
Submitted:
09 February 2026
Posted:
10 February 2026
You are already at the latest version
Abstract
Background: The aim of this study was to evaluate the clinical performance of an in-house qualitative CMV real-time PCR assay for the detection of cytomegalovirus (CMV) DNA in various non-plasma clinical sample types, in comparison with a commercial ref-erence method. Methods: In this prospective comparative study, 186 clinical sam-ples—including bronchoalveolar lavage fluid (BALF), stool, urine, colonoscopic biopsy, amniotic fluid, and intraocular fluid—were analyzed. A total of 166 samples with valid results from both test systems were included in the inter-method comparison. CMV DNA was detected using an in-house qualitative PCR assay in parallel with the reference method (artus® CMV QS-RGQ kit). Clinical performance was assessed using positive percent agreement (PPA), negative percent agreement (NPA), overall percent agreement (OPA), Cohen’s kappa coefficient (κ), and predictive values, in accordance with the CLSI EP12-A2 guideline for qualitative diagnostic tests. Results: When all clinical samples were evaluated collectively, good overall agreement was observed between the developed test and the reference method (κ = 0.66). High PPA, NPA, and kappa values were obtained for stool, urine, and invasive samples. In BALF samples, inter-method agreement was mod-erate, and the high NPV supported clinical exclusion of CMV infection. Predictive values varied according to the CMV DNA positivity rate of the analyzed sample groups. Conclusions: The laboratory-developed qualitative CMV PCR assay showed good inter-method agreement with a commercial reference method and appears suitable for qualitative CMV detection in non-plasma clinical specimens under routine laboratory conditions. The findings reflect comparative performance rather than gold-standard-based diagnostic accuracy or viral load quantification.
Keywords:
Cytomegalovirus
; real-time PCR
; qualitative PCR
; non-plasma clinical samples
; diagnostic agreement
1. Introduction
Cytomegalovirus (CMV) is a DNA virus belonging to the Betaherpesvirinae subgroup of the Herpesviridae family, and is widely prevalent worldwide. While CMV infection is mostly asymptomatic or mild in individuals with healthy immune systems, it can lead to serious clinical conditions in immunocompromised patients. Particularly, recipients of solid organ and hematopoietic stem cell transplants, individuals with inflammatory bowel disease, and newborns at risk for congenital infections are among the high-risk groups for CMV infection morbidity and mortality [1,2]. Early detection of CMV infection in these patient groups is critically important for timely initiation of antiviral therapy and prevention of clinical complications [2,3]. Therefore, the methods used in the laboratory diagnosis of CMV infection must be reliable in terms of sensitivity, specificity, and clinical applicability.
Nowadays, in the diagnosis of CMV, serological methods, virological culture, and molecular techniques are used. Polymerase chain reaction (PCR), a molecular method, is increasingly preferred in clinical practice due to its high sensitivity and specificity, reproducibility, ability to detect CMV DNA even at low viral loads, and rapid results. These methods enable the direct detection of the viral genome, making it possible to diagnose active infection and monitor viral load. They are widely used in routine clinical laboratory practice because they offer the possibility of quantitative or qualitative assessment [4,5,6,7,8].
While plasma samples are commonly used for diagnosing CMV infections, the analysis of non-plasma clinical samples such as stool, urine, bronchoalveolar lavage fluid (BALF), and various tissue biopsies is critical for assessing the location and spread of the infection. Most commercially available real-time PCR kits are optimized for standardized plasma samples, and data regarding their performance in different clinical specimen types and the clinical validity of laboratory-developed qualitative CMV PCR assays for non-plasma samples remain limited [9,10,11,12,13,14]. However, it has been shown that in some cases, CMV DNA can be detected in tissue samples even when it is negative in serum or plasma samples, which highlights the diagnostic potential of non-plasma samples [15].
However, the clinical performance of CMV PCR tests can vary depending on many factors, such as the test platform used, the targeted viral genome region, the nucleic acid extraction method, and the type of clinical sample analyzed [5,16]. The presence of PCR inhibitors, low viral load levels, and the heterogeneous distribution of CMV in tissue or body fluids, particularly in plasma-free clinical samples, can lead to inconsistent results between tests, highlighting the need for clinical validation studies for these sample types [16,17].
In the literature, studies comparing different commercial or laboratory-developed CMV PCR tests have frequently reported sample type-based performance differences, and inter-method agreement can vary depending on the sample matrix [18,19,20]. This situation highlights the importance of new CMV PCR tests undergoing comparative validation studies on different clinical sample types before being introduced into clinical use. The aim of this study was to comparatively evaluate a laboratory-developed CMV PCR assay against existing commercial methods across different non-plasma clinical specimen types and to assess its applicability under routine clinical laboratory conditions. In this respect, the original contribution of the study lies not in a technological innovation of PCR itself, but in providing a systematic, inter-method comparative evaluation across multiple specimen matrices.
2. Materials and Methods
2.1. Study Design and Clinical Specimens
This study is a prospective and comparative validation study designed to evaluate the clinical performance of a qualitative real-time PCR kit developed for the detection of cytomegalovirus (CMV) DNA in different clinical sample types. The kit evaluated in the study was developed by PharmaLine (PharmaLine Health Services Ind. Trade Inc., Istanbul, Türkiye).
