Submitted:
14 February 2026
Posted:
17 February 2026
You are already at the latest version
Abstract
Background Lipophilic environmental contaminants—including persistent organic pollutants (POPs), PFAS, PCBs, and PAHs - exert a long-term biological influence that cannot be explained by acute toxicity alone. Their extreme hydrophobicity drives high-affinity sequestration within lipid-rich tissues, such as adipose depots, myelin sheaths, and endocrine glands, creating "internal reservoirs" with biological half-lives measured in decades. These reservoirs fuel continuous, low-grade endogenous exposure and sequential absorption of hydrophilic species, persisting regardless of ongoing environmental contact. Scope of Review This article integrates toxicokinetic modeling with modern multi-omics evidence to update Zeliger’s model of lipophilicity-driven chronic disease. We examine how these diverse compounds activate a conserved set of biological injury pathways, regardless of their specific chemical structure. Specifically, we analyze the convergence of nuclear receptor disruption, mitochondrial dysfunction (amplified ROS production), calcium dysregulation, neuroimmune activation, and persistent epigenetic remodeling. Major Conclusions Lipophilic pollutants function as a unified category of systemic toxicants that reorganize cellular and metabolic systems. The identified mechanistic signatures provide a systems-level explanation for the epidemiological links between pollutant burdens and metabolic syndrome, cardiovascular morbidity, neurodegeneration, and cross-generational epigenetic effects. These findings validate the use of "total oxidative stress" as a predictor for non-communicable disease onset and support a paradigm shift toward mixture-based regulation and exposomic biomarkers for early detection.
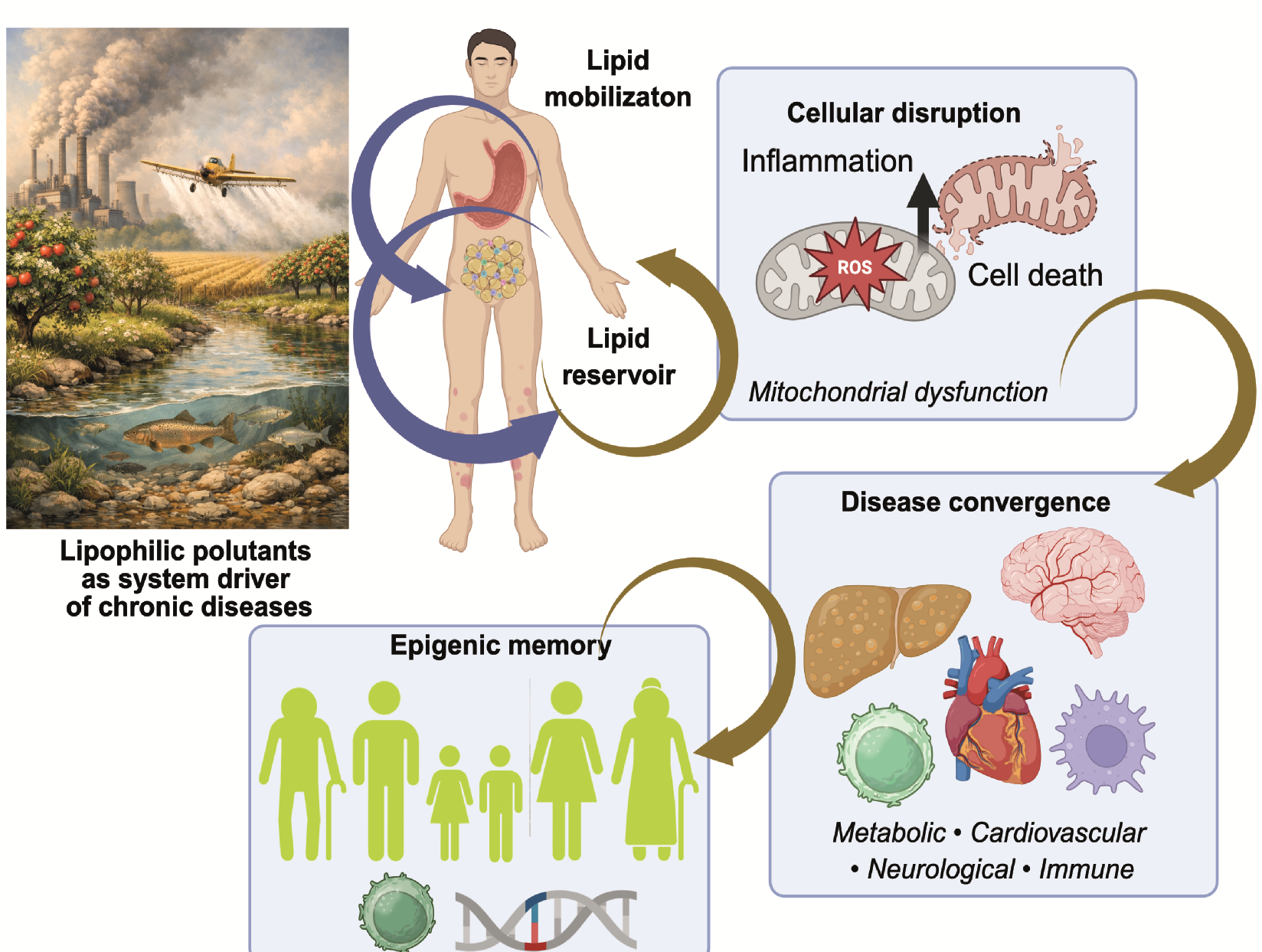
Keywords:
lipophilic toxicants
; persistent organic pollutants
; PFAS
; endocrine disruption
; mitochondrial dysfunction
; toxicokinetics
; exposmics
; systems toxicology
; chronic disease
; total oxidative stress
; human evolution
1. Introduction
Lipophilic-environmental chemicals are one of the most significant, yet least recognized, contributors to chronic disease in modern populations. Their defining characteristic, high hydrophobicity, drives their accumulation in lipid-rich tissues, creating long-term biological reservoirs that release toxicants into the bloodstream during routine metabolic processes [1]. Unlike hydrophilic xenobiotics, lipophilic pollutants such as PCBs, PFAS OCPs, PAHs, phthalates and bisphenols readily integrate into cell membranes, adipose tissue neural lipid structures and endocrine organs. This creates a toxicokinetic environment in which internal exposure substantially outlasts external exposure and may create unanticipated synergistic toxic effects when exposures are to chemical mixtures [2].
