
Submitted:
12 February 2026
Posted:
13 February 2026
You are already at the latest version
Abstract
Background
Cytokines are central regulators of inflammation and immune responses within the tumor microenvironment and have been implicated in cancer progression and prognosis. However, the prognostic value of coordinated cytokine-related transcriptional programs across cancer types has not been systematically explored.
Methods
Pan-cancer transcriptomic and clinical data were analyzed to construct a cytokine-related prognostic signature using LASSO Cox regression. Patients were stratified into high-risk and low-risk groups based on the derived risk score. Prognostic performance was evaluated in training and test cohorts, and biological relevance was assessed through survival analyses and pathway-level investigations.
Results
A 16-gene cytokine-related signature was established that consistently stratified patients into distinct prognostic groups across multiple cancer types. High cytokine-related risk scores were significantly associated with unfavorable survival outcomes and were linked to enhanced cell cycle activity, epithelial-mesenchymal transition, and extracellular matrix remodeling. Integration of the risk score with clinical variables improved individualized survival prediction. Immunohistochemical analyses further confirmed increased protein expression of representative risk-associated genes, including PANX1 and FRMD8, in multiple tumor tissues compared with corresponding normal tissues.
Conclusions
The cytokine-related prognostic signature captures key inflammatory and immune-related programs underlying tumor aggressiveness and provides a robust tool for pan-cancer risk stratification with potential clinical utility.
Keywords:
cytokines
; pan-cancer
; prognostic signature
; tumor microenvironment
; survival analysis
; inflammation
1. Introduction
Cytokines are central mediators of intercellular communication within the tumor microenvironment and play critical roles in regulating inflammation, immune responses, tissue remodeling, and cancer progression [1,2,3]. Dysregulated cytokine signaling has been widely implicated in tumor initiation, malignant transformation, immune evasion, and therapeutic resistance across diverse cancer types [1,4,5,6]. Rather than acting as isolated factors, cytokines function through complex and dynamic networks that integrate signals from tumor cells, immune cells, stromal components, and the extracellular matrix [7]. This intricate cytokine landscape contributes to substantial intertumoral and intratumoral heterogeneity, posing major challenges for accurate prognosis assessment and precision oncology [8,9].
Accumulating evidence suggests that cytokine-driven transcriptional programs are closely associated with aggressive tumor phenotypes, including enhanced proliferation, epithelial-mesenchymal transition, metastatic potential, and remodeling of the immune microenvironment [10,11,12]. Specific cytokines such as IL6, IL10, and IL1 family members have been linked to poor clinical outcomes in multiple malignancies, while others may exert context-dependent or even protective effects [13,14]. However, most prior studies have focused on individual cytokines or limited signaling axes, which may fail to capture the coordinated and system-level impact of cytokine networks on cancer biology. A comprehensive evaluation of cytokine-related gene expression patterns may therefore provide more robust and biologically meaningful prognostic information than single-gene biomarkers [15,16,17].
With the increasing availability of large-scale transcriptomic datasets, integrative computational approaches enable systematic dissection of cytokine-related signatures across cancer types. Regularized regression models, such as Least Absolute Shrinkage and Selection Operator Cox regression, allow the construction of parsimonious prognostic signatures while minimizing overfitting and multicollinearity [18,19,20]. When combined with pan-cancer analyses and independent validation cohorts, these strategies facilitate the identification of conserved cytokine-associated risk patterns and their relevance to patient survival. Moreover, linking cytokine-related signatures to hallmark malignant processes, including cell cycle progression, EMT, angiogenesis, and metabolic reprogramming, may provide mechanistic insights into how inflammatory signaling shapes tumor behavior [21,22,23].
In this study, we systematically investigated cytokine-related genes across pan-cancer cohorts to develop and validate a robust prognostic risk score. By integrating transcriptomic data with survival outcomes, we constructed a cytokine-related gene signature that stratifies patients into distinct risk groups with significantly different clinical prognoses. We further evaluated the distribution and prognostic performance of this signature across cancer types, assessed its clinical utility through nomogram modeling, and explored its biological relevance by correlating the risk score with malignant phenotypes and functional enrichment patterns. Together, our findings provide a comprehensive framework for understanding the prognostic and biological significance of cytokine dysregulation in cancer and highlight its potential value for pan-cancer risk stratification and personalized clinical management.
2. Results
2.1. Identification and Construction of a Cytokine-Related Prognostic Signature Using LASSO Cox Regression
To systematically characterize the prognostic relevance of cytokine-related genes across cancers, we applied LASSO Cox proportional hazards regression to the training cohort to construct a stable and interpretable prognostic signature. As illustrated in Figure 1A, coefficient trajectories demonstrated progressive shrinkage with increasing penalization, indicating effective control of multicollinearity among cytokine-associated candidates.
Using the one-standard-error criterion, an optimal lambda value of 9e-04 was selected, resulting in a parsimonious model comprising 16 cytokine-related genes with non-zero coefficients, including PANX1, FRMD8, GAPDH, MMP8, IL10, IL1A, NOD2, ZC3H12A, CLEC5A, S100A12, IL6, FERMT1, FN1, CX3CL1, CLEC9A, and ARG2. These genes collectively represent multiple facets of cytokine-driven tumor biology, encompassing pro-inflammatory signaling, innate immune sensing, leukocyte recruitment, extracellular matrix remodeling, and metabolic adaptation. Subsequent univariate Cox regression analysis revealed marked heterogeneity in prognostic effects among individual genes (Figure 1B), with PANX1, FRMD8, GAPDH, and MMP8 showing hazard ratios greater than 1, consistent with their reported roles in sustaining inflammatory microenvironments and facilitating tumor invasion, whereas ARG2, CLEC9A, and CX3CL1 exhibited protective associations, suggesting potential involvement in immune surveillance or anti-tumor immune modulation. Importantly, the coexistence of both risk-associated and protective cytokine-related genes within the same signature underscores the complex, context-dependent nature of cytokine signaling in cancer progression. To further evaluate redundancy and coordinated regulation within the model, pairwise Pearson correlation analysis was performed.
As shown in Figure 1C, moderate positive correlations were observed among subsets of inflammatory mediators such as IL6, IL10, CLEC5A, and NOD2, reflecting shared activation of innate immune and cytokine signaling pathways, while overall correlation coefficients remained below levels indicative of problematic collinearity, supporting the robustness and stability of the model. Collectively, these results demonstrate that the 16-gene cytokine-related signature integrates diverse yet complementary inflammatory and immune-regulatory signals, providing a biologically coherent framework for downstream prognostic stratification and functional interpretation in pan-cancer analyses.
2.2. Pan-Cancer Distribution and Prognostic Relevance of the Cytokine-Related Risk Score
Following construction of the 16-gene cytokine-related prognostic signature, we systematically investigated the distribution of the cytokine-related risk score across cancer types and its prognostic implications at the pan-cancer level. As shown in Figure 2A, the risk score exhibited pronounced heterogeneity among different tumor entities. Cancers characterized by chronic inflammation and immune activation, including head and neck squamous cell carcinoma, lung squamous cell carcinoma, bladder cancer, and multiple gastrointestinal malignancies, generally displayed higher risk score distributions, whereas endocrine-related and relatively indolent tumors, such as thyroid carcinoma and prostate adenocarcinoma, showed lower scores. This pattern suggests that cytokine-driven transcriptional programs are strongly shaped by tissue context and tumor-specific immune microenvironments. To further evaluate the prognostic impact of the cytokine-related risk score, univariate Cox regression analyses were conducted across cancer types for progression-free interval (PFI), disease-specific survival (DSS), and overall survival (OS). As illustrated by the bubble plots in Figure 2A, elevated risk scores were significantly associated with adverse survival outcomes in a wide spectrum of malignancies, with bubble size reflecting statistical significance and color intensity indicating effect magnitude. In the TCGA training cohort, forest plot analysis demonstrated that the cytokine-related risk score functioned predominantly as a hazard factor for survival in most cancer types (Figure 2B), with hazard ratios exceeding 1 for OS in multiple tumors, supporting its unfavorable prognostic role. Importantly, these findings were independently validated in the TCGA test cohort, where the majority of cancer types again showed consistent hazard effects associated with higher risk scores (Figure 2C), confirming the robustness and generalizability of the prognostic signal. To assess the clinical discriminative ability of the signature at the cohort level, patients in the TCGA training cohort were stratified into high-risk and low-risk groups according to the median cytokine-related risk score. Kaplan-Meier survival analyses revealed that high-risk patients experienced significantly worse DSS, OS, and PFI compared with low-risk patients (p < 0.001, Figure 2D).
This prognostic stratification was reproducibly observed in the TCGA test cohort, where high-risk patients consistently exhibited inferior survival outcomes across all endpoints (Figure 2E). Collectively, these results demonstrate that the cytokine-related risk score captures clinically relevant inflammatory and immune dysregulation signals and provides stable prognostic stratification across cancer types and independent cohorts, underscoring its potential value as a pan-cancer prognostic biomarker.
