Submitted:
14 February 2026
Posted:
26 February 2026
You are already at the latest version
Abstract
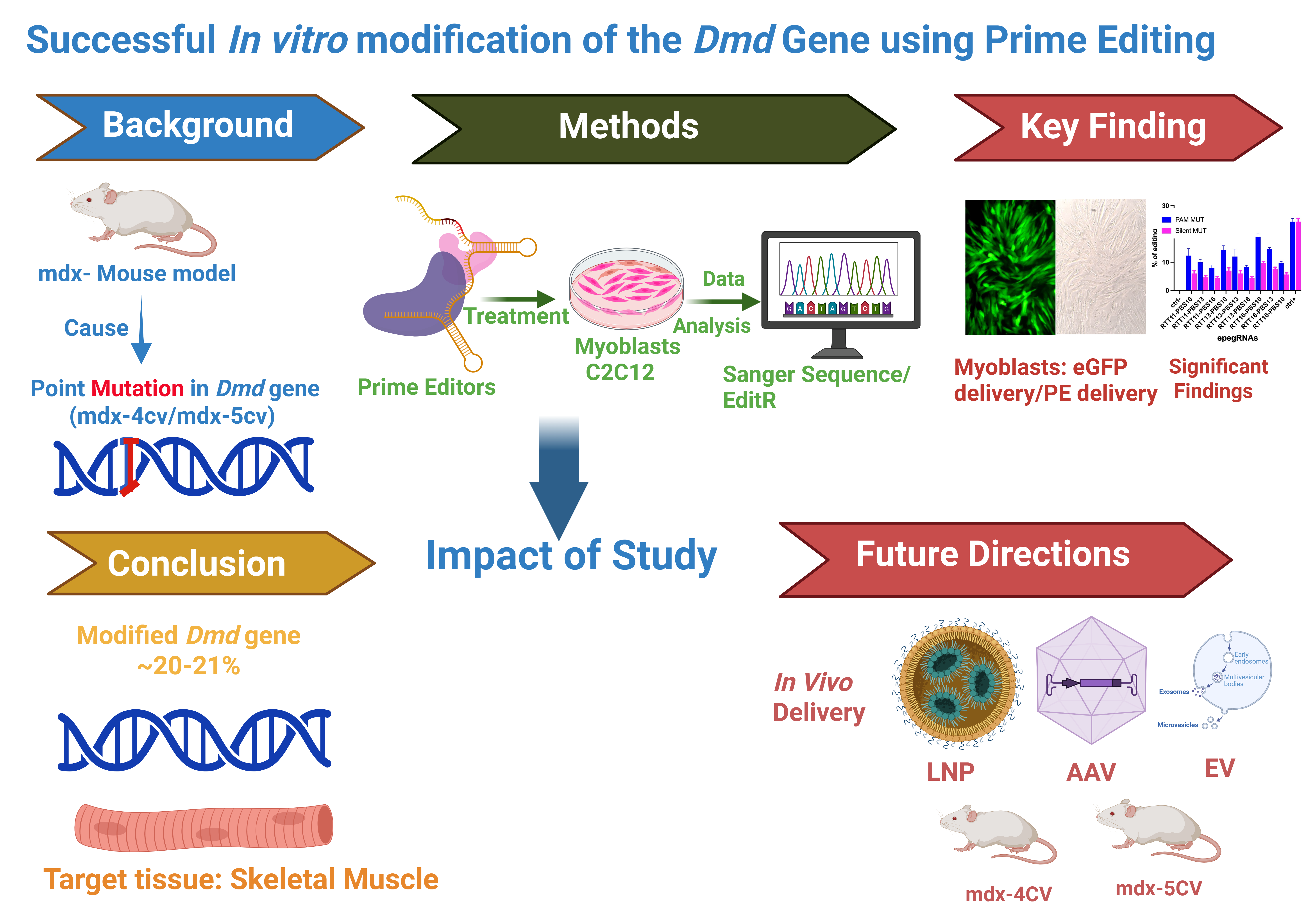
Duchenne muscular dystrophy (DMD) is a fatal X-linked neuromuscular disorder caused by mutations in the dystrophin gene. Prime editing is a versatile genome-editing technology capable of precise base correction without inducing double-strand DNA breaks, making it well suited for correction of point mutations in the DMD gene. Here, we applied prime editing to correct the mdx-4cv and mdx-5cv mutations in mouse myoblasts in vitro. Initial editing efficiencies were unexpectedly low and were traced to the presence of a 5′-TTCT-3′ motif within engineered prime editing guide RNAs (epegRNAs), consistent with premature RNA polymerase III–mediated transcription termination from the U6 promoter. Rational redesign of epegRNAs to eliminate this motif markedly improved editing efficiency, achieving up to 20% correction of the 4cv mutation and 21% editing at the 5cv locus using NGAG PAM. These results identify a critical epegRNA design constraint and establish an efficient prime editing framework for precise correction of DMD point mutations.
Keywords:
prime editing
; Duchenne muscular dystrophy
; Dmd gene
; myogenic cells
; CRISPR/Cas9
; RTT–PBS optimization
; point mutation
; genome editing
1. Introduction
Gene therapy has emerged as a transformative approach for the treatment of inherited genetic disorders by directly addressing pathogenic DNA variants rather than alleviating downstream symptoms [1,2]. Precise genome editing strategies offer the possibility of durable and potentially curative interventions through permanent modification of disease-causing mutations [3,4,5]. However, the clinical translation of genome editing technologies depends critically on editing precision, efficiency, and minimization of unintended genomic damage.
Early gene editing platforms, including zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), enabled locus-specific DNA cleavage but were limited by complex design requirements and the induction of double-strand DNA breaks (DSBs) [6,7]. The introduction of the CRISPR/Cas9 system in 2012 represented a major breakthrough, providing a simple and programmable platform for genome editing [8]. Nevertheless, conventional CRISPR/Cas9 editing relies on DSBs, which can trigger error-prone repair pathways and generate insertions, deletions, or chromosomal rearrangements.
To overcome these limitations, next-generation genome editing technologies were developed, including base editors capable of targeted nucleotide transitions without DSBs [9,10,11]. While base editing has enabled correction of many pathogenic variants, its applicability is restricted to specific base substitutions and constrained by editing windows and bystander edits.
Prime editing, first reported in 2019, represents a further advance by enabling targeted insertions, deletions, and all twelve possible base-to-base conversions without introducing DSBs or requiring donor DNA templates [12,13] (Figure 1).
Following nicking of the target strand, the primer binding site (PBS) anneals to the exposed DNA strand, allowing reverse transcription of the reverse transcription template (RTT) and installation of the edit through endogenous DNA repair pathways (Figure 1). Subsequent refinements, including the PE3 strategy and engineered pegRNAs (epegRNAs), have further improved editing efficiency by stabilizing guide RNAs and biasing DNA repair outcomes [12,14].
Duchenne muscular dystrophy (DMD) is a severe X-linked recessive neuromuscular disorder caused by mutations in the dystrophin gene (DMD) [15]. Loss of dystrophin compromises sarcolemmal stability, leading to progressive muscle degeneration, inflammation, fibrosis, and eventual cardiopulmonary failure [15,16,17] (Figure 2). Despite advances in supportive care and the approval of exon-skipping therapies and micro-dystrophin gene replacement strategies, DMD remains incurable, and existing treatments are mutation-specific or provide only partial functional benefit [17,18,19,20,21,22,23,24,25].
Importantly, approximately 30% of DMD cases arise from single-nucleotide variants, making them attractive candidates for precise genome correction strategies[17]. Prime editing is particularly well suited for correction of such mutations, as it enables exact restoration of the wild-type sequence without introducing frameshifts or large genomic alterations.
In this study, we investigated prime editing–mediated correction of two well-characterized DMD point mutations, mdx-4cv and mdx-5cv [26,27,28], using mouse myoblasts as an in vitro model. Since these models harbor single-nucleotide point mutations, we employed prime editing as a precise genome-editing strategy [29,30]. During the establishment of editing strategies for these loci, we identified a previously underappreciated constraint in engineered prime editing guide RNA (epegRNA) design involving a short transcription termination motif that markedly limited editing efficiency. Through rational redesign of epegRNAs, we achieved efficient correction of both mutations in vitro, highlighting key design considerations relevant to the future development of prime editing–based therapeutic strategies.
