
Submitted:
10 February 2026
Posted:
11 February 2026
You are already at the latest version
Abstract
Ferroptosis is an iron-dependent cell death driven by lipid peroxidation and failure of cellular antioxidant defenses. It is triggered by oxidative stress and can be aggravated by aging, inflammation, and dysregulation of iron homeostasis. In the central nervous system, iron dyshomeostasis, mitochondrial dysfunction, and membrane lipid remodeling can amplify oxidative injury and increase susceptibility to ferroptotic damage, particularly in vulnerable neurons. There is growing evidence that ferroptosis-related processes are linked to Alzheimer's disease, Parkinson's disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis. This review addresses novel approaches to track ferroptosis in vivo, such as imaging and biomarker techniques, and important molecular mechanisms linking iron metabolism, reactive oxygen species, and PUFA-driven lipid peroxidation to neuronal damage. We also explore upstream transcriptional control via NRF2, iron chelation and iron-handling modulation, inhibition of lipid peroxidation, and reinforcement of the System Xc-GSH-GPX4 and CoQ10-linked defense pathways. Subsequently, we highlight translational issues that need attention to further progress ferroptosis-targeted therapies for neurodegenerative disease.
Keywords:
ferroptosis
; neurodegeneration
; iron homeostasis
; lipid peroxidation
; GPX4
; oxidative stress
1. Introduction
Ferroptosis is a regulated form of cell death that is dependent on iron. It is mainly caused by extensive accumulation of lipid peroxides, particularly phospholipid hydroperoxides, which develop from oxidative stress and are distinct from other forms of cell death [1,2]. Ferroptosis plays a major role in the pathogenesis of various diseases such as cancer, ischemic organ injury, cardiac, and neurodegenerative diseases [3,4]. This oxidative-mediated cell death can exhibit mitochondrial abnormalities such as altered structure and impaired function due to lipid peroxidation-mediated membrane damage [5]. In normal cells, stress from physiological and pathological conditions can trigger an abnormal increase in the labile Fe2+ pool, particularly within endolysosomal compartments and other organelles, thereby promoting ferroptosis. Oxidative stress is characterized by an imbalance between the production of reactive oxygen species (ROS) and antioxidant defenses, which activate transcription factors and genes that alter immune cell signaling and increase pro-inflammatory cytokine release, leading to inflammation. One of the key proteins involved in ferroptosis is transferrin receptor 1 (TfR1), a cell surface receptor that facilitates iron uptake by binding iron-bound transferrin (Tf). Iron enters the neuron via clathrin-mediated endocytosis of TfR1 and exits endosomes as reduced forms of Fe2+ [2,6]. Ferroptosis is induced by lipid peroxidation and the accumulation of phospholipid hydroperoxides (PLOOH), with reactive aldehydes such as 4-hydroxynonenal (4-HNE) often serving as downstream markers [7]. In contrast, glutathione peroxidase 4 (GPX4) and glutathione (GSH) help maintain cellular redox homeostasis and suppress ferroptosis as key antioxidant defense mechanisms [1,2]. GPX4 converts active PLOOH to inactive PLOH and inhibits lipid peroxidation. Overexpression of GPX4 prevents RSL3-mediated ferroptosis while inhibition of GPX4 promotes it. As GSH is necessary for GPX4 activity, inhibition of system Xc-(SLC7A11) decreases GSH synthesis, cystine import, and GPX4-dependent detoxification of lipid peroxides, all of which are responsible for ferroptosis [8]. These lipid repair systems depend on a set of coordinated enzymatic reactions, such as PUFA activation and their incorporation into membrane phospholipids, which serve as important peroxidation substrates [9]. These key characteristics are significant contributors to neurodegenerative diseases.
Ferroptosis is increasingly recognized as a major contributor to neurodegeneration. It plays a similar role in diseases like Parkinson’s, Alzheimer’s, Huntington’s, and Amyotrophic Lateral Sclerosis (ALS), where iron imbalance, oxidative stress, mitochondrial dysfunction, and neuroinflammation often coexist. In this article, we will highlight the role of ferroptosis in neurodegeneration, available interventions, future therapeutics, and challenges involved in its regulation.
2. Ferroptosis Mechanistic Drivers
Mechanistically, ferroptosis reflects a failure of lipid peroxide control rather than a single linear pathway. The drivers can be grouped into processes that increase lipid peroxide formation and those that weaken their detoxification and repair.
2.1. Oxidative Stress and Lipid Peroxidation
Oxidative stress plays a vital role in the induction of ferroptosis. The imbalance between ROS generation and antioxidant levels leads to oxidative stress, triggering the activation of various transcription factors [2]. This oxidative stress often results in oxidation of polyunsaturated fatty acids (PUFA) in membrane phospholipids, which can alter membrane structure and increase membrane permeability [10]. Consequently, the plasma membrane may lose integrity due to the accumulation of lipid hydroperoxides [2]. These peroxides further promote the production of toxic aldehydes, which could inactivate cellular proteins and promote ferroptosis [2]. Oxidative stress and lipid peroxidation play a key role in driving ferroptosis, resulting in irreversible cell death.
Moreover, ferroptosis can also be triggered by increased intracellular iron availability, which expands the labile Fe2+ pool and accelerates ROS-driven lipid peroxidation [11,12]. The nuclear receptor coactivator 4 (NCOA4) mediates ferritinophagy, the autophagic degradation of ferritin, which is the primary cellular iron storage complex [13]. This process releases free iron, potentially increasing intracellular Fe2+ concentrations and initiating ferroptosis. Under conditions of oxidative stress, the Fenton reaction generates reactive intermediates that damage membrane lipids and facilitate PUFA peroxidation [12,14]. The Fenton reaction occurs when Fe2+ ions react with hydrogen peroxide, producing hydroxyl radicals, which are highly reactive ROS [15]. This production of membrane lipid peroxidation from ROS initiates iron toxicity [12]. Phospholipids containing PUFAs determine the levels of lipid peroxidation and cause subsequent cell death [12].
2.2. Antioxidant Defense System
Another mechanism involved in ferroptosis induction is system Xc-, which is a transport protein that facilitates the exchange of glutamate out of the cell for cysteine import. Cystine is reduced to cysteine, which then forms GSH by glutathione synthase (GSS) to support GPX4 activity [16]. Reduced cystine uptake or impaired GSH synthesis reduces GPX4 activity, leads to increased lipid ROS levels, and thereby promotes ferroptosis [17,18]. A schematic overview of the core ferroptosis circuitry in the CNS, integrating iron imbalance, lipid peroxidation, and the major protective systems (System Xc-GSH-GPX4 and CoQ10-linked defenses), is shown in Figure 1.
Figure 1. Core ferroptosis circuitry in the central nervous system. Ferroptosis arises from the convergence of labile iron (Fe²⁺), oxidative stress, and PUFA-containing membrane phospholipids, culminating in phospholipid hydroperoxide (PLOOH) accumulation and lethal membrane damage. Protective modules include the System Xc--GSH-GPX4 axis and GPX4-independent CoQ10 defenses (FSP1–CoQ10 at the plasma membrane and DHODH–CoQ10 in mitochondria). NRF2 integrates antioxidant and iron-homeostasis programs, whereas p53 and HO-1 modulate ferroptosis.
2.3. Iron Dysregulation and Labile Iron Pool
Iron regulation in the brain is essential as it supports cellular metabolism, mitochondrial respiration, neurotransmitter synthesis, and myelination of neurons [19]. However, excessive or improperly compartmentalized iron can be toxic [12,20]. Iron is transported into the brain via clathrin-mediated endocytosis of Transferrin receptor 1 (TfR1) at the blood-brain barrier (BBB) and on neural cells [21]. Within endosomes, iron is released from transferrin and transported into the cytosol as Fe2+ through the divalent metal transporter 1 (DMT1) [12,22]. TfR and transferrin (Tf) facilitate the transfer of Fe3+ into brain microvascular endothelial cells from the blood-facing (luminal) side through endocytosis, and the iron is then moved in a controlled manner to the brain-facing (abluminal) side [22]. Astrocytes are well-positioned to take iron from the circulation to redistribute to other cells, as they express machinery required for both iron influx and efflux during cell-to-cell iron transport [23,24]. Iron in the brain circulates in two forms: transferrin-bound (Tf-bound) and non-transferrin-bound [23]. Oligodendrocytes and other cell types can import iron through non-vesicular pathways, often involving DMT1. In contrast, microglia and neurons may take up transferrin-bound iron via transferrin receptors and mediate iron efflux through ferroportin [22]. An imbalance in iron homeostasis, whether from iron overload or impaired regulation, can promote lipid peroxidation and oxidative stress, thereby triggering ferroptosis [23]. Accordingly, iron uptake, efflux, storage, and turnover are critical processes that shape ferroptosis vulnerability [23]. Moreover, iron is primarily stored in ferritin as Fe3+ to mitigate toxicity, especially under conditions of excessive iron [25]. During iron deficiency, ferritin can be degraded by lysosomes to release stored iron and maintain cellular iron availability [12].
3. Ferroptosis Vulnerability Factors
Aging worsens ferroptosis, and oxidative stress increases with age; brain ferritin levels rise, often leading to cognitive problems and iron overload. The increase in total iron concentrations with aging is due to various factors, including increased permeability of the blood-brain barrier (BBB), inflammation, and changes in iron homeostasis [22,26]. The decrease in GSH is also associated with aging and promotes neurodegeneration [3,27]. The brain is particularly vulnerable to oxidative damage and ferroptosis due to its high lipid content and oxygen consumption. Iron accumulation with age can amplify these processes and may contribute to the progression of neurodegenerative disease symptoms [27,28]. Aging also affects neuromelanin in neurons, promoting the formation and accumulation of neuromelanin-iron complexes across different brain regions [28]. Neuromelanin is present in specific catecholamine neurons and plays a vital role in dopamine production [28]. Furthermore, dopamine’s role in modulating ferroptosis influences neurodegeneration [29]. For example, dopamine has been shown to reduce erastin-induced ferrous iron accumulation, glutathione depletion, and malondialdehyde production. It also stabilizes GPX4, a key enzyme that protects neurons from oxidative stress [29,30].
