Submitted:
12 February 2026
Posted:
13 February 2026
You are already at the latest version
Abstract
The development of stable electrocatalysts with ultralow noble metal loading is a paramount challenge for sustainable electrochemical technologies. This work reports a disruptive one-pot synthesis strategy for the preparation of Pd-based nanocomposites at the nanogram-scale, utilizing the tunable redox properties of electrogenerated hydrophilic carbons (EHC). We demonstrate that EHC generated in tartaric buffer (EHC@T) acts as a multi-functional platform, serving simultaneously as a conductive support and reducing agent. Crucially, the integration of a second component, EHC generated in phosphate buffer (EHC@P), provides an architectural synergy: beyond its oxygen-storing capacity, EHC@P is shown to be indispensable for the chemical and electrochemical stabilization of the ultralow Pd loadings The resulting Pd–EHC@T,P nanocomposite was tested for the oxygen reduction reaction (ORR) activity, showing a unique 'oxygen-memory' effect: the persistence of significant reduction current even in deoxygenated media. These findings open a new pathway for developing cost-effective, sustainable nanocatalysts and enables the design of electrochemical applications previously considered unfeasible.
Keywords:
palladium
; hydrophilic carbon
; ORR
; oxygen storage
; selective synthesis
1. Introduction
The stability of electrocatalysts with ultralow noble metal loading represents a critical challenge in electrocatalysis, as the limited number of active metal sites renders the material highly sensitive to metal dissolution, agglomeration, or detachment from the support. These catalysts are typically designed either as isolated metal atoms dispersed on a suitable support or as nanoparticles anchored onto high-surface-area materials, such as carbon-based supports [1,2,3]. These strategies aim to maximize metal utilization while maintaining high catalytic activity and long-term stability.
Several techniques can be employed to prepare these systems, including physical vapor deposition, sputter deposition and atomic layer deposition [4,5]. Alternatively, chemical wet methods based on the use of reducing agents are among the simplest and most widely adopted [6,7,8]. However, these solution-based approaches often lead to the formation of atom clusters and nanoparticle agglomerates, limiting the uniformity and dispersion of the active sites [9,10]. Therefore, it is crucial to develop preparation strategies that provide control over metal atom distribution, particle size, and metal–support interactions.
Taking advantage of the tunable redox properties that electrogenerated hydrophilic carbon (EHC) [11,12], this work investigates its ability to promote, in a one-pot synthesis, the formation of palladium and platinum species well-anchored to a carbonaceous support. EHC, released from a graphite electrode during the early stages of polarization, is structurally dominated by aromatic sp² carbon atoms arranged in a non-ordered network, forming conductive two-dimensional nanostructured layers [13,14]. Its redox behaviour depends strongly on the synthesis electrolyte: in tartaric, malic, or citric buffer solutions, EHC exhibits pronounced electron-donating properties [11,12], whereas EHC generated in phosphate buffer, it displays a weak electron-accepting ability and a high capacity to chemisorb and retain molecular oxygen [15].
In this study, two distinct EHC variants are explored: EHC generated in tartaric buffer (labeled as EHC@T) utilized for its reductive character, and EHC generated in phosphate buffer (labeled as EHC@P), noted for its oxygen-retention capacity. By combining these materials, this study aims to explore strategies for designing oxygen-related electrocatalysts with ultralow metal loadings of Pd and Pt. This approach seeks to contribute to the development of oxygen-related electrocatalysts with potential application in a wide range of electrochemical technologies, including fuel cells and electrochemical sensors. Particularly in fuel cells, the hydrophilic nature of EHC provides additional advantages. It can improve the wettability of carbon-based gas diffusion layers (typically carbon paper or carbon cloth), ensuring intimate contact between the electrocatalyst and the aqueous electrolyte. This facilitates efficient transport of ionic species, enhances mass transport to the active sites, and ultimately improves the utilization of the metal centers.
2. Materials and Methods
2.1. Preparation of the Nanocomposite
EHC nanomaterials were synthesized by an electrochemical approach, in which the material was collected from the anodic compartment of a three-compartment cell equipped with graphite rod electrodes after polarization at a constant current of 0.06 A (8.5 mA cm-2) for 1 h. Electrogeneration was carried out using either a tartaric buffer solution (0.11 M, pH 4.51) or a phosphate buffer solution (0.20 M, pH 5.99) as the supporting electrolyte. As previously mentioned, the obtained carbon-based nanomaterial is designated as EHC@T and EHC@P, respectively. The cell was connected to an Autolab PGSTAT100 potentiostat/galvanostat (EcoChimie B.V.), controlled by a GPES software. The same potentiostat/software was used for the electrochemical characterization of the nanocomposite.
Nanocomposite suspensions were obtained by combining 2 mL of freshly prepared EHC@T (1.78 mg mL-1) with 40 µL of the metal precursor solution (PdCl₂, 9.45 mM or K2PtCl6, 9.40 mM), followed by the addition of 2 mL of freshly prepared EHC@P (17.6 ± 2.6 µg C mL-1). A control nanocomposite was prepared under identical conditions, except that the EHC@P suspension was replaced with deionized water. After preparation, all nanocomposite suspensions were allowed to stand for 2 h and subsequently subjected to dialysis for 16 h (Spectra/Por®, MWCO: 3.5–5 kDa). The dialysed nanocomposite solutions were stored in dark glass vials at -18 ◦C. This storage condition was adopted because preliminary results indicated that, at room temperature, EHC suspensions undergo progressive physicochemical changes leading to the “passivation” of the Pd nanoparticles, i.e. the formation of a layer that hinders electrolyte access to the catalytic sites [16]. Before use, vials were brought to room temperature and sonicated for 30 s to ensure redispersion prior to the drop-casting process.
2.2. Characterization of the Nanocomposites
To prepare the working electrodes, 12 µL of the nanocomposite suspension were drop-cast onto a glassy carbon (GC) electrode and allowed to dry at room temperature. Anodic stripping voltammetry was conducted in 1.0 M HCl in a single-compartment cell configuration comprising a GC working electrode (3 mm diameter), an Ag/AgCl (3 M KCl) reference electrode, and a platinum foil counter electrode. Further electrochemical characterization was performed in a 0.1 M H2SO4 electrolyte using a double-junction Ag/AgCl (sat.) reference electrode. All the potentials are reported against Reversible Hydrogen Electrode (RHE) scale, except for the anodic stripping voltammograms, which were recorded against the Ag/AgCl (sat.) scale. Prior to measurements, the electrolyte solution was deaerated with argon for 30 min, and an argon atmosphere was maintained over the solution throughout the voltammetric experiments.