A total of 186 clinical samples collected between June 2025 and September 2025 were included in the study; however, 166 samples for which valid positive or negative results could be obtained with both test systems were included in the inter-method comparison analysis. All specimens included in the analysis were tested in parallel with both the laboratory-developed CMV PCR assay and the artus® CMV QS-RGQ reference method. A total of 20 samples were excluded from the analysis due to internal control failure or technical reasons. Most of the excluded samples consisted of specimen types with a high inhibition potential, such as BALF and stool, and this distribution is presented in Supplementary Tables S1 and S2.
The qualitative PCR kit evaluated in this study (PharmaLine Health Services Ind. Trade Inc., Istanbul, Türkiye) was developed in the laboratory and its results were analyzed by comparing them with a reference method.
2.2. Clinical Setting and Sample Collection
The clinical samples included in the study were obtained from pediatric and adult patients referred from gastroenterology, pulmonology, ophthalmology, perinatology, hematology, pediatric gastroenterology and hepatology, neonatology, and pediatric hematology and oncology clinics of Ankara Etlik City Hospital with suspected CMV infection. All clinical specimens were obtained from patients in whom CMV infection was considered in the differential diagnosis based on clinical, laboratory, and/or radiological findings, as part of routine diagnostic workup. Suspected CMV infection was defined based on clinical manifestations such as persistent or unexplained fever, gastrointestinal symptoms (e.g., diarrhea, abdominal pain, gastrointestinal bleeding), respiratory symptoms with radiological findings suggestive of pneumonia, cytopenia, elevated inflammatory markers, or organ-specific findings (e.g., visual symptoms, fetal abnormalities), particularly in immunocompromised patients. All samples were collected by the relevant clinics as part of the routine clinical diagnostic process, in appropriate sterile containers, and delivered to the Medical Microbiology Laboratory within a short period of time, in accordance with biosafety and pre-analytical quality criteria. Samples received in the laboratory were recorded, anonymized by concealing patient identification information, and stored frozen at -80 °C until analysis to preserve nucleic acid stability.
2.3. Inclusion and Exclusion Criteria
The study included clinical samples obtained from pediatric and adult patients referred from gastroenterology, pulmonology, ophthalmology, perinatology, and pediatric infectious diseases clinics for CMV infection testing and submitted to the Medical Microbiology Laboratory. The clinical specimens included BALF, stool, urine, colonoscopic biopsy, amniotic fluid, and intraocular fluid samples. Only samples that yielded valid positive or negative results with both the developed CMV PCR test and the reference method were included in the analysis.
Serum, plasma, and whole blood samples were not included in the study design. In addition, samples that failed the internal control during nucleic acid extraction or PCR, as well as samples that could not be evaluated by both test systems due to technical reasons, were excluded from the analysis.
2.4. Pre-Extraction Preparation of Clinical Samples
In this study, nucleic acid isolation was performed directly on urine and intraocular fluid samples without any pre-processing, using the volume recommended in the kit protocol. In contrast, tissue, stool, and BALF samples were pre-processed according to manufacturer protocols prior to extraction.
Stool samples were prepared with PharmaSEP buffer (Lot No: PHSP2024-50 / PHSP2024-100; Pharmaline Health Services Ind. Trade Inc., Istanbul, Türkiye); 100 mg of solid samples and 100 µL of liquid samples were added to 900 µL of buffer. Samples were vortexed, subjected to freeze-thaw treatment, and the supernatant obtained after centrifugation was used for extraction.
BALF samples were treated with 5× dithiothreitol (DTT, Sigma-Aldrich, USA) (DTT:sample = 1:4), and after incubation and homogenization, a volume of 300 µL was extracted. BALF samples were collected as part of the routine diagnostic evaluation upon clinicians’ request in patients with suspected CMV pneumonia or presumed pulmonary involvement in immunosuppressed individuals.
Tissue samples were prepared by homogenization according to the manufacturer's instructions for the Molecision Nucleic Acid Extraction Kit (SNIBE Co., Ltd., Shenzhen, China) and included in the extraction process.
2.5. Nucleic Acid Extraction
Total nucleic acid isolation from clinical samples was performed using the Molecision Fully Automated Nucleic Acid Purification System (MP-96) device. Extraction procedures were performed according to the manufacturer's instructions using the SNIBE Nucleic Acid Extraction Kit (Shenzhen New Industries Biomedical Engineering Co., Ltd., Shenzhen, Guangdong, China). The elution volume was standardized to 50 µL for all samples. The obtained nucleic acids were stored under appropriate conditions according to the manufacturer's recommendations until PCR analysis.
2.6. Real-Time PCR Design
The US17 gene, a conserved segment of the CMV genome, was targeted to detect CMV DNA. The human β-actin gene served as an internal control to assess extraction efficiency and PCR inhibition. Although the assay was qualitative in nature, the experimental design and reporting followed the relevant components of the MIQE guidelines. Accordingly, primer and probe sequences were fully reported, an internal control was included to monitor extraction efficiency and PCR inhibition, and replicate testing was not performed because the assay was qualitative.
