative stress thresholds [16,17,18]. ROS-mediated targeting has been proposed as a radical therapeutic approach exploiting this non-oncogene vulnerability [15].
In a multi–cell line survey (15 tumor vs 10 normal cell lines), Doskey et al. quantified the rate constant for extracellular H₂O₂ removal (k_cell) and found that normal cells had ~2× higher H₂O₂ removal capacity than tumor cells on average, and that ascorbate sensitivity (ED₅₀) correlated with k_cell and catalase activity [17].
4.2. Catalase Distribution
Glorieux and Calderon reviewed catalase as a potential cancer treatment axis, noting frequent decreases in catalase across many tumors with context-specific exceptions [19].
Practically, this means catalase is not a binary marker (“low vs high”) but a distributed trait that varies across tumors and can be treated as a stratification axis rather than a yes/no inclusion criterion (formalized below as CVI)[19].
4.3. Mechanistic Coherence of Pharmacologic Ascorbate
At pharmacologic plasma concentrations achievable only via intravenous administration, ascorbate can generate extracellular H2O2 in the presence of redox-active metal ions. Tumor cells with limited catalase capacity may experience cytotoxic oxidative stress, whereas normal tissues with higher detox capacity may be relatively protected, creating a redox threshold differential.
Exposure (what dose can achieve): Pharmacologic ascorbate is typically defined in this literature as plasma ascorbate on the order of ≥~20 mM, achievable via IV delivery [17].
Contrast with oral: Oral vitamin C is tightly constrained; the NIH ODS fact sheet notes that oral dosing gives typical plasma peaks ~135 µmol/L after ~1.25 g oral vitamin C and IV administration can yield plasma concentrations ~26 mM (30,000 µmol/L) [20].
Pharmacologic ascorbate achieves millimolar plasma concentrations not attainable through oral dosing, generating extracellular hydrogen peroxide in the presence of catalytic iron and disrupting tumor iron metabolism [12]. Differential susceptibility between tumor and normal cells has been demonstrated in preclinical systems, particularly in KRAS- and BRAF-mutant contexts [10].
Emerging preclinical data suggest that pharmacologic ascorbate resistance may be mediated through enhanced peroxide detoxification in some tumors, highlighting the importance of multi-parametric vulnerability stratification [21]
While intravenous vitamin C is not an FDA-approved cancer therapy, the U.S. National Cancer Institute’s PDQ summary notes that IV vitamin C has been generally well tolerated in clinical trials, and the NIH Office of Dietary Supplements emphasizes that individuals receiving vitamin C for medical treatment should be under the care of a physician [22].
4.4. Catalase Vulnerability Index (CVI)
CVI defined across tiers:
- Tier 1 — Functional peroxide clearance;
- Tier 2 — Catalase protein expression;
- Tier 3 — CAT mRNA expression.
CVI operates on a continuum. Thus, Redox Targetable Fraction (RTF) is phenotype-distributed and potentially broader than mutation-defined subsets.
In this framework, “redox targeting” is not defined by a rare mutation but by a quantifiable vulnerability axis (peroxide clearance capacity), which in vitro shows a systematic tumor–normal differential (~2× on average) and correlates with pharmacologic-ascorbate sensitivity [17].
5. Layer 3—Metabolic/Systemic Targeting
5.1. Metabolic Heterogeneity Clarified
Cancer metabolism is plastic. Not all tumors are OxPhos-deficient [6]. However, a pan-cancer TCGA analysis (9,668 patients; 33 tumor types) stratified tumors into glycolysis/OXPHOS subtypes using a double-score framework [23], demonstrating that metabolic phenotypes are widely distributed across tumor types.
5.2. Insulin–IGF Axis as Systemic Vulnerability
Insulin and IGF signaling activate PI3K/AKT/mTOR pathways across multiple malignancies, promoting anabolic growth and survival [13,14]. Hyperinsulinemia has been associated with increased cancer progression risk [13]. Systemic reduction of insulin signaling through carbohydrate restriction or ketogenic metabolic therapy therefore represents host-mediated pathway modulation rather than mutation-specific targeting [15,24].
Ketogenic metabolic therapy lowers systemic insulin, alters glucose availability, and modulates host growth signaling—representing host-mediated targeting rather than mutation-layer targeting.
5.3. Metabolic Targetability Index (MTI)
MTI may integrate glycolysis/OXPHOS transcriptomic scores, insulin resistance metrics, and substrate dependency markers. MTI conceptualizes metabolic vulnerability on a distributed scale.