2. Materials and Methods
2.1. Cell Lines
The mouse myoblast cell line C2C12 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). This cell line was originally derived from the skeletal muscle of a C3H mouse following a crush injury and is widely used as an in vitro model to study myogenesis, muscle regeneration, and neuromuscular disease mechanisms. C2C12 cells retain a stable diploid genome and are amenable to genetic manipulation, making them suitable for evaluating genome-editing strategies targeting muscle-associated genes such as Dmd. All experiments were performed using low-passage cells to minimize genetic drift and phenotypic variability.
2.2. Plasmids
Prime editing plasmids pCMV-PE2 (Addgene #132775), pCMV-PE6a (Addgene #207851), pCMV-PE2-VQR (Addgene #159982), and pCMV-PE2-SpG (Addgene #159978) were obtained from Addgene,Watertown, MA, USA (https://www.addgene.org/). These plasmids encode a fusion protein consisting of an engineered SpCas9 H840A nickase and an M-MLV reverse transcriptase, enabling precise genome editing without generating double-strand DNA breaks.
The pU6-tevopreq1-GG-acceptor plasmid (Addgene #174038), which allows U6 promoter–driven expression of enhanced prime editing guide RNAs (epegRNAs), was also obtained from Addgene. In our laboratory, this plasmid was further modified to include an additional U6 promoter and a cloning site for expression of a nicking single-guide RNA (nsgRNA) required for the PE3 strategy. This modified construct was designated pU6-epegRNA-nsgRNA.
Design of epegRNAs and nsgRNAs was performed according to the guidelines described by Anzalone et al. [12], with optimization of primer binding site (PBS) and reverse transcription template (RTT) lengths to maximize editing efficiency. Particular attention was given to avoiding premature transcriptional termination motifs within the epegRNA scaffold. All oligonucleotides used for cloning were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Cloning was performed using standard restriction enzyme digestion and ligation or Golden Gate assembly, followed by sequence verification.
2.3. Cell Culture Conditions
C2C12 myoblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Wisent Inc., Saint-Jean-Baptiste, QC, Canada) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (both from Wisent Inc.). Cells were maintained at 37 °C in a humidified incubator with 5% CO₂. Culture medium was replaced every 2–3 days, and cells were passaged at approximately 70–80% confluence using standard trypsinization procedures. All experiments were performed under sterile conditions.
2.4. Plasmid Electroporation in Mouse C2C12 Myoblasts
Plasmid delivery into C2C12 myoblasts was performed using the Neon™ Transfection System (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, 100,000 cells were resuspended in Neon resuspension buffer and electroporated with a total of 2 μg of plasmid DNA per reaction, consisting of 1 μg of a prime editor plasmid (PE2, PE6a, PE-VQR, or PE-SpG) and 1 μg of the corresponding epegRNA-nsgRNA plasmid (for PE3 experiments).
Electroporation was carried out using 10 μL Neon tips with the following parameters: 1300 V, 20 ms pulse width, and 2 pulses. Immediately after electroporation, the cells were transferred into one well of a 24-well plate containing 500 μL of pre-warmed culture medium. After 24 hours, the medium was replaced with 1 mL of fresh DMEM to remove cell debris and residual electroporation buffer. Cells were incubated for an additional 48 hours to allow sufficient time for prime editing to occur.
As a negative control, cells were electroporated with a plasmid expressing enhanced green fluorescent protein (eGFP) alone. Transfection efficiency was estimated by fluorescence microscopy and consistently exceeded 80%.
2.5. Genomic DNA Extraction and PCR Amplification
Genomic DNA was extracted directly from cultured C2C12 cells using the DirectPCR™ Lysis Reagent (Viagen Biotech Inc., Los Angeles, CA, USA). Cells were washed once with 500 μL phosphate-buffered saline (PBS), followed by addition of 100 μL lysis reagent supplemented with 1 μL proteinase K (20 mg/mL). Samples were incubated at 56 °C for 2 hours to ensure complete lysis and protein digestion, followed by enzyme inactivation at 85 °C for 45 minutes.
Lysates were centrifuged at 13,000 rpm for 5 minutes, and 1–2 μL of the supernatant was used as template DNA for PCR amplification of the target genomic regions. PCR was performed using Phusion™ High-Fidelity DNA Polymerase (Thermo Fisher Scientific) to minimize amplification errors. Cycling conditions consisted of an initial denaturation at 98 °C for 30 seconds, followed by 35 cycles of 98 °C for 10 seconds, 62 °C for 20 seconds, and 72 °C for 30 seconds, with a final extension step at 72 °C for 30 seconds. Primer sequences were designed to flank the edited region and generate amplicons suitable for Sanger sequencing (Figure 3).
2.6. Sanger Sequencing and Editing Analysis
PCR products were submitted for Sanger sequencing to the CHU de Québec Research Center sequencing platform (Université Laval, Québec, Canada) between March 2024 and February, 2025. Sequencing reactions were performed using BigDye™ Terminator v3.1 chemistry (Thermo Fisher Scientific) with internal primers to ensure high-quality base calling across the edited site (Figure 3).
Sequencing chromatograms were analyzed to quantify prime editing efficiencies using the EditR online tool (https://moriaritylab.shinyapps.io/editr_v10/ ) accessed from March, 2024 to February, 2025, which enables robust estimation of nucleotide substitution frequencies from mixed Sanger traces. Editing efficiency was calculated as the percentage of modified alleles relative to the total signal at the target nucleotide position.
2.7. Statistical Analysis
All quantitative data were analyzed using GraphPad Prism version 10.4.1 (GraphPad Software Inc., La Jolla, CA, USA). Comparisons between mean editing efficiencies across experimental groups were performed using the nonparametric Mann–Whitney U test, as data did not assume a normal distribution. Results are presented as mean ± standard error of the mean (SEM). A p-value < 0.05 was considered statistically significant, corresponding to a 5% confidence interval.
3. Results
3.1. Installation of the 4cv Nonsense Mutation in Exon 53 of the Dmd Gene in C2C12 Myoblasts Using Prime Editing
To validate the feasibility of using prime editing to precisely modify single nucleotides within the Dmd gene, we first sought to introduce the 4cv nonsense mutation into exon 53 of the Dmd gene in vitro. This approach served as a proof-of-concept to establish whether prime editing could efficiently generate a disease-relevant point mutation in myogenic cells prior to attempting mutation correction.
A target site within exon 53 harboring a proximal NGG PAM (5′-AGG-3′) was selected, enabling the use of the SpCas9 nickase (H840A) fused to an M-MLV reverse transcriptase. The prime editing strategy was designed to convert the wild-type GAA codon encoding glutamine into a TAA stop codon, thereby recreating the 4cv mutation observed in the mdx-4cv mouse model (Figure 4A).
To optimize editing efficiency, nine distinct engineered prime editing guide RNAs (epegRNAs) incorporating the tevopreQ1 motif at the 3′ end were designed to protect the PBS and RTT sequences from exonuclease-mediated degradation [14] were designed, differing in the lengths of their primer binding sites (PBS) and reverse transcription templates (RTT) (Figure 4B). In addition to introducing the nonsense mutation, a silent PAM-disrupting mutation was incorporated to prevent repeated Cas9 recognition following successful editing and to potentially enhance product stability.
Prime editing was performed by electroporating C2C12 mouse myoblasts, which carry a wild-type Dmd locus, with either pCMV-PE2 or pCMV-PE6a together with the corresponding pU6-pegRNA-GG-acceptor vector expressing individual epegRNAs. Negative control cells were electroporated with a plasmid encoding eGFP alone to monitor transfection efficiency and background editing.
Genomic DNA was extracted 72 hours post-electroporation, and editing outcomes were assessed by Sanger sequencing, followed by quantitative analysis using the EditR software [31]. Across all nine tested epegRNAs, only low-level editing efficiencies were observed, with a maximum of approximately 2% conversion of the target guanine to thymine and ~1% incorporation of the silent PAM mutation (Figure 4C).
Despite the favorable proximity of the nicking site to the intended edit (+10 nucleotides) and the inclusion of a PAM-disrupting mutation, editing efficiency remained unexpectedly low. To understand this limitation, we examined the epegRNA sequences in detail and identified the presence of a 5′-TTCT-3′ stretch within the RTT region of all nine constructs (Figure 4B). Previous studies have shown that such sequences can cause premature transcriptional termination of U6-driven guide RNAs, thereby severely reducing functional pegRNA expression and prime editing efficiency [32,33]. In the subsequent experiments, we did 9 epegRNA with optimatizations of the RTT sequence to remove this 5′-TTCT-3′ stretch.
3.2. Optimization of epegRNAs by Removal of the TTCT Stretch Enables Efficient Correction of the 4cv Mutation In Vitro
To overcome the transcriptional limitation identified in the initial designs, we next optimized epegRNAs to remove the 5′-TTCT-3′ stretch while maintaining precise correction ~20% of the 4cv nonsense mutation. The optimized strategy aimed to convert the TAA stop codon back to the wild-type GAA glutamine codon in exon 53 of the Dmd gene.