2.6.1. Primer and Probe Design
The primer and probe sequences used for the CMV target are as follows:
- CMV US17 forward primer: 5′-TCTCTGTACCTCCCGCAAAA-3′
- CMV US17 reverse primer: 5′-AGACAAACTCATCGCTTGGA-3′
- CMV US17 probe: 5′-FAM-TGACCTGGTTATCGTCACGCG-BHQ-3′
- Primer and probe sequences for the β-actin gene used as an internal control:
- β-actin forward primer: 5′-CACCATTGGCAATGAGCGGTT-3′
- β-actin reverse primer: 5′-TAGTTTCGTGGATGCCACAGG-3′
- β-actin probe: 5′-HEX-CACTCTTCCAGCCTTCCTTCCTGGG-BHQ-3′
2.6.2. PCR Reaction Components and Conditions
PCR reactions were prepared in a total volume of 20 µL. The reaction mixture consists of the following components:
- 10 µL RapidXFire™ qPCR Master Mix (2×)
- CMV primers: 500 nM (each)
- CMV probe: 125 nM
- β-actin primers: 250 nM (each)
- β-actin probe: 62.5 nM
- 5 µL of template nucleic acid
In each study, a negative control (NTC) and a reaction blank containing only the reaction components were included for contamination control purposes.
2.6.3. Thermal Cycling Conditions
The PCR amplification was carried out on a Bio-Rad CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA) under the following conditions: an initial denaturation step at 95 °C for 5 min, followed by 44 amplification cycles consisting of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 45 s.
The CMV PCR assay used in this study was designed as a qualitative test. Therefore, MIQE guideline criteria specific to quantitative analyses, such as generation of standard curves, calculation of PCR efficiency, and absolute quantification, were not applied. However, all MIQE components relevant to qualitative PCR were fully implemented, including target gene definition, primer and probe sequences, amplification conditions, nucleic acid extraction method, use of an internal control, positive and negative controls, and clearly defined criteria for result interpretation.
Fluorescence signal measurement was performed at the binding/extension step of each cycle. Although quantitative analysis was not the aim of the study, it was conducted by considering the methodological criteria included in the MIQE guidelines, which can be adapted to qualitative real-time PCR applications.
2.6.4. US17 Region and In Silico Analysis
To assess the conservation of the US17 target region, an in silico alignment analysis was performed on available CMV genomes, demonstrating the absence of significant mismatches within the primer and probe binding regions. This analysis is provided as supplementary material (Table S3).
2.7. Reference Method
The artus® CMV QS-RGQ kit (QIAGEN, Hilden, Germany) was used as the reference method in the comparative clinical performance evaluation. With the developed CMV PCR kit, the reference test was run in parallel on the same extraction product, and the results were classified qualitatively (positive/negative). Reference test results were interpreted in accordance with the manufacturer's recommendations.
2.8. Sample Size
In this study, the sample size was determined based on the inclusion of all clinical samples accessible during the study period that yielded valid results with both test systems. This approach is a widely used method in diagnostic performance studies that include different clinical sample types and assess inter-method agreement.
2.9. Statistical Analysis
All statistical analyses were performed using IBM SPSS Statistics v26.0 software. Clinical performance indicators such as positive percent agreement (PPA), negative percent agreement (NPA), and overall percent agreement (OPA) were calculated along with sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
These calculations are based on true positive (TP), false positive (FP), false negative (FN), and true negative (TN) results. Sensitivity was calculated using the formula TP / (TP + FN); specificity, TN / (TN + FP); PPV, TP / (TP + FP); NPV, TN / (TN + FN); PPA, TP / (TP + FN); NPA, TN / (TN + FP); and OPA, (TP + TN) / (TP + FP + FN + TN). Inter-method qualitative agreement was assessed using Cohen's Kappa coefficient (κ) [22], and the interpretation was based on the classification proposed by Altman [23]. The calculation and reporting of clinical performance metrics were performed in accordance with the Clinical and Laboratory Standards Institute (CLSI) EP12-A2 guideline recommended for qualitative diagnostic tests [24].
3. Results
3.1. Distribution of Clinical Specimens
A total of 166 non-plasma clinical specimens were included in the analysis. The distribution of specimen types and overall CMV DNA positivity rates are presented in Table 1, with CMV positivity varying across different specimen types.
3.2. Agreement between the Laboratory-Developed Qualitative CMV PCR Assay and the Reference Method by Specimen Type
The agreement between the laboratory-developed qualitative CMV PCR assay and the reference method was further evaluated according to specimen type. The percentage distribution of true-positive, true-negative, false-positive, and false-negative results by specimen type is illustrated in Figure 1.
The diagnostic concordance between the reference method and the laboratory-developed qualitative CMV PCR assay is shown in Table 2 according to sample type.
PPA in BALF samples was determined to be 45.5%, NPA 87.9%, and OPA 81.2%. The Cohen's kappa coefficient was 0.36, signifying a weak degree of concordance between the approaches. In the stool samples instances, a strong concordance was attained with PPA at 85.7%, NPA at 92.6%, OPA at 91.2%, and Cohen's κ at 0.79. PPA was detected in urine samples at 80.0%, NPA at 95.8%, and OPA at 93.1%. The Cohen's kappa coefficient was 0.74, signifying a substantial degree of agreement. In the cohort where colonoscopic biopsy, amniotic fluid, and intraocular fluid samples were concurrently assessed, the PPA was determined to be 100%, the NPA was 96.3%, and the OPA was 97.1%, indicating a substantial level of concordance (Cohen's κ = 0.93).