It is this toxicokinetic persistence that gives rise to the chronic nature of pollutant effects. Stored lipophilic pollutants re-enter the blood stream during lipid turnover, such as occurs during fasting, caloric restriction, illness, strenuous exertion, pregnancy or lactation. This generates endogenous pulses of exposure that may exceed the original environmental doses [3,4]. These fluctuations do not align well with traditional exposure assessment frameworks, which assume a linear relationship between environmental concentration and internal dose, and produce enhanced toxic effects.
Zeliger proposed that lipophilic chemicals, that are long-lived in body fat and can be absorbed in single doses or sequentially in multiple exposures over time, facilitate the cellular penetration of additional hydrophilic toxicants, that are not ordinarily absorbed through lipophilic barriers. Such absorption of produces low level effects; enhanced effects; and attacks on organs not targeted by either the lipophilic or hydrophilic species alone [1,5,6]. While this idea was ahead of its time, contemporary evidence reveals a broader phenomenon: lipophilic pollutants act as systemic biological disruptors whose persistent presence reorganizes cellular, metabolic, neuroimmune and epigenetic systems. Converging toxicological, epidemiological and omics evidence conforms that diverse lipophilic pollutants activate shared mechanistic pathways, including nuclear receptor interference, disrupted mitochondrial energetics, heightened oxidative stress, neuroimmune priming, inflammasome signaling and epigenetic remodeling.
As discussed below, epidemiological studies support these mechanistic insights, consistently linking pollutant burdens to an increased risk of metabolic disorders, cardiovascular disease, cognitive impairment, neurodevelopmental disorders, neurodegenerative disease, immune dysfunction and transgenerational health effects. These associations persist even when adjusted for diet, socioeconomic status, adiposity and lifestyle factors. This indicates that lipophilic pollutants exert an independent, systemic influence
These convergent findings highlight the need for a modern systems toxicology framework that can explain how lipophilic persistence drive mechanistic disruption across biological scales. This manuscript integrates toxicokinetic modeling, mechanistic evidence multi-omics data and epidemiological coherence to provide a comprehensive explanation of the pivotal role of lipophilic chemical accumulation in chronic disease.
2. Methods
This systems toxicology framework integrates evidence from toxicokinetic, mechanistic toxicology, multi-omics, developmental neurotoxicology, lipidomic and epidemiology. Because lipophilic chemicals produce persistent and multi-organ effects, a multi-layered analytical approach was required to map mechanistic convergence across biological domains.
Mechanistic evidence was identified through comprehensive searches of PubMed, Scopus, Web of Science and Embase using terms related to endocrine disruption, mitochondrial dysfunction, oxidative stress, neuroimmune signaling, inflammasome activation and epigenetic remodeling. Studies were included when they demonstrated biologically plausible mechanisms, at human-relevant doses and when toxicokinetic parameters supporter tissue distribution consistent with lipophilic persistence. Emphasis was placed on reproducibility across species, tissue types and pollutant classes.
Toxicokinetic insights were drawn from physiologically-based pharmacokinetic (PBPK) models, human biomonitoring studies (including NHANES and HBM4EU), depuration and lactational transfer studies and pollutant-specific half-life analyses. Key parameters included logK, lipid partitioning coefficients, adipose-serum release dynamics and re-mobilization kinetics during lipid turnover [2,24,35]. These data provided constraints for interpreting mechanistic pathways,
Mechanistic toxicology was evaluated across endocrine receptor biology, mitochondrial function, lipid metabolism, immune signaling and neuronal physiology. Nuclear receptive studies using ligand-binding assays, reporter-gene activation, ChlP-seq and transcriptional profiling were integrated with mitochondrial assays measuring electron transport chain activity, membrane potential, ATP synthesis and mitochondrial ROS generation [2,10,17,35,36,37]. Neurodevelopmental pathways were assessed through neuronal differentiation and oligodendrocyte maturation models, synaptic physiology, electrophysiology and microglial activation assays [11]. Immune and oxidative mechanisms were examined through cytokine profiling, NF-κB pathway analysis, NLRP3 inflammasome activation and ROS assays [10,17].
It should not be forgotten that metabolomics, the large-scale study of metabolites within organisms, tissues and cells, might be considered a fundamental tool for elucidating the associations between environmental pollutants and health, as well as the etiology of certain diseases [38,39].
Epigenomic evidence was drawn for DNA methylation studies, histone modification analyses, chromatin accessibility (ATAC-seq) and small RNA datasets with emphasis on long-term or transgenerational alterations induced by POPs, PFAS and related pollutants [22]. Epidemiological studies were evaluated for coherence with mechanistic findings, with special attention to prenatal exposures, longitudinal cohorts and chronic disease incidence [most of the references are concerned].