Ferroptosis has a profound impact on the blood-brain barrier (BBB), primarily through lipid peroxidation, which may disrupt the endothelial membranes [7]. In the BBB, lipid oxidation is promoted by several enzyme families, including cyclooxygenases (COXs), cytochrome P450 (CYP) enzymes, and lipoxygenases (LOX). These enzymes catalyze the conversion of PUFAs into lipid hydroperoxides, triggering iron-dependent lipid peroxidation. CYP enzymes play a regulatory role in astrocytes and function as a metabolic barrier that influences drug influx, vascular tone, and inflammatory signaling [7]. COXs are widely expressed in the central nervous system (CNS) and contribute to BBB disruption through inflammatory pathways and matrix metalloproteinases (MMPs). For instance, lipopolysaccharides (LPS) induce BBB destruction via a COX-dependent pathway, and tumor necrosis factor-alpha (TNF-α) has been shown to enhance BBB permeability by upregulating COX activity and elevating MMP levels. Activated MMPs then degrade tight junction proteins, which further disrupts BBB integrity [7].
p53 is a critical regulator of cellular stress responses and has been shown to influence BBB permeability [31]. It is located at the center of a signaling network that controls cellular proliferation and death and is mainly activated by DNA damage, nutritional deficiencies, hypoxia, or oxidative stress [32,33]. In a recent study using brain microvascular endothelial cells, p53 was found to support BBB integrity by reducing lipid peroxidation, preserving tight junctions, and limiting oxidative damage [34]. Conversely, high PUFA intake can enhance lipid peroxidation and free radical propagation, generating reactive aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) that accumulate and further damage the BBB [34]. p53 can regulate ferroptosis in both pro- and anti-ferroptotic directions, depending on the cell type and stress conditions context, through distinct signaling pathways. On one hand, p53 promotes ferroptosis by repressing SLC7A11 transcription, which reduces cystine import and GSH levels availability. This diminishes GPX4 activity, allowing phospholipid hydroperoxides to accumulate and drive ferroptotic cell death, particularly in stressed or malignant cells. On the other hand, p53 can inhibit ferroptosis in certain cellular contexts by promoting nuclear localization of dipeptidyl peptidase-4 (DPP4), which reduces lipid ROS levels and ferroptotic sensitivity [35].
4. Ferroptosis in Neurodegenerative Diseases
Iron is essential for mitochondrial respiration, myelin synthesis, and neurotransmitter metabolism, whereas excess redox-active Fe2+ can increase oxidative stress and lipid peroxidation, culminating in ferroptosis in neurodegeneration [12,20,36]. Ferroptosis is increasingly implicated in various neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS) [3]. A common cause of these diseases is iron deposition and lipid peroxidation, which contribute to neuronal damage and neurodegeneration, often accompanied by impaired GPX4-GSH antioxidant defenses [37]. In parallel, ROS also plays a significant role in BBB dysfunction by activating lipid-targeting enzymes and signaling cascades, as well as altering tight-junction proteins. Disruption of zonula occludens (ZO) increases BBB permeability, facilitating entry of harmful factors and amplifying neuroinflammation [38]. Experimental models of neurodegeneration have demonstrated that ferroptosis inhibitors and iron chelators can improve outcomes, emphasizing ferroptosis as a potential therapeutic target in disease progression [4,5]. Notably, regions most vulnerable to neuronal loss and atrophy often overlap with sites of iron accumulation, supporting an anatomical link between iron burden and neurodegeneration [26,39]. Excessive iron build-up is also correlated with an accelerated decline in cognitive performance [4]. Figure 2 summarizes shared ferroptosis-related pathways and key therapeutic targets across PD, AD, HD, and ALS.
4.1. Parkinson’s Disease
PD is the second most common neurodegenerative disorder, primarily affecting middle-aged and elderly individuals. It is characterized by a gradual decline in motor and non-motor functions due to a significant reduction in dopamine production; PD progresses slowly over time. A key risk factor for PD is iron accumulation in vulnerable brain regions that contributes to abnormal deposition. This iron overload induces oxidative stress, promoting the formation of Lewy Bodies and the aggregation of α-synuclein, a protein that plays an important role in the pathophysiology of PD [40]. The increase in oxidative stress in PD is partially attributed to decreased GSH levels, which increases susceptibility to lipid peroxidation. High oxygen demand by the brain further exacerbates its sensitivity to oxidative stress, ferroptosis, and neuronal damage [41]. A biomarker for PD progression is progressive iron accumulation in the substantia nigra pars compacta (SNpc). MRI scans show that elevated iron signals correlate with both cognitive and motor impairments. Iron accumulates in brain regions including globus pallidus, caudate nucleus, premotor cortex, prefrontal lobe, insula, cerebellum, and pons [42]. Also, iron metabolism dysfunction has been observed in the cerebrospinal fluid of PD patients with apathy and REM sleep behavior disorder [43,44].
Ferroptosis in PD has been tested in various models, both in vitro and in vivo, including differentiated human dopaminergic LUHMES neurons exposed to ferroptosis inducers such as erastin, which display characteristic ferroptosis features [45]. α-synuclein is abundant in the nervous system and a major component of Lewy Bodies, implicated in PD pathophysiology. It promotes ROS production, followed by lipid peroxidation in an iron-dependent manner, which subsequently increases calcium influx and results in cell death [29,46]. α-synuclein has also been reported to exhibit ferroreductase-like activity, converting Fe3+ to Fe2+ and binding Fe2+, thereby promoting misfolding and aggregation of α-syn, and further forming protein fibers that worsen PD progression [47].
4.2. Alzheimer’s Disease
In AD, dysregulated iron metabolism is a critical factor linking ferroptosis to disease progression [48]. Iron deposition in the brain is accompanied by increased ROS production, mitochondrial dysfunction, and neurodegeneration. High iron levels have been linked with AD severity and disease progression. Accumulated iron can interact with amyloid-β (Aβ) and tau, forming heme-associated complexes that promote oxidative stress and may favor ferroptosis [49]. Tau aggregation into neurofibrillary tangles is also associated with synaptic loss, neuroinflammation, and neuronal death [23]. Heme oxygenase-1 (1HO-1), an oxidative stress-responsive enzyme, has been implicated in iron handling and redox dysregulation in AD models [50]. In mice, HO-1 overexpression promotes iron loading and tau aggregation, thereby inducing AD-like pathological features [23,51]. AD pathology also involves increased lipid peroxides and lipid ROS, along with decreased cortical GSH, which together contribute to disease progression [52]. Consistent with this, several signaling pathways have been implicated in the regulation of ferroptosis in AD, including impaired iron transport and export, reduced GSH and GPX4 in antioxidant defenses, and enhanced lipid peroxidation and ROS generation within the lipid metabolism pathway [39,52].
4.3. Huntington’s Disease
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by an expanded cytosine-adenine-guanine (CAG) repeat in the huntingtin (HTT) gene, and emerging evidence suggests that ferroptosis contributes to disease progression [53]. In animal models of HD, excessive iron accumulation and oxidative stress have been directly linked to the initiation of ferroptosis [54]. Although no direct interaction is observed between iron and n-terminal HTT fragments, HTT may influence iron by disrupting iron homeostasis pathways [55]. Consistent with this, mutant huntington (mHTT) is associated with brain iron accumulation in HD, suggesting that mHTT-related metabolic and excitotoxic stress may be vital regulators of Fe status [56,57]. Reduced GSH levels have also been observed in HD, which may further contribute to the neuronal sensitivity to ferroptosis [58]. Increased iron levels were detected in early disease stages in the basal ganglia, occipital cortex, globus pallidus, and putamen by imaging techniques such as MRI and susceptibility mapping [59]. Additionally, arachidonate 5-lipoxygenase (ALOX5), an inducer of ferroptosis, has been implicated in HD models [60]. ALOX5 contributes to ACSL4-dependent ferroptosis and may be promoted by the expression of the N-terminal mHTT polyglutamine fragment (HTTQ94). Notably, loss of ALOX5 expression can prevent HTTQ94-mediated ROS stress and ferroptosis, which may serve as a potential new target for HD [57,61,62].
4.4. Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that primarily affects motor cells in the CNS, leading to progressive muscle weakness and paralysis [63]. A consistent feature observed in both animal models and patients is iron accumulation in affected regions, particularly the spinal cord and CNS [64,65]. The iron buildup is linked to an imbalance in iron regulation, with increased expression of key iron-handling proteins, including DMT1, TfR1, ferroportin (FPN), and ceruloplasmin (CP), especially in the spinal cord [66]. Oxidative stress plays a significant role in ALS progression, as evidenced by increased lipid peroxidation and protein oxidation markers, notably MDA, 4-HNE, and protein carbonyls in both experimental models and patients [67]. Moreover, neuronal loss of GPX4 leads to motor neuron degeneration and paralysis in animal models; however, its upregulation has been shown to slow ALS progression and improve motor function [68].
4.5. Other Diseases
Additionally, other neurodegenerative diseases that are triggered by ferroptosis include epilepsy, brain ischemia, and stroke [69]. In epilepsy, oxidative stress is a prominent pathogenic feature, and the disorder is defined by a persistent predisposition to seizures with neurobiological, cognitive, psychological, and social consequences [70]. Recent evidence shows that oxidative stress and iron dysregulation can act synergistically to exacerbate epileptic cell dysfunction. Thus, high concentrations of unbound Fe2+ can catalyze Fenton reactions, converting hydrogen peroxide into highly reactive ROS and thereby promoting neuronal injury [71].
