Submitted:
18 February 2026
Posted:
18 February 2026
You are already at the latest version
Abstract
Tumor hypoxia is a defining hallmark of solid cancers that profoundly influences tumor progression, genomic instability, and therapeutic response. Beyond its classical roles in angiogenesis and metabolic reprogramming, hypoxia has emerged as a central determinant of the tumor immune microenvironment (TME), promoting immune exclusion and resistance to immunotherapy. Our work has uncovered tumor cell–intrinsic mechanisms by which hypoxia drives immune escape. We identified hypoxia-induced autophagy as a key adaptive response that enables tumor cells to resist natural killer (NK) and cytotoxic T lymphocyte (CTL)–mediated killing. Under hypoxic stress, autophagy selectively degrades NK-derived granzyme B, neutralizing effector cytotoxicity, while genetic or pharmacologic inhibition of autophagy restores immune-mediated killing and enhances tumor regression in vivo. Furthermore, we demonstrated that Vps34 inhibition, a central regulator of autophagy and vesicular trafficking, converts poorly infiltrated “cold” tumors into inflamed “hot” tumors enriched in NK, CD4+, and CD8+ effector T cells, thereby potentiating the efficacy of PD‑1/PD‑L1 checkpoint blockade across multiple tumor models. Recently, we identified the atypical chemokine receptor ACKR2 as a hypoxia-inducible, HIF-1α–dependent checkpoint that restricts chemokine availability and limits immune infiltration. Targeting ACKR2 alleviates immune exclusion and synergizes with PD-1 blockades to induce tumor regression in otherwise refractory tumors. Collectively, these studies establish a coherent model in which hypoxia and its downstream stress-response pathways act as master regulators of tumor immune evasion. By rewiring autophagy and chemokine signaling, hypoxia shapes the immune landscape of solid tumors and defines responses to immunotherapy. Targeting these pathways represents a compelling strategy to overcome immune resistance and expand the clinical benefit of checkpoint inhibitor therapies.
Keywords:
tumor microenvironment
; hypoxia
; cancer immunity
; cancer immunotherapy
; autophagy
; inflammatory tumor microenvironment
; ACKR2
; cytokines chemokines
1. Introduction
Rapid tumor growth, aberrant vascular architecture, and disorganized blood perfusion result in generating regions of chronic and cycling hypoxia in most solid tumors [1]. Hypoxic stress stabilizes hypoxia-inducible factors (HIFs), which coordinate a broad transcriptional program in tumor cells and stromal components, promoting cell survival, angiogenesis, invasion, and profound metabolic rewiring [1]. Beyond these well-established effects, hypoxia has emerged as a central factor of the tumor microenvironment (TME), impairing the anti-tumor immunity [2].
Accumulating evidence indicates that hypoxia promotes immune suppression through multiple interconnected mechanisms, including metabolic reprogramming, accumulation of immunosuppressive metabolites such as adenosine, and disruption of chemokine networks. In parallel, hypoxia drives the recruitment, polarization, and functional education of suppressive myeloid and stromal cell populations [3]. Together, these hypoxia-driven processes contribute to immune tolerance and represent a major barrier to effective cancer immunotherapy.
Over the past years, our work has contributed to redefining hypoxia as a key regulator of immune evasion within the TME. In particular, we have elucidated how hypoxia-induced autophagy and hypoxia-regulated intracellular pathways in tumor cells impair cytotoxic lymphocyte function, thereby limiting tumor cell susceptibility to natural killer (NK) cell–mediated killing and shaping responses to immune checkpoint blockade. These findings place tumor cell–intrinsic adaptive responses to hypoxia at the center of resistance mechanisms to both innate and adaptive immunotherapies [2].
In this mini review, we provide a comprehensive overview of our contributions to understanding how hypoxia modulates anti-tumor immunity, with emphasis on emerging concepts and therapeutic implications. We also discuss emerging therapeutic strategies aimed at targeting hypoxia-driven pathways to restore immune effector function and enhance the efficacy of cancer immunotherapy.
Figure 1.