Mechanisms were incorporated into the final model only when supported by toxicokinetic feasibility, mechanistic reproducibility and multi-ohmic or epidemiologic validation. The resulting framework describes how lipophilic pollutant persistence, mixture synergy and mechanistic convergence collectively drive chronic disease across organ systems.
3. Results
The integrated evaluation of toxicokinetic, mechanistic and multi-omic evidence demonstrates that lipophilic environmental pollutants act through a set of convergent biological pathways that collectively disrupt metabolic, cardiovascular, neurological, immune and epigenetic systems. Despite the structural diversity among POPs, PFAS, PCBs, OCPs, PAHs, phthalates and bisphenols, their high lipophilicity drives similar patters of tissue accumulation and mechanistic injury [7]. All of these species chronically elevate OS as free radicals formed in them are stabilized by substitution on the radical carbon, resonance and having the radical α- to a π-bond with which it can overlap [8]. These shared patterns reflect a unifying systems-level architecture in which persistent internal reservoirs of pollutants continually perturb cellular regulation and physiological homeostasis.
A central finding is the profound disruption of metabolic and endocrine systems. Nuclear receptor interference has emerged as a dominant mechanism, with pollutants consistently impairing PPARy, RXR, Erα/β, CAR, PXR and AhR signaling networks [1,2,9,10,11,12] (Table 1).
These receptors regulate core metabolic functions – including lipid uptake, adipocyte differentiation, glucose homeostasis, bile acid metabolism and xenobiotic processing. Their perturbation produces the metabolic inflexibility characteristic of pollutant-associated insulin resistance, dyslipidemia and hepatic steatosis [1,2,3]. Effectively, chronic liver diseases, including metabolic dysfunction-associated steatosic liver disease (MASLD) and hepatocellular carcinoma (HCC), are on the rise, potentially due to daily exposure to complex mixtures of chemical compounds forming part of the exposome [13].
Experimental studies demonstrate that pollutant-induced suppression of PPARy and RXR disrupts adipogenic transcriptional programs, while endocrine disruptors such as phthalates and bisphenol alter estrogen receptor-dependent metabolic pathways, compounding the metabolic burden [11]. These mechanistic patterns align closely with epidemiological associations linking circulating POP and PFAS levels to metabolic syndrome, type 2 diabetes and NAFLD/NASH [1,3,11].
Adipose tissue plays a dual role as both a pollutant reservoir and an active endocrine organ. Lipophilic pollutants accumulate in adipocytes at concentrations exceeding those found in blood or lean tissues [3,4]. This accumulation triggers chronic local accumulation: M1 polarization, NF-kB activation and elevated secretion of TNF-α, IL-6 and Ilβ have been observed in pollutant-exposed adipose tissue [1,2,20,21]. Lipidomic analyses reveal that pollutant exposure increases ceremide and phospholipid levels, biochemicals signatures closely linked to insulin resistance and lipotoxicity [1,2]. Pollutants therefore shift adipose tissue from a metabolic buffer to an inflammatory driver, amplifying systemic metabolic dysfunction.
Toxicokinetic models demonstrate that pollutant burdens in adipose tissue also propagate metabolic disease through episodic re-mobilization of stored toxicants during periods of lipid turnover. Fasting, illness, pregnancy, lactation and rapid weight loss all promote the release of POP and PFAS into circulation, creating transient spikes in internal exposure that may exceed levels at the time of original uptake [2,3]. These internal exposure cycles explain why metabolic diseases remain strongly associated with POP and PFAS levels even in populations with declining environmental exposures.
Cardiovascular and vascular tissues exhibit a similarly coherent mechanistic profile. Endothelial cells exposed to lipophilic pollutants show OS-driven dysfunction, reduced nitric oxide availability, impaired vasodilation and increased expression of adhesion molecules associated with vascular inflammation [3,4,5]. Mitochondrial dysfunction plays a central role: pollution exposure impairs electron transport chain activity, increases mitochondrial ROS generation and disrupts mitochondrial membrane potential [3,4,5]. These deficits compromise endothelial metabolism and induce vascular stiffness, laying the groundwork for hypertension and atherosclerosis.
The vascular injury observed experimentally aligns with the strong epidemiological associations between circulating POP and PFAS burdens and increased risk for hypertension, carotid-media thickness, coronary artery disease and ischemic stroke [3,5]. Oxidized LDL and foam cell formation are consistently elevated in pollutant exposure models, linking POPs and PAHs mechanistically to atherogenesis. Pollutant induced matrix metalloproteinase activation further contributes to plaque instability providing a rationale for increases in cardiovascular events in exposed populations [3,5].
The central nervous system (CNS) presents even greater vulnerability due to its high lipid content, extended cellular lifespans and limited regenerative capacity. Human and animal studies have repeatedly shown that prenatal and early-life exposure to PCBs, PFAS, OCPs, phthalates and bisphenols is associated with impaired cognitive function, reduced executive functioning, behavioral dysfunction and neurodevelopmental disorders such as ASD and ADHD [8,11,15]. These findings are consistent with mechanistic evidence demonstrating that lipophilic pollutants disrupt neuronal differentiation, synaptogenesis, and oligodendrocyte maturation. Calcium imaging studies reveal pollutant-induced dysregulation of Ca2+ signaling and increased excitotoxicity, while electrophysiological recordings document reduced synaptic strength and altered neurotransmission [7].
Figure 1.
System biology Framework.