Upon evaluating all samples collectively, the overall concordance between the test and reference methodologies was determined to be PPA 73.3%, NPA 91.9%, OPA 88.6%, and Cohen's κ = 0.66, signifying a substantial degree of overall agreement.
3.3. Analytical Sensitivity
The LoD95 for the CMV qPCR test, as determined by probit regression analysis, was 63.8 copies/µL, corresponding to a 95% probability of detection (Table 3, Figure 2). Considering the extraction and elution volumes, this value corresponds to approximately 1.6 × 10⁴ copies/mL of plasma.
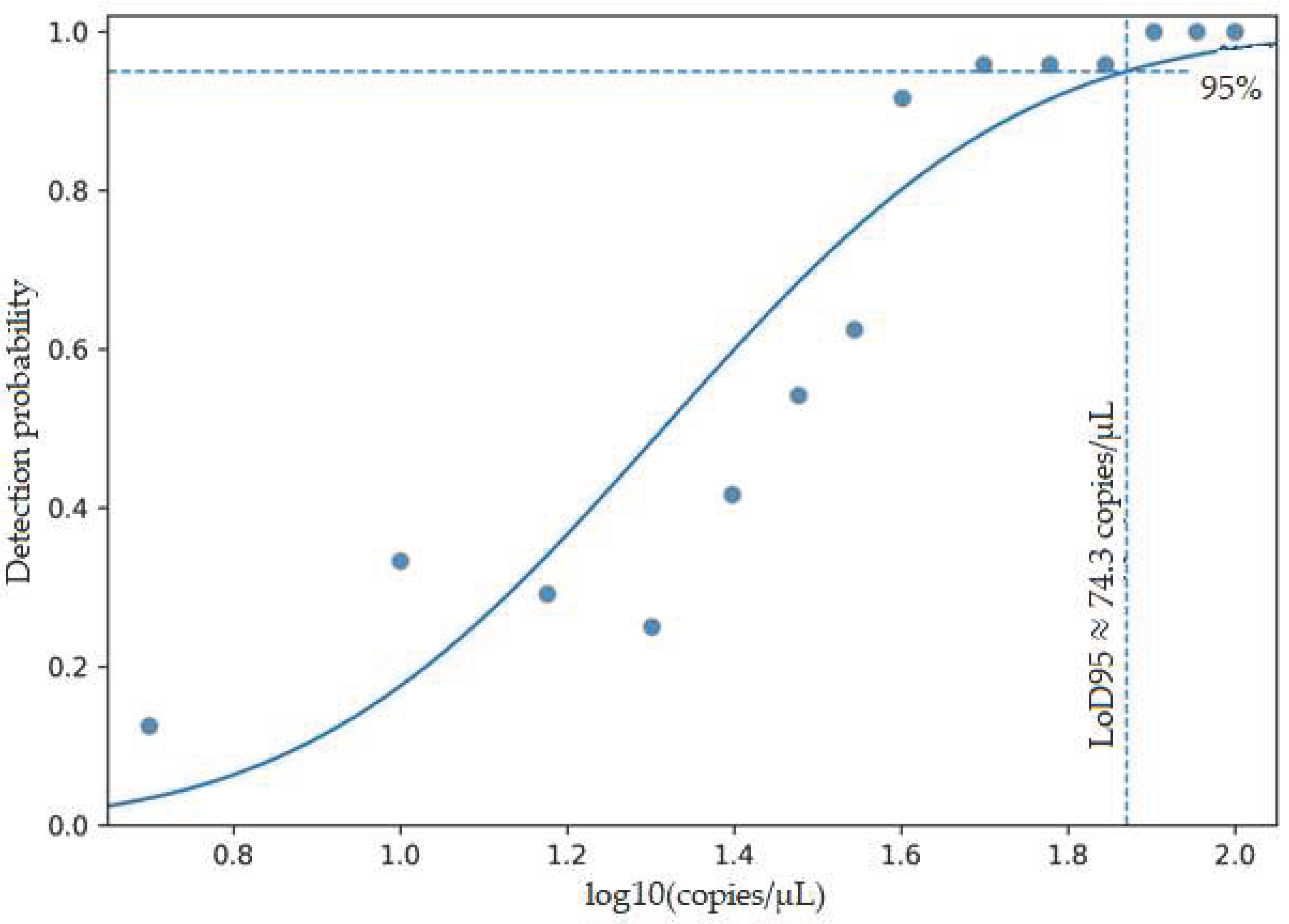
Figure 1.
Probit dose-response curve for determining the LoD95.
The detection probabilities (n_pos/n_total) obtained from 24 replicates for each concentration level were plotted against concentration in log10 (copies/µL) and fitted with a probit regression model. The dashed horizontal line indicates a 95% detection probability, while the dashed vertical line shows the calculated LoD95 value (63.80 copies/µL) corresponding to this threshold.
Analytical sensitivity was assessed to characterize the detection capability of the assay.
However, the assay was designed as a qualitative test and was not intended for clinical viral load quantification.
3.4. Performance Characteristics of the Laboratory-Developed Qualitative CMV PCR Assay
The diagnostic performance characteristics of the laboratory-developed qualitative CMV PCR assay, including sensitivity, specificity, and predictive values, are summarized by specimen type in Table 4.
In BALF samples, sensitivity was 45.5% (95% CI: 26.9–65.3), specificity was 87.9% (95% CI: 78.5–96.0), PPV was 41.7% (95% CI: 24.5–61.2), and NPV was 89.5% (95% CI: 78.9–95.1).