Hypoxia-driven tumor cell–intrinsic pathways coordinate immune escape and shape responses to cancer immunotherapy. Rapid tumor growth and abnormal vascularization generate poorly oxygenated regions within solid tumors. Hypoxic stress stabilizes HIF-1α, initiating transcriptional programs that promote tumor adaptation and immune suppression. (A) Under hypoxic conditions, HIF-1α–driven autophagy is activated in tumor cells, conferring resistance to cytotoxic lymphocyte–mediated killing. Autophagy promotes the degradation of NK cell–derived granzyme B within autophagosomes and autolysosomes, thereby impairing NK cell and cytotoxic T lymphocyte (CTL) effector function. In addition, hypoxia-induced autophagy suppresses immune cell trafficking by limiting the production of T cell–attracting chemokines. Pharmacological inhibition of autophagy, particularly through targeting Vps34/Beclin1 or ATG5, reprograms poorly infiltrated “cold” tumors into inflamed “hot” tumors. Vps34 inhibition activates STAT1/IRF7-dependent inflammatory signaling and induces interferon-regulated chemokines such as CCL5 and CXCL10, resulting in enhanced recruitment of NK cells and CD8+ T cells and increased sensitivity to PD-1/PD-L1 immune checkpoint blockade. Targeting Beclin1 reduces PP2A phosphatase activity, leading to activation of the JNK/c-JUN signaling pathway. This signaling cascade promotes CCL5 expression, thereby enhancing NK cell recruitment to the tumor microenvironment. (B) Genetic deletion of the HIF-1α domain required for dimerization with HIF-1β abrogates its transcriptional activity and leads to the induction of CCL5 through a mechanism that remains incompletely understood. This suggests that specific HIF-1α structural domains may differentially regulate chemokine expression independently of canonical transcriptional activity. (C) Hypoxia also induces ACKR2 expression in a HIF-1α–dependent manner. ACKR2 functions as a chemokine scavenger receptor that reduces the availability of pro-inflammatory chemokines, limits immune cell infiltration, and promotes an immune-cold tumor phenotype. Targeting HIF-1α reduces ACKR2 expression and prevents the scavenging of chemokines such as CCL5, thereby restoring chemokine gradients, enhancing NK and T-cell recruitment, and synergizing with PD-1 blockade.
Figure 1.
Hypoxia-driven tumor cell–intrinsic pathways coordinate immune escape and shape responses to cancer immunotherapy. Rapid tumor growth and abnormal vascularization generate poorly oxygenated regions within solid tumors. Hypoxic stress stabilizes HIF-1α, initiating transcriptional programs that promote tumor adaptation and immune suppression. (A) Under hypoxic conditions, HIF-1α–driven autophagy is activated in tumor cells, conferring resistance to cytotoxic lymphocyte–mediated killing. Autophagy promotes the degradation of NK cell–derived granzyme B within autophagosomes and autolysosomes, thereby impairing NK cell and cytotoxic T lymphocyte (CTL) effector function. In addition, hypoxia-induced autophagy suppresses immune cell trafficking by limiting the production of T cell–attracting chemokines. Pharmacological inhibition of autophagy, particularly through targeting Vps34/Beclin1 or ATG5, reprograms poorly infiltrated “cold” tumors into inflamed “hot” tumors. Vps34 inhibition activates STAT1/IRF7-dependent inflammatory signaling and induces interferon-regulated chemokines such as CCL5 and CXCL10, resulting in enhanced recruitment of NK cells and CD8+ T cells and increased sensitivity to PD-1/PD-L1 immune checkpoint blockade. Targeting Beclin1 reduces PP2A phosphatase activity, leading to activation of the JNK/c-JUN signaling pathway. This signaling cascade promotes CCL5 expression, thereby enhancing NK cell recruitment to the tumor microenvironment. (B) Genetic deletion of the HIF-1α domain required for dimerization with HIF-1β abrogates its transcriptional activity and leads to the induction of CCL5 through a mechanism that remains incompletely understood. This suggests that specific HIF-1α structural domains may differentially regulate chemokine expression independently of canonical transcriptional activity. (C) Hypoxia also induces ACKR2 expression in a HIF-1α–dependent manner. ACKR2 functions as a chemokine scavenger receptor that reduces the availability of pro-inflammatory chemokines, limits immune cell infiltration, and promotes an immune-cold tumor phenotype. Targeting HIF-1α reduces ACKR2 expression and prevents the scavenging of chemokines such as CCL5, thereby restoring chemokine gradients, enhancing NK and T-cell recruitment, and synergizing with PD-1 blockade.