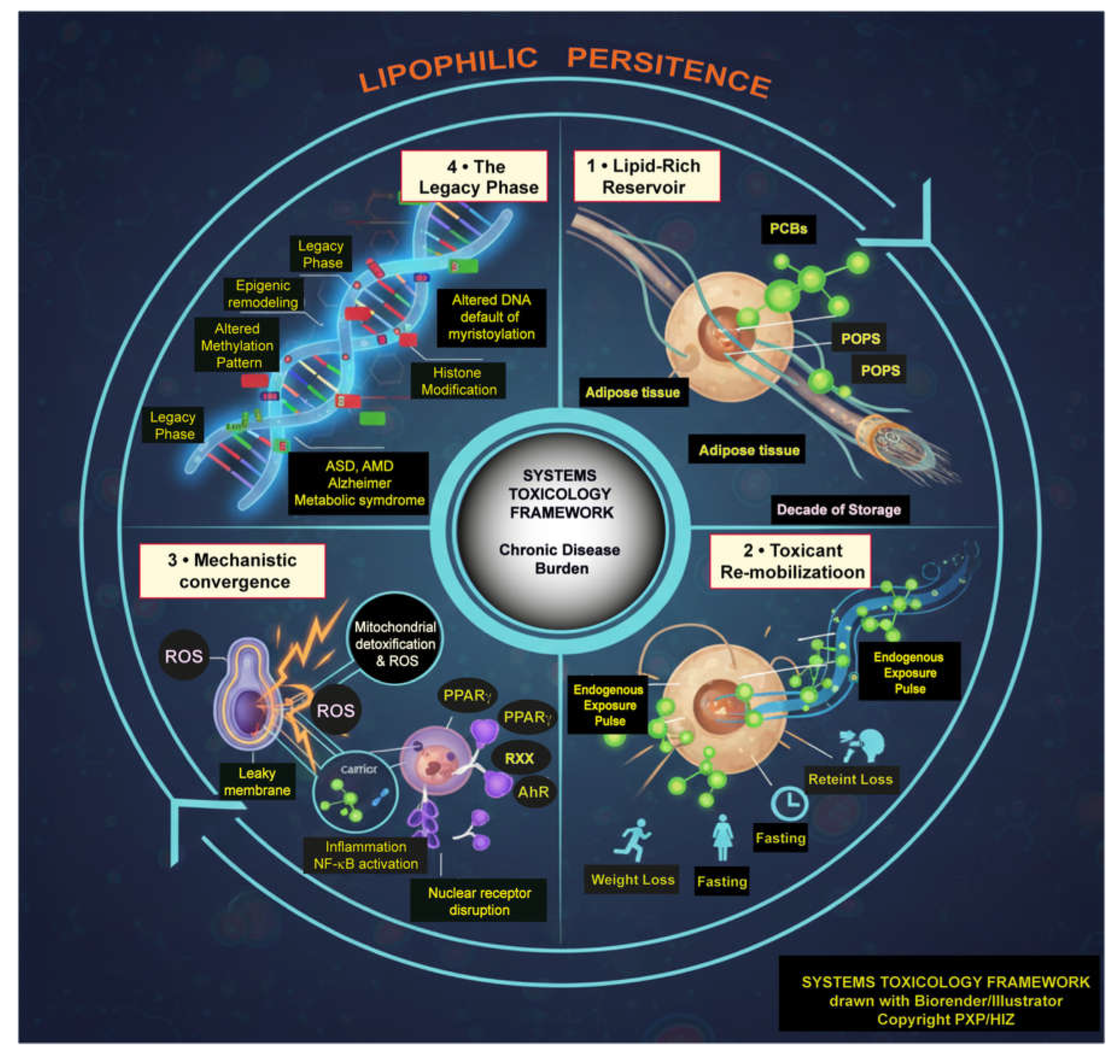
Neuroimmune interactions emerged as a powerful unifying pathway linking pollutant exposure to both neurodevelopmental and neurodegenerative outcomes. Pollutant-exposed microglia consistently exhibit a primed inflammatory state characterized by exaggerated cytokine responses, increased ROS production and impaired synaptic pruning [7,11,15]. This priming lowers the threshold for neural injury throughout life, aligning with epidemiological evidence linking cumulative burdens to accelerated cognitive decline and risk of Alzheimer’s and Parkinson’s disease [11,12]. Experimental models show pollutant-induced tau phosphorylation, amyloidogenic processing and α-synuclein aggregation, suggesting a direct molecular link between pollutant exposure and hallmark features of neurodegenerative pathology.
Across immune issues, lipophilic pollutants activate a persistent inflammatory state that bridges multiple chronic diseases. NF-kB signaling is reliably elevated in pollutant-exposed immune cells, accompanied by increased secretion of TNF-α, IL-6 and IL-1β [10,17]. The NLRP3 inflammasome is repeatedly activated pollutant-induced mitochondrial ROS, oxidized lipids and mitochondrial DNA leakage, leading to caspase-1 activation and release of IL-1β, and IL-18 [10,17]. These innate immune disturbances reproduce clinical inflammatory states linked to metabolic syndrome, cardiovascular disease, and autoimmune disorders.
Perhaps the most groundbreaking mechanistic insight is the pervasive epigenomic remodeling induced by lipophilic pollutant exposure. Genome-wide methylation studies in both humans and experimental models demonstrate global hypomethylation. Promotor-specific hypermethylation in metabolic and inflammatory genes, as well as altered methylation patterns in neurodevelopmental pathways [22]. Histone modifications and chromatin accessibility further indicate pollutant-induced reprogramming of transcriptional landscapes, often persisting long after external exposure ends. In prenatal and early-life contexts, these epigenetic alterations shape developmental trajectories, aligning with DOHaD principles [11].