Stroke can be broadly classified as ischemic and hemorrhagic [72]. Ischemic stroke is caused by interruptions of the cerebral blood supply, which accounts for approximately 80% of stroke cases [73]. Hemorrhagic stroke is caused by cerebral vascular rupture and is divided into subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and intraventricular hemorrhage (IVH) [74]. Both in vitro and in vivo studies support a role of ferroptosis in early brain damage after SAH [75]. After hemorrhage or ischemia, iron released from blood products, together with disrupted iron, can increase brain iron burden. Additionally, BBB disruption can allow iron-rich proteins and ferritin to enter the brain parenchyma, increasing ROS production through Fenton reactions and promoting ferroptotic damage to proteins, membranes, and nucleic acids [76,77].
5. In Vivo Detection of Ferroptosis
Ferroptosis in the brain can be visualized using imaging techniques, such as positron emission tomography (PET), magnetic resonance imaging (MRI), and fluorescence-based approaches in both mouse models and humans [78,79]. Because ferroptosis is a molecular process, these modalities primarily measure upstream or downstream correlations of iron burden, labile Fe2+ availability, ROS, lipid peroxidation, and antioxidant depletion. Such imaging techniques are most informative when combined with biochemical validation [80].
PET is non-invasive and enables repeated, quantitative molecular imaging of tracer uptake over time [81]. It can probe ferroptosis-related processes by reporting on redox-active iron pools and oxidative stress pathways. For example, the radiotracer 18F-TRX is used to monitor the labile iron pool (LIP), which correlates with the iron-mediated cell death pathways [82]. Another tracer, 18F-labeled dihydromethidine (18F-FDHM), can cross the BBB and react with intracellular ROS, enabling experimental mapping of ROS-rich regions [83].
MRI is widely used in clinical practice and research to monitor the progression of neurodegenerative diseases [84]. MRI technologies use proton transverse relaxation rates to quantify brain iron, but these measures are indirect and not specific to ferroptosis [85]. To improve sensitivity to iron deposition, approaches such as relaxation time mapping and magnetic field correlation (MFC) imaging have been used to detect iron associated with ferritin and hemosiderin [79]. However, distinguishing Fe2+ from Fe3+ in routine clinical settings remains challenging, and iron-related signals can be influenced by factors such as myelin changes or calcification [79].
Thirdly, fluorescence imaging is a non-invasive optical imaging method to detect intracellular metabolites, parameters, and biomolecules related to oxidative homeostasis in ferroptosis [86]. Fluorescence probes used in preclinical studies, including in vivo and ex vivo experiments, reported intracellular Fe2+ accumulation, ROS generation, lipid peroxidation, and antioxidant status, providing a mechanistic context that complements PET/MRI [87]. One widely used design is the N-oxide reduction strategy, which produces Fe2+-fluorescence “turn-on” response [83,88]. Another probe, BODIPY (4,4-difluoroboradiazaindacene), can specifically detect GSH and image the changes in GSH in cells and tissues during ferroptosis [89].
6. Therapeutic Strategies for Neurodegenerative Diseases
Dopamine has a significant impact on PD as it results from low dopamine levels in the brain. Some of the common dopamine-based therapies include levodopa and dopamine receptor agonists [90]. Levodopa is a dopamine precursor that restores dopamine levels and improves motor symptoms [91]. Moreover, non-oxidative dopamine has been shown to inhibit erastin-induced ferroptosis, resulting in GPX4 stabilization and suppression of ferroptosis [92]. Iron chelation therapy is a potential treatment strategy for PD to treat iron overload and protect against neuronal injury. It also prevents dopaminergic neuronal loss in the SNpc and treats motor deficits [93]. Various studies on MPTP-induced (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a neurotoxin) Parkinsonian models show that mitochondrial ferritin can protect neurons and inhibit cellular iron accumulation and oxidative stress [93].
Furthermore, glutathione depletion is linked to dopaminergic neuronal death and progressive motor imbalance in PD [94]. Clinical trials have shown that glutathione administration can restore glutathione levels for mild therapeutic benefits [93]. The enzyme GPX4 also plays a protective role against neurodegeneration by regulating ferroptosis. Loss of GPX4 in dopaminergic neurons has been associated with increased anxiety and diminished spontaneous locomotor activity [95]. A pharmacological inhibitor of ferroptosis uses ferrostatin 1 (Fer-1) derivatives and iron chelators, which have shown improvements in motor control and delays in neurodegeneration. Fer-1 can inhibit 1-methyl-4-phenylpyridinium (MPP+), which kills dopaminergic neurons [96].
Neuroprotection in AD is often accompanied by strategies that stabilize antioxidant defenses, including production and metabolism of GSH and glutamine, respectively [97]. The GSH molecule consists of glutamic acid, cysteine, and glycine, which play a critical role in reducing oxidative stress [97]. A deficiency in these amino acids causes increased susceptibility to oxidative stress. In addition, aromatic amine antioxidants like Fer-1 and liproxstatin-1 (Lip-1) are potential inhibitors of ferroptosis that markedly reduce ROS production and lipid peroxidation [98]. α-tocopherol is a form of vitamin E that exerts its antioxidant capacity by interrupting the chain of lipid oxidation [99]. The levels of α-tocopherol in the brain are regulated by the tocopherol transfer protein (TTP), while a deficiency in this protein or vitamin E can exacerbate oxidative stress [99]. Clinical studies have shown that vitamin E supplements can slow cognitive decline and oxidative stress in AD [99]. Nitroxides such as iron (II) citrate can cross the BBB, suggesting therapeutic potential for oxidative stress-induced diseases [23]. Additionally, double selenium nanospheres also have the potential to cross the BBB and ameliorate AD [100]. The small selenium sphere binds to the BBB receptor and targets the αβ42 fibrils in the brain [100]. It transforms αβ42 fibrils into αβ42 oligomers and inhibits tau protein phosphorylation. This treatment reduces neuronal toxicity and ferroptosis and can also be used for other neuroinflammatory diseases [101].
In HD mouse models, iron-selective chelation by using deferoxamine (DFO) or deferiprone (DFP) alleviates disease symptoms [102,103]. Administration of 3-nitropropionic acid (3-NP) can reduce GSH levels, while cysteamine supplementation reverses 3-NP-induced striatal neuronal death by upregulating GSH [103,104]. Fer-1 treatment shows a significant inhibition in lipid peroxidation and iron-induced cell death in HD [103]. In contrast, ALS treatments include ferroptosis inhibitors as potential therapeutic agents. An iron chelator such as DFP is known to improve lifespan in mouse models and has demonstrated potential efficacy in human patients [65,102]. Other iron chelators, such as DFO and SIH, reduce pathological iron accumulation, improve motor neuronal survival, and restore motor function [65]. In ALS, the ferroptosis inhibitor Cull (atsm) prevents lipid peroxidation, and the free radical scavenger, edaravone is found to protect motor neurons and inhibit ferroptosis [105]. Currently, there is no therapeutic intervention available to cure or prevent HD and ALS.
7. Ferroptosis-Targeted Neuroprotection
Iron chelators play a critical role in preventing ferroptosis by removing excess iron from the body. Common iron chelators include DFO, DFP, and DFX, which help regulate iron levels and are safer alternatives that cross the BBB. Iron chelators transfer iron from DFP-Fe to transferrin, which helps regulate iron levels [107]. DFP is a safer alternative to DFO, as it can easily cross the BBB. Conversely, DFX offers protection against kidney and neuronal damage. Some newer iron chelators, CN128 and ciclopirox olamine, are still being studied for broader applications [108].
Another component in the regulation of ferroptosis is TFR-1. Blocking TFR-1 by RNA interference prevents ferroptosis, whereas increasing TFR1 expression stimulates ferroptosis through the NF2-YAP signaling pathways [109]. TFR-1 is also used as a marker for cells that undergo ferroptosis and regulates iron efflux via hepcidin agonists. Additionally, ferroptosis is also targeted by reductive-oxidative pathways through the enzyme HO-1. The enzyme HO-1, encoded by the gene HMOX1, catalyzes heme to produce carbon monoxide and free iron and plays a cytoprotective role against ferroptosis [110]. High HMOX1 can be toxic; it can cause excessive heme breakdown and promote an iron pool. The regulation of ferroptosis by HO-1 is influenced by both HO-1-catalyzed heme metabolites and Nrf2 proteins [111]. In reductive-oxidative responses, gene transcription is activated by NRF2, where the KEAP1-NRF2 axis could be a viable strategy for modulating ferroptosis.
Moreover, lipid metabolism plays a vital role in ferroptosis. Acetyl-CoA synthetase long-chain family member 4 (ACSL4) promotes phospholipid-PUFA synthesis. Exogenous monounsaturated fatty acids also protect against ferroptosis by reducing lipid ROS accumulation in the plasma membrane and removing PUFAs from their cellular sites [112].
Another micronutrient is selenium that can modulate ferroptosis. It is essential for producing selenocysteine, which acts as the active site for GPX4 [113]. Selenium-containing peptides tat SelPep, can cross the BBB and help protect against ferroptosis-mediated tissue damage.