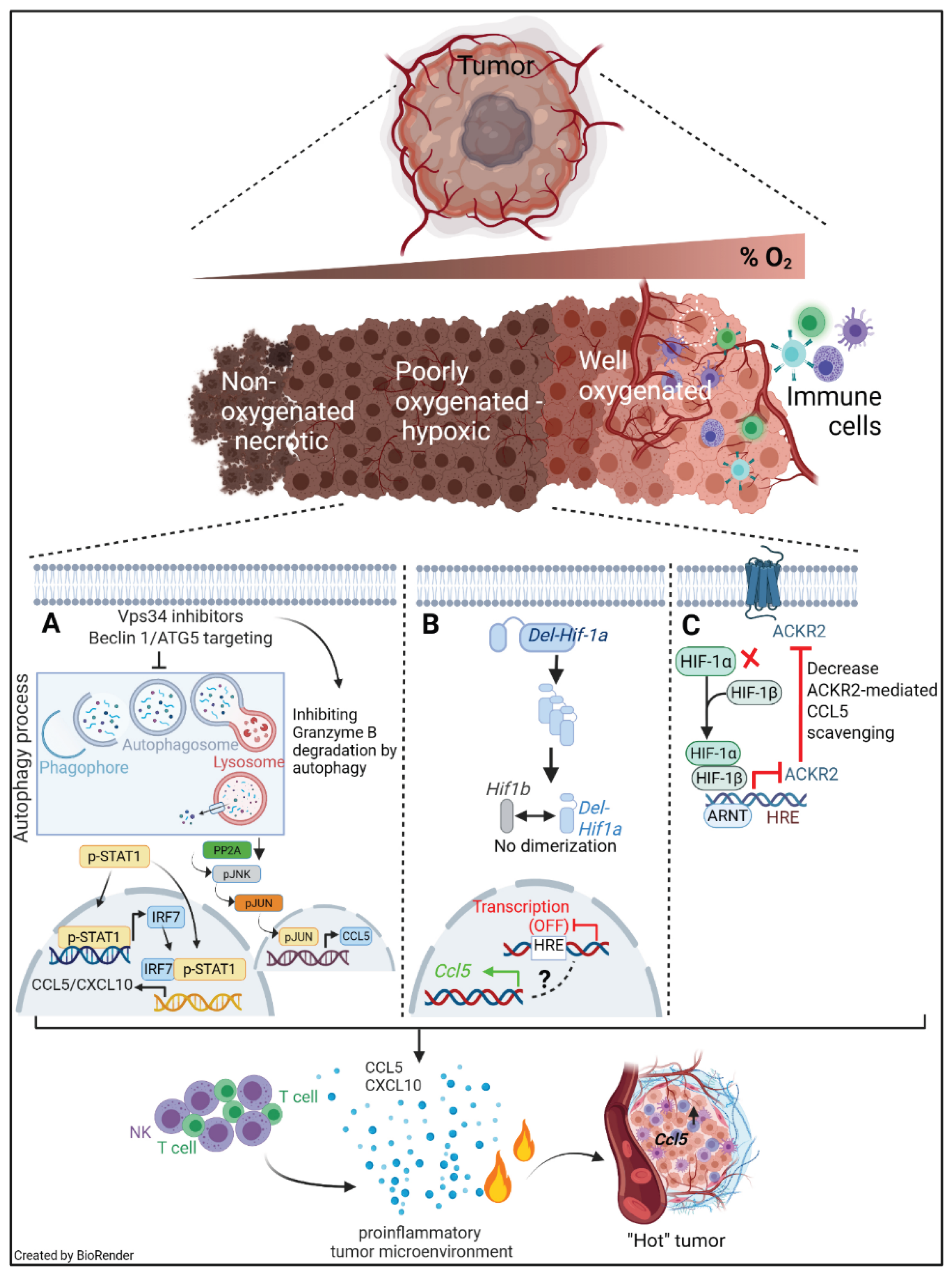
Collectively, this figure illustrates how hypoxia-driven stress-response pathways and chemokine trafficking mechanisms converge to enforce tumor immune evasion. Therapeutic targeting of autophagy, Vps34, HIF-1α, or ACKR2 has the potential to remodel the tumor immune microenvironment and improve the efficacy of cancer immunotherapy.