Together, these results show that lipophilic pollutants form a unified mechanistic class whose toxicokinetic persistence and mechanistic convergence explain their broad epidemiological associations with chronic disease. The resulting framework significantly strengthens Zeliger’s hypothesis regarding lipophilicity-driven systemic toxicity [23]
4. Discussion
The integrated results presented here reveal a coherent and biologically robust mechanism through which lipophilic environmental pollutants contribute to the global burden of chronic disease. Despite structural diversity among PFAS, POPs, PCBs, OCPs, PAHs, phthalates and bisphenols, their toxicokinetic persistence and lipid affinity drive accumulation in adipose tissue, neuronal membranes, endocrine organs and hepatic lipid pools, creating a prolonged internal exposure state independent of ongoing environmental contact [2,26,27]. This persistence forms the basis for a systemic and multi-organ pattern of biological disruption.
Zeliger’s foundational hypothesis – that lipophilic chemicals act as carriers facilitating toxicity of multiple compounds – accurately anticipated the modern systems toxicology perspective [24]. Yet, current evidence indicates that lipophilic pollutants act not just as carriers, but as biological integrators, coordinating mechanistic disruptions across metabolic, cardiovascular, neurological, immune and epigenetic domains. This evolution in understanding is driven by advancements in exposomics, toxicogenomics, lipidomics and developmental neurobiology that collectively illuminate the breath of pathways altered by lipophilic toxicity.
A key unifying mechanism is nuclear receptor disruption. Pollutants interfere with PPARy, RXR, Erα/β, CAR, PXR and AhR signaling pathways, impairing metabolic regulation, lipid homeostasis, bile acid metabolism and endocrine disruption [24,28,29,30].
These receptor interactions produce a metabolic signaling bottleneck that aligns closely with the phenotype of metabolic syndrome, insulin resistance and NAFLD/NASH circulating POP levels [24,28,29]. The coherence of mechanistic and epidemiological evidence strengthens the causal inference that lipophilic pollutants contribute directly to metabolic disease.
Mitochondrial dysfunction is another cross-issue mechanism central to pollutant toxicity. By integrating into mitochondrial membranes, lipophilic toxicants inhibit electron transport chain complexes, depolarize mitochondrial membranes, reduce ATP synthesis and promote ROS [17,18] (Figure 2). Mitochondrial ROS serve as signaling amplifiers that propagate injury to nuclear pathways such as NF-kB and inflammasome activation [11,17,18]. This mitochondrial inflammatory axis links pollutant exposure to cardiovascular disease, adipose inflammation, immune dysregulation and neurodegeneration.
The neurodevelopmental and neurodegenerative findings highlight a lifespan perspective on pollutant toxicity. Prenatal exposure alters neuronal differentiation, synaptic architecture, oligodendrocyte maturation and microglial function, creating neurodevelopmental trajectories associated with ASD, ADHD and cognitive impairment [6,12]. These early-life disruptions establish conditions favorable to later-life neurodegenerative processes. Experimental evidence of pollutant-induced phosphorylation, amyloidogenic processing and α-synuclein aggregation [15] supports a mechanistic continuum linking developmental exposures to age-related neurodegeneration.
The immune system also plays a crucial integrating role. Chronic pollutant exposure induces sustained activation of innate immune pathways, particularly through
NF-kB signaling and the NLRP3 inflammasome [12,31]. These immune signatures produce systemic inflammation that fosters metabolic dysfunction, endothelial injury and CNS vulnerability. The mechanistic overlap between pollutant-induced inflammation and chronic disease biology helps clarify long-standing epidemiological associations across diverse conditions.
Perhaps the most durable and far-reaching effect of lipophilic pollutant exposure lies in epigenomic programming DNA methylation drift, altered histone modification patterns and changes in chromatin accessibility persist long after pollutant removal, especially when exposure occurs prenatally or early in life [11,19]. These changes encode molecular “memory” of exposure that shapes long-term health trajectories, and in some cases, extends to future generations [22,32]. This epigenic evidence strongly supports the DOHaD paradigm and suggests that lipophilic pollutants play a significant role in transgenerational disease risk.
Overall, the results of this integrative review suggest that lipophilic pollutants form a systems-disrupting toxicological class whose effects are rooted in persistence, toxicokinetic cycling, mixture interactions and multi-organ mechanistic convergence. Addressing these pollutants requires rethinking environmental regulation to incorporate mixture toxicity, long-term internal exposure and early-life sensitivity. The findings justify the adoption of exposomic biomarkers, systems biology models and mixture-aware risk assessments in both environmental policy and clinical practice.
5. Conclusion
This systems toxicology framework demonstrates that lipophilic environmental chemicals represent a unified mechanistic class of chronic disease drivers whose effects stem from toxicokinetic persistence and multi-organ mechanistic convergence. Their extreme hydrophobicity insured long-term sequestration within adipose deposits, hepatic lipid pools, neuronal membranes, myelin sheaths and endocrine issues resulting in half-d internal reservoir that releases pollutants during lipid mobilization, generating recurrent endogenous exposure spikes that can exceed original intake levels [1,2].