Nine newly designed epegRNAs were generated, all targeting the same NGG PAM (5′-AGG-3′). To eliminate the 5′-TTCT-3′ motif and simultaneously improve experimental readout, we introduced two silent mutations within the PAM, converting AGG to CGC, both of which encode arginine (Figure 5A,B). Importantly, this modification served two purposes:
a) Removal of the 5′-TTCT-3′ sequence to restore proper epegRNA transcription.
b) Introduction of a traceable silent mutation, allowing editing efficiency to be quantified even in wild-type C2C12 cells lacking the pathogenic stop codon.
Prime editing experiments were conducted in wild-type C2C12 myoblasts using PE3 and PE6a prime editing techniques, and editing efficiency was evaluated by quantifying incorporation of the silent PAM mutation. An epegRNA targeting the Atp2a2 gene was included as a positive control to confirm overall prime editing activity.
Among the nine optimized epegRNAs, epegRNA-16-10, containing a 16 nucleotides RTT and a 10 nucleotides PBS, demonstrated the highest editing efficiency. Following a single electroporation, this construct achieved 20% editing with PE3 and 6% editing with PE6a (Figure 5C). In comparison, the positive control Atp2a2 epegRNA resulted in 23% editing with PE3 and 19% with PE6a, while no detectable background editing was observed in negative control samples.
These results demonstrate that removal of the 5′-TTCT-3′ motif markedly improves prime editing efficiency, validating the optimized epegRNAs design and confirming the feasibility of correcting the 4cv mutation in vitro.
3.3. In Vitro Prime Editing of the 5cv Mutation Using an NGAG PAM
We next extended our prime editing strategy to the 5cv mutation in the Dmd gene. This mutation involves a single-nucleotide substitution in exon 10, where the wild-type GGA glycine codon is converted to GGT, creating an aberrant splice donor site that leads to dystrophin dysfunction.
To target this locus, we designed nine epegRNAs with variable RTT and PBS lengths (Figure 6A,B). Because no suitable NGG PAM was present near the mutation, we selected the SpCas9-VQR nickase variant, which recognizes a 5′-NGAG-3′ PAM, enabling precise targeting of the 5cv locus [34].
Since C2C12 cells contain the wild-type GGA codon and do not harbor the disease mutation, a silent PAM mutation was incorporated into the design to allow detection of prime editing events independently of mutation correction. C2C12 myoblasts were electroporated with PE-VQR and individual epegRNAs, while negative controls received an eGFP reporter plasmid. Transfection efficiency was consistently high, exceeding 80%.
Editing outcomes were assessed by Sanger sequencing. Across the nine epegRNAs tested, editing efficiencies ranged from 2% to 6% at the target locus. In contrast, the Atp2a2 positive control achieved 29% editing, confirming robust prime editing activity under the same experimental conditions (Figure 6C). Background editing in negative controls remained negligible.
Sequence analysis of the epegRNAs revealed that, similar to the initial 4cv designs, all nine constructs contained a 5′-TTCT-3′ motif within the RTT (Figure 6B), likely accounting for the limited editing efficiency. This finding reinforced the importance of avoiding transcription-terminating sequences in epegRNA design and motivated further optimization [32]. In the subsequent experiments, we did 9 epegRNA optimatizations of the RTT and PBS to remove this 5′-TTCT-3′ stretch.
3.4. Removal of the TTCT Stretch Enhances Prime Editing Efficiency for Correction of the 5cv Mutation In Vitro
To improve prime editing efficiency at the 5cv mutation site, epegRNAs were redesigned to disrupt a 5′-TTCT-3′ sequence located within the reverse transcription template (RTT). This was achieved by introducing a nucleotide substitution with a conservative amino acid change, converting glutamic acid to glutamine (E→Q). This substitution preserves the physicochemical properties of the protein while effectively eliminating the 5′-TTCT-3′ sequence from the RTT (Figure 7A,B).
The optimized epegRNAs were designed to target either 5′-NGAG-3′ PAMs using the VQR variant [34] or 5′-NGN-3′ PAMs using the SpG variant [35]. All constructs were cloned into the pU6-epegRNA-GG-acceptor plasmid and delivered into C2C12 cells by electroporation together with plasmids coding either for pCMV-PE2-VQR or pCMV-PE2-SpG [35]. Negative controls expressing eGFP confirmed transfection efficiencies of at least 80%.
Following optimization, prime editing efficiency increased substantially. Editing at the 5cv target site reached 21% with PE-VQR and 9% with PE-spG (Figure 7C). In parallel, the Atp2a2 positive control again achieved 29% editing, confirming consistent prime editing performance across experiments.
Collectively, these results demonstrate that strategic elimination of transcription-terminating motifs within epegRNAs is essential for achieving efficient prime editing and establish optimized reagents for future in vivo correction of the mdx-4cv and mdx-5cv mutations.
4. Discussion
In this study, we establish efficient prime editing–mediated correction of two point mutations and define key guide RNA design parameters that govern editing efficiency in myogenic cells. By systematically optimizing engineered prime editing guide RNAs (epegRNAs) for the mdx-4cv and mdx-5cv loci, we achieved editing efficiencies of up to 20–21%, levels that are within a biologically meaningful range for dystrophin restoration in muscle tissue.
Prime Editing Efficiency Is Highly Sensitive to RTT–PBS Configuration
Our data demonstrate that prime editing outcomes at the Dmd locus are strongly influenced by the combined length and composition of the reverse transcription template (RTT) and primer binding site (PBS). In the case of the mdx-4cv mutation, nine initial epegRNA designs spanning multiple RTT and PBS lengths resulted in uniformly low editing efficiencies (≤2%), despite optimal PAM proximity and high transfection efficiency (Figure 4C). These results underscore that favorable genomic targeting alone is insufficient to ensure productive prime editing.
Following redesign to eliminate the 5′-TTCT-3′ motif, editing efficiencies increased markedly and revealed clear differences among RTT–PBS configurations. Notably, the epegRNA-16-10 construct, containing a 16-nucleotide RTT and a 10-nucleotide PBS, consistently outperformed other designs, achieving ~20% editing with PE3 and ~6% with PE6a (Figure 5C). This observation is consistent with prior reports indicating that intermediate-length PBS and RTT combinations often provide optimal primer annealing and reverse transcription efficiency without compromising flap resolution or DNA repair outcomes.
Importantly, the lower editing efficiency observed with PE6a compared to PE3 at the same locus suggests that nicking of the non-edited strand remains a significant contributor to productive editing at the mdx-4cv site. This is in agreement with previous studies demonstrating that PE3 frequently outperforms PE2- or PE6-based strategies at loci where mismatch repair bias favors reversion of the edited strand.
Identification of a Cryptic Transcription Termination Constraint in epegRNAs
A central finding of this work is the identification of a short 5′-TTCT-3′ motif within the RTT that severely limits prime editing efficiency when epegRNAs are expressed under a U6 promoter. While RNA polymerase III termination has classically been associated with poly-T tracts (≥4 thymidines), accumulating evidence indicates that shorter thymidine-rich motifs can also impair transcriptional processivity or guide RNA stability. Our data extend this concept by demonstrating that the 5′-TTCT-3′ motif, although shorter than canonical terminators, is sufficient to reduce functional epegRNA output in a biologically relevant context.
Crucially, removal of this motif alone — without altering the prime editing chemistry — was sufficient to restore editing efficiencies from ≤2% to ~20% at the mdx-4cv locus and from ≤6% to ~21% at the mdx-5cv locus. This effect was reproducible across multiple prime editor architectures, indicating that the observed improvement reflects enhanced epegRNA performance rather than editor-specific effects.
Prime Editing of the mdx-5cv Mutation Highlights the Importance of PAM Expansion and Guide Optimization
Correction of the mdx-5cv mutation posed additional challenges due to the absence of a nearby NGG PAM. By employing the SpCas9-VQR variant, which recognizes NGAG PAMs, we were able to access this clinically relevant locus and achieve initial editing efficiencies of 2–6% across nine epegRNA designs (Figure 6C). The relatively modest editing levels observed at this stage mirrored those seen in the initial mdx-4cv experiments and again correlated with the presence of the 5′-TTCT-3′ motif in all RTT designs.