2. Hypoxia-Induced Autophagy as a Mechanism of Immune Escape
2.1. Hypoxia, Autophagy and CTL and NK Cell Cytotoxicity
In close collaboration with our group, Noman et al. were the first to establish a causal link between hypoxia-induced autophagy in tumor cells and resistance to cytotoxic T lymphocyte (CTL)-mediated killing. Under hypoxic stress, tumor cells activate autophagy in association with increased HIF-1α stabilization and STAT3 phosphorylation. Genetic inhibition of key autophagy regulators (Beclin 1 or ATG5) restored tumor cell susceptibility to CTL-mediated lysis, demonstrating that autophagy limits antigen-specific cytotoxicity. In vivo, targeting autophagy, either through genetic depletion of Beclin 1 or pharmacological inhibition using hydroxychloroquine, enhanced immune-mediated tumor regression and increased tumor cell apoptosis in melanoma models [4]. Together, these findings identified hypoxia-induced autophagy as a tumor-intrinsic mechanism of immune evasion and highlighted autophagy inhibition as a potential strategy to enhance anti-tumor immunity.
Autophagy is a conserved catabolic process that enables cells to recycle cytoplasmic components and adapt to metabolic and oxidative stress. Although initially considered a tumor-suppressive mechanism, autophagy in established tumors frequently promotes survival and resistance to therapy, particularly within hypoxic niches [5]. However, its role in regulating natural killer (NK) cell–mediated anti-tumor immunity in the context of hypoxia has only recently begun to be elucidated.
We provided direct evidence that hypoxia-induced autophagy reduces tumor cell susceptibility to NK cell–mediated lysis. Under hypoxic conditions, breast cancer cells exhibited robust activation of autophagy, as demonstrated by increased LC3-II accumulation and autophagosome formation. Functional cytotoxicity assays showed that hypoxia markedly impaired NK-mediated tumor cell killing despite preserved NK cell recognition and degranulation, revealing a tumor cell–intrinsic resistance mechanism. Mechanistically, we demonstrated that hypoxia-induced autophagy drives the selective degradation of NK-derived granzyme B (GzmB) within autophagosomes and autolysosomes of tumor cells. Pharmacological or genetic inhibition of autophagy, including targeting Beclin 1, restored intracellular granzyme B levels in hypoxic tumor cells and rescued NK-mediated cytotoxicity in vitro. Importantly, inhibition of autophagy in hypoxic tumors in vivo enhanced NK-dependent tumor cell elimination and promoted tumor regression, strongly establishing hypoxia-induced autophagy as a critical mechanism of immune escape [6,7]. Collectively, this study defined autophagy as a unique protection that hypoxic tumor cells deploy to neutralize granzyme B and evade NK-mediated killing and suggested that autophagy inhibition could improve the efficacy of NK-based immunotherapies against solid tumors.
In a follow-up study we investigated how inhibition of autophagy influences the recruitment and function of natural killer (NK) cells within the tumor microenvironment. While autophagy inhibition had been previously shown to reduce tumor cell survival, its impact on immune cell trafficking was unclear. Using genetic ablation of the autophagy initiation gene Beclin 1 (BECN1) in B16-F10 melanoma models, our study revealed that autophagy-deficient tumors not only exhibited reduced growth but also displayed markedly increased infiltration of functional NK cells into the tumor bed. Mechanistically, the enhanced NK infiltration was driven by upregulation of the chemokine CCL5 by autophagy-defective tumor cells. Cytokine profiling showed that loss of Beclin 1 induced transcriptional overexpression and secretion of CCL5, a potent chemoattractant for NK cells. Silencing CCL5 in BECN1-deficient tumors abrogated both NK recruitment and tumor suppression, directly implicating CCL5 in this process.