2.3. Cancer-Specific Prognostic Performance of the Cytokine-Related Risk Score in Representative Tumor Types
To further delineate the cancer-type-specific prognostic value of the cytokine-related risk score, Kaplan-Meier survival analyses were performed in multiple representative malignancies using disease-specific survival (DSS), overall survival (OS), and progression-free interval (PFI) as clinical endpoints. As shown in Figure 3A, patients with bladder urothelial carcinoma (BLCA) stratified into the high-risk group exhibited significantly poorer DSS and OS compared with those in the low-risk group, whereas the difference in PFI did not reach statistical significance. These findings suggest that cytokine-driven transcriptional programs in BLCA are more closely linked to mortality risk rather than early disease progression. In pancreatic adenocarcinoma (PAAD), a highly aggressive and inflammation-associated malignancy, the cytokine-related risk score demonstrated strong and consistent prognostic power across all three endpoints (Figure 3B). High-risk patients showed markedly reduced DSS, OS, and PFI (p < 0.001), highlighting the critical role of dysregulated cytokine signaling in driving rapid disease progression and poor outcomes in PAAD. In lung adenocarcinoma (LUAD), elevated risk scores were also associated with significantly worse DSS, OS, and PFI (Figure 3C), indicating that cytokine-related inflammatory and immune-regulatory pathways contribute to both survival and disease progression in this tumor type. By contrast, in cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), the prognostic impact of the cytokine-related risk score appeared endpoint-dependent (Figure 3D).
While high-risk patients experienced significantly inferior DSS and OS, no significant difference was observed for PFI, suggesting that cytokine-related dysregulation in CESC may preferentially influence long-term survival rather than short-term progression dynamics. Collectively, these cancer-specific analyses demonstrate that although the cytokine-related risk score consistently stratifies patient survival in multiple tumor types, its association with disease progression varies across cancers, reflecting underlying biological heterogeneity and context-dependent roles of cytokine signaling within distinct tumor microenvironments.
2.4. Development and Validation of a Cytokine-Related Nomogram for Individualized Survival Prediction
To enhance the clinical applicability of the cytokine-related risk score and provide an intuitive tool for individualized survival prediction, we integrated the risk score with key clinical variables to construct a comprehensive prognostic nomogram. As shown in Figure 4A, the nomogram incorporates the cytokine-related risk score, patient age, and cancer type, with each variable assigned a weighted point value proportional to its contribution to overall survival risk. By summing the total points for an individual patient, the nomogram enables direct estimation of 1-, 3-, 5-, and 10-year overall survival probabilities, thereby facilitating personalized risk assessment in routine clinical settings. Calibration analysis demonstrated a high degree of concordance between nomogram-predicted and observed survival outcomes. As illustrated in Figure 4B, the calibration curve for overall survival showed close alignment with the 45-degree reference line, indicating that the nomogram provided accurate survival probability estimates with minimal systematic bias. To further quantify predictive performance, time-dependent receiver operating characteristic analyses were performed. The ROC curves at 5 years (Figure 4C) revealed that the nomogram consistently outperformed the cytokine-related risk score alone in both the TCGA training and test cohorts, achieving higher area under the curve values, thereby confirming the added prognostic value of integrating clinical factors with molecular information. This advantage was maintained across time, as demonstrated by the dynamic AUC profiles in Figure 4D, where the nomogram exhibited superior and more stable discrimination ability over long-term follow-up compared with the risk score alone. Beyond statistical performance, the potential clinical utility of the nomogram was evaluated using decision curve analysis.
As shown in Figure 4E and Figure 4F, the nomogram yielded a higher standardized net benefit than the cytokine-related risk score across a wide range of threshold probabilities in both the training and test cohorts. Importantly, the nomogram consistently outperformed default strategies of treating all or no patients, indicating that its application could improve clinical decision-making by optimizing the balance between benefit and harm. Collectively, these results demonstrate that the cytokine-related nomogram provides accurate, robust, and clinically meaningful survival prediction, supporting its potential role as a practical decision-support tool for risk stratification and personalized management across diverse cancer types.
2.5. Association Between the Cytokine-Related Risk Score and Malignant Biological Features Across Pan-Cancer
To explore the biological implications underlying the cytokine-related risk score, we systematically examined its relationship with key malignant features, including angiogenesis, epithelial-mesenchymal transition (EMT), and cell cycle activity, using pathway-level scores across the pan-cancer cohort. As shown in Figure 5A-C, at the global pan-cancer level, the cytokine-related risk score exhibited a significant positive correlation with EMT score and cell cycle score, indicating that tumors with higher cytokine-related risk scores tend to display enhanced mesenchymal characteristics and proliferative capacity. In contrast, a weak but statistically significant negative correlation was observed between the risk score and angiogenesis score, suggesting that cytokine-driven risk may not uniformly reflect angiogenic activity and may instead capture alternative inflammatory or immune-mediated malignant programs.
To further delineate tumor-specific patterns, stratified correlation analyses were performed across individual cancer types. As illustrated in Figure 5D, the relationship between the cytokine-related risk score and angiogenesis score varied markedly across cancers. Positive correlations were observed in selected tumor types such as COAD, DLBC, and READ, whereas weak or negative correlations predominated in other cancers, highlighting substantial inter-tumoral heterogeneity in cytokine-angiogenesis coupling. In contrast, the association between the risk score and EMT score was consistently positive across the majority of cancer types (Figure 5E), including BLCA, BRCA, COAD, KIRC, LGG, LUAD, and STAD, supporting the notion that cytokine-related transcriptional programs are broadly linked to EMT activation and invasive phenotypes. Similarly, correlation analyses between the risk score and cell cycle score revealed widespread positive associations across multiple malignancies (Figure 5F), particularly in highly proliferative tumors such as LGG, LUAD, PRAD, and THCA, indicating that elevated cytokine-related risk is closely aligned with enhanced cell cycle progression and tumor growth dynamics. Collectively, these findings demonstrate that the cytokine-related risk score is not merely a prognostic indicator but also reflects fundamental malignant biological behaviors, particularly EMT activation and proliferative signaling, while its relationship with angiogenesis appears to be context-dependent and tumor-specific. These results provide mechanistic insights into how cytokine dysregulation may drive aggressive tumor phenotypes across diverse cancer types.
2.6. Functional Characterization of Cytokine-Related Risk Groups Reveals Distinct Biological Programs
To further elucidate the biological mechanisms underlying the prognostic stratification defined by the cytokine-related risk score, we performed differential expression analysis between high-risk and low-risk groups followed by comprehensive functional enrichment analyses. Gene Ontology and KEGG pathway enrichment results demonstrated that genes upregulated in the high-risk group were strongly associated with fundamental malignant processes (Figure 6A). At the biological process level, high-risk tumors showed significant enrichment of cell cycle-related terms, including nuclear division, mitotic nuclear division, chromosome segregation, and sister chromatid segregation, indicating enhanced proliferative activity. Consistently, cellular component analysis highlighted enrichment in chromosomal regions and condensed chromosomes, further supporting active mitosis and genomic instability in cytokine-high tumors. Molecular function terms were predominantly related to extracellular matrix structural constituents, integrin binding, proteoglycan binding, and microtubule binding, suggesting coordinated activation of extracellular matrix remodeling and cytoskeletal reorganization. Notably, KEGG pathway analysis revealed enrichment of ECM-receptor interaction and IL-17 signaling pathway, implicating cytokine-driven inflammatory signaling in promoting invasive and proliferative tumor phenotypes. In contrast, genes upregulated in the low-risk group exhibited a markedly different functional profile (Figure 6B). Biological processes were enriched for transmembrane transport-related activities, including anion and carboxylic acid transport, vascular transport, and transport across the blood-brain barrier, reflecting a more differentiated and metabolically regulated cellular state. Cellular component terms were dominated by plasma membrane-associated structures, such as apical, basal, and basolateral plasma membranes, as well as neuronal cell bodies, suggesting preserved cellular polarity and tissue-specific functions. Molecular function enrichment further emphasized active and organic anion transmembrane transporter activity. KEGG pathways enriched in the low-risk group included adrenergic signaling in cardiomyocytes, thyroid hormone synthesis, bile secretion, and one-carbon pool by folate, indicating engagement of endocrine, metabolic, and homeostatic signaling rather than aggressive malignant programs.
Collectively, these results demonstrate that high cytokine-related risk tumors are characterized by hyperproliferation, extracellular matrix remodeling, and inflammatory signaling, whereas low-risk tumors preferentially maintain transport, metabolic, and differentiated cellular functions. This functional divergence provides mechanistic insight into how cytokine dysregulation contributes to tumor aggressiveness and poor clinical outcomes.