6. Comparative Targetable Fraction Summary
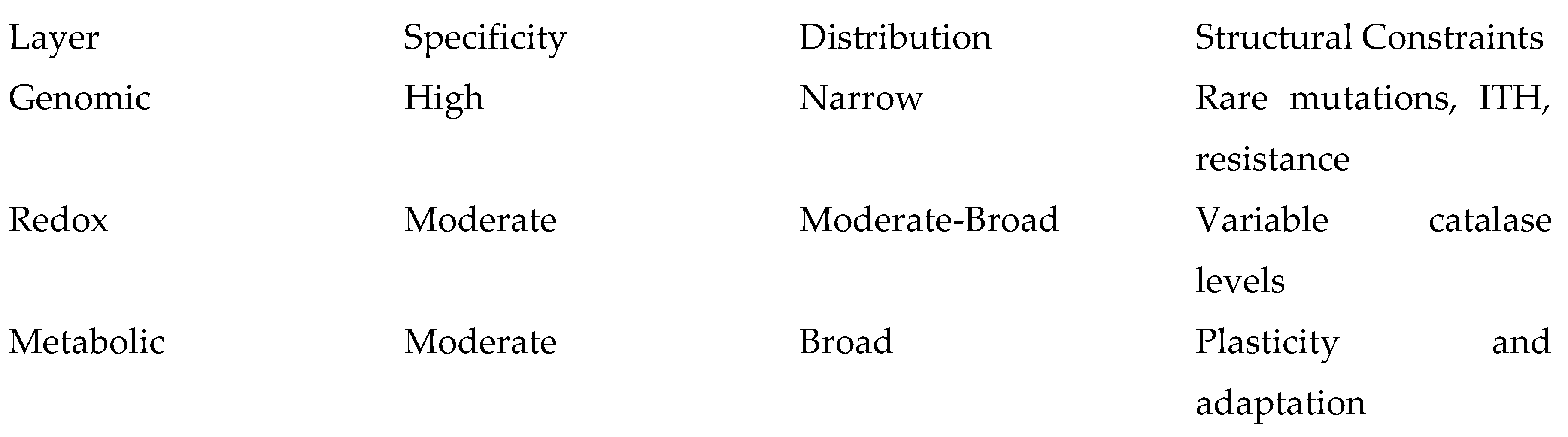
Plasticity and adaptation
Genomic targeting is precise but structurally narrow. Redox and metabolic targeting are less node-specific but broader in phenotypic distribution.
7. Counterarguments
- Redox and metabolic vulnerabilities are heterogeneous → Stratification (CVI/MTI) is necessary.
- Tumors adapt metabolically → Layered targeting may outperform single-layer approaches.
- Clinical trial evidence for HDIVC and ketogenic therapy is limited → This paper establishes conceptual legitimacy, not clinical equivalence.
8. Limitations
- CVI and MTI require prospective validation.
- GETC is conceptual rather than definitive.
- Redox and metabolic clinical trial data remain emerging.
- This framework is theoretical and integrative.
9. Global Vulnerability Architecture
Hierarchical targeting model: GENOMIC → REDOX → METABOLIC → HOST SYSTEM. Each higher layer affects broader biological domains.
10. Conclusions
Genomic targeted therapies represent an important advance. However, they do not constitute a general theory of cancer targeting. Redox and metabolic targeting address distributed biological vulnerabilities that may span broader tumor fractions and operate at higher systemic levels. A comprehensive oncology doctrine should integrate all layers of vulnerability rather than equating targeting exclusively with mutation-specific pharmacology. This manuscript establishes a formal conceptual framework for such integration. In this framework, genomic, redox, and metabolic targeting are not competitors but interoperable layers; combining them is the logical systems strategy for improving coverage across heterogeneous tumor ecosystems. For patients and clinicians seeking systems-level strategies beyond mutation-matched pharmacology, redox and metabolic targeting represent rational, biologically grounded extensions of the targeting concept rather than departures from it.
Author Contributions
R.Z.C. conceived the conceptual framework, performed the literature synthesis, and wrote the manuscript.
Funding
This work received no external funding.
Institutional Review Board Statement
Not applicable. This manuscript is a conceptual synthesis of published literature and does not involve new human or animal subject research.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new datasets were generated or analyzed. All information presented is derived from published and publicly available sources cited in the manuscript.
Conflicts of Interest
Richard Z. Cheng, M.D., Ph.D., is a Board Director of the Riordan Clinic, an institution that conducts research and clinical services related to intravenous vitamin C. The present manuscript is a conceptual and literature-based analysis and does not report proprietary data, commercial products, or patient-level outcomes. No financial compensation was received for the preparation of this manuscript.