Small-molecule inducers play a significant role in modulating ferroptosis. Ferroptosis was initially defined through small molecules known as RAS-selective lethal compounds (RSLs) [114]. Nanoparticles contain compounds to directly modulate ferroptosis by depleting GSH and/or inhibiting GPX4. For instance, arginine-rich manganese silicate nanoparticles are known to induce ferroptosis by depleting GSH and inactivating GPX4 [115]. Erastin mediates ferroptosis without morphological changes or biochemical processes by inhibiting system Xc and RSL3 [116]. Ferritin-bound erastin and rapamycin decrease GPX4 activity while enhancing lipid peroxidation. Another nanoparticle, SRF@FeIIITA, is formed by Fe3+ and a network-like tannic acid corona that contains the kinase inhibitor sorafenib (SRF) [117]. SRF@FeIIITA releases SRF and reduces GSH levels. Other inducers, including buthionine sulfoximine and cisplatin, may induce synthetic lethality like that caused by GSH depletion [117]. Targeting these molecules by ferroptosis inhibitors such as vitamin E, Trolox, DFO, DFX, Zileuton, Ferrostatin-1, and liproxstatin-1 suppresses oxidative stress and prevents ferroptosis cell death [93]. Table 1 summarizes inhibitors and inducers of ferroptosis in neurodegenerative diseases.
8. Nutritional Neuroroprotection Against Ferroptosis
Diet plays a vital role in preventing ferroptosis by taking multivitamins and consuming antioxidant-rich foods [118].
8.1. Multivitamins
Multivitamins have substantial benefits in maintaining physiological functions and lowering toxic effects. Some vitamins have anti-ferroptosis properties, making them an important agent for neuroprotection. Vitamins A, B, C, D, E, and K have been shown to inhibit ferroptosis [118]. Vitamin A is an essential vitamin that provides retinol, retinoic acids, and carotenoids from animal and plant sources. Retinol can resist ferroptosis by directly capturing anti-peroxides. In contrast, carotenoids also prevent ferroptosis by promoting Nrf2 and can mediate signal transduction via retinoic acid receptors [119]. B vitamins are known for their roles in brain function, energy production, NA synthesis, and repair [120]. Vitamin B6 is used to compensate for impaired GSH levels and to restore GPX4 expression, thereby inhibiting ferroptosis. It can also enhance Nrf2 expression and production of antioxidant enzymes [121]. Vitamin C plays its role in oxygenated enzymes, such as the oxidation of Fe2+ to Fe3+ [121]. It has antioxidant properties that prevent ferroptosis; however, higher concentrations of vitamin C can induce ferroptosis by decreasing GPX4 levels [121]. Additionally, vitamin D is vital in binding to its target organ to complete metabolism and reabsorption. It can inhibit ferroptosis by upregulating the production of anti-ferroptosis proteins and reducing iron accumulation [122]. Vitamin E has lipophilic antioxidants that may protect against excessive degradation. Vitamin E has been shown to protect neurons from oxidative stress-induced damage, positively influencing the prevention and progression of neurodegenerative diseases [123]. Lastly, vitamin K is vital in maintaining bone and mineralization in the body [124]. Vitamin K also prevents ferroptosis by reducing lipid peroxidation radicals and maintaining GPX4 activity [125,126].
8.2. Jucara Fruit Extract
Jucara fruit extracts exert neuroprotection against glutamate-induced oxidative stress in HT22 cells [127]. These cells are immortalized and are used to study the neuroprotective effects against glutamate-induced oxidative stress. Jucara extract contains high concentrations of antioxidants. The phenolic compounds were found in fractions of crude extracts as well as in hexane, dichloromethane, ethyl acetate, and butanol. These fractions tested the viability of HT22 cells during co-treatment. The results showed that dichloromethane and hexane fractions from fruit extraction protect HT22 cells through their phenolic compounds. The results are still being studied to determine the neuroprotective dosing regimen of Jucara fruits [127,128].
8.3. Flavonoids
Flavonol fisetin is effective in preventing ferroptosis in preclinical models [129]. There is evidence that 2 out of the 30 flavonoids can maintain GSH levels in oxidative stress (129). Fisetin is abundant in fruits and vegetables; high amounts are found in strawberries (160µg/g), and low amounts in apples, persimmons, kiwis, peaches, grapes, tomatoes, onions, and cucumbers. The bioavailability of flavonoid compounds is yet to be studied [130]. Experiments on mice have been conducted to determine the effects of Fisetin on neurodegenerative diseases. In AD, fisetin consistently prevents cognitive decline [131]. It also maintains synaptic proteins and decreases markers of inflammation and oxidative stress. In PD, fisetin improves motor function and reduces rotenone-mediated decrease in dopamine levels and immune reactivity [132,133]. Moreover, fisetin also improves mitochondrial function and markers of oxidative stress in the midbrain. In HD, mice on a fisetin-based diet had a slower decline in motor function than those on a regular diet; however, further research on the underlying effects and observations is still needed [134]. Thus, flavonoids are promising neuroprotective compound that requires additional research to confirm these effects [133].
9. Future Technologies and Challenges
Although multiple techniques have been proposed to mitigate ferroptosis, there are still research gaps to be bridged across mechanistic insights to safe translational approaches. New technologies and methodologies are being explored to enhance the understanding of ferroptosis in neurodegeneration.
Targeted protein degradation is an emerging therapeutic approach that eliminates “undrugged” targets and other difficult-to-treat proteins [135]. Three major classifications of protein degraders are: proteolysis-targeting chimeras (PROTACs), monomeric targeted protein degraders, and molecular glues (MGs). PROTACs are small heterobifunctional molecules that have a target-binding ligand and an E3 ubiquitin ligase-binding ligand connected by a linker to promote target ubiquitination and protein degradation [135]. On the other hand, monomeric degraders have a lower molecular weight and can easily cross the BBB. MGs induce proximity between the target protein and the ubiquitin ligase, which causes protein degradation. Future studies are needed to determine the efficacy and safety of monomeric targeted protein degraders or MG-based degraders to target ferroptosis-related proteins [135].
Artificial intelligence (AI) has the potential to revolutionize drug discovery by improving predictive accuracy and expediting drug development [136]. Rapid machine-based decision-making using artificial neural networks can serve as a cost-effective platform to identify new drugs [136]. AI can integrate bioinformatics and pharmacological networks to accelerate the discovery of new therapeutics to target ferroptosis.
Melatonin, which is produced in the pineal glands, has anti-aging, anti-inflammatory, and anti-cancer properties [137]. It was recently determined that melatonin can affect anti-ferroptosis pathways, including Nrf2 antioxidants, GPX4, HO-1, and NCOA4 [138]. It can inhibit ferroptosis by activating intracellular defense regulatory pathways. Melatonin can suppress inflammation and ROS generation and can aid in regulating autophagy and apoptosis pathways. Further research is needed to elucidate the exact mechanisms by which melatonin can inhibit ferroptosis and be a potential therapeutic target [138,139].
RNA-based therapies represent a rapidly expanding field primarily using messenger RNA (mRNA), RNAi, single-stranded antisense oligonucleotides, aptamers, ribozymes, and CRISPR-Cas endonuclease-mediated gene editing [140]. These technologies are used to develop vaccines and are now being explored for the treatment of various diseases, including ferroptosis. RNA-based therapies could offer a new therapeutic approach to prevent ferroptosis; however, the research in this area remains in its early stages [140].
Some future challenges include understanding the regulatory mechanisms underlying ferroptosis in neurological disorders [141]. Second, balancing therapeutic strategies is vital as inhibiting ferroptosis may be beneficial for neurodegenerative diseases but could exacerbate other conditions, such as cancer, presenting a double-edged sword that needs careful management [16,142]. Another challenge is identifying specific biomarkers to enable accurate prediction of ferroptosis [141]. Lastly, recent data on ferroptosis are mostly from experimental studies and not supported by clinical applications. Through clinical research, it will be easier to determine the efficacy of ferroptosis-targeting therapies.
10. Conclusion
Ferroptosis is an iron-dependent cell death characterized by excessive lipid peroxide accumulation. With the current understanding of oxidative stress and its impact on neurodegeneration, it is determined that ferroptosis plays a significant role in causing neurodegenerative diseases like PD, AD, HD, and ALS. However, current solutions and implications require more specification, validation, and an efficient delivery route to reach a particular brain region to preserve iron metabolism while limiting toxic lipid peroxidation.
Author Contributions
All authors contributed to the conception and design of the review. The first draft was written by A.J. All authors commented on previous versions and approved the final manuscript for the term explanation.