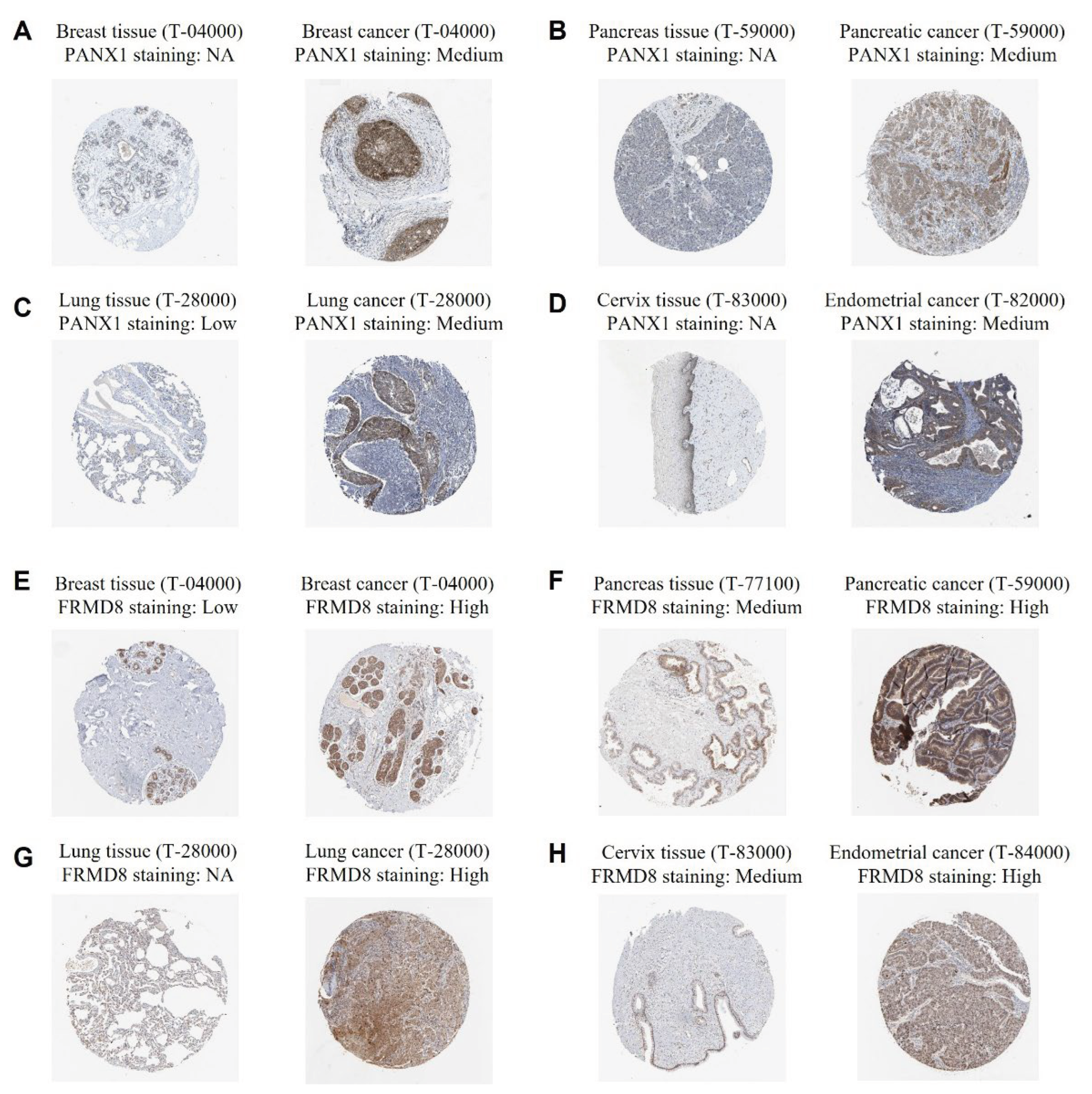
2.7. Immunohistochemical Validation of PANX1 and FRMD8 Expression in Normal and Malignant Tissues
To further support the transcriptomic findings and validate the protein-level expression of key genes within the cytokine-related prognostic signature, immunohistochemical staining data for representative signature genes were examined in normal and tumor tissues. As shown in Figure 7A-D, PANX1 protein expression was generally absent or undetectable in corresponding normal tissues, including breast, pancreas, lung, and cervix, whereas moderate cytoplasmic staining was consistently observed in the matched cancer tissues. This differential expression pattern suggests that PANX1 is upregulated during malignant transformation across multiple tissue types and supports its role as a risk-associated gene within the cytokine-related signature. Consistent with these observations, FRMD8 exhibited low or undetectable expression in normal breast and lung tissues, while markedly increased staining intensity was observed in breast cancer, pancreatic cancer, lung cancer, and endometrial cancer specimens (Figure 7E-H). Notably, FRMD8 showed high and diffuse cytoplasmic expression in several tumor types, indicating broad activation at the protein level in malignant tissues. Together, these immunohistochemical results corroborate the transcriptomic analyses and demonstrate that PANX1 and FRMD8 are upregulated at the protein level in diverse cancers, providing additional biological and translational support for their inclusion in the cytokine-related prognostic signature.
3. Discussion
In the present study, we systematically characterized cytokine-related transcriptional programs across pan-cancer cohorts and developed a robust prognostic signature based on cytokine-associated genes. By integrating large-scale transcriptomic data with survival outcomes, we identified a 16-gene cytokine-related risk score derived from an initial pool of 131 cytokine-related genes curated from GSEA and MSigDB. This signature consistently stratified patients into distinct risk groups with significantly different clinical outcomes across multiple cancer types and independent cohorts, underscoring the broad prognostic relevance of cytokine dysregulation in cancer. Unlike single-cytokine biomarkers, the composite nature of this signature captures the coordinated and context-dependent effects of inflammatory and immune signaling networks within the tumor microenvironment [24].
The cytokine-related risk score demonstrated pronounced heterogeneity across tumor types, reflecting tissue-specific immune landscapes and inflammatory states. Tumors characterized by chronic inflammation or strong immune cell infiltration tended to exhibit higher risk scores and poorer prognoses, whereas hormonally regulated or relatively indolent cancers displayed lower scores and attenuated prognostic effects. Importantly, the risk score acted predominantly as a hazard factor in both training and test cohorts, indicating robust generalizability [25]. Cancer-specific analyses further revealed that while the signature consistently predicted survival, its association with disease progression varied across malignancies, suggesting that cytokine-driven programs may differentially influence tumor growth, invasion, and metastatic dissemination depending on biological context [26].
Beyond its prognostic value, the cytokine-related risk score was closely linked to fundamental malignant phenotypes [27]. Correlation analyses revealed strong associations between elevated risk scores and enhanced epithelial-mesenchymal transition and cell cycle activity across most cancer types, supporting a role for cytokine dysregulation in promoting invasive and proliferative tumor behavior [28]. Functional enrichment analyses further demonstrated that high-risk tumors were characterized by activation of cell cycle progression, chromosome segregation, extracellular matrix remodeling, and inflammatory signaling pathways, including IL-17-related processes [29]. In contrast, low-risk tumors preferentially enriched pathways related to transmembrane transport, metabolic regulation, and tissue-specific homeostatic functions [1]. Together, these findings provide mechanistic insight into how cytokine-related transcriptional programs may drive tumor aggressiveness and adverse clinical outcomes [3].
To facilitate clinical translation, we integrated the cytokine-related risk score with clinical variables to construct a prognostic nomogram, which showed improved predictive accuracy and clinical net benefit compared with the molecular score alone [30]. This integrated model enables individualized survival estimation and highlights the potential utility of cytokine-based signatures as decision-support tools in precision oncology [31]. Nevertheless, several limitations should be acknowledged. First, the study is based on retrospective transcriptomic data, and prospective validation in independent clinical cohorts is warranted [32]. Second, although the signature reflects coordinated cytokine activity, it does not directly capture post-transcriptional regulation or protein-level cytokine dynamics within the tumor microenvironment [33]. Finally, functional experiments are required to delineate the causal roles of individual genes within the signature [34]. Despite these limitations, our findings provide a comprehensive framework linking cytokine dysregulation to pan-cancer prognosis and tumor biology, and they support further exploration of cytokine-related signatures as biomarkers and potential therapeutic targets in cancer.
4. Materials and Methods
4.1. Data Collection and Preprocessing
Transcriptomic expression profiles and corresponding clinical annotations were obtained from The Cancer Genome Atlas pan-cancer cohort through the UCSC Xena platform. Only primary tumor samples with complete survival information were included. Overall survival, disease-specific survival, and progression-free interval were defined according to TCGA standardized clinical endpoints [35]. Gene expression matrices were log2-transformed and subsequently standardized by z-score normalization within each dataset to reduce technical variability. Samples were randomly divided into a training cohort and an internal test cohort at a ratio of 7:3 for model development and validation [36].
4.2. Cytokine-Related Gene Curation Based on GSEA and MSigDB
To comprehensively define cytokine-related genes, we systematically curated gene sets associated with cytokine signaling and cytokine-mediated immune responses from Gene Set Enrichment Analysis resources and the Molecular Signatures Database (MSigDB; URL: https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp). Specifically, cytokine-related gene sets were retrieved from the MSigDB collections, including Hallmark, C2 curated gene sets, and Gene Ontology biological process categories related to cytokine activity, cytokine-mediated signaling pathways, inflammatory response, interleukin signaling, and immune regulation [37]. After removing redundant entries and consolidating overlapping genes across gene sets, a total of 131 cytokine-related genes were identified and used as the initial candidate pool for downstream prognostic modeling [38,39].