The electrocatalytic activity towards the oxygen reduction reaction (ORR) was evaluated in an O2-saturated 0.1 M H2SO4 solution. For these measurements, the nanocomposite coating was covered with 3 μl of 5 wt% Nafion® solution (Aldrich).
Transmission electron microscope (TEM) was performed using a Hitachi STEM HD2700 at 200 kV. A drop of the particle suspension was deposited onto a continuous carbon grid, which was dried prior to introduction into the microscope.
3. Results and Discussion
3.1. Electrochemical Characterization of the Metal Nanocomposites
The initial assessment of whether Pd(II) and Pt(IV) species could be successfully reduced by the EHC@T solution was performed using anodic stripping voltammetry (ASV) on the modified glassy carbon electrodes (GCE) with the respective Pd-EHC@T and Pt-EHC@T solutions. As shown in Figure 1a, a well-defined anodic stripping peak was observed for the Pd-based sample, providing clear evidence of metallic Pd formation via the electron-donating properties of EHC@T. To verify if Pd(II) ions were reduced by the tartaric buffer itself, rather than by the carbon nanomaterial, a control experiment was conducted in which the tartaric buffer was mixed with the palladium salt to form a blank solution, no Pd oxidation peaks were observed. Regarding the electrode modified with Pt-EHC@T, no oxidation signal was detected for platinum, suggesting that the Pt(IV) species from the [PtCl6]2- precursor were not reduced to Pt0 by the EHC@T. This was further confirmed by TEM/EDS analysis, which showed no evidence of platinum nanoparticles or atomic clusters. Even though the standard reduction potential for the [PtCl62-]/Pt couple (E0=0.717V) is lower than that of Pd2+/Pd (E0= 0.915 V) [17], making the reduction of Pd species thermodynamically more favourable, this selective reduction may also be attributed to the sluggish multi-electron transfer kinetics required for the Pt(IV) to Pt0 transformation.
Interestingly, when Pd0 is formed in the presence of EHC@P (sample Pd-EHC@T,P), Figure 1b, a significant increase in the anodic peak height is observed compared to Pd–EHC@T. Integration of the ASV anodic peaks allowed for a comparative evaluation of the respective palladium loadings. The results indicate that the loading in the Pd–EHC@T,P nanocomposite is approximately twice that of Pd–EHC@T (91.6 ng cm⁻² and 51.5 ng cm⁻², respectively). This suggests that the inclusion of EHC@P plays a synergistic role in the immobilization and retention of Pd species, possibly through additional anchoring sites or oxygen-containing functional groups that stabilize the metal centers.
Notably, these values are several orders of magnitude lower than those typically categorized as 'ultra-low' in the literature, which often fall within the 1.4-50 μg cm-2 range [5,18]. While achieving nanogram-scale loading is desirable for cost-efficiency, it raises serious concerns regarding long-term stability, a challenge that requires robust anchoring strategies.
In this context, the stability of these nanocomposites was evaluated using different approaches. In one such approach, the Pd loading of the original stock solution of Pd–EHC@T,P and Pd–EHC@T solutions was monitored regularly over a period of 75 days, being defrosted for periodic electrochemical analysis and subsequently refrozen at −18 °C. For the Pd–EHC@T,P nanocomposite, the anodic stripping charge (measured via anodic oxidation in 1.0 M HCl) remained remarkably stable, changing from 11.8 μC to 10.3 μC (Figure 2), which represents a minimal variation of 12.7%. This was accompanied by a minor anodic shift of only 15 mV.
In contrast, the Pd–EHC@T material exhibited a more pronounced decrease in the stripping charge, from 6.64 μC to 4.94 μC (a 25.6% reduction), alongside a noticeable decrease in the full width at half maximum (FWHM) of the stripping peak. Rather than a chemical loss of the metal, this decline may be attributed to a gradual loss of palladium accessibility. In the absence of the EHC@P stabilizer, structural changes or adsorption of residual species may hinder the electrolyte’s ability to reach and oxidize the palladium centers. This trend is consistent with our preliminary findings [15], where storage at room temperature led to a near-total loss (~100%) of the stripping response. These results further highligh the critical role of both the EHC@P support and low-temperature storage in preserving the electrochemical availability of the metal under rigorous storage conditions.
The synergistic effect of EHC@P and EHC@T on the stability of palladium was further demonstrated by recording consecutives cyclic voltammograms in 0.1 M H2SO4 using a glassy carbon electrode (GCE) modified with the nanocomposites, Figure 3.
For the Pd–EHC@T modified electrode, the currents associated with the hydrogen adsorption/desorption peaks and the palladium oxide reduction peak decrease rapidly, becoming undetectable after few scans. In contrast, the Pd–EHC@T,P electrode also showed a decrease in current, but the decay was significantly less pronounced. This current reduction is likely a reflection of palladium dissolution at the highly anodic potentials of the scan. Consequently, these results reinforce the conclusion that EHC@P is essential for effectively anchoring the palladium nanoparticles, thereby mitigating metal dissolution and surface area loss during potential cycling. The enhanced stability of Pd–EHC@T,P suggests a stronger interaction between palladium species and the modified support, which will be further corroborated by morphological analysis.
Given the different metal loadings of the immobilized materials, the cyclic voltammograms recorded in 0.1 M H2SO4 were normalized by both the electrode geometric area and the catalyst mass (Figure 4). The mass-normalized voltammograms revealed that the electrochemical activity of Pd–EHC@T,P is superior to that of Pd–EHC@T, as evidenced by the enhanced voltammetric features for the hydrogen adsorption/desorption and palladium oxide formation/reduction regions. This indicates that the synergistic interaction between the two EHC supports not only increases the amount of retained palladium, but also ensures a higher dispersion of the metal, keeping the active sites more accessible for electrocatalytic reactions.