Mechanistic evidence reveals that lipophilic pollutants consistently disrupt nuclear receptor networks – particularly PPARy, RXR, ERα/β, CAR, PXR and non-AhR-altering metabolic regulation, adipogenesis, lipid homeostasis, glucose signaling and hormonal balance [2,11,16]. These receptor-level disturbances are reinforced by mitochondrial dysfunction, including impaired electron chain activity, reduced ATP synthesis and pathological elevation of mitochondrial ROS [3,4,31]. ROS amplification activates NF-kB signaling and NLRP3 inflammasome establishing a chronic inflammatory milieu associated with metabolic disease, endothelial dysfunction and immune dysregulation [17,18]. In the cardiovascular syste[11,16m, these mechanisms manifest as endothelial oxidative stress, diminished nitric oxide bioavailability, impaired vasodilation, increased vascular stiffness and accelerated atherogenesis [13,17]. These injury patterns explain epidemiological associations between POP and PFAS burdens as well as heightened risk of hypertension, coronary heart disease and stroke [17,18].
The central nervous system shows pronounced vulnerability due to its lipid richness and long-lived neuronal populations. Prenatal and early-life exposures to PCBs, PFAS, OCPs, bisphenols and phthalates are strongly associated with impaired neurodevelopment and increased risk of ASD, ADHD and cognitive deficits [11,16] Mechanisms include excitotoxic calcium dysregulation, disrupted oligodendrocyte maturation, impaired synaptogenesis and microglial priming [11,32]. These early-life alterations persist through adulthood and converge with pollutant-induced tau phosphorylation, amyloidogenic processing and α-synuclein sensitization, linking developmental exposures to neurodegenerative pathways associated with Alzheimer’s and Parkinson’s disease [11].
Epigenomic remodeling is perhaps the most enduring consequence of lipophilic toxicant exposure. Across human cohort studies and experimental models, pollutants induce stable changes in DNA methylation, histone modifications, chromatin accessibility and non-coding RNA networks [5,11]. These changes provide a molecular memory of exposure that aligns with the Developmental Origins of Health and Disease paradigm [9]. Transgenerational studies reveal inheritance of pollutant-associated epigenomic marks across multiple generations [5,11].
Collectively, this framework significantly extends Zeliger’s original hypothesis that lipophilic chemicals act as a systemic driver of chronic illness [23].
Our findings demonstrate that lipophilic pollutants function not as isolated toxicants but as biological integrators whose persistence, internal cycling and multi-organ mechanistic convergence render them central contributors to chronic disease etiology. Effective public health
Responses must, therefore, shift from single-chemical risk assessment to mixture-aware toxicokinetic-informed frameworks, incorporating exposomic biomarkers and systems-level models of biological disruption.
There is work that still needs to be done. As discussed above, exposures to lipophilic toxicants lead to disease onsets in multiple organs and make it virtually impossible to predict which NCD will strike first or next in one exposed to these toxicants. Multi-morbidities almost always follow the onset of an initial NCD, and bidirectionality between most NCD pairs have been reported [8]. Also, genomic alterations attributed to lipophilic toxicants leave one to wonder how future human evolution will be impacted [33,34].
Finally, it has been demonstrated that highly lipophilic exosomes (30-150 nm in diameter) derived from stem cells, readily penetrate the blood brain barrier and offer a therapeutic role in reversing neurodegenerative diseases that include AD, PD and ALS [24,25]. This suggests that other lipophilic species can similarly be investigated as therapeutic agents for other NCDs.
Author Contributions
P.X.P. and H.I.Z. conceived and wrote the manuscript.
Funding
This work has been entirely funded by C.N.R.S. and I.N.S.E.R.M common support. The funders had no role in study design, data collection and analysis,.
Acknowledgments
The authors thank M. Murphy (Medical Research Council Dunn Human Nutrition Unit, Wellcome Trust/MRC Building, Hills Road, C[33,34ambridge CB2 2XY, United Kingdom) and VP Skulachev† (Belozersky Institute of Physico- Chemical Biology, Lomonosov Moscow State University, Vorob’evy Gory 1/40, 119992 Moscow, Russia) for their gift and advices concerning the mitochondrially targeted anti-oxidants MitoQ10 and SKQ1 respectively.
Competing Interests
The authors declare that they have no competing interests.
Abbreviations
AhR, aryl hydrocarbon receptor; ATP, Adenosine triphosphate; CNS, Central nervous system; DOHaD, Developmental Origins of Health and Disease; ETC, Electron transport chain, GR, Glucocorticoid receptor; IL-1β/IL-6 /IL-18, Interleukin 1β / Interleukin 6 / Interleukin-18; MMP, Matrix metalloproteinase; NAFLD, Nash, Non-alcoholic fatty liver disease / Non-alcoholic steatohepatitis; NCD, non-communicative disease; NF-kB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NOD-like receptor family pyrin domain-coating 3; PAHs, polynuclear aromatic hydrocarbons; PBPK, Physiologically based pharmacokinetic; PCBs, Polychlorinated biphenyls; PFAS, Per- and polyfluoroalkyl substances; OCPs, Oral contraceptive pills; PPARy, OS, oxidative stress; Peroxisome proliferator-activated receptor gamma; PXR/CAR, Pregnane X receptor/Constitutive androstane receptor; ROS, Reactive oxygen species; RXR, Retinoid X receptor; TCDD, Dioxin, TNF-α, Tumor necrosis factor alpha.