Funding
This research was funded by Canadian Institutes of Health Research (CIHR-Research Grant MB).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no new datasets were generated or analyzed.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang Y, Li H, He Q, Zou R, Cai J, Zhang L. Ferroptosis: underlying mechanisms and involvement in neurodegenerative diseases. Apoptosis. 2024 Feb;29(1):3-21. [CrossRef]
- Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, Liu Y, Zhao X, Qian L, Liu P, Xiong Y. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021 Jul 26;7(1):193. [CrossRef]
- Fei Y, Ding Y. The role of ferroptosis in neurodegenerative diseases. Front Cell Neurosci. 2024 Oct 15; 18:1475934. [CrossRef]
- Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Targeted Ther. 2023 Sep 21;8(1):372. [CrossRef]
- Ryan SK, Ugalde CL, Rolland AS, Skidmore J, Devos D, Hammond TR. Therapeutic inhibition of ferroptosis in neurodegenerative disease. Trends Pharmacological Sci. 2023 Oct 1;44(10):674-88. [CrossRef]
- Xu W, Guo ZN, Shao A. Ferroptosis in stroke, neurotrauma and neurodegeneration, volume II. Front Cell Neurosci. 2023 Jul 5; 17:1238425. [CrossRef]
- Nguyen TP, Alves F, Lane DJ, Bush AI, Ayton S. Triggering ferroptosis in neurodegenerative diseases. Trends Neurosci. 2025 Jul 21. [CrossRef]
- Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021 Sep 2;17(9):2054-81. [CrossRef]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014 Oct 1;13(10):1045-60. [CrossRef]
- Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021 Apr;22(4):266-82. [CrossRef]
- Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, Lei P. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021 Feb 3;6(1):49. [CrossRef]
- Ji Y, Zheng K, Li S, Ren C, Shen Y, Tian L, Zhu H, Zhou Z, Jiang Y. Insight into the potential role of ferroptosis in neurodegenerative diseases. Front Cell Neurosci. 2022 Oct 27; 16:1005182. [CrossRef]
- Xie Z, Zhou Q, Qiu C, Zhu D, Li K, Huang H. Inaugurating a novel adjuvant therapy in urological cancers: Ferroptosis. Cancer Pathog Ther. 2023 Apr 30;1(02):127-40. [CrossRef]
- Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016 Mar 1;41(3):274-86. [CrossRef]
- He YJ, Liu XY, Xing L, Wan X, Chang X, Jiang HL. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020 May 1;241:119911. [CrossRef]
- Wang Y, Wu S, Li Q, Sun H, Wang H. Pharmacological inhibition of ferroptosis as a therapeutic target for neurodegenerative diseases and strokes. Adv Sci. 2023 Aug;10(24):2300325. [CrossRef]
- Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019 Mar 1;133:144-52. [CrossRef]
- Poltorack CD, Dixon SJ. Understanding the role of cysteine in ferroptosis: progress & paradoxes. The FEBS J. 2022 Jan;289(2):374-85. [CrossRef]
- Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020 May 20;152:175-85. [CrossRef]
- Alrouji M, Anwar S, Venkatesan K, Shahwan M, Hassan MI, Islam A, Shamsi A. Iron homeostasis and neurodegeneration in the ageing brain: Insight into ferroptosis pathways. Ageing Res Rev. 2024 Dec 1; 102:102575. [CrossRef]
- Gao Q, Zhou Y, Chen Y, Hu W, Jin W, Zhou C, Yuan H, Li J, Lin Z, Lin W. Role of iron in brain development, aging, and neurodegenerative diseases. Ann Med. 2025 Dec 31;57(1):2472871. [CrossRef]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014 Oct 1;13(10):1045-60. [CrossRef]
- Ma H, Dong Y, Chu Y, Guo Y, Li L. The mechanisms of ferroptosis and its role in alzheimer’s disease. Front Mol Biosci. 2022 Aug 26;9:965064. [CrossRef]
- Qian ZM, Ke Y. Brain iron transport. Biol Rev Camb Philos Soc. 2019 Oct;94(5):1672-84. [CrossRef]
- Cheli VT, Correale J, Paez PM, Pasquini JM. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro. 2020 Sep;12. [CrossRef]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014 Oct 1;13(10):1045-60. [CrossRef]
- Benarroch E. What is the role of ferroptosis in neurodegeneration?. Neurology. 2023 Aug 15;101(7):312-9. [CrossRef]
- Liu C, Pan J, Bao Q. Ferroptosis in senescence and age-related diseases: pathogenic mechanisms and potential intervention targets. Mol Biol Rep. 2025 Dec;52(1):238. [CrossRef]
- Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PD, Levy D, Bydlowski SP. Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci. 2020 Nov 20;21(22):8765. [CrossRef]
- Cerasuolo M, Di Meo I, Auriemma MC, Trojsi F, Maiorino MI, Cirillo M, Esposito F, Polito R, Colangelo AM, Paolisso G, Papa M. Iron and ferroptosis more than a suspect: beyond the most common mechanisms of neurodegeneration for new therapeutic approaches to cognitive decline and dementia. Int J Mol Sci. 2023 Jun 1;24(11):9637. [CrossRef]
- Xu R, Wang W, Zhang W. Ferroptosis and the bidirectional regulatory factor p53. Cell Death Discov. 2023 Jun 29;9(1):197. [CrossRef]
- Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022 May;29(5):895-910. [CrossRef]
- Lei L, Lu Q, Ma G, Li T, Deng J, Li W. P53 protein and the diseases in central nervous system. Front Genet. 2023 Jan 9; 13:1051395. [CrossRef]
- Chen, X., Pang, X., Yeo, A. J., Xie, S., Xiang, M., Shi, B., Yu, G., & Li, C. (2022). The molecular mechanisms of ferroptosis and its role in blood-brain barrier dysfunction. Front Cell Neurosci. [CrossRef]
- Ji H, Wang W, Li X, Han X, Zhang X, Wang J, Liu C, Huang L, Gao W. p53: A double-edged sword in tumor ferroptosis. Pharmacol Res. 2022 Mar 1; 177:106013. [CrossRef]
- Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci. 2013 Aug;14(8):551-64. [CrossRef]
- Ayton S, Moreau C, Devos D, Bush AI. Iron on trial: recasting the role of iron in neurodegeneration. Brain. 2025 Dec;148(12):4241-7. [CrossRef]
- Shuvalova M, Dmitrieva A, Belousov V, Nosov G. The role of reactive oxygen species in the regulation of the blood-brain barrier. Tissue Barriers. 2025 Apr 3;13(2):2361202. [CrossRef]
- Chen K, Jiang X, Wu M, Cao X, Bao W, Zhu LQ. Ferroptosis, a potential therapeutic target in Alzheimer’s disease. Front Cell Dev Biol. 2021 Aug 5; 9:704298. [CrossRef]
- Chen Y, Luo X, Yin Y, Thomas ER, Liu K, Wang W, Li X. The interplay of iron, oxidative stress, and α-synuclein in Parkinson’s disease progression. Mol Med. 2025 Apr 26;31(1):154. [CrossRef]
- Zhou M, Xu K, Ge J, Luo X, Wu M, Wang N, Zeng J. Targeting ferroptosis in Parkinson’s disease: mechanisms and emerging therapeutic strategies. Int J Mol Sci. 2024 Dec 4;25(23):13042. [CrossRef]
- Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral iron deposition in neurodegeneration. Biomolecules. 2022 May 17;12(5):714. [CrossRef]
- Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006 Sep 1;49(4):379-86. [CrossRef]
- Xu Y, Huang X, Geng X, Wang F. Meta-analysis of iron metabolism markers levels of Parkinson’s disease patients determined by fluid and MRI measurements. J Trace Elem Med Biol. 2023 Jul 1; 78:127190. [CrossRef]
- Tong ZB, Kim H, El Touny L, Simeonov A, Gerhold D. LUHMES dopaminergic neurons are uniquely susceptible to ferroptosis. Neurotox Res. 2022 Oct;40(5):1526-36. [CrossRef]
- Riederer P, Nagatsu T, Youdim MB, Wulf M, Dijkstra JM, Sian-Huelsmann J. Lewy bodies, iron, inflammation and neuromelanin: pathological aspects underlying Parkinson’s disease. J Neural Transm. 2023 May;130(5):627-46. [CrossRef]
- Tofaris GK. Initiation and progression of α-synuclein pathology in Parkinson’s disease. Cell Mol Life Sci. 2022 Apr;79(4):210. [CrossRef]
- Wu L, Xian X, Tan Z, Dong F, Xu G, Zhang M, Zhang F. The role of iron metabolism, lipid metabolism, and redox homeostasis in Alzheimer’s disease: from the perspective of ferroptosis. Mol Neurobiol. 2023 May;60(5):2832-50. [CrossRef]
- Pal A, Cerchiaro G, Rani I, Ventriglia M, Rongioletti M, Longobardi A, Squitti R. Iron in Alzheimer’s disease: from physiology to disease disabilities. Biomolecules. 2022 Sep 6;12(9):1248. [CrossRef]
- Wu YH, Hsieh HL. Roles of heme oxygenase-1 in neuroinflammation and brain disorders. Antioxidants. 2022 May 8;11(5):923. [CrossRef]
- Perluigi M, Di Domenico F, Butterfield DA. Oxidative damage in neurodegeneration: roles in the pathogenesis and progression of Alzheimer disease. Physiol Rev. 2024 Jan 1;104(1):103-97. [CrossRef]
- Maheshwari S. Ferroptosis signaling pathways: Alzheimer’s disease. Horm Metab Res. 2023 Dec;55(12):819-26. [CrossRef]
- Vega AJ, Hernandez GV, O’Malley PA, Robin CJ, Parra AN, Varrassi G, Shekoohi S, Kaye AD, Malley PA, Robin C. Overview of Huntington’s Disease and Emerging Treatment Strategies: A Narrative Review. Cureus. 2025 Dec 1;17(12). [CrossRef]
- Li J, Jia B, Cheng Y, Song Y, Li Q, Luo C. Targeting molecular mediators of ferroptosis and oxidative stress for neurological disorders. Oxid Med Cell Longev. 2022; Jul 22;2022(1):3999083. [CrossRef]
- Long H, Zhu W, Wei L, Zhao J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm. 2023 Aug;4(4):e298. [CrossRef]