The electrochemical surface area (ECSA) was estimated from the charge associated with the PdO reduction peak. However, a rigorous ECSA determination was hindered by the inherent instability of the palladium surface at high anodic potentials. It was observed that the reduction charge increased with the anodic scan limit up to approximately 1.41 V vs. RHE (1.15 V vs. Ag/AgCl), but began to decrease at more positive potentials, suggesting the onset of Pd dissolution. Consequently, while the experimental charge for a complete PdO monolayer could not be precisely determined, the values obtained at a scan limit of 1.26 V vs. RHE (1.0 V vs. Ag/AgCl) were used for comparative purposes. The resulting apparent ECSA values were 74.2 m2 g-1 for Pd–EHC@T,P and 58.5 m2 g-1 for the Pd–EHC@T material. This ~27% increase in mass-specific area confirms that the addition of EHC@P effectively minimizes the formation of large clusters and maximizes the exposure of active sites, a crucial factor for achieving high catalytic efficiency at ultralow metal loadings. The calculated ECSA values are consistent with those reported in the literature for commercial Pd/C [18,19] and palladium nanoparticles supported on carbon-based materials such as graphene, reduced graphene oxide and carbon nanotubes [20,21], despite the significantly higher metal loadings typically employed in those studies.
To evaluate the size, morphology, and dispersion of the palladium entities, TEM/HRTEM and STEM analyses were performed using different imaging modes (Figure 5). The micrographs, obtained via transmitted and secondary electrons, show that in both samples the nanoparticles appear to be intimately embedded in or anchored to the carbon matrix. For the Pd–EHC@T sample, the images reveal the presence of relatively large aggregates of palladium nanoparticles (Pd NPs) that are heterogeneously distributed over the EHC support (Figure 5a´and 5c´), with individual particles diameters ranging between 5 and 22 nm. In contrast, the Pd–EHC@T,P nanocomposite exhibits a highly uniform distribution of smaller nanoparticles across the entire carbon framework, with a much narrower particle size distribution (3 to 5 nm). The crystalline nature of these nanoparticles was further confirmed by high-resolution TEM (Figure 5d and Figure 5d´), where lattice fringes are clearly visible. These morphological and structural observations are in excellent agreement with the electrochemical data. The superior dispersion and smaller particle size of the Pd–EHC@T,P effectively increase the available surface area, which explains its enhanced electrochemical activity. Furthermore, the smaller and well-anchored nanoparticles in Pd–EHC@T,P are less susceptible to the metal dissolution that led to the more pronounced current decay observed in consecutive voltammograms in Pd–EHC@T sample.
3.2. Electrochemical Activity for the Oxygen Reduction
The electrocatalytic performance of the Pd–EHC@T and Pd–EHC@T,P nanocomposites toward the oxygen reduction reaction (ORR) was evaluated in acidic medium. Linear sweep voltammetry (LSV) measurements were carried out from the open-circuit potential (OCP) to −0.15 V vs. RHE in an oxygen-saturated 0.1 M H₂SO₄ solution. As shown in Figure 6a and summarized in Table 1, the Pd–EHC@T,P catalyst exhibits improved ORR performance relative to Pd–EHC@T, as reflected by a more positive onset potential (0.669 V vs. RHE versus 0.605 V for Pd–EHC@T) and higher mass and specific activities.
Following the initial ORR characterization (Figure 6a), the influence of a Nafion overlayer, commonly employed in the fabrication of membrane electrode assemblies (MEAs), was subsequently investigated (Figure 6b). For the Pd–EHC@T electrode, the presence of Nafion leads to a pronounced decrease in current density in the diffusion-limited region. Notably, this effect is not observed in the kinetic region. Since intrinsic catalytic descriptors are extracted from the kinetically controlled region, these results indicate that the addition of Nafion does not alter the intrinsic ORR activity of the Pd active sites in the Pd–EHC@T system. The reduction in current density observed at high overpotentials can therefore be ascribed to additional mass transport resistance introduced by the Nafion layer, most likely associated with hindered oxygen diffusion through the ionomer film. In contrast, the Pd–EHC@T,P electrode displays nearly identical current densities across the examined potential range. Although a detailed mechanistic interpretation is beyond the scope of this study, this behaviour suggests a more favourable interaction between the Pd–EHC@T,P catalyst layer and the ionomer, effectively mitigating mass transport limitations.
To investigate the oxygen-retention properties of the nanocomposites, the electrochemical response was monitored in an O2-free 0.1 M H2SO4 solution after pre-exposing the electrodes to an O2-saturated medium. It is important to note that the presence of the Nafion overlayer was found to be essential for observing any sustained current, without the ionomer, the ORR signal dropped immediately upon transfer, even for the modified electrode with Pd–EHC@T,P. For the Pd–EHC@T,P/Nafion system (Figure 7a), a significant ORR current is observed, even 5 minutes after transfer, maintaining a significant limit current density of -0.22 mA cm⁻². This result provides direct evidence that the EHC@P support possesses an intrinsic ability to store oxygen within its porous structure. In this case, the Nafion film successfully traps the O2 that was previously adsorbed or absorbed by the phosphorus-templated carbon matrix, allowing it to be gradually reduced at the Pd active sites. In contrast, the Pd–EHC@T/Nafion system (Figure 7b) fails to show significant oxygen retention. Under identical conditions, the limit current density at 5 minutes is nearly negligible (~ -0.01 mA cm⁻²). Even with the Nafion 'seal' in place, the lack of a current signal confirms that the EHC@T support is unable to store oxygen. These findings demonstrate that the synthesis of the carbon support in phosphate buffer (EHC@P) creates a specialized architecture capable of oxygen sequestration. While the Nafion layer is necessary to reveal this effect by preventing rapid degassing, the experiment proves that only the Pd–EHC@T,P nanocomposite functions as a self-sustaining electrocatalytic system in oxygen-depleted conditions, whereas Pd–EHC@T lacks this storage functionality.
4. Conclusions
This work successfully demonstrates a sustainable and efficient strategy for the one-pot preparation of electrocatalysts with nanogram-scale noble metal loading using electrogenerated hydrophilic carbon (EHC). We established that the redox properties of the EHC support, dictated by the synthesis electrolyte, play a decisive role in the formation and stabilization of the active sites. Specifically, EHC generated in tartaric buffer (EHC@T) exhibited a selective reducing ability, successfully forming Pd species while remaining inactive towards Pt(IV) reduction.