In stool samples, sensitivity was found to be 85.7% (95% CI: 48.7–97.4), specificity 92.6% (95% CI: 76.6–97.9), PPV 75.0%, and NPV 96.2%. The sensitivity in urine samples was determined to be 80.0% (95% CI: 37.6–96.4), the specificity 95.8% (95% CI: 79.8–99.3), the PPV 80.0%, and the NPV 95.8%.
Colonoscopic biopsy, amniotic fluid, and intraocular fluid groups showed no false negative results, with a sensitivity of 100.0% (95% CI: 64.6–100.0), specificity of 96.3% (95% CI: 81.7–99.3), PPV of 87.5%, and NPV of 100.0%.
When all samples were considered together, the overall sensitivity of the test was 73.3% (95% CI: 57.0–84.6), specificity was 91.9% (95% CI: 88.5–97.3), PPV was 66.7% (95% CI: 49.6–80.2), and NPV was 94.0% (95% CI: 88.6–96.9).
4. Discussion
In this study, the qualitative performance of a CMV PCR kit developed in a laboratory setting was evaluated in comparison to a reference method using different clinical sample types, and the results were analyzed on a sample type basis. The study design and reporting process were conducted in accordance with the recommended MIQE guidelines for PCR experiments and the CLSI EP12-A2 recommendations for the evaluation of qualitative diagnostic tests [21,22,23,24]. Although the artus® CMV QS-RGQ assay was used as the comparator method in this study, it is not a universally clinically validated gold standard for all non-plasma specimen types. Therefore, the findings should be interpreted as an evaluation of inter-method agreement and comparative performance rather than true diagnostic accuracy.
The results obtained from BALF samples showed a moderate level of agreement between the developed test and the reference method. The latent presence of CMV in the respiratory tract and subclinical replication suggest that CMV DNA detected by PCR in BALF samples may not always reflect invasive CMV pneumonia. Indeed, Boeckh and his colleagues emphasized that the relationship between BALF and plasma viral loads can be variable and that BALF PCR results should be interpreted carefully in clinical context [25].
CMV PCR positivity in BAL samples alone is not sufficient for the diagnosis of CMV pneumonia and should be interpreted in conjunction with the clinical presentation, radiological findings, and, when available, histopathological data. Likewise, research involving lung transplant recipients advocates for the assessment of BALF CMV PCR results in conjunction with histopathological and clinical data [26].
In our study, the false positive and false negative results observed in BALF samples may be related to the biological heterogeneity and rich composition of PCR inhibitors in this sample type. It has been shown previously in detail that inhibitors can suppress amplification in respiratory samples [17].
In BALF samples, the high NPV (89.5%) together with the relatively low PPV (41.7%) indicates that negative CMV PCR results are reliable for clinical exclusion, whereas positive results should be interpreted cautiously. This situation may be related to the biological heterogeneity of respiratory samples, the presence of PCR inhibitors, and the latent presence of CMV in the respiratory tract. These findings support the interpretation of BALF PCR results in conjunction with clinical, radiological, and histopathological findings [25,26].
The high concordance rates obtained from gastrointestinal system and stool samples indicate that the laboratory-developed qualitative CMV PCR assay exhibits reliable qualitative performance in these sample types. It is known that CMV can cause active infection and colitis in the gastrointestinal system, especially in immunocompromised patients [27]. Michel and his colleagues reported that the detection of CMV DNA in stool samples could be a valuable approach in excluding intestinal CMV disease [28].
Although stool samples create a matrix rich in PCR inhibitors, it has been shown that this effect can be largely reduced with appropriate nucleic acid extraction protocols and internal control strategies [17,18]. In our study, the high combined PPV and NPV, along with the particularly high negative percent agreement in stool samples, suggests that the test performs well enough for clinical use in screening and excluding intestinal CMV infection.
The good agreement and high PPV and NPV obtained in urine samples indicate that the laboratory-developed qualitative CMV PCR assay performs reliably in this sample type. The urinary excretion of CMV is a well-defined virological characteristic, and Revello and Gerna's comprehensive review emphasizes that urine and other sterile body fluids are reliable clinical materials for CMV diagnosis [28]. It has been shown that urine PCR tests have high diagnostic value, especially in congenital CMV infections [30]. In our study, the high compliance and predictive values observed in urine samples were found to be consistent with the literature [28,29].
In this group, which includes colonoscopy biopsies, amniotic fluid, and intraocular fluid samples, the high predictive values and excellent agreement among the methods, as well as the reliable qualitative performance of the laboratory-developed qualitative CMV PCR assay in invasive and sterile body fluids, are noteworthy. It has been reported in the literature that CMV can reflect direct tissue invasion in invasive samples such as gastrointestinal tissue, amniotic fluid, and ocular fluids, and that molecular methods can provide faster and more sensitive diagnostic information compared to histopathological approaches in these sample types. However, due to the limited number of samples in this group, the findings need to be validated in larger patient series [18,28,30,31].