References
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Benefit From Genome-Driven Oncology. JAMA Oncol 2018, 4, (8), 1093–1098. [CrossRef]
- Haslam, A.; Kim, M.S.; Prasad, V. Updated Estimates of Eligibility for and Response to Genome-Targeted Oncology Drugs among US Cancer Patients, 2006-2020. Ann Oncol 2021, 32, (7), 926–932. [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med 2012, 366, (10), 883–892. [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017, 376, (22), 2109–2121. [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018, 378, (2), 113–125. [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med 2021, 384, (25), 2371–2381. [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019, 381, (7), 626–636. [CrossRef]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, (7381), 306–313. [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, (4), 613–628. [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; et al. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, (6266), 1391–1396. [CrossRef]
- Welsh, J.L.; Wagner, B.A.; Erve, T.J. van’t; et al. Pharmacological Ascorbate with Gemcitabine for the Control of Metastatic and Node-Positive Pancreatic Cancer (PACMAN): Results from a Phase I Clinical Trial. Cancer Chemother Pharmacol 2013, 71, (3), 765–775. [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; et al. O2⋅- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, (4), 487-500.e8. [CrossRef]
- Pollak, M. Targeting Oxidative Phosphorylation: Why, When, and How. Cancer Cell 2013, 23, (3), 263–264. [CrossRef]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, Insulin, and Cancer. Endocrinology 2011, 152, (7), 2546–2551. [CrossRef]
- Fine, E.J.; Segal-Isaacson, C.J.; Feinman, R.D.; et al. Targeting Insulin Inhibition as a Metabolic Therapy in Advanced Cancer: A Pilot Safety and Feasibility Dietary Trial in 10 Patients. Nutrition 2012, 28, (10), 1028–1035. [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu Rev Cancer Biol 2017, 1, 79–98. [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; et al. Tumor Cells Have Decreased Ability to Metabolize H2O2: Implications for Pharmacological Ascorbate in Cancer Therapy. Redox Biol 2016, 10, 274–284. [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat Rev Drug Discov 2009, 8, (7), 579–591. [CrossRef]
- Glorieux, C.; Buc Calderon, P. Targeting Catalase in Cancer. Redox Biol 2024, 77, 103404. [CrossRef]
- Office of Dietary Supplements - Vitamin C. Available online: https://ods.od.nih.gov/factsheets/VitaminC-HealthProfessional/ (accessed 27 November 2025).
- Pope, A.; O’Leary, B.; Du, J.; et al. Pharmacologic Ascorbate Resistant Pancreatic Cancer Demonstrates Enhanced Metastatic Potential. Redox Biol 2025, 84, 103694. [CrossRef]
- National Cancer Institute Intravenous Vitamin C (PDQ®)–Health Professional Version. Available online: https://www.cancer.gov/about-cancer/treatment/cam/hp/vitamin-c-pdq (accessed 11 October 2025).
- Bi, G.; Bian, Y.; Liang, J.; et al. Pan-Cancer Characterization of Metabolism-Related Biomarkers Identifies Potential Therapeutic Targets. J Transl Med 2021, 19, (1), 219. [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; et al. Cancer as a Metabolic Disease: Implications for Novel Therapeutics. Carcinogenesis 2014, 35, (3), 515–527. [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat Cell Biol 2014, 16, (1), 2–9. [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat Rev Cancer 2015, 15, (5), 276–289. [CrossRef]
- Takahashi, N.; Kim, S.; Schultz, C.W.; et al. Replication Stress Defines Distinct Molecular Subtypes across Cancers. Cancer Res Commun 2022, 2, (6), 503–517. [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, (5), 1060–1072. [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cell Biol 2021, 22, (4), 266–282. [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, (5), 646–74. Available online: http://linkinghub.elsevier.com/retrieve/pii/S0092867411001279 http://www.ncbi.nlm.nih.gov/pubmed/21376230. [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell 2009, 136, (5), 823–837. [CrossRef]
- Lee, H.Y.; Parkinson, E.I.; Granchi, C.; et al. Reactive Oxygen Species Synergize To Potently and Selectively Induce Cancer Cell Death. ACS Chem Biol 2017, 12, (5), 1416–1424. [CrossRef]
- Bodeker, K.L.; Smith, B.J.; Berg, D.J.; et al. A Randomized Trial of Pharmacological Ascorbate, Gemcitabine, and Nab-Paclitaxel for Metastatic Pancreatic Cancer. Redox Biol 2024, 77, 103375. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