The most significant finding, however, lies in the synergistic effect between EHC@T and EHC generated in phosphate buffer (EHC@P) for the stability of remarkably low palladium loadings (nanogram scale). The inclusion of EHC@P not only doubled the palladium loading but also proved essential for the material's long-term integrity. While Pd–EHC@T showed rapid degradation, the Pd–EHC@T,P nanocomposite remained stable for over 75 days, even when subjected to multiple freeze-thaw cycles. This robustness indicates a strong metal-support interaction, likely mediated by oxygen-containing functional groups in the EHC@P network that act as anchoring sites. Furthermore, the nanocomposites achieved electrochemical surface area (ECSA) values comparable to state-of-the-art catalysts with significantly higher loadings, while exhibiting a unique oxygen-retention capability that sustains ORR activity even in deoxygenated environments.
The hydrophilic nature and stability of these nanocomposites, combined with their unprecedently low metal content, position them as promising candidates for electrochemical devices where minimizing noble metal usage under transient oxygen supply is a critical challenge.
Funding
This research was funded by the Fundação para a Ciência e Tecnologia, grant number UIDB/00616/2025, UIDP/00616/2025 and by the RISE Project - Smart Cities, Sustainable Energy and Materials, under operation code NORTE2030-FEDER-02723100, funded by the European Regional Development Fund (ERDF/FEDER), through Incentives for Research, Development and Innovation, under the North Regional Program 2021-2027 [NORTE2030].
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Islam, M; Basha, A.; Kollath, V.; Soleymani, A.; Jankovic, J.; Karan, K. Designing fuel cell catalyst support for superior catalytic activity and low mass-transport resistance. Nature Communications, 2022, 13, 6157. [CrossRef]
- Lokteva, E. ; Golubina, E. Metal-support interactions in the design of heterogeneous catalysts for redox processes. Pure Appl. Chem., 2019, 91 (4) 609-631. [CrossRef]
- Ren, XJ ; Quan, YH ; Yang, W ; Zhao, JX ; Shi, RA ; Ren, J. Highly efficient super activated carbon supported ultra-low loading copper catalyst for the oxidative carbonylation of methanol to dimethyl carbonate, Molecular Catalysis 2022, 531, 112694. [CrossRef]
- Guo, F; Macdonald, T; Sobrido, A.; Liu, L.; Feng, J and He, G. Recent Advances in Ultralow-Pt-Loading Electrocatalysts for the Efficient Hydrogen Evolution. Adv. Sci. 2023, 10, 2301098. [CrossRef]
- Lori, O.; Elbaz, L. Recent Advances in Synthesis and Utilization of Ultra-low Loading of Precious Metal-based Catalysts for Fuel Cells. ChemCatChem 2020,12,3434–3446. [CrossRef]
- Kharissova, O; Dias, H ; Kharisov, B ; Pérez, B ; Pérez, V. The greener synthesis of nanoparticles. Trends in Biotechnology 2013, 31(4) 240-249. [CrossRef]
- Saldan, I; Semenyuk, Y; Marchuk, I; Reshetnyak, O. Chemical synthesis and application of palladium nanoparticles. Mater Sci. 2015 50:2337–2354. [CrossRef]
- Chen A.; Ostrom; C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999-12044. [CrossRef]
- Xia, Y.; Xiong, Y.; Lim, B.; Skrabalak. Shape-Controlled Synthesis of Metal Nanocrystals: Simple Chemistry Meets Complex Physics?. A New. Chem. Int. Ed. 2009, 48, 60, 103. [CrossRef]
- Zadick, A.; Dubau, L.; Demirci, Umit; Chatenet, M. Effects of Pd Nanoparticle Size and Solution Reducer Strength on Pd/C Electrocatalyst Stability in Alkaline Electrolyte. J. Electrochem. Soc. 2016, 163, F781-F787. [CrossRef]
- Veloso, A. D.; Videira, R. A.; Oliveira, M. C. An Electrochemical Strategy for Synthesising Carbon-Based Nanomaterials with Tuned Redox Properties. Electrochimica Acta 2022, 404, 139734. [CrossRef]
- Veloso, A. D.; Ferraria, A. M.; Videira, R. A.; Oliveira, M. C. Advances in the Understanding of the Electron-Donating Properties of Carbon-Based Nanomaterials Electrogenerated from Graphite. Electrochimica Acta 2025, 525, 146071. [CrossRef]
- Oliveira, M. C.; Viana, A. S.; Botelho do Rego, A. M.; Ferraria, A. M.; Tavares, P. B.; Veloso, A. D.; Videira, R. A. Dual Behaviour of Amorphous Carbon Released Electrochemically from Graphite. ChemistrySelect 2016, 1 (13), 4126–4130. [CrossRef]
- Vieira, R., Fernandes, A., Oliveira, M. Electrochemical behaviour of electrogenerated hydrophilic carbon nanomaterials. Electrochimica Acta 2018, 260, 338e34. [CrossRef]
- Veloso, A. D.; Ferraria, A. M.; Botelho Do Rego, A. M.; Viana, A. S.; Fernandes, A. J. S.; Fielding, A. J.; Videira, R. A.; Oliveira, M. Structural Effects Induced by Dialysis-Based Purification of Carbon Nanomaterials. Nano Mater. Sci. 2024, 6 (4), 475–483. [CrossRef]
- Ouanzi, I.; and Oliveira M. Characterization of Pd Nanocomposites for the Oxygen-Reduction Reaction. Proceedings 2025, 133, 1- 4. [CrossRef]
- A. J. Bard, R. Parsons, and J. Jordan. Standard Potentials in Aqueous Solution (New York: Marcel Dekker, 1985).
- Tiwari, J.; Sultan, S.; Myung, C.; Yoon, T.; Li, N.; Ha, M.; Kim, K. et al. Multicomponent electrocatalyst with ultralow Pt loading and high hydrogen evolution activity. Nature Energy 2018, 3 773-782. [CrossRef]
- Wang, Q.; Liao, Y.; Zhang, H.; Li, J.; Zhao, W.; Chen, S.; One-pot synthesis of carbon-supported monodisperse palladium nanoparticles as excellent electrocatalyst for ethanol and formic acid oxidation. J. Power Source 2015, 292 72-77. [CrossRef]
- Zhao, J.; Liu, Z.; Li, H.; Hu, W.; Zhao, C.; Zhao, P.; Shi, D.; Development of a Highly Active Electrocatalyst via Ultrafine Pd Nanoparticles Dispersed on Pristine Graphene. Langmuir 2015, 31, 2576−2583. [CrossRef]
- Zhao, H.; Yang, J.; Wang, L.; Tian, C.; Jiang, B.; Fu, H. Fabrication of a palladium nanoparticle/graphene nanosheet hybrid via sacrifice of a copper template and its application in catalytic oxidation of formic acid. Chem. Commun. 2011, 47 (7), 2014−2016.