4.3. Survival Analysis and Pan-Cancer Validation
Patients were stratified into high-risk and low-risk groups according to the median cytokine-related risk score within each cohort. Kaplan-Meier survival analyses were performed to compare survival outcomes between groups, and statistical significance was assessed using the log-rank test. Univariate Cox regression analyses were conducted across cancer types to evaluate the prognostic impact of the risk score on overall survival, disease-specific survival, and progression-free interval [24]. The robustness and generalizability of the signature were independently validated in the TCGA test cohort [40].
4.4. Nomogram Construction and Performance Evaluation
To improve clinical applicability, a prognostic nomogram was developed by integrating the cytokine-related risk score with key clinical variables, including age and cancer type, using multivariable Cox regression analysis. Calibration curves were generated to assess agreement between predicted and observed survival probabilities [30]. Time-dependent receiver operating characteristic analyses were used to evaluate predictive accuracy over time, and dynamic area under the curve values were compared between the risk score alone and the nomogram model. Decision curve analysis was performed to quantify the clinical net benefit of each model across a range of threshold probabilities [41,42].
4.5. Functional Enrichment and Pathway Analysis
Differentially expressed genes between high-risk and low-risk groups were identified using predefined statistical thresholds. Gene Ontology enrichment analyses covering biological process, cellular component, and molecular function categories, as well as Kyoto Encyclopedia of Genes and Genomes pathway analyses, were performed using the clusterProfiler package in R [43]. In addition, pathway-level scores for angiogenesis, epithelial-mesenchymal transition, and cell cycle activity were calculated, and Pearson correlation analyses were conducted to assess associations between these malignant features and the cytokine-related risk score at both pan-cancer and cancer-specific levels [44].
4.6. Immunohistochemistry (IHC) Analysis
To validate the protein-level expression of representative cytokine-related signature genes, immunohistochemistry analyses were performed using publicly available tissue microarray-based staining data. Representative IHC images for PANX1 and FRMD8 in normal and corresponding tumor tissues were obtained from a curated human protein expression database [45]. Multiple tissue types, including breast, pancreas, lung, cervix, and endometrium, were examined to assess expression patterns across different malignancies. IHC staining intensity was annotated according to the database-defined scoring system and categorized as not available (NA), low, medium, or high based on staining strength and the proportion of positively stained cells. Protein expression levels in tumor tissues were qualitatively compared with those in matched normal tissues to evaluate differential expression associated with malignant transformation. All IHC images were independently reviewed to ensure consistency of staining patterns across tissue types [46]. These analyses were used to corroborate transcriptomic findings and to provide translational evidence supporting the involvement of PANX1 and FRMD8 in cytokine-related tumor biology.
4.7. Immune Infiltration Analysis
All statistical analyses were performed using R (4.4.1) software. Two-sided p values less than 0.05 were considered statistically significant unless otherwise specified. Continuous variables were summarized using appropriate descriptive statistics, and correlations were evaluated using Pearson correlation coefficients. All analyses followed reproducible research principles.
5. Conclusions
In summary, this study systematically delineates the prognostic, biological, and translational significance of cytokine-related gene expression across pan-cancer cohorts. By integrating transcriptomic profiles with survival data, we established and validated a robust 16-gene cytokine-related prognostic signature that consistently stratifies patients into distinct risk groups with significantly different clinical outcomes across multiple cancer types and independent cohorts. The cytokine-related risk score was strongly associated with key malignant phenotypes, including enhanced epithelial-mesenchymal transition, cell cycle activation, and extracellular matrix remodeling, providing mechanistic insight into its unfavorable prognostic impact. Importantly, immunohistochemical validation further confirmed the upregulation of representative risk-associated genes, PANX1 and FRMD8, at the protein level in multiple tumor tissues compared with corresponding normal tissues, reinforcing the biological credibility and translational relevance of the signature. Moreover, integration of the cytokine-related risk score with clinical variables into a nomogram improved individualized survival prediction and demonstrated potential clinical utility. Collectively, these findings highlight cytokine-related transcriptional programs as critical determinants of tumor aggressiveness and patient prognosis and support their further development as clinically relevant biomarkers for pan-cancer risk stratification and precision oncology.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Developed the concept and designed the study: S.-C.C., Y.-K.L., C.-Y.W. and H.-P.L.; Performed data analysis and interpretation: K.-F.C., C.-C.C., C.-H.L., C.-H.C., H.-R.L., C.-J.W., C.-H. Y., S.K., D.-T.-M. X., N.P., D.-D.S., A. F., and H.-Y. L. All authors read and approved the final version of this manuscript.
Funding
This research was funded by the National Science and Technology Council (NSTC) of Taiwan, grant nos. 114-2320-B-393 -003, 114-2320-B-393 -004, 114-2314-B-038-133-MY3 and 114-2811-B-038-046, and by Kaohsiung Armed Forces General Hospital grant nos. KAFGH_D_115016 and KAFGH_D_115018. The APC was funded by Kaohsiung Armed Forces General Hospital. This work was financially supported by the Higher Education Sprout Project of the Ministry of Education (MOE) in Taiwan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data involved in this study are available from the corresponding author on request. We employed the Molecular Signatures Database (MSigDB; URL: https://www.gsea-msigdb.org/gsea/msigdb/human/collections.jsp), using the keyword “cytokine” for analysis in this study.
Acknowledgments
We would like to acknowledge Yi-Ting Wu, Yun-Yu Lin, and Yueh-Yuan Shieh for their excellent technical support at the Laboratory of Research and Medical Education and Research Center, Kaohsiung Armed Forces General Hospital.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AUC | area under the curve |
| BLCA | bladder urothelial carcinom |
| CESC | cervical squamous cell carcinoma and endocervical adenocarcinoma |
| COAD | colon adenocarcinoma |
| DCA | decision curve analysis |
| DSS | disease-specific survival |
| EMT | epithelial-mesenchymal transition |
| FRMD8 | FERM domain containing 8 |
| GSEA | gene set enrichment analysis |
| IHC | immunohistochemistry |
| IL | interleukin |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KM | Kaplan-Meier |
| LASSO | least absolute shrinkage and selection operator |
| LUAD | lung adenocarcinoma |
| MSigDB | Molecular Signatures Database |
| OS | overall survival |
| PAAD | pancreatic adenocarcinoma |
| PFI | progression-free interval |
| ROC | receiver operating characteristic |
| TCGA | The Cancer Genome Atlas |
References
- Habanjar, O. Crosstalk of Inflammatory Cytokines within the Breast Tumor Microenvironment . Int J Mol Sci 2023, 24(4). [Google Scholar] [CrossRef]
- Pradhan, R.; Kundu, A.; Kundu, C.N. The cytokines in tumor microenvironment: from cancer initiation-elongation-progression to metastatic outgrowth . Crit Rev Oncol Hematol 2024, 196, 104311. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.J. Cytokine-driven cancer immune evasion mechanisms . Int Rev Cell Mol Biol 2025. 396, 1–54. [Google Scholar]
- Kureshi, C.T.; Dougan, S.K. Cytokines in cancer . Cancer Cell 2025, 43(1), 15–35. [Google Scholar] [CrossRef] [PubMed]
- Yin, X. The “double-edged sword” effect of cytokines in cancer: coexisting opportunities and challenges . Front Immunol 2025, 16, 1701405. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy . Nat Rev Clin Oncol 2022, 19(4), 237–253. [Google Scholar] [CrossRef]
- Manickasamy, M.K. Cytokine symphony: Deciphering the tumor microenvironment and metastatic axis in oral cancer . Cytokine Growth Factor Rev 2025. 85, 1–10. [Google Scholar] [CrossRef]