At the molecular level, the increased CCL5 expression was linked to enhanced activation of the transcription factor c-Jun, driven by increased phosphorylation through JNK signaling. This was associated with impaired activity of the phosphatase PP2A in autophagy-defective cells, suggesting that disruption of autophagy alters key signaling pathways controlling chemokine transcription. Importantly, the clinical relevance of this mechanism was supported by human melanoma data showing a positive correlation between tumor CCL5 levels and infiltration by NK cells (NKp46+) in patient biopsies, and that high CCL5 expression was associated with improved overall survival [8,9]. This work thus provided foundational evidence that autophagy actively suppresses immune infiltration by limiting chemokine release. Inhibition of autophagy disrupts this immunosuppressive barrier, promoting NK cell trafficking and tumor suppression, and highlighting CCL5 induction as a key mediator of improved anti-tumor immunity
3. Targeting Hypoxia Signaling to Reprogram the Immune Landscape
3.1. Hypoxia and Resistance to Immune Checkpoint Blockade
Immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 and CTLA-4 have transformed the treatment of multiple malignancies. However, a substantial fraction of patients exhibits primary or acquired resistance, frequently associated with non-inflamed, “cold” tumor phenotypes. Hypoxia has been implicated in this resistance by limiting effector T-cell infiltration, promoting immunosuppressive cell populations, and inducing inhibitory molecules. Nevertheless, direct genetic evidence demonstrates that modulation of HIF activity in tumor cells can reshape the tumor immune contexture and influence responsiveness to ICIs has remained limited.
In a recent study, we used a highly hypoxic, PD-1-resistant melanoma model to assess the contribution of HIF-1α/β transcriptional activity to immune exclusion and ICI failure. We showed that disrupting HIF-1α/β heterodimerization in tumor cells reduced hypoxic stress signatures and resulted in increased infiltration of CD45+ immune cells, including NK cells, CD4+ and CD8+ T lymphocytes, into the tumor microenvironment. Importantly, this reprogrammed, inflamed TME translated into a markedly improved response to immunotherapy, establishing hypoxia signaling as a determinant of primary resistance to immunotherapy [10,11]. These findings supported a model in which targeting HIF activity in cancer cells can “unlock” tumor immunogenicity and convert a cold, poorly infiltrated tumor into a hot, T cell–inflamed tumor that is more responsive to checkpoint blockade.
3.2. Vps34 Inhibition: Turning Cold Tumors Hot and Boosting Anti–PD-1/PD-L1 Therapy
Using preclinical melanoma (B16-F10) and colorectal (CT26, MC38) models with poor immune infiltration, we demonstrated that genetic or pharmacologic inhibition of the autophagy-related gene Vps34 reprograms cold tumors into highly inflamed tumors. Vps34 inhibition triggered a robust pro-inflammatory transcriptional program characterized by STAT1 and IRF7 activation, resulting in elevated expression and secretion of interferon-regulated chemokines, including CCL5 and CXCL10. Consequently, Vps34-targeted tumors exhibited markedly increased infiltration of NK cells, CD8+ T cells, CD4+ effector T cells, dendritic cells, and M1-like macrophages into the tumor microenvironment, converting immunologically “cold” tumors into “hot” tumors.
Consistent with the establishment of a T cell–inflamed TME, we observed increased expression of PD-1 on tumor-infiltrating lymphocytes and PD-L1 on tumor and myeloid cells following Vps34 inhibition. Importantly, combining Vps34 inhibitors with anti–PD-1 or anti–PD-L1 antibodies significantly improved tumor control and prolonged survival in multiple tumor models compared with either monotherapy. These data provided preclinical proof-of-concept that pharmacologic targeting of Vps34 can convert immune-desert tumors into inflamed lesions and sensitize them to checkpoint blockade [12,13,14].
Together with our previous studies on hypoxia-induced autophagy, these results highlight how stress-response and intracellular trafficking pathways in hypoxic tumors can be exploited to remodel the immune landscape and overcome resistance to immunotherapy.