Figure 1.
- Anodic stripping voltammograms in 0.1 M HCl solution of GC electrodes modified with: a) Pd-EHC@T (_____), Pd blank (- - - ), Pt-EHC (____), b) Pd-EHC@T,P (____), ν = 5 mV s−1.
Figure 1.
- Anodic stripping voltammograms in 0.1 M HCl solution of GC electrodes modified with: a) Pd-EHC@T (_____), Pd blank (- - - ), Pt-EHC (____), b) Pd-EHC@T,P (____), ν = 5 mV s−1.
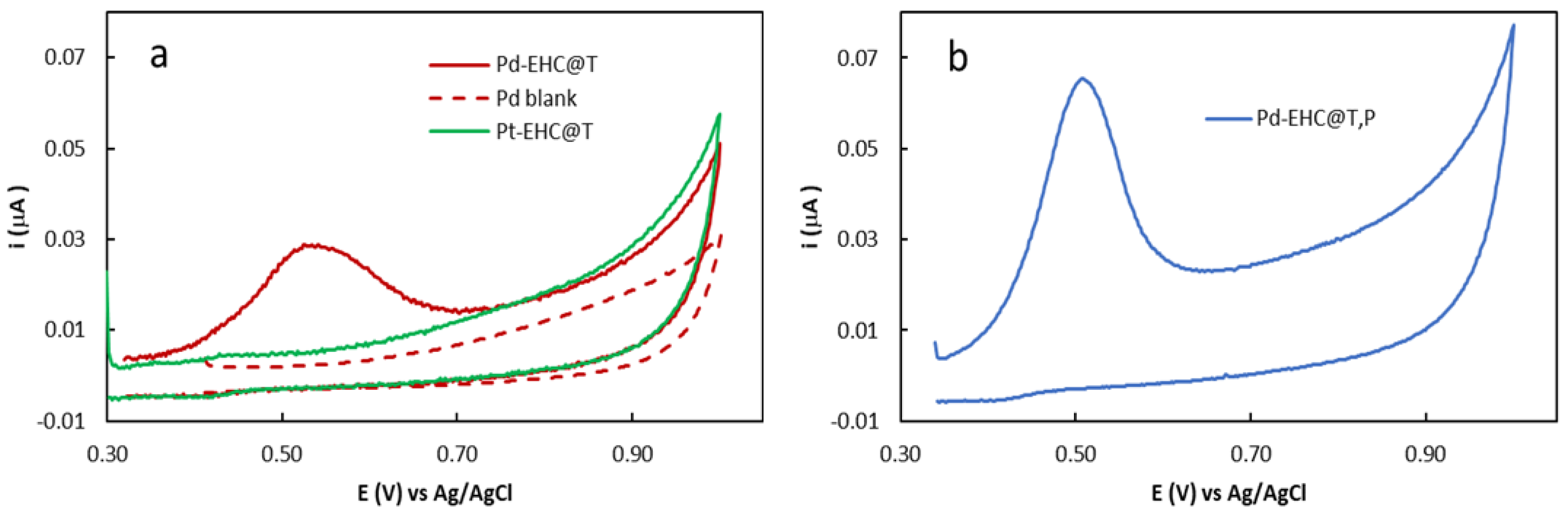
Figure 2.
Anodic stripping voltammograms in 0.1 M HCl solution of GC electrodes modified with: a) Pd-EHC@T,P and b) Pd-EHC@T nanocomposites, recorded after different storage periods (bold- as prepared, dashed- stored for 75 days), ν = 5 mV s−1.
Figure 2.
Anodic stripping voltammograms in 0.1 M HCl solution of GC electrodes modified with: a) Pd-EHC@T,P and b) Pd-EHC@T nanocomposites, recorded after different storage periods (bold- as prepared, dashed- stored for 75 days), ν = 5 mV s−1.
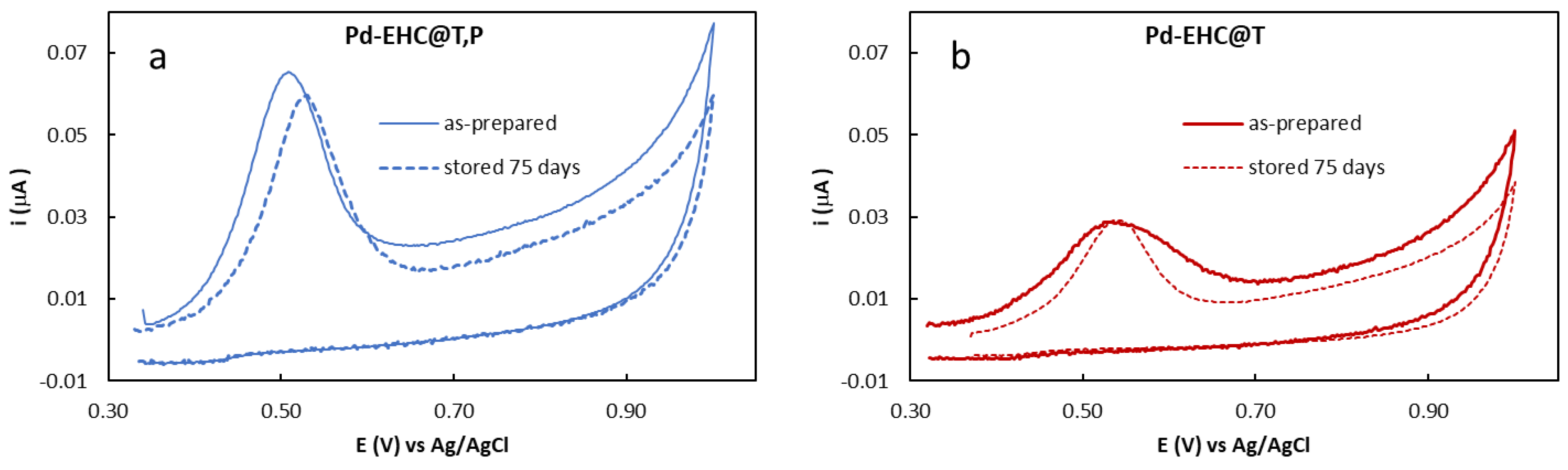
Figure 3.