- Christodoulou, M.I.; Zaravinos, A. Single-Cell Analysis in Immuno-Oncology . Int J Mol Sci 2023, 24(9). [Google Scholar] [CrossRef]
- Wolf, Y.; Samuels, Y. Intratumor Heterogeneity and Antitumor Immunity Shape One Another Bidirectionally . Clin Cancer Res 2022, 28(14), 2994–3001. [Google Scholar] [CrossRef]
- Abaurrea, A.; Araujo, A.M.; Caffarel, M.M. The Role of the IL-6 Cytokine Family in Epithelial-Mesenchymal Plasticity in Cancer Progression . Int J Mol Sci 2021, 22(15). [Google Scholar] [CrossRef]
- Chen, Y. Breast neoplasm epithelial-mesenchymal transition and cytokines: a systematic review . Cancer Cell Int 2025, 25(1), p. 323. [Google Scholar] [CrossRef]
- Oh, A. NF-κB signaling in neoplastic transition from epithelial to mesenchymal phenotype . Cell Commun Signal 2023, 21(1), p. 291. [Google Scholar] [CrossRef]
- Mohamed, A.H. Interleukin-6 serves as a critical factor in various cancer progression and therapy . Med Oncol 2024, 41(7), 182. [Google Scholar] [CrossRef]
- Bao, C. Cytokine profiles in patients with newly diagnosed diffuse large B-cell lymphoma: IL-6 and IL-10 levels are associated with adverse clinical features and poor outcomes . Cytokine 2023, 169, 156289. [Google Scholar] [CrossRef]
- Han, Z. Identification and Characterization of Cytokine Genes in Breast Cancer for Predicting Clinical Outcomes . Mediators Inflamm 2025, 8441796. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.H. Integrated Multi-Omics Analysis Reveals Cytokine Network Dynamics and Prognostic Signatures in Hepatitis B Virus-Associated Hepatocellular Carcinoma . Appl Biochem Biotechnol 2026. [Google Scholar] [CrossRef] [PubMed]
- Yabuno, A. Identification of serum cytokine clusters associated with outcomes in ovarian clear cell carcinoma . Sci Rep 2020, 10(1), p. 18503. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W. Integration of gene interaction information into a reweighted Lasso-Cox model for accurate survival prediction . Bioinformatics 2021, 36(22-23), 5405–5414. [Google Scholar] [CrossRef] [PubMed]
- Jardillier, R. Prognosis of lasso-like penalized Cox models with tumor profiling improves prediction over clinical data alone and benefits from bi-dimensional pre-screening . BMC Cancer 2022, 22(1), 1045. [Google Scholar] [CrossRef]
- Hou, Y. Comprehensive Analysis of a Cancer-Immunity Cycle-Based Signature for Predicting Prognosis and Immunotherapy Response in Patients With Colorectal Cancer . Front Immunol 2022, 13, 892512. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, M.; Qu, H. Identification of cytokine-induced cell communications by pan-cancer meta-analysis . PeerJ 2023, 11, p. e16221. [Google Scholar] [CrossRef]
- Turanli, B. Decoding Systems Biology of Inflammation Signatures in Cancer Pathogenesis: Pan-Cancer Insights from 12 Common Cancers . Omics 2023, 27(10), 483–493. [Google Scholar] [CrossRef]
- Heimes, A.S. Prognostic Impact of Acute and Chronic Inflammatory Interleukin Signatures in the Tumor Microenvironment of Early Breast Cancer . Int J Mol Sci 2024, 25(20). [Google Scholar] [CrossRef]
- Liu, X.; Zhang, L.; Chen, L. Establishment of a novel cytokine-related 8-gene signature for distinguishing and predicting the prognosis of triple-negative breast cancer . Front Med (Lausanne) 2023, 10, 1189361. [Google Scholar] [CrossRef]
- Pei, J.; Gao, Y.; Wu, A. An inflammation-related subtype classification for analyzing tumor microenvironment and clinical prognosis in colorectal cancer . Front Immunol 2024, 15, 1369726. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y. Systematic assessment of the clinicopathological prognostic significance of tissue cytokine expression for lung adenocarcinoma based on integrative analysis of TCGA data . Sci Rep 2019, 9(1), p. 6301. [Google Scholar] [CrossRef]
- Sun, C. Risk Signature of Cancer-Associated Fibroblast-Secreted Cytokines Associates With Clinical Outcomes of Breast Cancer . Front Oncol 2021, 11, 628677. [Google Scholar] [CrossRef] [PubMed]
- Liang, J. The Correlation Between the Immune and Epithelial-Mesenchymal Transition Signatures Suggests Potential Therapeutic Targets and Prognosis Prediction Approaches in Kidney Cancer . Sci Rep 2018, 8(1), p. 6570. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Integrated analysis reveals survival-related genes promoting cholangiocarcinoma progression via G2/M cell cycle, with validation of cyclin B1 in tumor proliferation. Sci Rep 2025, 15(1), p. 41352. [Google Scholar] [CrossRef]
- Cheng, Q. Prognostic nomogram incorporating cytokines for overall survival in patients with newly diagnosed multiple myeloma . Int Immunopharmacol 2021, 99, 108016. [Google Scholar] [CrossRef]
- Yin, X. Potential predictive biomarkers in antitumor immunotherapy: navigating the future of antitumor treatment and immune checkpoint inhibitor efficacy . Front Oncol 2024, 14, 1483454. [Google Scholar] [CrossRef]
- Hoekstra, M.E. Distinct spatiotemporal dynamics of CD8(+) T cell-derived cytokines in the tumor microenvironment . Cancer Cell 2024, 42(1), 157–167.e9. [Google Scholar] [CrossRef]
- Wong, H.S. Characterization of cytokinome landscape for clinical responses in human cancers . Oncoimmunology 2016, 5(11), p. e1214789. [Google Scholar] [CrossRef]
- Przybyla, L.; Gilbert, L.A. A new era in functional genomics screens . Nat Rev Genet 2022, 23(2), 89–103. [Google Scholar] [CrossRef]
- Lim, A.M. Assessment of endpoint definitions in curative-intent trials for mucosal head and neck squamous cell carcinomas: Head and Neck Cancer International Group consensus recommendations . Lancet Oncol 2024, 25(7), e318–e330. [Google Scholar] [CrossRef]
- Dubray-Vautrin, A. Internal validation strategy for high dimensional prognosis model: A simulation study and application to transcriptomic in head and neck tumors . Comput Struct Biotechnol J 2025. 27, 3792–3802. [Google Scholar] [CrossRef]
- Jiang, P. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels . Nat Methods 2021, 18(10), 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Santoso, C.S. Comprehensive mapping of the human cytokine gene regulatory network . Nucleic Acids Res 2020, 48(21), 12055–12073. [Google Scholar] [CrossRef]
- Cui, A. Dictionary of immune responses to cytokines at single-cell resolution . Nature 2024, 625(7994), 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z. Single-cell RNA analysis to identify five cytokines signaling in immune-related genes for melanoma survival prognosis . Front Immunol 2023, 14, 1148130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L. Development and validation of a prognostic nomogram for early stage non-small cell lung cancer: a study based on the SEER database and a Chinese cohort . BMC Cancer 2022, 22(1), 980. [Google Scholar] [CrossRef]
- Li, H. A comprehensive nomogram for assessing the prognosis of non-small cell lung cancer patients receiving immunotherapy: a prospective cohort study in China . Front Immunol 2024, 15, 1487078. [Google Scholar] [CrossRef]
- Muley, V.Y. Functional Insights Through Gene Ontology, Disease Ontology, and KEGG Pathway Enrichment . Methods Mol Biol 2025. 2927, 75–98. [Google Scholar]
- Oshi, M. Intra-Tumoral Angiogenesis Is Associated with Inflammation, Immune Reaction and Metastatic Recurrence in Breast Cancer . Int J Mol Sci 2020, 21(18). [Google Scholar] [CrossRef] [PubMed]
- Li, H. Immunohistochemistry Microarrays . Anal Chem 2017, 89(17), 8620–8625. [Google Scholar] [CrossRef]
- Tachikawa, M. Targeted Proteomics-Based Quantitative Protein Atlas of Pannexin and Connexin Subtypes in Mouse and Human Tissues and Cancer Cell Lines . J Pharm Sci 2020, 109(2), 1161–1168. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Construction of a cytokine-related prognostic signature. (A) LASSO Cox regression coefficient trajectories of cytokine-related genes with the optimal lambda selected by the one-standard-error criterion. (B) Forest plot showing hazard ratios and 95% confidence intervals of the genes included in the final cytokine-related signature. (C) Pearson correlation heatmap of the signature genes, illustrating their expression correlations.
Figure 1.
Construction of a cytokine-related prognostic signature. (A) LASSO Cox regression coefficient trajectories of cytokine-related genes with the optimal lambda selected by the one-standard-error criterion. (B) Forest plot showing hazard ratios and 95% confidence intervals of the genes included in the final cytokine-related signature. (C) Pearson correlation heatmap of the signature genes, illustrating their expression correlations.