4. ACKR2: A Hypoxia-Regulated Chemokine Checkpoint in Tumor Immune Evasion
Atypical chemokine receptor 2 (ACKR2, also known as D6) is a member of the atypical chemokine receptor family that functions primarily as a chemokine scavenger, rather than a conventional signaling receptor. ACKR2 is widely expressed in lymphatic endothelial cells, stromal cells. Unlike canonical chemokine receptors, ACKR2 does not activate typical G-protein–mediated signaling upon ligand binding; instead, it binds, internalizes, and directs inflammatory chemokines for lysosomal degradation, thereby regulating chemokine availability and gradients in tissues.
Our recent studies have identified ACKR2 as a critical hypoxia-inducible regulator of immune infiltration in the tumor microenvironment. We first demonstrated that ACKR2 is a direct transcriptional target of HIF-1α. Using silico promoter analysis, we identified hypoxia response elements (HREs) in both human and murine ACKR2 promoters. Consistent with these findings, multiple cancer cell types, including colorectal, melanoma, and breast cancer cells, upregulated ACKR2 under hypoxic conditions in a HIF-1α–dependent manner. Mechanistically, chromatin immunoprecipitation (ChIP) assays confirmed HIF-1α binding to the ACKR2 promoter, and silencing HIF-1α abolished hypoxia-induced ACKR2 expression. At the functional levels, ACKR2 acts as a decoy receptor for inflammatory chemokines, limiting chemokine availability in the tumor microenvironment and thereby restricting recruitment of effector T cells and NK cells [15]. This study established ACKR2 as a hypoxia-responsive checkpoint that contributes to immune cold tumor phenotypes.
Based on this mechanistic insight, we explored the therapeutic potential of targeting ACKR2 in vivo. Using a syngeneic melanoma mouse model, both genetic knockdown and pharmacologic inhibition of ACKR2 enhanced tumor infiltration by CD8+ T cells, NK cells, and dendritic cells. Importantly, ACKR2 blockade sensitized tumors to anti-PD-1 therapy, resulting in reduced tumor growth, improved survival, and increased markers of T-cell activation. These effects were particularly pronounced in immunologically “cold” tumors that are typically resistant to immune checkpoint blockade, highlighting ACKR2 as a targetable mechanism of Immune checkpoint blockade resistance in hypoxic tumors [16]
Together, these studies define ACKR2 as a hypoxia-regulated chemokine scavenger that limits immune infiltration and promotes immune evasion. The work establishes a mechanistic link between hypoxia, HIF-1α–dependent transcription, and impaired anti-tumor immunity, and it identifies ACKR2 inhibition as a promising strategy to reshape the tumor microenvironment, convert cold tumors into hot tumors, and enhance the efficacy of immunotherapies. Targeting ACKR2 could therefore complement existing strategies that aim to alleviate hypoxia or modulate autophagy and chemokine networks to overcome tumor immune resistance.
5. Discussion and Future Directions
Our studies position tumor hypoxia as a central factor of immune evasion, integrating both tumor cell–intrinsic and extrinsic mechanisms that shape the tumor immune microenvironment. Hypoxia triggers cell-intrinsic stress responses, such as autophagy, which allow tumor cells to neutralize cytotoxic lymphocytes, exemplified by the selective degradation of NK-derived granzyme B. Simultaneously, hypoxia reshapes the extrinsic immune landscape through HIF-1α–dependent induction of ACKR2, limiting chemokine availability and thereby restricting recruitment of NK cells, CD8+ T cells, and other effector populations. Together, these pathways generate immune cold tumors that are resistant to checkpoint blockades and other immunotherapies.
Importantly, these mechanisms are interconnected and targetable. Inhibition of autophagy (e.g., by targeting Beclin 1 or Vps34) restores NK and T-cell cytotoxicity and converts poorly infiltrated tumors into inflamed, T cell–enriched microenvironments. Similarly, targeting ACKR2 alleviates the scavenging of important proinflammatory chemokine enhances effector cell recruitment, and synergizes with PD-1 blockade to improve tumor control. This integrated model shows how hypoxia dictates both the functional capacity of immune effectors and the spatial organization of immune cells within the tumor, revealing actionable strategies for therapeutic intervention.