Following redesign of the RTT to eliminate the 5′-TTCT-3′ motif through a conservative glutamic acid–to–glutamine substitution, editing efficiency increased substantially. Using PE-VQR, optimized epegRNAs achieved ~21% editing, whereas PE-spG resulted in ~9% editing at the same locus (Figure 7C). These results demonstrate that PAM expansion alone is not sufficient to ensure efficient prime editing and that epegRNA sequence integrity remains a dominant determinant of editing success.
The difference in performance between PE-VQR and PE-SpG further highlights locus-specific constraints in prime editing, likely reflecting differences in PAM binding affinity, nick positioning, and downstream repair outcomes.
5. Implications for Therapeutic Prime Editing of DMD
From a therapeutic perspective, the editing efficiencies achieved in this study are notable. Previous work has demonstrated that partial restoration of dystrophin expression can confer substantial functional benefit in Duchenne muscular dystrophy (DMD), with dystrophin levels in the range of approximately 10–20% often sufficient to produce measurable improvements in muscle pathology and function in both animal models and patients [36,37,38]. Genome editing studies have further shown that incomplete correction leading to partial dystrophin recovery can significantly ameliorate disease phenotypes [39,40]. Although the present study is limited to in vitro correction in myoblasts, the optimized epegRNA designs and editor configurations established here provide a strong foundation for subsequent in vivo investigations.
More broadly, our findings highlight that epegRNA design must consider not only target sequence selection and RTT–PBS optimization, but also transcriptional constraints imposed by RNA polymerase III promoters. Pol III– driven guide RNAs are known to be sensitive to short thymidine-rich termination motifs, which can lead to premature transcriptional termination [41]. The identification of a 5′-TTCT-3′ motif as a functional design pitfall in this study therefore has practical implications and is likely relevant to a wide range of prime editing applications beyond DMD.
6. Conclusions
In summary, we demonstrate efficient prime editing–mediated edition of the mdx-4cv and mdx-5cv mutations and identify a critical, previously underappreciated guide RNA design constraint that governs editing efficiency. By systematically optimizing RTT–PBS configurations and eliminating a cryptic transcription termination motif, we achieved robust editing efficiencies of up to 20–21% across multiple prime editor variants. These findings provide important design principles for therapeutic prime editing and support the feasibility of precise genome correction for Duchenne muscular dystrophy.
Supplementary Materials
The supporting information can be downloaded at Preprints.org.
Author Contributions
Author Contributions: Conceptualization, A.S., F.E.H., J.R. and J.P.T. methodology, A.S., F.E.H., and J.R.; validation, A.S., F.E.H.,; formal analysis, A.S., F.E.H.,; investigation, A.S., F.E.H.,; writing—original draft preparation, A.S., F.E.H.,; writing—review and editing, A.S., F.E.H.,; visualization, A.S., F.E.H.,; supervision, J.R. and J.P.T.; funding acquisition, J.P.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Defeat Duchenne Foundation and the grant number is 53320215. A.S. is the recipient of a Vanier Canada Graduate Scholarship (Vanier CGS) funded by the Canadian Institutes of Health Research (CIHR; Application No. 492510).
Institutional Review Board Statement
Not applicable. This study did not involve human participants or live animals.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ginn, S.L., et al., Gene therapy clinical trials worldwide to 2017: An update. The journal of gene medicine, 2018. 20(5): p. e3015.
- Doudna, J.A. and E. Charpentier, The new frontier of genome engineering with CRISPR-Cas9. Science, 2014. 346(6213): p. 1258096. [CrossRef]
- Porteus, M.H., A new class of medicines through DNA editing. New England Journal of Medicine, 2019. 380(10): p. 947-959. [CrossRef]
- Villiger, L., et al., CRISPR technologies for genome, epigenome and transcriptome editing. Nature Reviews Molecular Cell Biology, 2024. 25(6): p. 464-487. [CrossRef]
- Pacesa, M., O. Pelea, and M. Jinek, Past, present, and future of CRISPR genome editing technologies. Cell, 2024. 187(5): p. 1076-1100. [CrossRef]
- Urnov, F.D., et al., Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics, 2010. 11(9): p. 636-646. [CrossRef]
- Sun, N. and H. Zhao, Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnology and bioengineering, 2013. 110(7): p. 1811-1821. [CrossRef]
- Sander, J.D. and J.K. Joung, CRISPR-Cas systems for editing, regulating and targeting genomes. Nature biotechnology, 2014. 32(4): p. 347-355. [CrossRef]
- Komor, A.C., et al., Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 2016. 533(7603): p. 420-424. [CrossRef]
- Gaudelli, N.M., et al., Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature, 2017. 551(7681): p. 464-471. [CrossRef]
- Kurt, I.C., et al., CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nature biotechnology, 2021. 39(1): p. 41-46. [CrossRef]
- Anzalone, A.V., et al., Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 2019. 576(7785): p. 149-157. [CrossRef]
- Anzalone, A.V., L.W. Koblan, and D.R. Liu, Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature biotechnology, 2020. 38(7): p. 824-844. [CrossRef]
- Nelson, J.W., et al., Engineered pegRNAs improve prime editing efficiency. Nature biotechnology, 2022. 40(3): p. 402-410. [CrossRef]
- EP, H., Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 1987. 51: p. 919-928.
- Ryder, S., et al., The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet journal of rare diseases, 2017. 12(1): p. 79. [CrossRef]
- Okubo, M., et al., Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. Journal of human genetics, 2016. 61(6): p. 483-489.
- Ricotti, V., et al., NorthStar Clinical Network. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry, 2013. 84(6): p. 698-705. [CrossRef]
- Kourakis, S., et al., Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: Can we do better? Orphanet Journal of Rare Diseases, 2021. 16(1): p. 117. [CrossRef]
- Gloss, D., et al., Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy [RETIRED] Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology, 2016. 86(5): p. 465-472.
- Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. The Lancet Neurology, 2018. 17(4): p. 347-361.
- Hoffman, E.P., et al., Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology, 2019. 93(13): p. e1312-e1323. [CrossRef]
- Wilton-Clark, H. and T. Yokota, Antisense and gene therapy options for Duchenne muscular dystrophy arising from mutations in the N-terminal hotspot. Genes, 2022. 13(2): p. 257. [CrossRef]
- Komaki, H., et al., Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Science Translational Medicine, 2018. 10(437): p. eaan0713.
- Aartsma-Rus, A., et al., Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human mutation, 2009. 30(3): p. 293-299. [CrossRef]
- Yucel, N., et al., Humanizing the mdx mouse model of DMD: the long and the short of it. NPJ Regenerative medicine, 2018. 3(1): p. 4.
- Im, W.B., et al., Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Human molecular genetics, 1996. 5(8): p. 1149-1153. [CrossRef]
- Beastrom, N., et al., mdx5cv mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx mice. The American journal of pathology, 2011. 179(5): p. 2464-2474.
- Murray, J.B., P.T. Harrison, and J. Scholefield, Prime editing: therapeutic advances and mechanistic insights. Gene Therapy, 2025. 32(2): p. 83-92. [CrossRef]
- Fu, Y., et al., Prime editing: current advances and therapeutic opportunities in human diseases. Science bulletin, 2023. 68(24): p. 3278-3291.
- Kluesner, M.G., et al., EditR: a method to quantify base editing from Sanger sequencing. The CRISPR journal, 2018. 1(3): p. 239-250. [CrossRef]
- Ui-Tei, K., S. Maruyama, and Y. Nakano, Enhancement of single guide RNA transcription for efficient CRISPR/Cas-based genomic engineering. Genome, 2017. 60(6): p. 537-545. [CrossRef]
- Doman, J.L., et al., Designing and executing prime editing experiments in mammalian cells. Nature protocols, 2022. 17(11): p. 2431-2468.
- Anders, C., K. Bargsten, and M. Jinek, Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Molecular cell, 2016. 61(6): p. 895-902.
- Walton, R.T., et al., Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science, 2020. 368(6488): p. 290-296.
- Arechavala-Gomeza, V., et al., Revertant fibres and dystrophin traces in Duchenne muscular dystrophy: implication for clinical trials. Neuromuscular Disorders, 2010. 20(5): p. 295-301. [CrossRef]
- Happi Mbakam, C., et al., Prime editing strategies to mediate exon skipping in DMD gene. Frontiers in Medicine, 2023. 10: p. 1128557.
- Wang, Q., et al., Selection-free precise gene repair using high-capacity adenovector delivery of advanced prime editing systems rescues dystrophin synthesis in DMD muscle cells. Nucleic Acids Research, 2024. 52(5): p. 2740-2757.
- Long, C., et al., Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science, 2016. 351(6271): p. 400-403.
- Nelson, C.E., et al., In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science, 2016. 351(6271): p. 403-407.