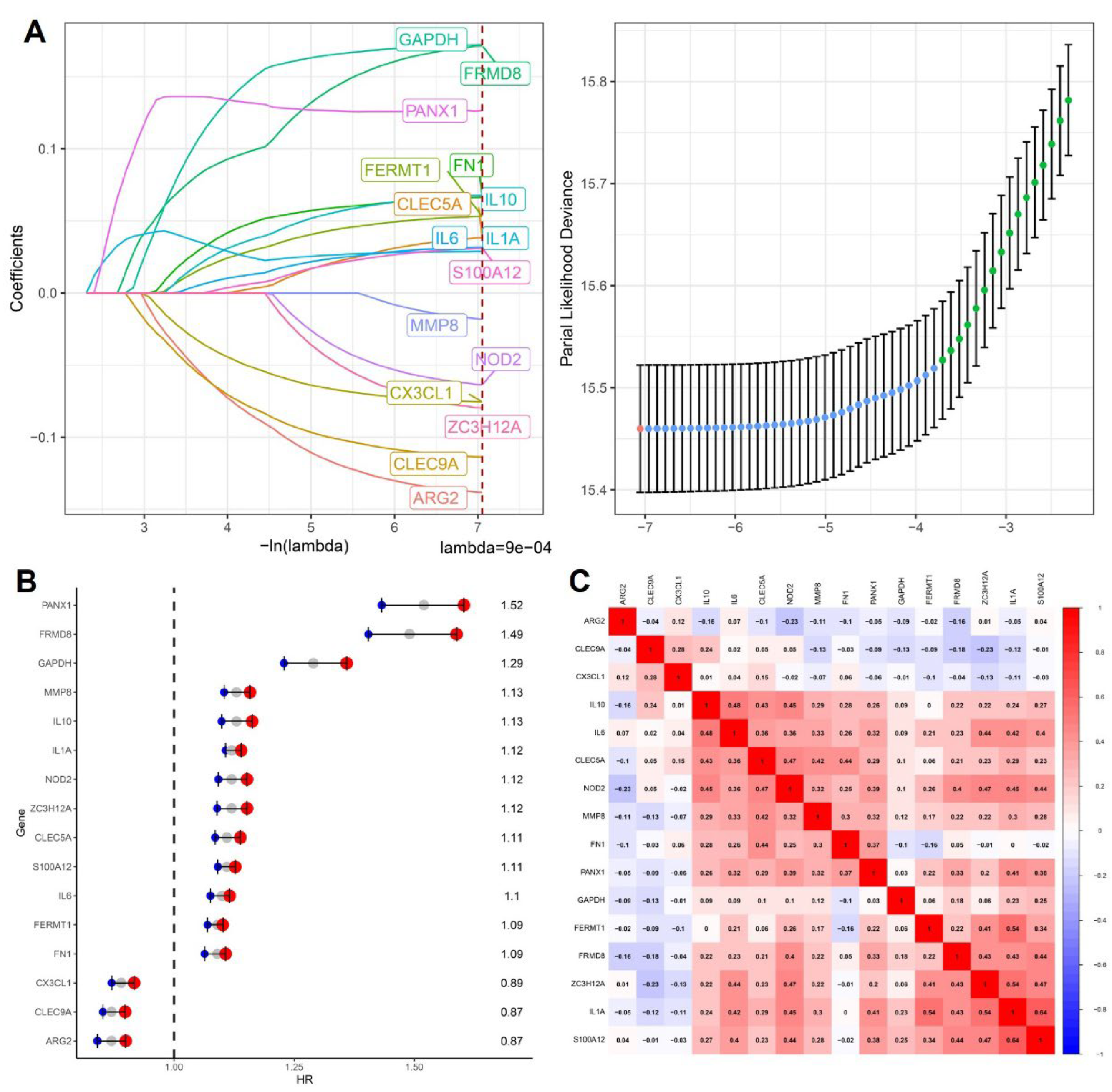
Figure 2.
Pan-cancer prognostic performance of the cytokine-related risk score. (A) Distribution of cytokine-related risk scores across cancer types and their associations with PFI, DSS, and OS. (B) Forest plot showing hazard ratios of the risk score for OS across cancer types in the TCGA training cohort. (C) Forest plot showing hazard ratios of the risk score for OS across cancer types in the TCGA test cohort. (D) Kaplan-Meier curves for DSS, OS, and PFI in the TCGA training cohort stratified by cytokine-related risk score. (E) Kaplan-Meier validation of prognostic stratification in the TCGA test cohort.
Figure 2.
Pan-cancer prognostic performance of the cytokine-related risk score. (A) Distribution of cytokine-related risk scores across cancer types and their associations with PFI, DSS, and OS. (B) Forest plot showing hazard ratios of the risk score for OS across cancer types in the TCGA training cohort. (C) Forest plot showing hazard ratios of the risk score for OS across cancer types in the TCGA test cohort. (D) Kaplan-Meier curves for DSS, OS, and PFI in the TCGA training cohort stratified by cytokine-related risk score. (E) Kaplan-Meier validation of prognostic stratification in the TCGA test cohort.
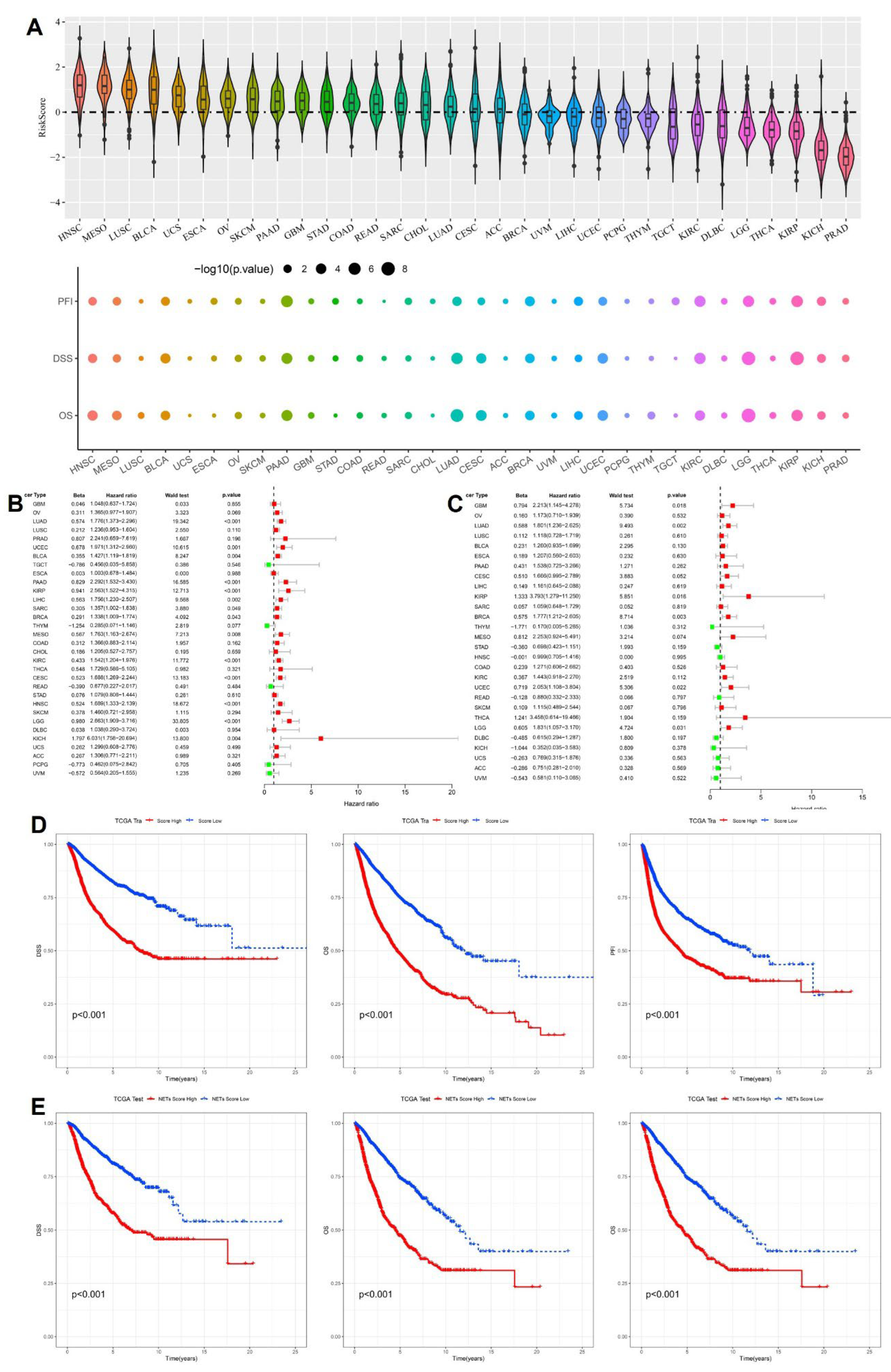
Figure 3.
Cancer-specific survival analyses based on the cytokine-related risk score. Kaplan-Meier curves comparing high-risk and low-risk groups stratified by the cytokine-related risk score in (A) BLCA, (B) PAAD, (C) LUAD, and (D) CESC. Disease-specific survival (DSS), overall survival (OS), and progression-free interval (PFI) are shown for each cancer type, with p values determined by the log-rank test.
Figure 3.
Cancer-specific survival analyses based on the cytokine-related risk score. Kaplan-Meier curves comparing high-risk and low-risk groups stratified by the cytokine-related risk score in (A) BLCA, (B) PAAD, (C) LUAD, and (D) CESC. Disease-specific survival (DSS), overall survival (OS), and progression-free interval (PFI) are shown for each cancer type, with p values determined by the log-rank test.