References
- Zeliger, HI. Toxic effects of chemical mixtures. Arch Environ Health 2003, 58, 23–9. [Google Scholar] [CrossRef]
- Lee, YM; Jacobs, DR, Jr.; Lee, DH. Persistent Organic Pollutants and Type 2 Diabetes: A Critical Review of Review Articles. Front Endocrinol (Lausanne) 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Lind, PM; Lee, DH; Jacobs, DR; Salihovic, S; van Bavel, B; Wolff, MS; Lind, L. Circulating levels of persistent organic pollutants are related to retrospective assessment of life-time weight change. Chemosphere 2013, 90, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Rainey, NE; Saric, A; Leberre, A; Dewailly, E; Slomianny, C; Vial, G; Zeliger, HI; Petit, PX. Synergistic cellular effects including mitochondrial destabilization, autophagy and apoptosis following low-level exposure to a mixture of lipophilic persistent organic pollutants. Sci Rep 2017, 7, 4728. [Google Scholar] [CrossRef]
- Zeliger, HI. Lipophilic chemical exposure as a cause of cardiovascular disease. Interdiscip Toxicol 2013, 6, 55–62. [Google Scholar] [CrossRef]
- Zeliger, HI. Exposure to lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip Toxicol 2013, 6, 103–10. [Google Scholar] [CrossRef]
- Lolescu, BM; Furdui-Linta, AV; Ilie, CA; Sturza, A; Zara, F; Muntean, DM; Blidisel, A; Cretu, OM. Adipose tissue as target of environmental toxicants: focus on mitochondrial dysfunction and oxidative inflammation in metabolic dysfunction-associated steatotic liver disease. Mol Cell Biochem 2025, 480, 2863–2879. [Google Scholar] [CrossRef] [PubMed]
- Zeliger, HI. Co-morditities of environmental diseases: A common cause. Interdiscip Toxicol 2014, 7, 117–22. [Google Scholar] [CrossRef]
- Heindel, JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci 2003, 76, 247–9. [Google Scholar] [CrossRef]
- Heindel, JJ; Newbold, R; Schug, TT. Endocrine disruptors and obesity. Nat Rev Endocrinol 2015, 11, 653–61. [Google Scholar] [CrossRef]
- Grandjean, P; Landrigan, PJ. Developmental neurotoxicity of industrial chemicals. Lancet 2006, 368, 2167–78. [Google Scholar] [CrossRef]
- Ing, C; Bellinger, DC. Long-term cognitive and behavioral outcomes following early exposure to general anesthetics. Curr Opin Anaesthesiol 2022, 35, 442–447. [Google Scholar] [CrossRef]
- Choi, MA; Rose, S; Langouet, S. Per- and polyfluoroalkyl substances as potentiators of hepatotoxicity in an exposome framework: Current challenges of environmental toxicology. Toxicology 2025, 515, 154167. [Google Scholar] [CrossRef]
- Lolescu, BM; Furdui-Linta, AV; Ilie, CA; Sturza, A; Zara, F; Muntean, DM; Blidisel, A; Cretu, OM. Correction to: Adipose tissue as target of environmental toxicants: focus on mitochondrial dysfunction and oxidative inflammation in metabolic dysfunction-associated steatotic liver disease. Mol Cell Biochem 2025, 480, 3927. [Google Scholar] [CrossRef]
- Grandjean, P; Kishi, R; Kogevinas, M. International Society for Environmental E Prevention of Developmental Neurotoxicity. Epidemiology 2017, 28, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Forns, J; Stigum, H; Hoyer, BB; Sioen, I; Sovcikova, E; Nowack, N; Lopez-Espinosa, MJ; Guxens, M; Ibarluzea, J; Torrent, M; Wittsiepe, J; Govarts, E; Trnovec, T; Chevrier, C; Toft, G; Vrijheid, M; Iszatt, N; Eggesbo, M. Prenatal and postnatal exposure to persistent organic pollutants and attention-deficit and hyperactivity disorder: a pooled analysis of seven European birth cohort studies. Int J Epidemiol 2018, 47, 1082–1097. [Google Scholar] [CrossRef]
- Hamzavi, SF; Elahi Vahed, I; Samadi Shams, A; Nozari, F; Gamzeh Latava, B; Mardukhi, S; Sabaghi, B; Hosseini, ZS; Masoumi Shahr, EBZ; Ahrari, S; Keshavarzian, A; Rahmanian, M. Association between polychlorinated biphenyls and hypertension risk: a systematic review and meta-analysis. Front Cardiovasc Med 2025, 12, 1529431. [Google Scholar] [CrossRef]
- Lind, PM; Salihovic, S; Stubleski, J; Karrman, A; Lind, L. Association of Exposure to Persistent Organic Pollutants With Mortality Risk: An Analysis of Data From the Prospective Investigation of Vasculature in Uppsala Seniors (PIVUS) Study. JAMA Netw Open 2019, 2, e193070. [Google Scholar] [CrossRef]
- Zeliger, HI. Lipophilic chemical exposure as a cause of type 2 diabetes (T2D). Rev Environ Health 2013, 28, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg Lind, Y; Lind, PM; Salihovic, S; van Bavel, B; Lind, L. Circulating levels of persistent organic pollutants (POPs) are associated with left ventricular systolic and diastolic dysfunction in the elderly. Environ Res 2013, 123, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Roos, V; Ronn, M; Salihovic, S; Lind, L; van Bavel, B; Kullberg, J; Johansson, L; Ahlstrom, H; Lind, PM. Circulating levels of persistent organic pollutants in relation to visceral and subcutaneous adipose tissue by abdominal MRI. Obesity (Silver Spring) 2013, 21, 413–8. [Google Scholar] [CrossRef]
- Reddy, SR; Bangeppagari, M; Lee, SJ. Immune-Epigenetic Effects of Environmental Pollutants: Mechanisms, Biomarkers, and Transgenerational Impact. Curr Issues Mol Biol 2025, 47. [Google Scholar] [CrossRef]