Future research should define how hypoxia-driven pathways interplay with metabolic constraints, adenosine accumulation, and myeloid cell–mediated suppression to fully understand hypoxia-dependent immune escape. Optimized combination therapies that integrate autophagy and ACKR2 inhibition with immune checkpoint blockade will be critical for improving efficacy across diverse tumor types. Identifying predictive biomarkers, such as HIF-1α activity, ACKR2 expression, or autophagy signatures, will enable rational patient selection and therapy monitoring. In summary, these studies establish a framework in which hypoxia and its downstream stress-response pathways act as master regulators of tumor immune escape. By rewiring autophagy and chemokine signaling, hypoxia shapes both the function and distribution of immune effectors, offering actionable strategies to convert cold tumors into hot, therapy-responsive tumors and enhance the clinical benefit of immunotherapy.
Funding
This research was funded by Roche (Belgium) grant number Roche, Luxembourg National Research Fund grant number INTER/PerMed24/18906356/Hi-ROC and European Union grant number EP-PerMed, JTC2024 Hi-ROC.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable.
Acknowledgments
This work was funded by Roche and the Luxembourg National Research Fund (FNR; grant reference INTER/PerMed24/18906356/Hi-ROC) and was co-funded by the European Union within the framework of EP-PerMed (European Partnership for Personalized Medicine), JTC2024 Hi-ROC.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Chouaib, S. The Promise of Targeting Hypoxia to Improve Cancer Immunotherapy: Mirage or Reality? Front Immunol 2022, 13, 880810. [Google Scholar] [CrossRef] [PubMed]
- Abou Khouzam, R.; Janji, B.; Thiery, J.; Zaarour, R.F.; Chamseddine, A.N.; Mayr, H.; Savagner, P.; Kieda, C.; Gad, S.; Buart, S.; et al. Hypoxia as a potential inducer of immune tolerance, tumor plasticity and a driver of tumor mutational burden: Impact on cancer immunotherapy. Semin Cancer Biol 2023, 97, 104–123. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Lattime, E.C.; Guo, J.Y. Autophagy Regulates Stress Responses, Metabolism, and Anticancer Immunity. Trends Cancer 2021, 7, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Baginska, J.; Berchem, G.; Noman, M.Z.; Medves, S.; Chouaib, S.; Janji, B. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014, 10, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving Natural Killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, e1452583. [Google Scholar] [CrossRef] [PubMed]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Van Moer, K.; Hasmim, M.; Benoit, A.; Bosseler, M.; Viry, E.; Arakelian, T.; Berchem, G.; et al. Targeting HIF-1 alpha transcriptional activity drives cytotoxic immune effector cells into melanoma and improves combination immunotherapy. Oncogene 2021, 40, 4725–4735. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Chouaib, S. Suffocating" tumors by blocking adaptation to hypoxia: a new headway in melanoma immunotherapy. Oncoimmunology 2021, 10, 1968611. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Hasmim, M.; Parpal, S.; Berchem, G.; Noman, M.Z. Firing up the cold tumors by targeting Vps34. Oncoimmunology 2020, 9, 1809936. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv 2020, 6, eaax7881. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Hasmim, M.; Parpal, S.; De Milito, A.; Berchem, G.; Noman, M.Z. Lighting up the fire in cold tumors to improve cancer immunotherapy by blocking the activity of the autophagy-related protein PIK3C3/VPS34. Autophagy 2020, 16, 2110–2111. [Google Scholar] [CrossRef] [PubMed]
- Benoit, A.; Lequeux, A.; Harter, P.; Berchem, G.; Janji, B. Atypical chemokine receptor 2 expression is directly regulated by hypoxia inducible factor-1 alpha in cancer cells under hypoxia. Sci Rep 2024, 14, 26589. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Szpakowska, M.; Xiao, M.; Gao, R.; Van Moer, K.; Kumar, A.; Ollert, M.; Berchem, G.; Chevigne, A.; Janji, B. Targeting the atypical chemokine receptor 2 (Ackr2) improves the benefit of anti-PD-1 immunotherapy in melanoma mouse model. Oncoimmunology 2025, 14, 2494426. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