- Briner, A.E., et al., Guide RNA functional modules direct Cas9 activity and orthogonality. Molecular cell, 2014. 56(2): p. 333-339.
Figure 1.
Prime editing mechanism. A) The spacer sequence of the epegRNA binds to its complementary genomic sequence, directing the Prime Editor to the target site. Cas9 nickase (Cas9n) recognizes the PAM sequence and introduces a single strand cut 3 nucleotides upstream. B) The primer binding site (PBS) anneals to the complementary sequence on the nicked DNA strand. C) The reverse transcription template (RTT) serves as a template to synthesize the edited DNA strand. D) The resulting DNA flap is resolved, and any mismatches are repaired, completing the edit. Image created with Created in BioRender.com.
Figure 1.
Prime editing mechanism. A) The spacer sequence of the epegRNA binds to its complementary genomic sequence, directing the Prime Editor to the target site. Cas9 nickase (Cas9n) recognizes the PAM sequence and introduces a single strand cut 3 nucleotides upstream. B) The primer binding site (PBS) anneals to the complementary sequence on the nicked DNA strand. C) The reverse transcription template (RTT) serves as a template to synthesize the edited DNA strand. D) The resulting DNA flap is resolved, and any mismatches are repaired, completing the edit. Image created with Created in BioRender.com.
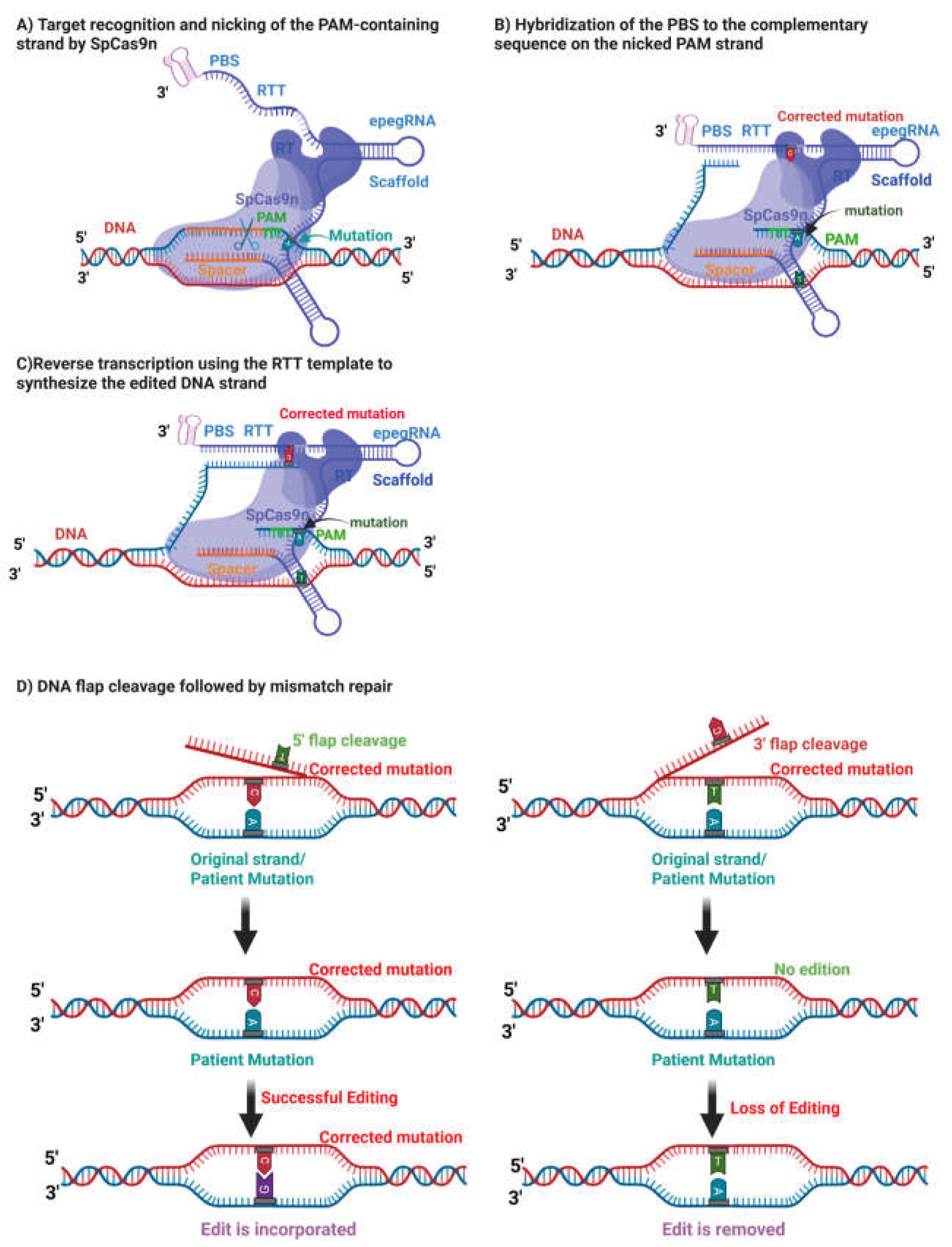
Figure 2.
Duchenne Muscular Dystrophy: cause and symptoms. Image created with BioRender.com.
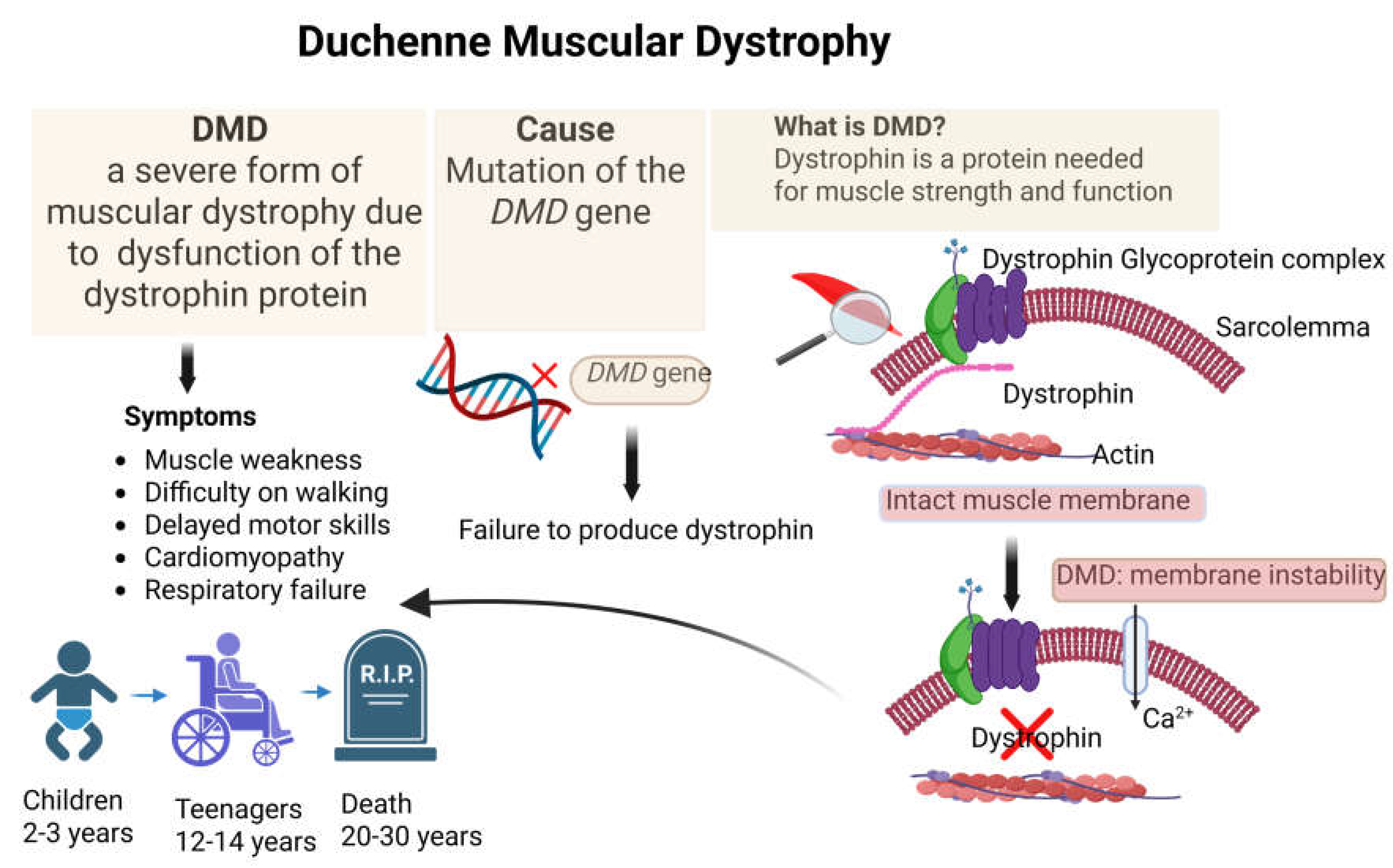
Figure 3.