- Maity S, Komal P, Kumar V, Saxena A, Tungekar A, Chandrasekar V. Impact of ER stress and ER-mitochondrial crosstalk in Huntington’s disease. Int J Mol Sci. 2022 Jan 11;23(2):780. [CrossRef]
- Ma J, Liu J, Chen S, Zhang W, Wang T, Cao M, Yang Y, Du Y, Cui G, Du Z. Understanding the Mechanism of Ferroptosis in Neurodegenerative Diseases. Front Biosci. 2024 Aug 20;29(8):291. [CrossRef]
- Tucci P, Lattanzi R, Severini C, Saso L. Nrf2 pathway in huntington’s disease (hd): What is its role?. Int J Mol Sci. 2022 Dec 3;23(23):15272. [CrossRef]
- Zhou CH, Zhu YC. Imaging of cerebral iron as an emerging marker for brain aging, neurodegeneration, and cerebrovascular diseases. Brain Sci. 2025 Aug 29;15(9):944. [CrossRef]
- Song S, Su Z, Kon N, Chu B, Li H, Jiang X, Luo J, Stockwell BR, Gu W. ALOX5-mediated ferroptosis acts as a distinct cell death pathway upon oxidative stress in Huntington’s disease. Genes Dev. 2023 Mar 1;37(5-6):204-17. [CrossRef]
- Fang W, Hu Z, Shen B, Zeng X, Chen S, Wang S, Xie S, Deng W. Downregulation of Alox5 Inhibits Ferroptosis to Improve Doxorubicin-Induced Cardiotoxicity via the P53/SLC7A11 Pathway. J Cell Mol Med. 2025 Jun;29(11):e70641. [CrossRef]
- Liu M, Zhao J, Xue C, Yang J, Ying L. Uncovering the ferroptosis related mechanism of laduviglusib in the cell-type-specific targets of the striatum in Huntington’s disease. BMC Genomics. 2024 Jun 25;25(1):633. [CrossRef]
- Verma A, DM M. Clinical manifestation and management of amyotrophic lateral sclerosis. Exon Publications. 2021 Jul 25:1-4. [CrossRef]
- Verma S, Khurana S, Vats A, Sahu B, Ganguly NK, Chakraborti P, Gourie-Devi M, Taneja V. Neuromuscular junction dysfunction in amyotrophic lateral sclerosis. Mol Neurobiol. 2022 Mar;59(3):1502-27. [CrossRef]
- Wang L, Fang X, Ling B, Wang F, Xia Y, Zhang W, Zhong T, Wang X. Research progress on ferroptosis in the pathogenesis and treatment of neurodegenerative diseases. Front Cell Neurosci. 2024 Mar 7; 18:1359453. [CrossRef]
- Dong K, Liu B, Cheng G, Li Y, Xie F, Zhang J, Qian L. Stress-Induced Dysregulation of Brain Iron Metabolism and Its Links to Neurological Disorders. Biology. 2025 Nov 11;14(11):1575. [CrossRef]
- Park HR, Yang EJ. Oxidative stress as a therapeutic target in amyotrophic lateral sclerosis: opportunities and limitations. Diagnostics. 2021 Aug 26;11(9):1546. [CrossRef]
- Tu LF, Zhang TZ, Zhou YF, Zhou QQ, Gong HB, Liang L, Hai LN, You NX, Su Y, Chen YJ, Mo XK. GPX4 deficiency-dependent phospholipid peroxidation drives motor deficits of ALS. J Adv Res. 2023 Jan 1; 43:205-18. [CrossRef]
- Tian HY, Huang BY, Nie HF, Chen XY, Zhou Y, Yang T, Cheng SW, Mei ZG, Ge JW. The interplay between mitochondrial dysfunction and ferroptosis during ischemia-associated central nervous system diseases. Brain Sci. 2023 Sep 25;13(10):1367. [CrossRef]
- Parsons AL, Bucknor EM, Castroflorio E, Soares TR, Oliver PL, Rial D. The interconnected mechanisms of oxidative stress and neuroinflammation in epilepsy. Antioxidants. 2022 Jan 14;11(1):157. [CrossRef]
- Sun H, Li X, Guo Q, Liu S. Research progress on oxidative stress regulating different types of neuronal death caused by epileptic seizures. Neurol Sci. 2022 Nov;43(11):6279-98. [CrossRef]
- Ohashi SN, DeLong JH, Kozberg MG, Mazur-Hart DJ, Van Veluw SJ, Alkayed NJ, Sansing LH. Role of inflammatory processes in hemorrhagic stroke. Stroke. 2023 Feb;54(2):605-19. [CrossRef]
- Qin C, Yang S, Chu YH, Zhang H, Pang XW, Chen L, Zhou LQ, Chen M, Tian DS, Wang W. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2022 Jul 6;7(1):215. [CrossRef]
- Morais Filho AB, Rego TL, Mendonca LD, Almeida SS, Nóbrega ML, Palmieri TD, Giustina GZ, Melo JP, Pinheiro FI, Guzen FP. The physiopathology of spontaneous hemorrhagic stroke: a systematic review. Rev Neurosci. 2021 Aug 26;32(6):631-58. [CrossRef]
- Li Y, Liu Y, Wu P, Tian Y, Liu B, Wang J, Bihl J, Shi H. Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage in vitro and in vivo via reduction of lipid peroxidation. Cell Mol Neurobiol. 2021 Mar;41(2):263-78. [CrossRef]
- Huang S, Li S, Feng H, Chen Y. Iron metabolism disorders for cognitive dysfunction after mild traumatic brain injury. Front Neurosci. 2021 Mar 16; 15:587197. [CrossRef]
- Zhang Y, Lu X, Tai B, Li W, Li T. Ferroptosis and its multifaceted roles in cerebral stroke. Front Cell Neurosci. 2021 Jun 3; 15:615372. [CrossRef]
- Zeng F, Nijiati S, Tang L, Ye J, Zhou Z, Chen X. Ferroptosis detection: from approaches to applications. Angew Chem Int Ed Engl. 2023 Aug 28;135(35):e202300379. [CrossRef]
- Wang C, Wen L, Wang K, Wu R, Li M, Zhang Y, Gao Z. Visualization of ferroptosis in brain diseases and ferroptosis-inducing nanomedicine for glioma. Am J Nucl Med Mol Imaging. 2023 Oct 20;13(5):179. PMC10656630.
- Chen X, Tsvetkov AS, Shen HM, Isidoro C, Ktistakis NT, Linkermann A, Koopman WJ, Simon HU, Galluzzi L, Luo S, Xu D. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy. 2024 Jun 2;20(6):1213-46. [CrossRef]
- Meikle SR, Sossi V, Roncali E, Cherry SR, Banati R, Mankoff D, Jones T, James M, Sutcliffe J, Ouyang J, Petibon Y. Quantitative PET in the 2020s: a roadmap. Phys Med Biol. 2021 Mar 12;66(6):06RM01. [CrossRef]
- Mayr E, Rotter J, Kuhrt H, Winter K, Stassart RM, Streit WJ, Bechmann I. Detection of molecular markers of ferroptosis in human Alzheimer’s disease brains. J Alzheimers Dis. 2024 Dec;102(4):1133-54. [CrossRef]
- Dubey Y, Kanvah S. Fluorescent N-oxides: applications in bioimaging and sensing. Org Biomol Chem. 2024 Sep;25;22(37):7582-7595. [CrossRef]
- Zhang J, Han L, Wu H, Zhong Y, Shangguan P, Liu Y, He M, Sun H, Song C, Wang X, Liu Y. A Brain-targeting NIR-II ferroptosis system: effective visualization and oncotherapy for orthotopic glioblastoma. Adv Sci. 2023 May;10(13):2206333. [CrossRef]
- Shepherd TM, Dogra S. Clinical Translation of Integrated PET-MRI for Neurodegenerative Disease. J Magn Reson Imaging. 2026 Jan;63(1):60-78. [CrossRef]
- Welton T, Saw WT, Zhou Z, Sun Q, Mai AS, Teo TW, Chan LL, Tan LC, Tan EK. Identification of genes underlying nigrostriatal iron accumulation: transcriptome-wide association study of iron-sensitive brain MRI. EBioMedicine. 2026 Jan 1;123. [CrossRef]
- Gong J, Liu XR, Liu X, Sun R, Ge JF. A rapid-response NIR fluorescent probe for imaging Cys/Hcy in ferroptosis and inflammation models. Spectrochim Acta Part A Mol Biomol Spectrosc. 2026 Jan 24:127519. [CrossRef]
- Hirayama T, Tsuboi H, Niwa M, Miki A, Kadota S, Ikeshita Y, Okuda K, Nagasawa H. A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem Sci. 2017 Jul 1;8(7):4858-4866. [CrossRef]
- Flores CA, Yang WS. Detection of Ferroptosis by Liperfluo and BODIPY™ 581/591 C11. Immune Mediators in Cancer: Methods Mol Biol. 2026 Jan 2 (pp. 135-142). New York, NY: Springer US. [CrossRef]
- Bano I, Jeevanandam J, Tsenov G. Advances in therapeutic hybrid neuromodulation for Parkinson’s disease. 3 Biotech. 2026 Jan;16(1):58. [CrossRef]
- Jin Y, Wang L, Lin R, He J, Liu D, Liu Y, Deng Y. Synergistic Effects of Plant Polysaccharides and Probiotics: A Novel Dietary Approach for Parkinson’s Disease Intervention. Pharmaceuticals. 2026 Jan 15;19(1):157. [CrossRef]
- Liu Z, Wang R, Shen M, Lan X, Yan W, Wang S, Jiang M, Li R, Zhao J, Wang Q, Xv X. A LNK–CBL–HNRPA2B1–GPX4 Signaling Axis Mediates Dopaminergic Neuron Vulnerability to Ferroptosis in Parkinson’s disease. Redox Biol. 2026 Jan 23:104039. [CrossRef]
- Hristov BD. The role of glutathione metabolism in chronic illness development and its potential use as a novel therapeutic target. Cureus. 2022 Sep 28;14(9). [CrossRef]
- Mahoney-Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog Neurobiol. 2021 Jan 1; 196:101890. [CrossRef]
- Kumari M. Nutritional Strategies for Neuroprotection: Vitamins, Minerals, and Beyond. Nourishing the Brain: Diet and Nutrition Strategies in Managing Neurological Disorders 2026 Jan 8 (pp. 83-114). Singapore: Springer Nature Singapore. [CrossRef]
- Liu Z, Zhang H, Wan B, Yin S, Yue R. Advances and Therapeutic Potential of Anthraquinone Compounds in Neurodegenerative Diseases: A Comprehensive Review. Drug Des Devel Ther. 2026 Dec 31:1-31. [CrossRef]
- Chen XL, Lv JP, Li RH, Ge CB, Wang YY, Liu SC, Zhang GJ. Alkaloid Components From Orychophragmus violaceus and Their Ferroptosis Inhibition Activity. Chem Biodivers. 2026 Jan;23(1):e03337. [CrossRef]
- Tuo QZ, Bush AI, Lei P. Ferroptosis in neurological diseases: moving towards therapeutic intervention. Mol Psychiatry. 2026 Jan 19:1-9. [CrossRef]
- Sonkar P, Saif M, Sonkar A. Dementia and Diet: How Nutrients Can Influence Cognitive Decline. InNourishing the Brain: Diet and Nutrition Strategies in Managing Neurological Disorders 2026 Jan 8 (pp. 255-273). Singapore: Springer Nature Singapore. [CrossRef]