Consecutive voltammogrmas (ten scans) recorded in 0.1 M H2SO4 solution using glassy carbon electrodes modified with Pd-EHC@T,P or Pd-EHC@T, ν = 50 mV s−1.
Figure 3.
Consecutive voltammogrmas (ten scans) recorded in 0.1 M H2SO4 solution using glassy carbon electrodes modified with Pd-EHC@T,P or Pd-EHC@T, ν = 50 mV s−1.
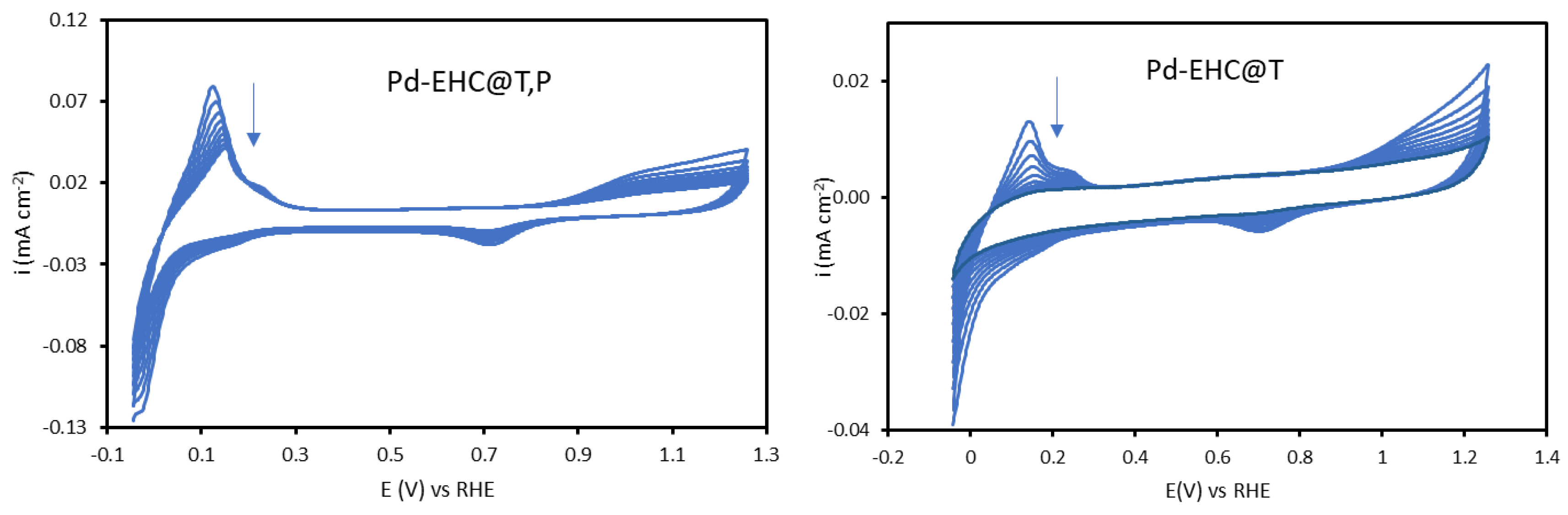
Figure 4.
- Mass-normalized voltammograms (corresponding to second scan) recorded in 0.1 M H2SO4 solution using glassy carbon electrodes modified with Pd-EHC@T,P and Pd-EHC@T after identical storage period, ν = 50 mV s−1.~.
Figure 4.
- Mass-normalized voltammograms (corresponding to second scan) recorded in 0.1 M H2SO4 solution using glassy carbon electrodes modified with Pd-EHC@T,P and Pd-EHC@T after identical storage period, ν = 50 mV s−1.~.
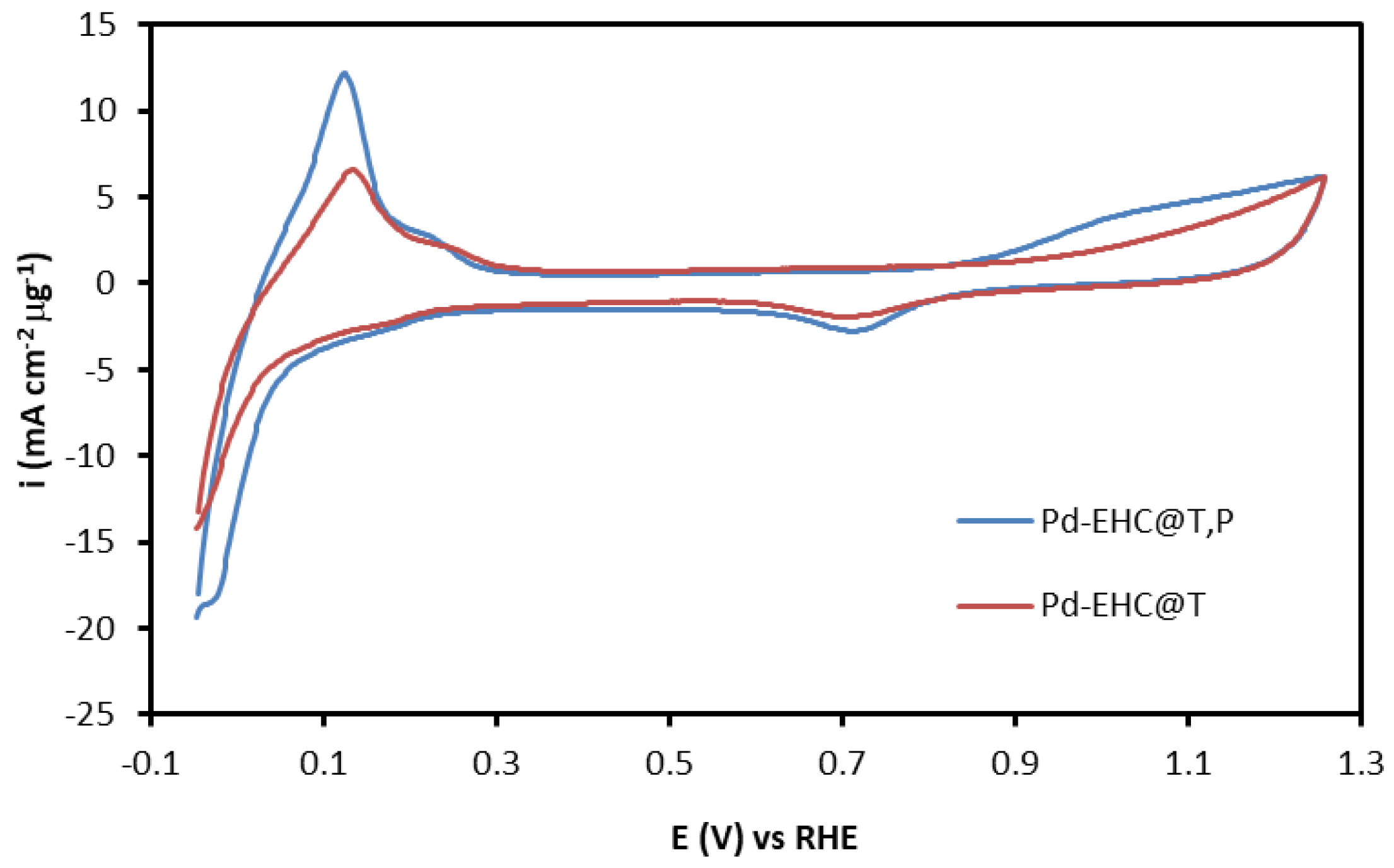
Figure 5.