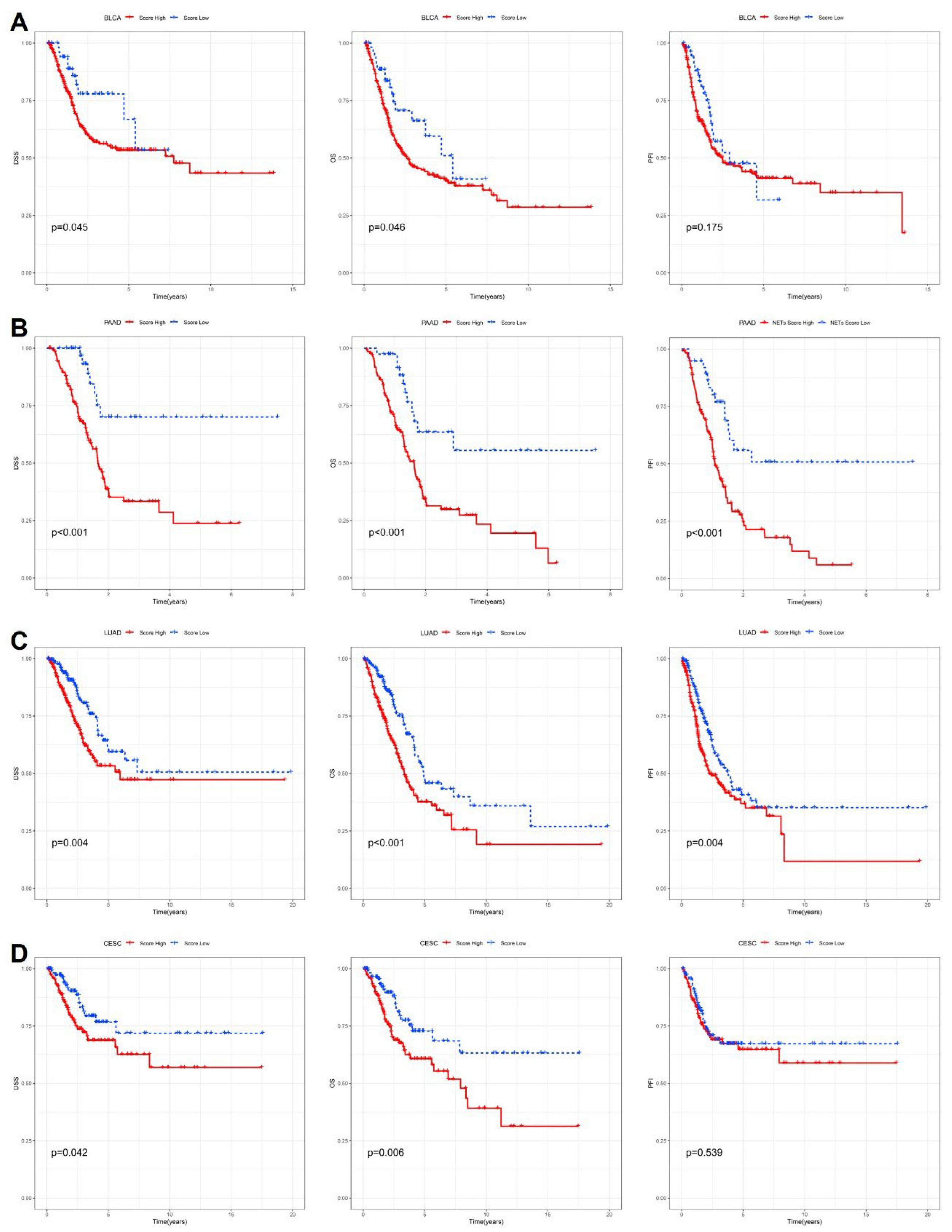
Figure 4.
Construction and evaluation of a cytokine-related prognostic nomogram. (A) Nomogram integrating the cytokine-related risk score, age, and cancer type to predict 1-, 3-, 5-, and 10-year overall survival. (B) Calibration curve comparing nomogram-predicted and observed overall survival probabilities. (C) Time-dependent ROC curves at 5 years for the risk score and nomogram in the TCGA training and test cohorts. (D) Dynamic AUC profiles over time comparing predictive performance of the risk score and nomogram. (E-F) Decision curve analyses illustrating the standardized net benefit of the nomogram and risk score in the TCGA training and test cohorts, respectively.
Figure 4.
Construction and evaluation of a cytokine-related prognostic nomogram. (A) Nomogram integrating the cytokine-related risk score, age, and cancer type to predict 1-, 3-, 5-, and 10-year overall survival. (B) Calibration curve comparing nomogram-predicted and observed overall survival probabilities. (C) Time-dependent ROC curves at 5 years for the risk score and nomogram in the TCGA training and test cohorts. (D) Dynamic AUC profiles over time comparing predictive performance of the risk score and nomogram. (E-F) Decision curve analyses illustrating the standardized net benefit of the nomogram and risk score in the TCGA training and test cohorts, respectively.
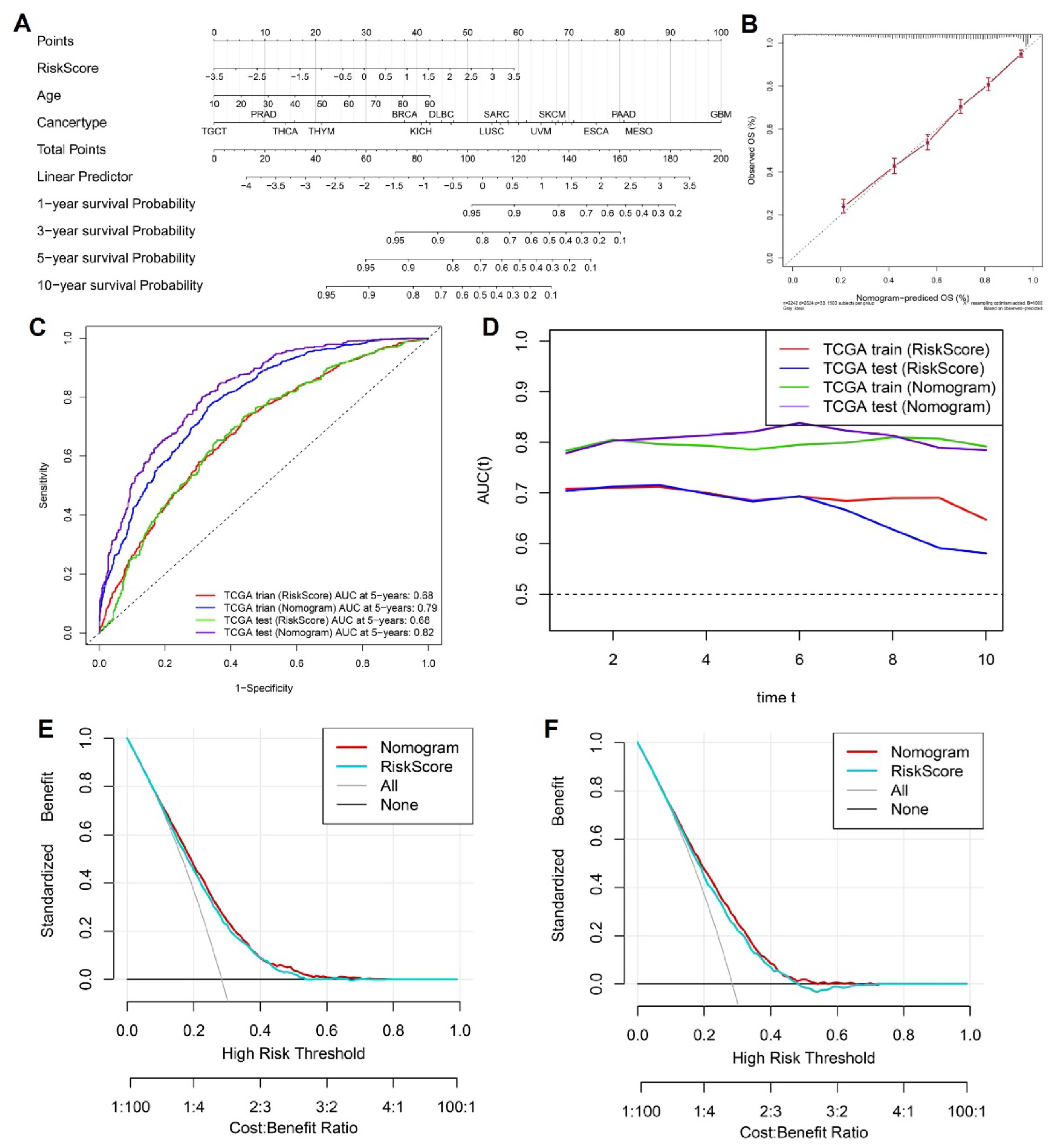
Figure 5.
Correlation between the cytokine-related risk score and malignant biological features. (A-C) Scatter plots showing pan-cancer correlations between the cytokine-related risk score and angiogenesis score, EMT score, and cell cycle score, respectively. (D-F) Cancer-type-specific correlations between the risk score and angiogenesis score (D), EMT score (E), and cell cycle score (F). Pearson correlation coefficients and corresponding p values are indicated in each panel.
Figure 5.