- Du ZR, Li C, Cai MY, Liu X, Chen TQ, Sun CC, Chen WF, Zhou SN, Yuan SX, Zhao ZC, Wu JY. Selenium nanoparticles functionalized by mushroom polysaccharides potentiated neuroprotection of selenium against Parkinson’s disease with lower toxicity. Food Sci Hum Wellness. 2026 Jan 15. [CrossRef]
- Topalis V, Voros C, Ziaka M. Targeting Inflammation in Alzheimer’s Disease: Insights Into Pathophysiology and Therapeutic Avenues-A Comprehensive Review. J Geriatr Psychiatry Neurol. 2026 Mar;39(2):115-44. [CrossRef]
- Sharma V, Verma R, Sharma P, Singh TG. Programmed cell death pathways in huntington’s disease: spotlight on ferroptosis and pyroptosis. Neurol Sci. 2026 Jan;47(1):83. [CrossRef]
- Zhou RP, Chen Y, Wei X, Yu B, Xiong ZG, Lu C, Hu W. Novel insights into ferroptosis: implications for age-related diseases. Theranostics. 2020 Oct 26;10(26):11976. [CrossRef]
- Samaddar S, Redhwan MA, Eraiah MM. Exploring the Efficacy of Natural Anti-inflammatory Compounds in Neurological Disorders. InNourishing the Brain: Diet and Nutrition Strategies in Managing Neurological Disorders 2026 Jan 8 (pp. 175-200). Singapore: Springer Nature Singapore. [CrossRef]
- Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, Barnham KJ, Crouch PJ, Ayton S, Donnelly PS, Bush AI. CuII (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020 Feb;177(3):656-667. [CrossRef]
- Chen J, Lu X, Liu S, Hu W, Zhou X, Wang Z. Betaine Inhibits Ferroptosis After Intracerebral Hemorrhage by Activating the Nrf2/HO-1 Pathway. Antioxidants. 2026 Jan 21;15(1):135. [CrossRef]
- Entezari S, Haghi SM, Norouzkhani N, Sahebnazar B, Vosoughian F, Akbarzadeh D, Islampanah M, Naghsh N, Abbasalizadeh M, Deravi N. Iron Chelators in Treatment of Iron Overload. J Toxicol. 2022 May 5;2022:4911205. [CrossRef]
- Zhu K, Zhuang Y, Lu H, Song C, Cao X, Cai F, Liu A. Ferroptosis in Mycotoxin-Induced Toxicity: Molecular Mechanisms, Intervention Implications, and Future Directions. J Agri Food Chem. 2026 Jan 10. [CrossRef]
- Niu L, Yang Z, Zeng Y, Huang Z, Yang T, Liu S, Zhong H, Ye M, Lei S, Gao L, Tan P. Moscatilin alleviates severe acute pancreatitis by activating the NRF2/HO-1 pathway to inhibit ferroptosis. Int Immunopharmacol. 2026 Jan 15; 169:115972. [CrossRef]
- Taiwei Jiao, Yiman Chen, Haiyan Sun, Lina Yang. Targeting ferroptosis as a potential prevention and treatment strategy for aging-related diseases, Pharmacol Res, Oct 208; 107370, 2024. [CrossRef]
- Xiang Y, Song X, Long D. Ferroptosis regulation through Nrf2 and implications for neurodegenerative diseases. Arch Toxicol. 2024 Mar;98(3):579-615. [CrossRef]
- Fei Y, Leng Q. Research Progress of Ferroptosis in Cerebral Infarction. Brain Behav. 2026 Jan;16(1):e71192. [CrossRef]
- Kumar A, Mondal T, Ramesh M, Bhoi J, Samanta S, Govindaraju T. Griess’s Reagent-Based Azo Compounds Ameliorate Multifaceted Toxicity and Ferroptosis in Alzheimer’s Disease. ACS Chem Neurosci. 2026 Jan 28. [CrossRef]
- Namdev Dhas, Ritu Kudarha, Ruchi Tiwari, Gaurav Tiwari, Neha Garg, Praveen Kumar, Sanjay Kulkarni, Jahnavi Kulkarni, Soji Soman, Aswathi R. Hegde, Jayvadan Patel, Atul Garkal, Anam Sami, Deepanjan Datta, Viola Colaco, Tejal Mehta, Lalitkumar Vora, Srinivas Mutalik. Recent advancements in nanomaterial-mediated ferroptosis-induced cancer therapy: Importance of molecular dynamics and novel strategies, Life Sci, 346, 122629, 2024. [CrossRef]
- Zhang X, Sun Y, Zhao C, Zou H, Hu L, Wen L, Tang Q. Panax notoginseng Saponins Alleviate OSF by Inhibiting Ferroptosis Through GPX4 Activation. J Oral Pathol Med. 2026 Jan;55(1):107-19. [CrossRef]
- Lu M, Zhou Y, Sun L, Shafi S, Ahmad N, Sun M, Dong J. The molecular mechanisms of ferroptosis and its role in glioma progression and treatment. Front Oncol. 2022 Aug 16;12:917537. [CrossRef]
- Pathak K, Kumari T, Aggarwal L, Singh V. Preventive dietary and lifestyle strategies for neurodegenerative diseases: a comprehensive review. Nutr Neurosci. 2026 Jan 22:1-26. [CrossRef]
- Jakaria M, Belaidi AA, Bush AI, Ayton S. Vitamin A metabolites inhibit ferroptosis. Biomed Pharmacother. 2023 Aug 1; 164:114930. [CrossRef]
- Kumar S, Mishra DK, Usmani N, Kumar H. Vitamins and Their Role in Brain Function. Optimizing Brain Health and Performance Through Essential Nutrients and Lifestyle Changes. 2024:83-97. [CrossRef]
- Zhang M, Chen X, Zhang Y. Mechanisms of vitamins inhibiting ferroptosis. Antioxidants. 2024 Dec 20;13(12):1571. [CrossRef]
- Gulcin İ, Alwasel SH. Fe3+ reducing power as the most common assay for understanding the biological functions of antioxidants. Processes. 2025 Apr 24;13(5):1296. [CrossRef]
- Zhang W, Yu S, Jiao B, Zhang C, Zhang K, Liu B, Zhang X. Vitamin D3 Attenuates Neuropathic Pain via Suppression of Mitochondria-Associated Ferroptosis by Inhibiting PKCα/NOX4 Signaling Pathway. CNS Neurosci Ther. 2024 Sep;30(9):e70067. [CrossRef]
- da Cunha Germano BC, de Morais LC, Idalina Neta F, Fernandes AC, Pinheiro FI, do Rego AC, Araújo Filho I, de Azevedo EP, de Paiva Cavalcanti JR, Guzen FP, Cobucci RN. Vitamin E and its molecular effects in experimental models of neurodegenerative diseases. Int J Mol Sci. 2023 Jul 7;24(13):11191. [CrossRef]
- Zhang M, Zhang Q, Du P, Chen X, Zhang Y. Roles of vitamin K-dependent protein in biomineralization. Int J Mol Med. 2023 Nov 24;53(1):6. [CrossRef]
- Mishima E, Wahida A, Seibt T, Conrad M. Diverse biological functions of vitamin K: from coagulation to ferroptosis. Nat Metab. 2023 Jun;5(6):924-32. [CrossRef]
- Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M, Henkelmann B. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022 Aug 25;608(7924):778-83. [CrossRef]
- Winter, A. N., & Bickford, P. C. (2019). Anthocyanins and Their Metabolites as Therapeutic Agents for Neurodegenerative Disease. Antioxidants, 8(9), 333. [CrossRef]
- Schulz M, Gonzaga LV, de Souza V, Farina M, Vitali L, Micke GA, Costa AC, Fett R. Neuroprotective effect of juçara (Euterpe edulis Martius) fruits extracts against glutamate-induced oxytosis in HT22 hippocampal cells. Food Res Int. 2019 Jun 1; 120:114-23. [CrossRef]
- Yang H, Hong Y, Gong M, Cai S, Yuan Z, Feng S, Chen Q, Liu X, Mei Z. Fisetin exerts neuroprotective effects in vivo and in vitro by inhibiting ferroptosis and oxidative stress after traumatic brain injury. Front Pharmacol. 2024 Nov 20;15:1480345. [CrossRef]
- Chahal SK, Kabra A, Saeedan AS, Ansari MN. Fisetin: a multitarget flavonol bridging oxidative stress, inflammation, aging, and cancer. Naunyn Schmiedebergs Arch Pharmacol. 2026 Jan 15:1-23. [CrossRef]
- Nabizadeh F. Neuroprotective role of Fisetin in Alzheimer’s disease: An overview of potential mechanism and clinical findings. Neurol Lett. 2024 Jun 20;3:14-25. [CrossRef]
- Varshney H, Siddique YH. Effect of flavonoids against Parkinson’s disease. Cent Nerv Syst Agents Med. 2024 Aug 1;24(2):145-65. [CrossRef]
- Maher P. Preventing and treating neurological disorders with the flavonol fisetin. Brain Plast. 2020 Dec;6(2):155-66. [CrossRef]
- Jiang Y, Tang X, Deng P, Jiang C, He Y, Hao D, Yang H. The neuroprotective role of fisetin in different neurological diseases: A systematic review. Mol Neurobiol. 2023 Nov;60(11):6383-94. [CrossRef]
- Samarasinghe KTG, Crews CM. Targeted protein degradation: A promise for undruggable proteins. Cell Chem Biol. 2021 Jul 15;28(7):934-951. [CrossRef]
- Tiwari PC, Pal R, Chaudhary MJ, Nath R. Artificial intelligence revolutionizing drug development: Exploring opportunities and challenges. Drug Dev Res. 2023 Dec;84(8):1652-63. [CrossRef]
- Das J, Ghosh S, Ghosh B, Sarkar S, Sarkar S. Exploring the Role of Melatonin in Disease Prevention and Anti-Aging. Biotech Miracles: Harnessing the Power of Microbes. 2024;14:888292. [CrossRef]
- Yehia A, Abulseoud OA. Melatonin: a ferroptosis inhibitor with potential therapeutic efficacy for the post-COVID-19 trajectory of accelerated brain aging and neurodegeneration. Mol Neurodegener. 2024 Apr 19;19(1):36. [CrossRef]
- Pourhanifeh MH, Hosseinzadeh A, Koosha F, Reiter RJ, Mehrzadi S. Therapeutic effects of Melatonin in the regulation of ferroptosis: a review of current evidence. Curr Drug Targets. 2024 Jun 1;25(8):543-57. [CrossRef]
- Mollica L, Cupaioli FA, Rossetti G, Chiappori F. An overview of structural approaches to study therapeutic RNAs. Front Mol Biosci. 2022 Oct 28; 9:1044126. [CrossRef]
- Viktorinova A. Future perspectives of oxytosis/ferroptosis research in neurodegeneration diseases. Cell Mol Neurobiol. 2023 Aug;43(6):2761-8. [CrossRef]
- Ali A, Babar Q, Saeed A, Sergi D, Naumovski N, Mohamed Ahmed IA, Shamlan G, Hafiz HA, Manzoor MF, Trafialek J, Madilo FK. Emerging Links Between Ferroptosis and Neurodegeneration: Implications for Disease Mechanisms and Nutraceutical Interventions. Food Sci Nutr. 2025 Jun;13(6):e70385. [CrossRef]
Figure 2.