– TEM and HRTEM analysis of Pd-EHC@T and Pd-EHC@T,P samples recorded using transmitted electrons (a, b, d, a´, d´) and secondary electrons (c, c´).
Figure 5.
– TEM and HRTEM analysis of Pd-EHC@T and Pd-EHC@T,P samples recorded using transmitted electrons (a, b, d, a´, d´) and secondary electrons (c, c´).
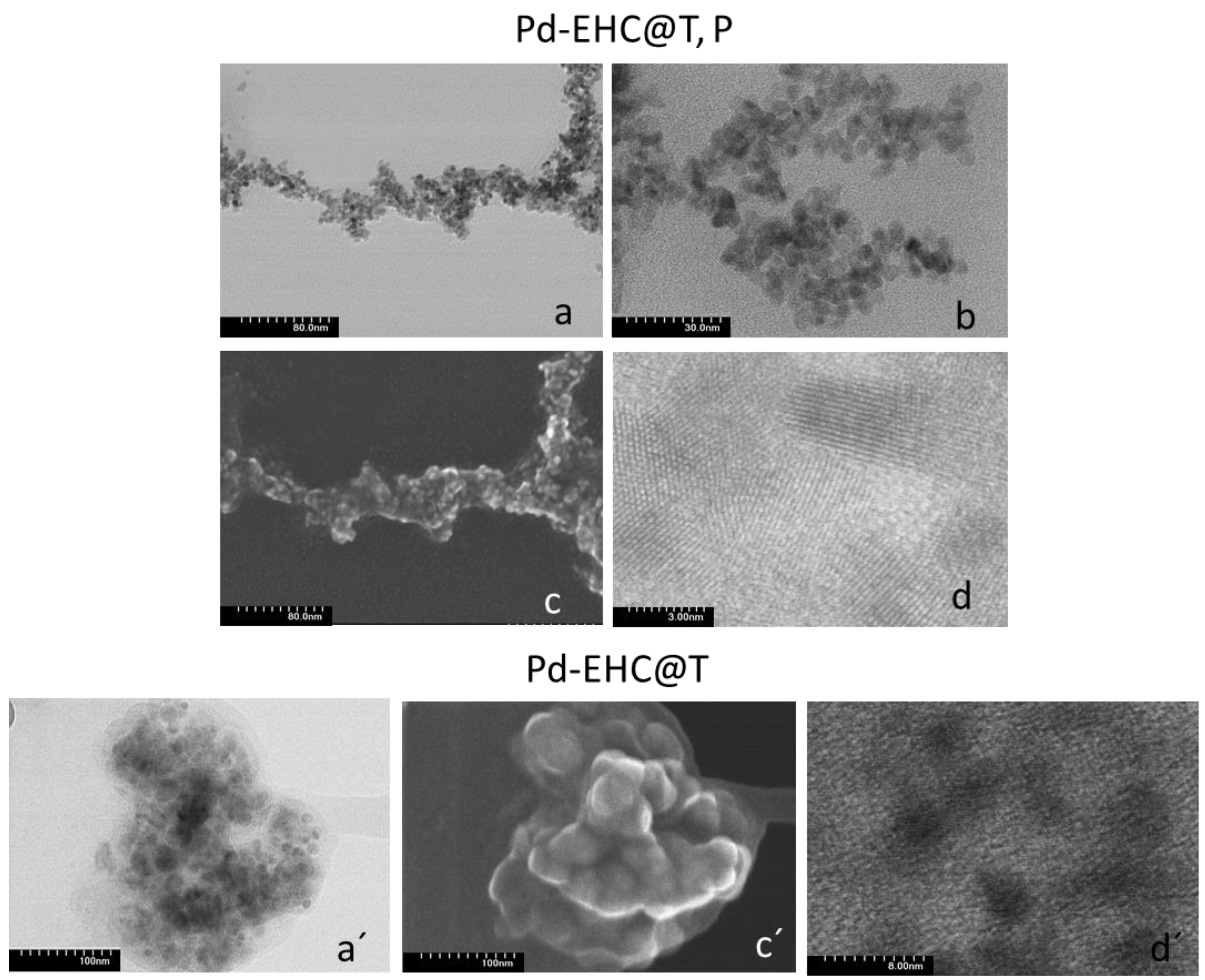
Figure 6.
– Linear voltammograms of GC electrode modified with: a) Pd-EHC@T and Pd-EHC@T,P and b) Pd-EHC@T/Nafion and Pd-EHC@T,P/Nafion in O2-saturated 0.1 M H2SO4 solution. ν=50 mV s-1.
Figure 6.
– Linear voltammograms of GC electrode modified with: a) Pd-EHC@T and Pd-EHC@T,P and b) Pd-EHC@T/Nafion and Pd-EHC@T,P/Nafion in O2-saturated 0.1 M H2SO4 solution. ν=50 mV s-1.