Methodology. Plasmids encoding PE and epegRNA were first constructed by cloning. The plasmids were then transfected into C2C12 cells by Neon electroporation. 72 h after transfecting the cells, the gDNA of these cells was extracted. Part of the targeted gene was amplified by PCR, then sequenced by Sanger sequencing. The percentage of editing obtained was determined using EditR software. Image created with BioRender.com.
Figure 3.
Methodology. Plasmids encoding PE and epegRNA were first constructed by cloning. The plasmids were then transfected into C2C12 cells by Neon electroporation. 72 h after transfecting the cells, the gDNA of these cells was extracted. Part of the targeted gene was amplified by PCR, then sequenced by Sanger sequencing. The percentage of editing obtained was determined using EditR software. Image created with BioRender.com.
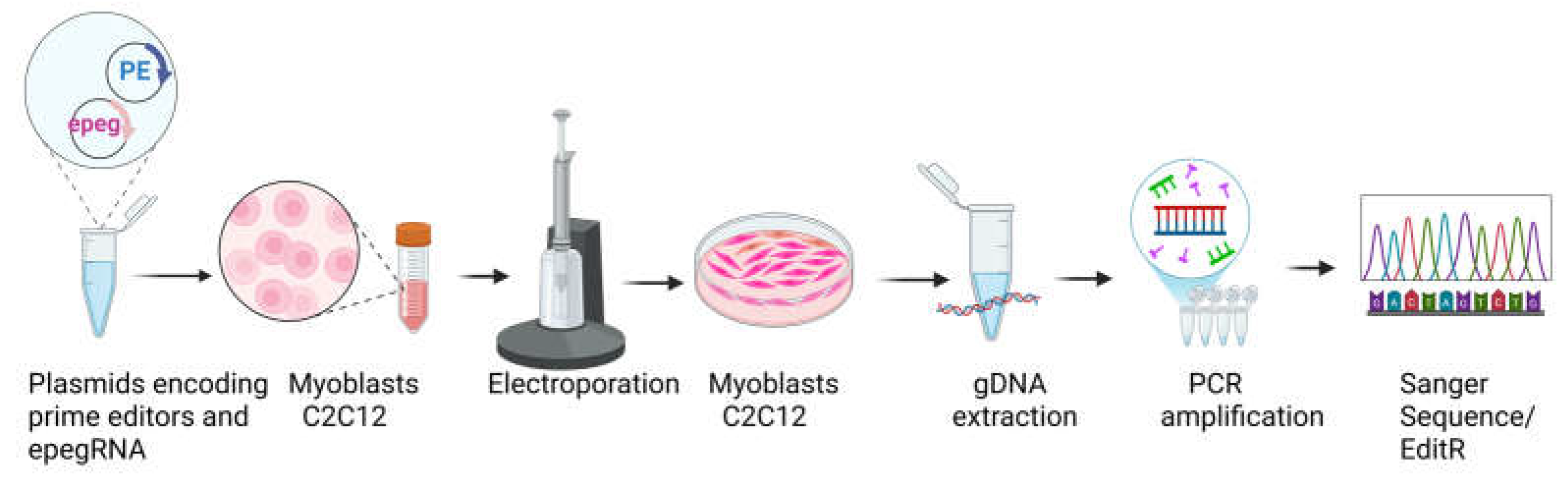
Figure 4.
Generation of the mdx-4cv mutation in Dmd exon 53 by prime editing and identification of epegRNA design constraints. (A) Schematic representation of the prime editing strategy used to introduce the mdx-4cv nonsense mutation (GAA → TAA) in exon 53 of the Dmd gene using SpCas9 nickase (H840A) recognizing a nearby NGG (5′-AGG-3′) PAM. The epegRNA design included a silent PAM-disrupting mutation to prevent re-editing. (B) Design of nine epegRNAs differing in reverse transcription template (RTT) and primer binding site (PBS) lengths. All constructs were found to contain an unintended TTCT sequence within the RTT. (C) Quantification of editing efficiency in C2C12 myoblasts three days post-electroporation, assessed by Sanger sequencing and EditR analysis. Limited editing (~2%) was detected at the target nucleotide, with minimal PAM modification, indicating suboptimal prime editing efficiency.
Figure 4.
Generation of the mdx-4cv mutation in Dmd exon 53 by prime editing and identification of epegRNA design constraints. (A) Schematic representation of the prime editing strategy used to introduce the mdx-4cv nonsense mutation (GAA → TAA) in exon 53 of the Dmd gene using SpCas9 nickase (H840A) recognizing a nearby NGG (5′-AGG-3′) PAM. The epegRNA design included a silent PAM-disrupting mutation to prevent re-editing. (B) Design of nine epegRNAs differing in reverse transcription template (RTT) and primer binding site (PBS) lengths. All constructs were found to contain an unintended TTCT sequence within the RTT. (C) Quantification of editing efficiency in C2C12 myoblasts three days post-electroporation, assessed by Sanger sequencing and EditR analysis. Limited editing (~2%) was detected at the target nucleotide, with minimal PAM modification, indicating suboptimal prime editing efficiency.
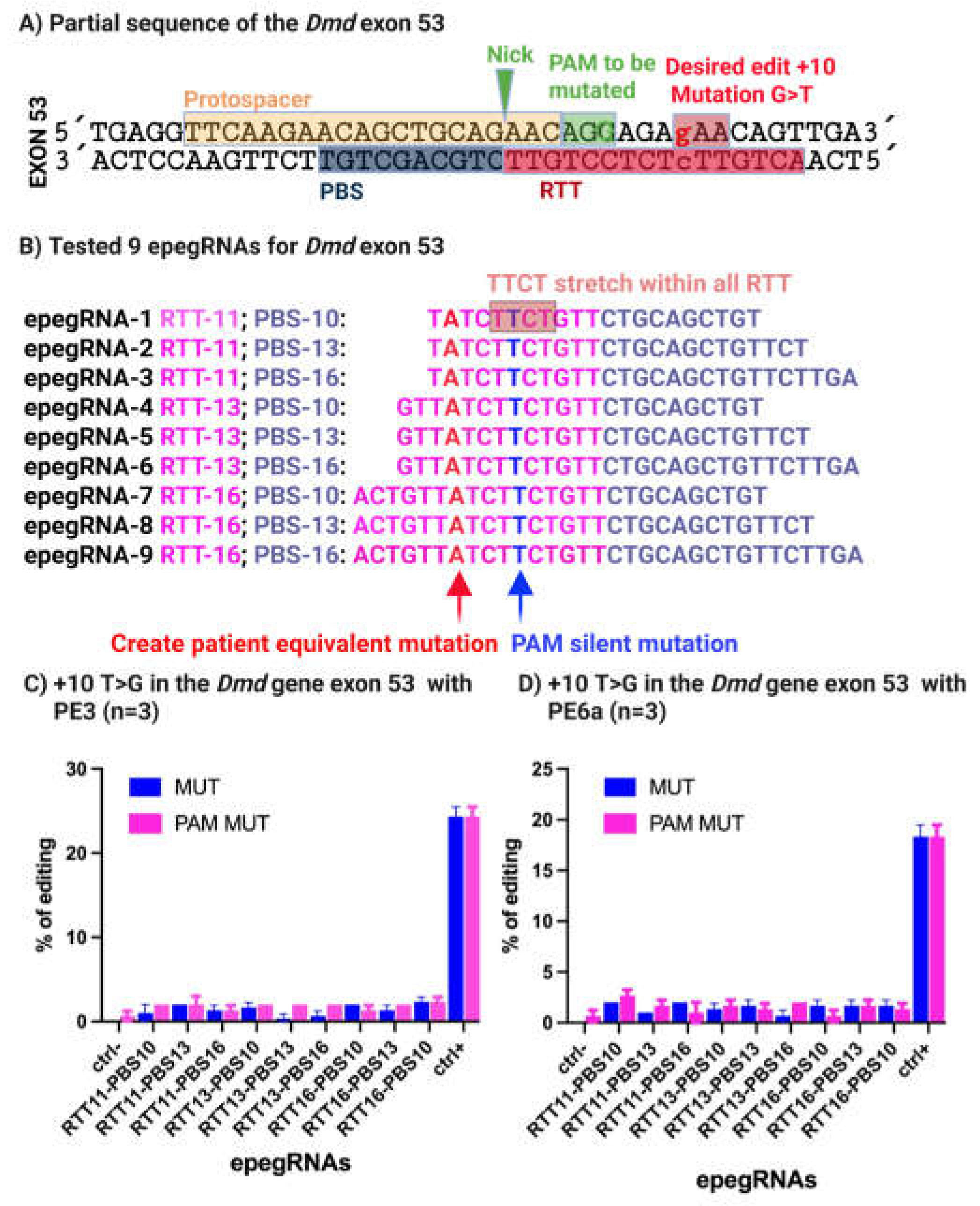
Figure 5.