Convergent ferroptosis-associated axes across neurodegenerative diseases. PD, AD, HD, and ALS share overlapping determinants of ferroptotic susceptibility, including iron dyshomeostasis, impairment of GSH/GPX4-linked antioxidant defense, PUFA-driven lipid remodeling and peroxidation, mitochondrial ROS amplification, and permissive neuroinflammatory milieu. Disease context shapes the dominant nodes and vulnerable regions, but the shared axes highlight common therapeutic leverage points that can be targeted to reduce ferroptosis-associated injury.
Figure 2.
Convergent ferroptosis-associated axes across neurodegenerative diseases. PD, AD, HD, and ALS share overlapping determinants of ferroptotic susceptibility, including iron dyshomeostasis, impairment of GSH/GPX4-linked antioxidant defense, PUFA-driven lipid remodeling and peroxidation, mitochondrial ROS amplification, and permissive neuroinflammatory milieu. Disease context shapes the dominant nodes and vulnerable regions, but the shared axes highlight common therapeutic leverage points that can be targeted to reduce ferroptosis-associated injury.
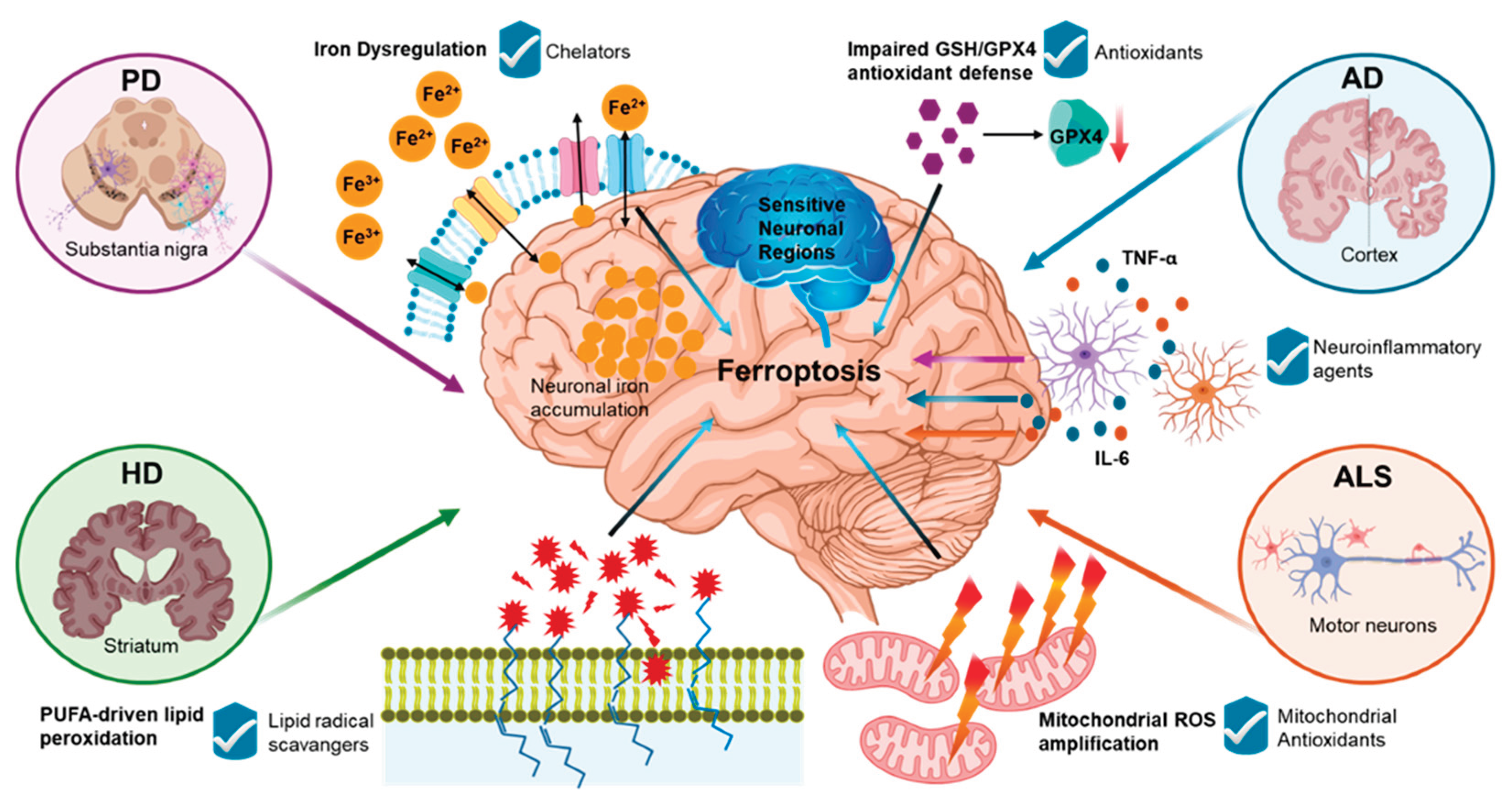

Table 1.
Inhibitors and inducers of ferroptosis in neurodegenerative diseases.
| Mechanisms | Purpose | Method Examples | Role of Action | References |
|---|---|---|---|---|
| Dopamine-based therapies | Inhibitor | Levodopa, a dopamine receptor agonist | In PD, these drugs can cross the BBB, facilitate the removal of excess iron from the brain, and stabilize GPX4. | [91] |
| TfR 1 regulators | Inhibitor | Hepcidin agonist | In AD, regulating TfR 1 mediates cellular iron uptake and maintains iron homeostasis in neuronal cells. | [110] |
| Antioxidants | Inhibitor | Fer-1, Lip-1 |
It targets lipid peroxidation and slows cognitive decline in patients with mild-to-moderate AD. It blocks ROS and effectively fixes AB-induced neuronal death | [98] |
| ROS free radicals | Inhibitor | Edaravone | In ALS patients, it reduces motor neuron damage and inhibits ferroptosis. | [105] |
| Iron Chelators | Inhibitor | DFP, DFO, DFE |
In PD, it protects against neuronal injury through inhibiting ferroptosis. In AD, DFO inhibits erastin-induced ROS accumulation. In ALS, it is shown to improve motor neuron survival and restore motor function. In HD, IV administration of DFO shown to relieve symptoms in mouse models. |
[102,103,107] |
| Vitamin E | Inhibitor | A-tocopherol | It targets lipid peroxidation and slows cognitive decline in patients with mild-to-moderate AD. It destroys the chain reaction of automatic oxidation. | [99] |
| Nitroxides | Inhibitor | NOX2 mediated ROS, Iron (II) citrate |
In neurogenerative diseases, nitroxides can cross the BBB and target lipid peroxidation. In AD, nitroxides positively induce neuroplasticity and neuroprotection. |
[23] |
| Selenium | Inhibitor | Selenocysteine, Tat SelPep | It acts as the active site of GPX4 and can cross the BBB to help protect against ferroptosis. | [100] |
| Zileuton | Inhibitor | 5-lipoxygenase (LOX) | It protects cells from lipid peroxidation by down-regulating LOX. | [93] |
| Erastin | Inducer | System Xc- | It mediates ferroptosis via inhibiting system Xc-. | [116] |
| Glutamate | Inducer | cystine | It mediates ferroptosis through cystine uptake inhibition of system Xc-. | [16] |
| Sulfasalazine | Inducer | System Xc- | It mediates ferroptosis via inhibiting system Xc-. | [16] |
| Sorafenin | Inducer | cystine | It mediates ferroptosis through cystine uptake inhibition of system Xc-. | [14] |
| RSL3 | Inducer | Selenocysteine | It blocks the activity of GSH and GPX4 at the active site selenocysteine. | [8] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