When all clinical sample types were evaluated together, it was observed that the laboratory-developed qualitative CMV PCR assay showed a high degree of overall agreement with the reference method and good to very good inter-method agreement according to Cohen's kappa coefficient. The high NPV (94.0%) and moderate PPV (66.7%) observed when all specimens were evaluated together, together with the high predictive values in urine, stool, and invasive samples, support the use of the test as a reliable qualitative screening and exclusion tool in routine clinical practice. In contrast, the high NPV and relatively low PPV in BALF samples support the idea that CMV DNA detected in BALF samples may not always reflect active or invasive infection, and that the results should be interpreted in conjunction with clinical, radiological, and histopathological data [25,26].
It is known that in the evaluation of qualitative diagnostic tests, PPV and NPV are dependent on the prevalence of CMV in the population where the test is applied. Therefore, it should be kept in mind that the predictive values obtained in our study may vary across different patient groups and clinical scenarios [4,20,24]. However, the consistent performance observed in sample type-based analyses supports the potential of the evaluated CMV PCR kit to be a suitable qualitative diagnostic tool for routine laboratory use in different clinical materials.
In the evaluation of inter-method agreement, the choice of the targeted viral genome region is also an important factor affecting test performance. In studies comparing the clinical performance of real-time PCR approaches targeting different regions of the cytomegalovirus genome, conserved gene regions such as US17 have been reported to be reliable targets. Sanghavi and her colleagues used real-time PCR targeting the US17 and UL54 regions to assess CMV load in organ transplant recipients, and the resulting viral load results were consistent with pp65 antigenemia [32]. This study supports the reliability of the PCR target regions used for clinical monitoring. Additionally, it has been reported that in studies conducted by NIST for the development of reference material for CMV, different gene regions, including US17, were evaluated sequentially and analytically as qPCR targets [33]. In light of these findings, the preference for PCR design targeting the US17 region in our study can be considered one of the potential determinants of the overall concordance observed across different clinical sample types.
The main strengths of this study are the comprehensive sample type assessment, methodological robustness, and multifaceted performance analysis for clinical application. This study has the potential to make significant contributions to the literature by providing a comprehensive and sample-type-based assessment of the clinical performance of the laboratory-developed qualitative CMV PCR assay in CMV diagnosis. A particularly notable strength of the study is the comparative evaluation of the test in a wide range of non-plasma clinical samples (stool, urine, bronchoalveolar lavage, colonoscopic biopsy, amniotic fluid, and intraocular fluid). This comprehensive example type assessment expands the potential clinical use of the test and makes a significant contribution to the diagnosis of CMV infections in different locations. From a methodological perspective, the prospective and comparative design of the study, its execution in accordance with CLSI EP12-A2 and MIQE guidelines, and the parallel analysis of all samples with the reference method using a consistent nucleic acid extraction protocol support the reliability and transparency of the findings. Additionally, the high concordance rates obtained with the reference method, particularly in groups of stool, urine, and invasive samples, indicate the feasibility of using the evaluated CMV PCR test in routine clinical laboratory practices.
The CMV PCR assay used in this study was designed as a qualitative test and was not intended for viral load quantification. Since numerical Cq/Ct values were not retrospectively archived in a standardized manner within the routine diagnostic workflow, a quantitative analysis based on Cq values could not be performed. Nevertheless, to facilitate the interpretation of samples showing inter-method discordance, a descriptive “Cq category” classification was applied to discordant samples, and the details of this classification are presented in Supplementary Tables S2 and S3.
However, our study also has some limitations. First, the assessments were performed on qualitative results obtained from non-plasma clinical samples, which did not allow for a detailed evaluation of viral load levels specific to different specimen types. In addition, non-plasma clinical samples were not paired with concurrent plasma specimens, which limits the direct translation of the findings into clinical diagnostic interpretation and precludes comparisons between viral compartments. Although colonoscopic biopsy, amniotic fluid, and intraocular fluid specimens differ in their biological and clinical characteristics, statistically meaningful subgroup analyses could not be performed due to the limited number of samples available for each specimen type. Therefore, these specimens were evaluated descriptively under a single ‘invasive specimen’ group. The sample size of the study was not determined based on a priori statistical power calculations but was limited to the clinical specimens accessible during the study period. As a result, subgroup analyses, particularly for invasive specimens, were underpowered. Additionally, the relatively low number of samples for certain specimen types further limited the statistical power of subgroup analyses. Finally, the single-center design of the study restricts the generalizability of the findings. Future prospective studies evaluating paired plasma and non-plasma samples in larger, multicenter cohorts are warranted to strengthen clinical correlation and validate these results.