Correlation between the cytokine-related risk score and malignant biological features. (A-C) Scatter plots showing pan-cancer correlations between the cytokine-related risk score and angiogenesis score, EMT score, and cell cycle score, respectively. (D-F) Cancer-type-specific correlations between the risk score and angiogenesis score (D), EMT score (E), and cell cycle score (F). Pearson correlation coefficients and corresponding p values are indicated in each panel.
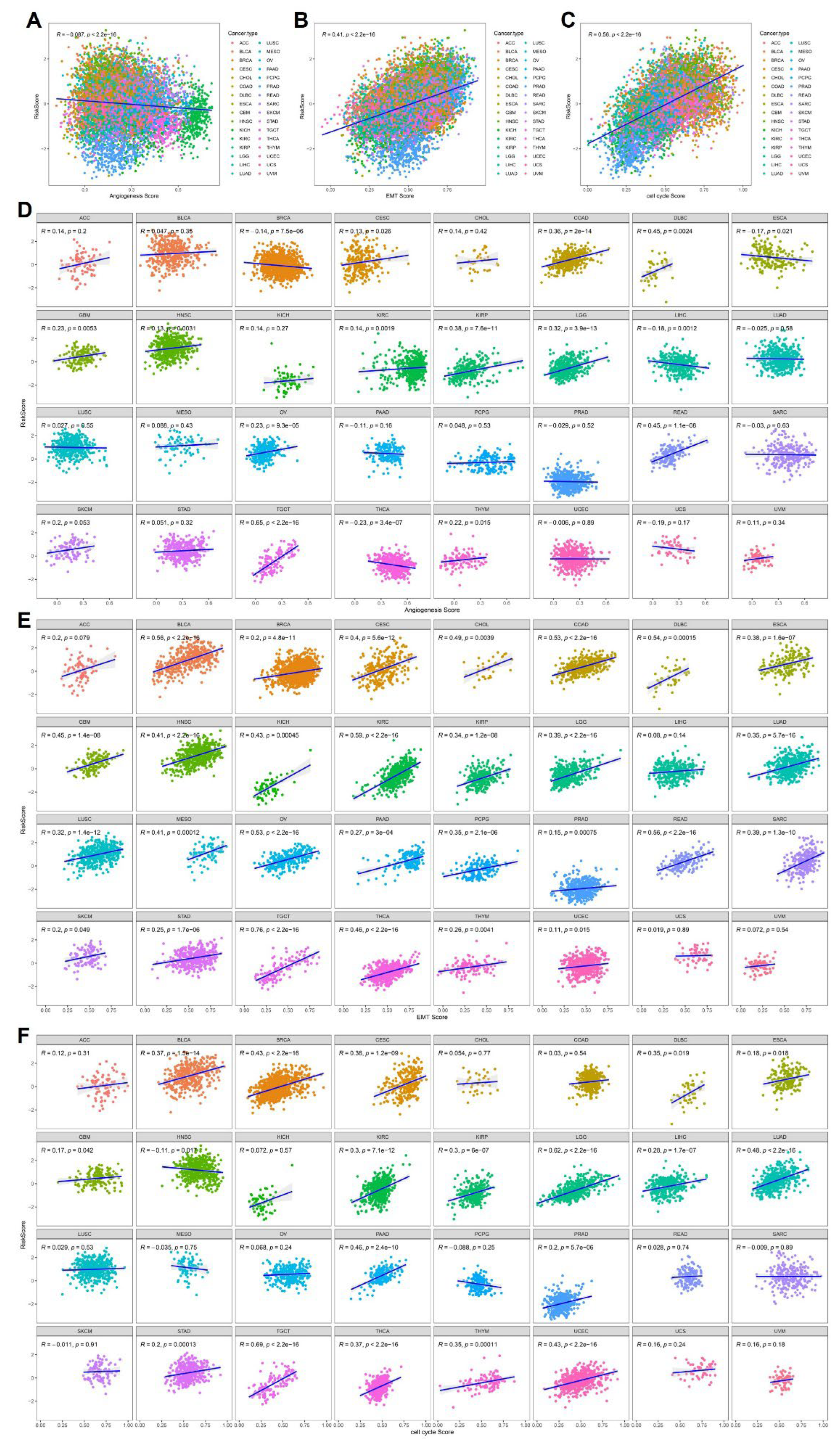
Figure 6.
Functional enrichment analysis of cytokine-related risk groups. (A) GO and KEGG enrichment results for genes upregulated in the high-risk group. (B) GO and KEGG enrichment results for genes upregulated in the low-risk group.
Figure 6.
Functional enrichment analysis of cytokine-related risk groups. (A) GO and KEGG enrichment results for genes upregulated in the high-risk group. (B) GO and KEGG enrichment results for genes upregulated in the low-risk group.
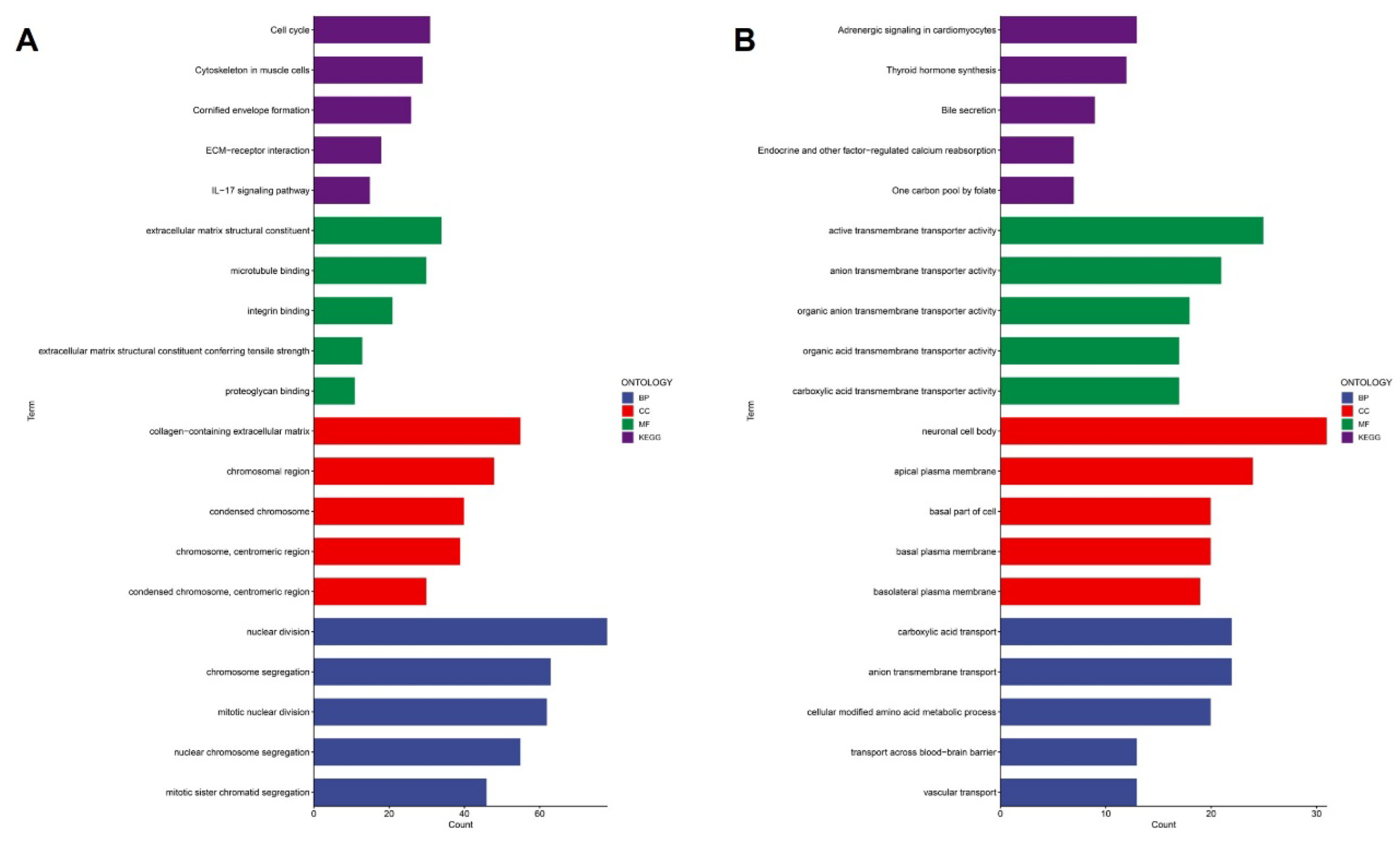
Figure 7.
Immunohistochemical validation of PANX1 and FRMD8 expression in normal and cancer tissues. (A-D) Representative immunohistochemical images showing PANX1 expression in normal tissues and corresponding cancers. (E-H) Representative immunohistochemical images showing FRMD8 expression in normal tissues and corresponding cancers.
Figure 7.
Immunohistochemical validation of PANX1 and FRMD8 expression in normal and cancer tissues. (A-D) Representative immunohistochemical images showing PANX1 expression in normal tissues and corresponding cancers. (E-H) Representative immunohistochemical images showing FRMD8 expression in normal tissues and corresponding cancers.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