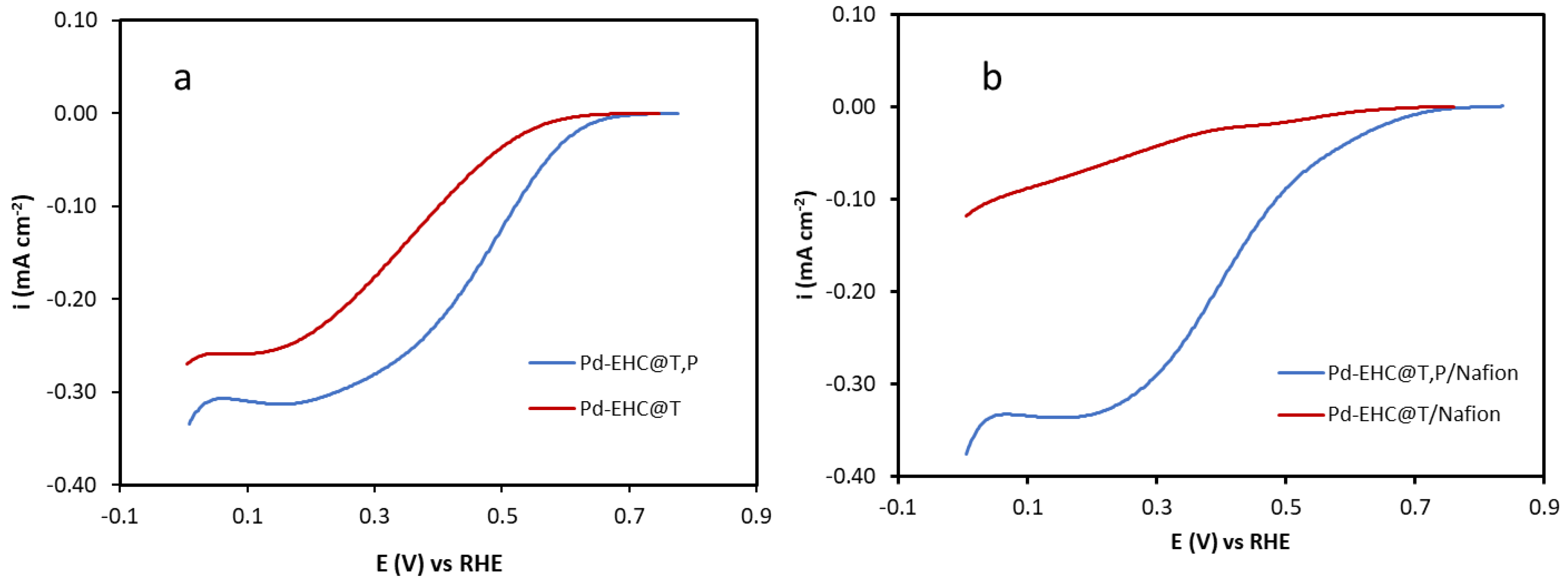
Figure 7.
Electrochemical response of glassy carbon electrodes modified with: a) Pd-EHC@T,P/Nafion, b) Pd-EHC@T/Nafion, in deoxygenated 0.1 M H2SO4 solution after x minutes of conditioning in an O₂-saturated solution.
Figure 7.
Electrochemical response of glassy carbon electrodes modified with: a) Pd-EHC@T,P/Nafion, b) Pd-EHC@T/Nafion, in deoxygenated 0.1 M H2SO4 solution after x minutes of conditioning in an O₂-saturated solution.
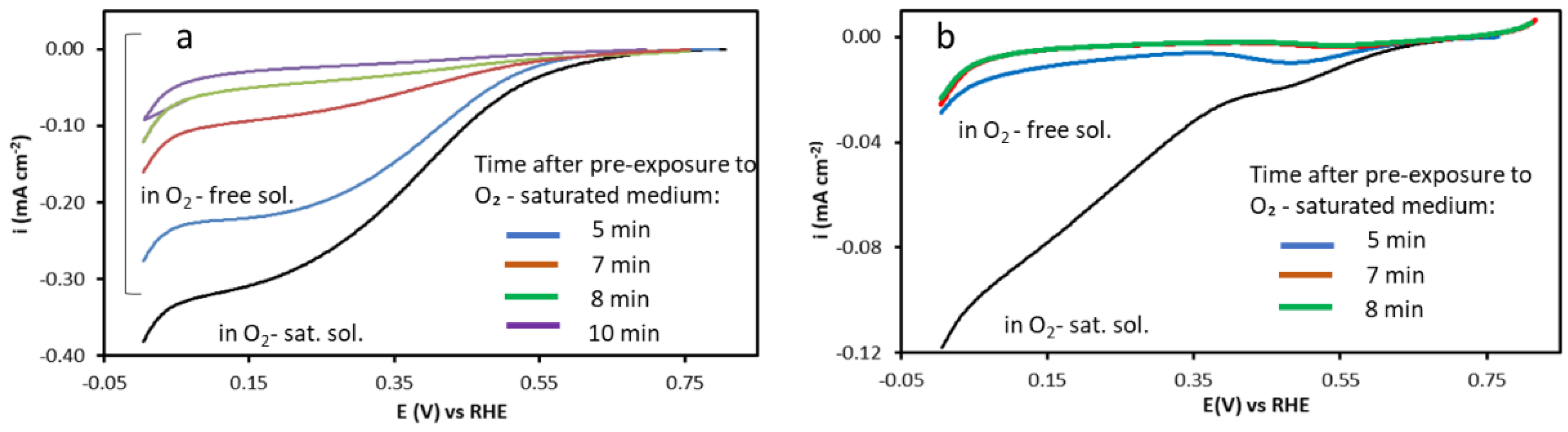

Table 1.
Catalytic activity descriptors for the oxygen reduction reaction (ORR) of GCE modified with Pd-EHC@T or Pd-EHC@T,P solutions in O₂-saturated 0.1 M H₂SO₄ solution.
Table 1.
Catalytic activity descriptors for the oxygen reduction reaction (ORR) of GCE modified with Pd-EHC@T or Pd-EHC@T,P solutions in O₂-saturated 0.1 M H₂SO₄ solution.
| Modified electrodes | Eonset at -5.0 μA cm-2 (V vs RHE) |
Mass activity at 0.55 V (A g-1) |
Specific activity at 0.55 V (mA cm-2) |
| Pd-EHC@T | 0.605 | 0.323 | 0.554 |
| Pd-EHC@T,P | 0.669 | 0.759 | 1.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