5. Conclusions
In conclusion, the laboratory-developed qualitative CMV real-time PCR assay demonstrated good overall inter-method agreement with the artus® CMV QS-RGQ reference method across different non-plasma clinical specimen types. The high concordance and predictive values observed particularly in stool, urine, and invasive samples support the use of the assay as a reliable qualitative CMV screening and diagnostic tool in routine clinical microbiology laboratories. In BALF samples, the high negative predictive value indicates that negative PCR results are useful for clinical exclusion, whereas positive results should be interpreted cautiously and in conjunction with clinical, radiological, and, when available, histopathological findings. As the evaluated assay was designed as a qualitative test, the findings reflect inter-method agreement and comparative performance rather than gold-standard-based diagnostic accuracy or viral load quantification. Overall, these results support the applicability of the laboratory-developed qualitative CMV PCR assay for the detection of CMV DNA in non-plasma clinical specimens under routine laboratory conditions, while highlighting the importance of clinical context in result interpretation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
Conceptualization, M.A., A.B. and C.Ç.; methodology, M.A. and A.B.; software, M.A., A.B., C.Ç., E.T.G., D.Ö.; validation, M.A., A.B., C.Ç., E.T.G., G.C.Y., M.F.K. and M.M.; formal analysis, M.A.; investigation, M.A., C.Ç., E.T.G., D.Ö., M.F.K., G.C.Y., M.M. and Y.Ü.; resources, M.A., Y.Ü., H.E., Ş.Ç., E.B.K., S.A.D., A.N.K., F.Ö.H., A.T., N.E., F.K., N.H., Y.E., A.K.G., G.Y.T. and E.O.; data curation, Y.Ü., H.E., Ş.Ç., E.B.K., S.A.D., A.N.K., F.Ö.H., A.T., N.E., F.K., N.H., Y.E., A.K.G., G.Y.T., G.C.Y., M.F.K., D.Ö., M.M., and E.O.; writing—original draft preparation, M.A., A.B., E.T.G; writing—review and editing, M.A., A.B., E.T.G., supervision, M.A. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Scientific Research Evaluation and Ethics Committee of Ankara Etlik City Hospital (Approval No: AEŞH-BADEK-2025-0119, 26 February 2025).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study
Data Availability Statement
Data supporting the findings of this study are available from the corresponding author, Murat Aral, upon reasonable request.
Acknowledgments
The authors would like to thank Pharmaline Health Services Ind. Trade Inc. (Istanbul, Türkiye) for providing the PCR kit used in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AF | Amniotic fluid |
| BALF | Bronchoalveolar lavage fluid |
| CB | Colonoscopic biopsy |
| CI | Confidence interval |
| CLSI | Clinical and Laboratory Standards Institute |
| CMV | Cytomegalovirus |
| FN | False negative |
| FP | False positive |
| κ | Cohen’s kappa coefficient |
| MIQE | Minimum Information for Publication of Quantitative Real-Time PCR Experiments |
| NIST | National Institute of Standards and Technology |
| NPV | Negative predictive value |
| NPA | Negative percent agreement |
| NTC | No-template control |
| OPA | Overall percent agreement |
| PCR | Polymerase chain reaction |
| PPA | Positive percent agreement |
| PPV | Positive predictive value |
| qPCR | Quantitative real-time polymerase chain reaction |
| TN | True negative |
| TP | True positive |
| WHO | World Health Organization |
References
- Ljungman, P.; Boeckh, M.; Hirsch, H.H.; Josephson, F.; Lundgren, J.; Nichols, G.; Pikis, A.; Razonable, R.R.; Miller, V.; Griffiths, P.D. Disease Definitions Working Group of the Cytomegalovirus Drug Development Forum.Definitions of Cytomegalovirus Infection and Disease in Transplant Patients for Use in Clinical Trials. Clin. Infect. Dis. 2017, 64, 87–91. [Google Scholar] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A.; The Transplantation Society International CMV Consensus Group. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-Organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef] [PubMed]
- Emery, V.C.; Sabin, C.A.; Cope, A.V.; Gor, D.; Hassan-Walker, A.F.; Griffiths, P.D. Application of Viral-Load Kinetics to Identify Patients Who Develop Cytomegalovirus Disease after Transplantation. Lancet 2000, 355, 2032–2036. [Google Scholar] [CrossRef] [PubMed]
- Caliendo, A.M.; Schuurman, R.; Yen-Lieberman, B.; Spector, S.A.; Andersen, J.; Manjiry, R.; Crumpacker, C.; Lurain, N.S.; Erice, A. CMV Working Group of the Complications of HIV Disease RAC, AIDS Clinical Trials Group. Comparison of Quantitative and Qualitative PCR Assays for Cytomegalovirus DNA in Plasma. J. Clin. Microbiol. 2001, 39, 1334–1338. [Google Scholar] [CrossRef]
- Myers, J.B.; Amsterdam, D. The Laboratory Diagnosis of Cytomegalovirus Infections. Immunol. Investig. 1997, 26, 383–394. [Google Scholar] [CrossRef]
- Cobo, F. Application of Molecular Diagnostic Techniques for Viral Testing. Open Virol. J. 2012, 6, 104–114. [Google Scholar] [CrossRef]