Optimization of epegRNA design enables efficient correction of the mdx-4cv mutation in Dmd exon 53. (A) Strategy for correcting the mdx-4cv mutation (TAA → GAA) using redesigned epegRNAs targeting the same NGG PAM site. Two synonymous PAM mutations (AGG → CGC) encoding arginine were introduced to remove the TTCT stretch and enable detection of editing in wild-type cells. (B) Updated epegRNA architectures showing optimized RTT and PBS configurations lacking the TTCT motif. (C, D) Editing efficiencies obtained in wild-type C2C12 myoblasts using PE3 and PE6a. The epegRNA-16-10 construct achieved the highest correction efficiency (20% with PE3 and 6% with PE6a). An Atp2a2-targeting epegRNA served as a positive control, while no background editing was observed in negative controls.
Figure 5.
Optimization of epegRNA design enables efficient correction of the mdx-4cv mutation in Dmd exon 53. (A) Strategy for correcting the mdx-4cv mutation (TAA → GAA) using redesigned epegRNAs targeting the same NGG PAM site. Two synonymous PAM mutations (AGG → CGC) encoding arginine were introduced to remove the TTCT stretch and enable detection of editing in wild-type cells. (B) Updated epegRNA architectures showing optimized RTT and PBS configurations lacking the TTCT motif. (C, D) Editing efficiencies obtained in wild-type C2C12 myoblasts using PE3 and PE6a. The epegRNA-16-10 construct achieved the highest correction efficiency (20% with PE3 and 6% with PE6a). An Atp2a2-targeting epegRNA served as a positive control, while no background editing was observed in negative controls.
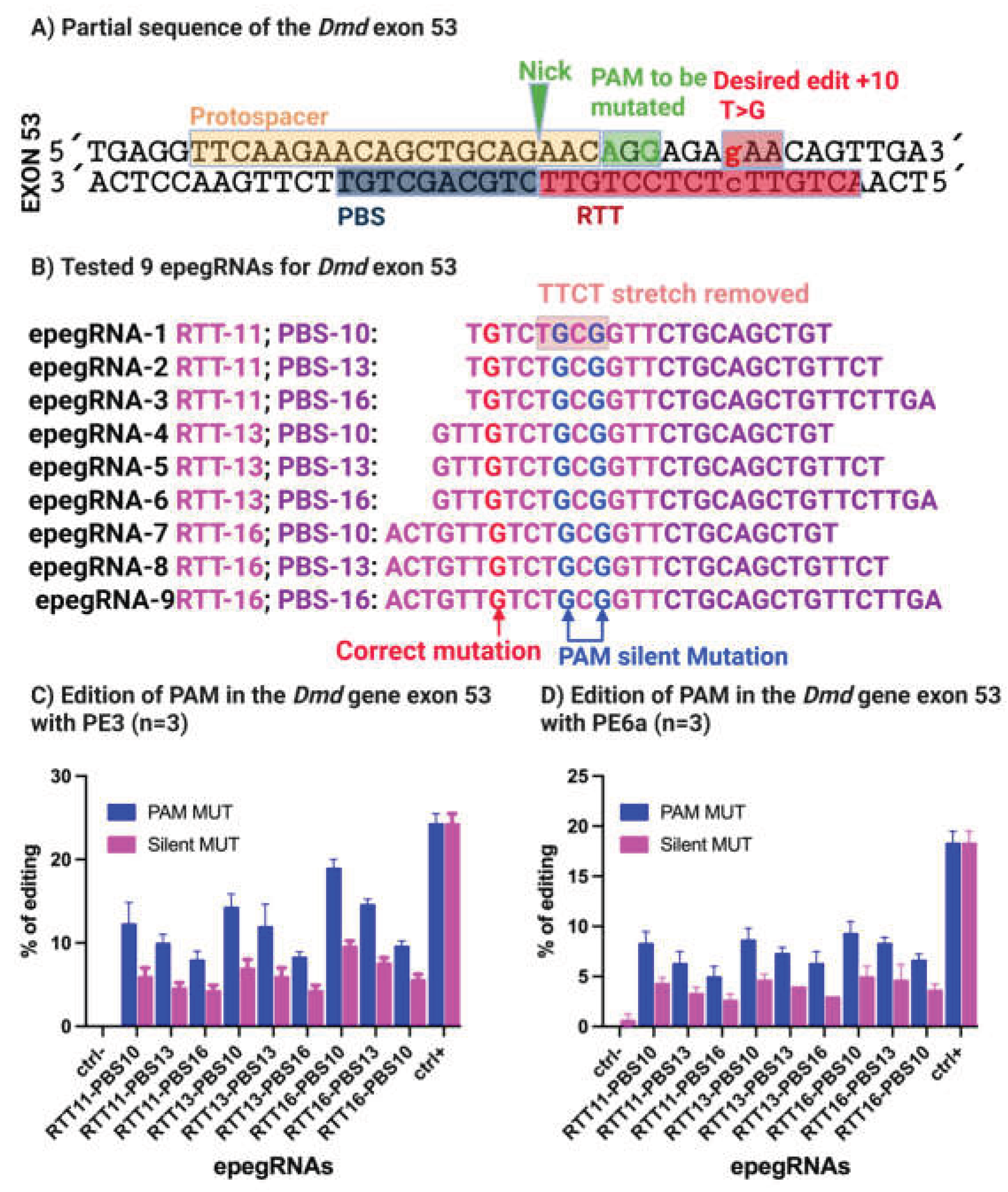
Figure 6.
Prime editing–mediated targeting of the mdx-5cv mutation using an NGAG PAM and SpCas9-VQR. (A) Illustration of the mdx-5cv mutation in exon 10, where a synonymous GGA → GGT substitution creates a cryptic splice donor site. (B) Design of nine epegRNAs targeting the mutation using SpCas9-VQR recognizing a 5′-NGAG-3′ PAM. RTT and PBS lengths were systematically varied, and a silent PAM mutation was included to allow detection of editing in wild-type cells. All constructs contained a TTCT stretch. (C) Editing efficiencies measured in C2C12 myoblasts following electroporation. Targeted editing ranged from 2–6%, whereas the Atp2a2 positive control reached 29% efficiency. Transfection efficiency exceeded 80%, as confirmed by eGFP expression.
Figure 6.
Prime editing–mediated targeting of the mdx-5cv mutation using an NGAG PAM and SpCas9-VQR. (A) Illustration of the mdx-5cv mutation in exon 10, where a synonymous GGA → GGT substitution creates a cryptic splice donor site. (B) Design of nine epegRNAs targeting the mutation using SpCas9-VQR recognizing a 5′-NGAG-3′ PAM. RTT and PBS lengths were systematically varied, and a silent PAM mutation was included to allow detection of editing in wild-type cells. All constructs contained a TTCT stretch. (C) Editing efficiencies measured in C2C12 myoblasts following electroporation. Targeted editing ranged from 2–6%, whereas the Atp2a2 positive control reached 29% efficiency. Transfection efficiency exceeded 80%, as confirmed by eGFP expression.