- Zeliger, HI; Lipinski, B. Physiochemical basis of human degenerative disease. Interdiscip Toxicol 2015, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zeliger, HI. Predicting disease onset in clinically healthy people. Interdiscip Toxicol 2016, 9, 39–54. [Google Scholar] [CrossRef] [PubMed]
- zeliger, HI. Oxidative stress indes (OSI). condensed questionnaire. Eur. J. Med. Sci 2020, 2, 163. [Google Scholar] [CrossRef]
- Lind, PM; van Bavel, B; Salihovic, S; Lind, L. Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly. Environ Health Perspect 2012, 120, 38–43. [Google Scholar] [CrossRef]
- Lind, PM; Salihovic, S; van Bavel, B; Lind, L. Circulating levels of perfluoroalkyl substances (PFASs) and carotid artery atherosclerosis. Environ Res 2017, 152, 157–164. [Google Scholar] [CrossRef]
- Lee, YM; Bae, SG; Lee, SH; Jacobs, DR, Jr.; Lee, DH. Persistent organic pollutants and hyperuricemia in the U.S. general population. Atherosclerosis 2013, 230, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P; Harari, R; Barr, DB; Debes, F. Pesticide exposure and stunting as independent predictors of neurobehavioral deficits in Ecuadorian school children. Pediatrics 2006, 117, e546-56. [Google Scholar] [CrossRef]
- Le Magueresse-Battistoni, B; Vidal, H; Naville, D. Lifelong consumption of low-dosed food pollutants and metabolic health. J Epidemiol Community Health 2015, 69, 512–5. [Google Scholar] [CrossRef]
- Zeliger, HI. Air pollution, obesity and disease. Euro J Med and Health Sci 2024, 6, 2093. [Google Scholar] [CrossRef]
- Le Magueresse-Battistoni, B; Multigner, L; Beausoleil, C; Rousselle, C. Effects of bisphenol A on metabolism and evidences of a mode of action mediated through endocrine disruption. Mol Cell Endocrinol 2018, 475, 74–91. [Google Scholar] [CrossRef]
- Pienavani, T. Imperfection. A natural history; MIT press: Cambridge Massachusetts, USA, 2022. [Google Scholar]
- Lind, L; Zethelius, B; Salihovic, S; van Bavel, B; Lind, PM. Circulating levels of perfluoroalkyl substances and prevalent diabetes in the elderly. Diabetologia 2014, 57, 473–9. [Google Scholar] [CrossRef]
- Bolognesi, G; Bacalini, MG; Pirazzini, C; Garagnani, P; Giuliani, C. Evolutionary Implications of Environmental Toxicant Exposure. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Lind, L; Salihovic, S; Lampa, E; Lind, PM. Mixture effects of 30 environmental contaminants on incident metabolic syndrome-A prospective study. Environ Int 2017, 107, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Le Magueresse-Battistoni, B; Vidal, H; Naville, D. Environmental Pollutants and Metabolic Disorders: The Multi-Exposure Scenario of Life. Front Endocrinol (Lausanne) 2018, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J; Boccard, J; Codesido, S; Gagnebin, Y; Joshi, A; Picard, D; Gonzalez-Ruiz, V; Rudaz, S. Implementation of liquid chromatography-high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal Chim Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, X; Gui, T; Yang, S; Jin, S; Qiao, J; Xie, Y; Wang, J; Ouyang, W; Song, G; Yi, X; Liu, C; Pan, M; Liu, W; Qiao, N; Dai, Y; Tao, Y; Xu, J; Yin, T; Fang, H; Mi, J; Chen, SJ. Metabolic biomarkers for predicting onset and severity of CAR-T therapy-induced cytokine release syndrome in multiple myeloma. In Front Med; 2025. [Google Scholar] [CrossRef]
Figure 2.
Schematic interpretation of the biology events associated to the low exposure with two powerful pollutants (Endosufan + TCDD “dioxin”).
Figure 2.
Schematic interpretation of the biology events associated to the low exposure with two powerful pollutants (Endosufan + TCDD “dioxin”).


Table 1.
Description of the pollutant classes, their primary target and the key mechanisms that underlies health outcomes.
Table 1.
Description of the pollutant classes, their primary target and the key mechanisms that underlies health outcomes.
| Pollutant Class |
Primary Target | Key Mechanisms & Health Outcomes | Refs |
|---|---|---|---|
| PFAS | Adipose tissue, Liver, CNS, Vascular system | Linked to Metabolic Syndrome, Type 2 Diabetes, and NAFLD/NASH. Strongly associated with hypertension, coronary artery disease, and stroke. Connected to neurodevelopmental disorders (ASD, ADHD). |
[1,2,3,5,7,10,11,14,15,16] |
| PCBs | Adipose tissue, Myelin sheaths, CNS | Associated with impaired cognitive function, reduced executive functioning, and neurodevelopmental disorders. Disrupts neuronal differentiation and synaptogenesis. Linked to hypertension. |
[7,11,15,16,17,18] |
| OCPs (Pesticides) | Adipose tissue, CNS | Readily integrate into cell membranes and neural lipid structures. Linked to neurodevelopmental impairments and cognitive deficits. |
[7,11,15,16] |
| PAHs | Vascular tissue, Adipose tissue | Mechanistically linked to atherogenesis, oxidized LDL formation, and plaque instability. Contributes to cardiovascular events and vascular inflammation. |
[2,19] |
| Phthalates & Bisphenols | Endocrine organs, CNS | Act as endocrine disruptors altering estrogen receptor-dependent metabolic pathways. Associated with ADHD, cognitive impairment, and behavioral dysfunction. |
[7,10,11,15,16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