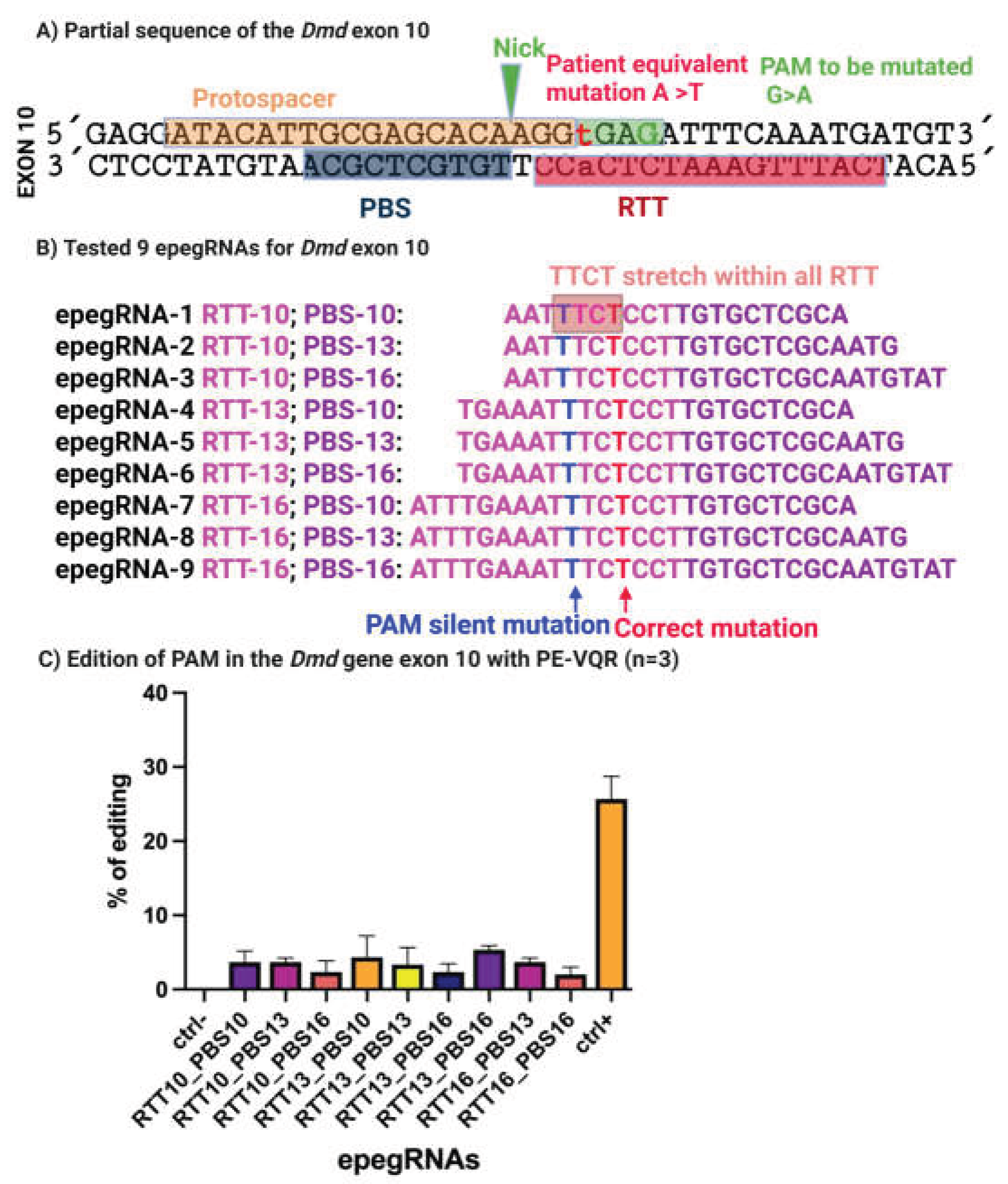
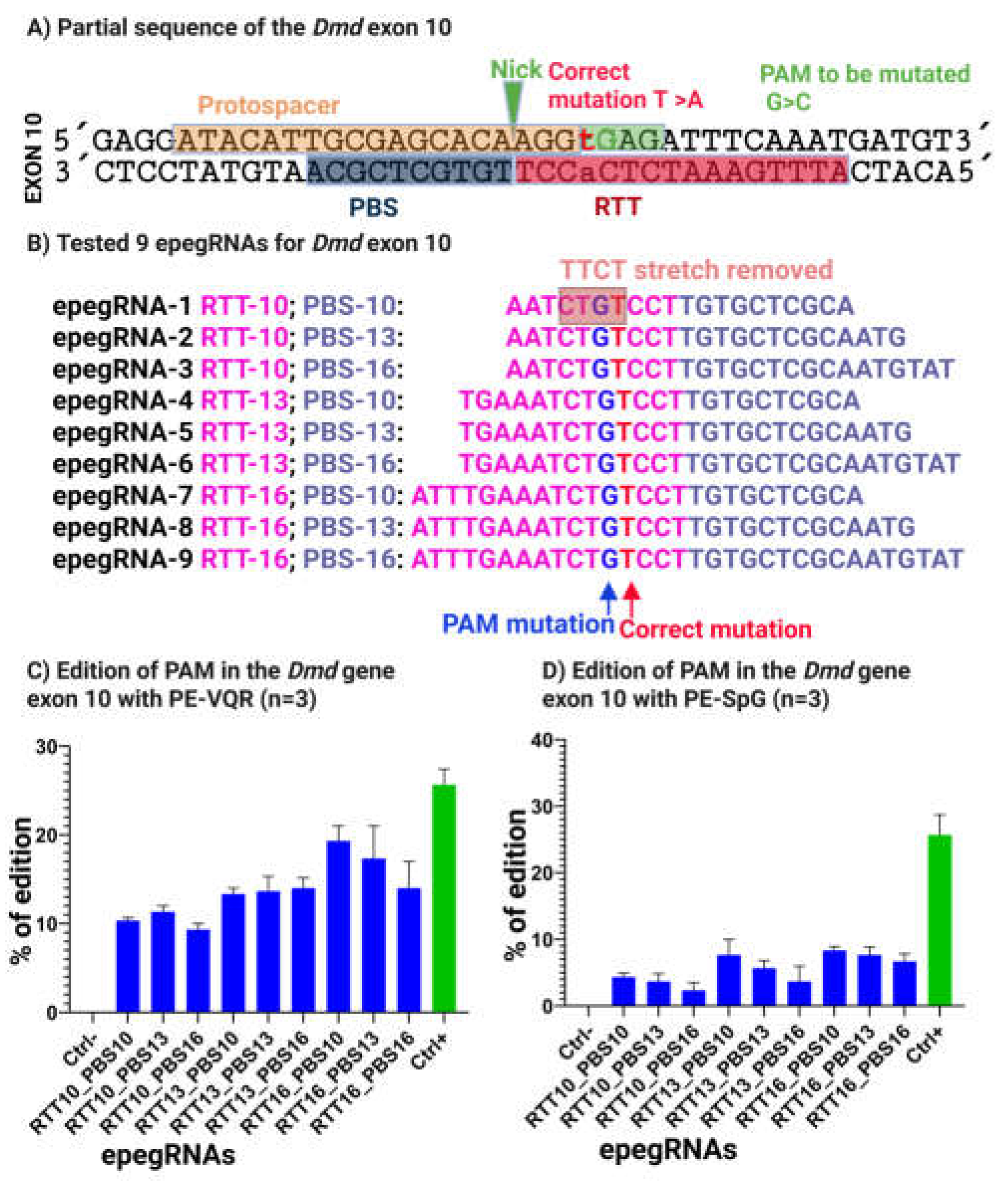
Figure 7.
Removal of the TTCT stretch markedly improves prime editing efficiency for correction of the mdx-5cv mutation. (A) Schematic of the optimization strategy in which a PAM-disrupting nucleotide substitution was introduced to eliminate the TTCT motif, resulting in a conservative amino-acid change (glutamic acid to glutamine) while preserving protein function and preventing Cas9 re-binding to the edited allele. (B) Final optimized epegRNA designs targeting NGAG protospacer adjacent motifs (PAMs) recognized by SpCas9-VQR or NGN PAMs recognized by SpCas9-spG. (C, D) Quantitative analysis of prime-editing outcomes in C2C12 myoblasts. Optimized epegRNAs achieved editing efficiencies of up to 21% using PE-VQR and 9% using PE-spG. Editing at the Atp2a2 locus served as a positive control and reached 29% efficiency, confirming robust prime-editing activity.
Figure 7.
Removal of the TTCT stretch markedly improves prime editing efficiency for correction of the mdx-5cv mutation. (A) Schematic of the optimization strategy in which a PAM-disrupting nucleotide substitution was introduced to eliminate the TTCT motif, resulting in a conservative amino-acid change (glutamic acid to glutamine) while preserving protein function and preventing Cas9 re-binding to the edited allele. (B) Final optimized epegRNA designs targeting NGAG protospacer adjacent motifs (PAMs) recognized by SpCas9-VQR or NGN PAMs recognized by SpCas9-spG. (C, D) Quantitative analysis of prime-editing outcomes in C2C12 myoblasts. Optimized epegRNAs achieved editing efficiencies of up to 21% using PE-VQR and 9% using PE-spG. Editing at the Atp2a2 locus served as a positive control and reached 29% efficiency, confirming robust prime-editing activity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