- Engstrom-Melnyk, J.; Rodriguez, P.L.; Peraud, O.; Hein, R.C. Clinical Applications of Quantitative Real-Time PCR in Virology. Methods Microbiol. 2015, 42, 161–197. [Google Scholar]
- Pitt, S.J.; Phillips, D.I. Diagnostic Virology and Patient Care: From Vaguely Interesting to Vitally Important. Br. J. Biomed. Sci. 2017, 74, 16–23. [Google Scholar] [CrossRef]
- Bustin, S.A.; Mueller, R. Real-Time Reverse Transcription PCR (qRT-PCR) and Its Potential Use in Clinical Diagnosis. Clin. Sci. 2005, 109, 365–379. [Google Scholar] [CrossRef]
- Raymaekers, M.; Smets, R.; Maes, B.; Cartuyvels, R. Checklist for Optimization and Validation of Real-Time PCR Assays. J. Clin. Lab. Anal. 2009, 23, 145–151. [Google Scholar] [CrossRef]
- Apfalter, P.; Reischl, U.; Hammerschlag, M.R. In-House Nucleic Acid Amplification Assays in Research: How Much Quality Control Is Needed before One Can Rely upon the Results? J. Clin. Microbiol. 2005, 43, 5835–5841. [Google Scholar] [CrossRef] [PubMed]
- Rzepka, M.; Depka, D.; Gospodarek-Komkowska, E.; Bogiel, T. Whole Blood versus Plasma Samples—How Does the Type of Specimen Collected for Testing Affect the Monitoring of Cytomegalovirus Viremia? Pathogens 2022, 11, 1384. [Google Scholar] [CrossRef] [PubMed]
- Suganda, S.; Tang, L.; Carr, J.; Sun, Y.; Pounds, S.; Hayden, R. Comparative Evaluation of Whole Blood versus Plasma for Quantitative Detection of Cytomegalovirus Using an Automated System. Diagn. Microbiol. Infect. Dis. 2016, 85, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Toohey-Kurth, K.; Reising, M.M.; Tallmadge, R.L.; Goodman, L.B.; Bai, J.; Bolin, S.R.; Pedersen, J.C.; Bounpheng, M.A.; Pogranichniy, R.M.; Christopher-Hennings, J.; et al. Suggested Guidelines for Validation of Real-Time PCR Assays in Veterinary Diagnostic Laboratories. J. Vet. Diagn. Invest. 2020, 32, 802–814. [Google Scholar] [CrossRef]
- Çınar, C.; Karaahmet, S.; Tanrıverdi Çaycı, Y.; Gür Vural, D.; Bilgin, K.; Bırıncı, A. Investigation of the Diagnostic Value of CMV DNA Positivity in Different Clinical Samples. J. Exp. Clin. Med. 2025, 42, 274–278. [Google Scholar]
- Aspin, M.M.; Gallez-Hawkins, G.M.; Giugni, T.D.; Tegtmeier, B.; Lang, D.J.; Schmidt, G.M.; Forman, S.J.; Zaia, J.A. Comparison of Plasma PCR and Bronchoalveolar Lavage Fluid Culture for Detection of Cytomegalovirus Infection in Adult Bone Marrow Transplant Recipients. J. Clin. Microbiol. 1994, 32, 2266–2269. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR Inhibitors—Occurrence, Properties and Removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Michel, D.; Marre, E.; Hampl, W.; Roczkos, J.; Müller, S.; Hertenstein, B.; Kern, P.; Heymer, B.; Salzberger, B.; Arasteh, K.; et al. Intestinal Cytomegalovirus Disease in Immunocompromised Patients May Be Ruled Out by Search for Cytomegalovirus DNA in Stool Samples. J. Clin. Microbiol. 1995, 33, 3064–3067. [Google Scholar] [CrossRef]
- Fernholz, E.C.; Vidal-Folch, N.; Hasadsri, L. Rapid and Direct Detection of Congenital Cytomegalovirus Using a Commercial Real-Time PCR Assay. J. Clin. Microbiol. 2023, 61, e01781–22. [Google Scholar] [CrossRef]
- Razonable, R.R.; Hayden, R.T. Clinical Utility of Viral Load in Management of Cytomegalovirus Infection after Solid Organ Transplantation. Clin. Microbiol. Rev. 2013, 26, 703–727. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G. Practical Statistics for Medical Research, 1st ed.; Chapman and Hall/CRC: New York, NY, USA, 1990. [Google Scholar]
- Landis, J.R.; Koch, G.G. The Measurement of Observer Agreement for Categorical Data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). User Protocol for Evaluation of Qualitative Test Performance; Approved Guideline—Second Edition (EP12-A2). Available online: https://clsi.org/standards/products/method-evaluation/documents/ep12/ (accessed on 2 January 2026).
- Boeckh, M.; Stevens-Ayers, T.; Bowden, R.A. Plasma and Bronchoalveolar Lavage Fluid Viral Load Measurements for Cytomegalovirus Disease. J. Infect. Dis. 2004, 190, 714–722. [Google Scholar]
- Lodding, I.P.; Schultz, H.H.; Jensen, J.U.; Kirkby, N.; Perch, M.; Andersen, C.; Lundgren, J.D.; Iversen, M. Cytomegalovirus Viral Load in Bronchoalveolar Lavage to Diagnose Lung Transplant–Associated CMV Pneumonia. Transplantation 2018, 102, 326–332. [Google Scholar] [CrossRef]
- Ljungman, P. CMV Infections of the Gastrointestinal Tract in Immunocompromised Patients. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 861–873. [Google Scholar]
- Revello, M.G.; Gerna, G. Diagnosis and Management of Human Cytomegalovirus Infection in the Mother, Fetus, and Newborn Infant. Clin. Microbiol. Rev. 2002, 15, 680–715. [Google Scholar] [CrossRef]
- Ross, S.A.; Ahmed, A.; Palmer, A.L.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; Tolan, R.W., Jr.; Novak, Z.; Chowdhury, N.; Fowler, K.B.; et al. Detection of Congenital Cytomegalovirus Infection by Real-Time Polymerase Chain Reaction Analysis of Saliva or Urine Specimens. JAMA 2011, 306, 2595–2602. [Google Scholar] [CrossRef]
- Binnicker, M.J. Multiplex Molecular Panels for Diagnosis of Gastrointestinal Infection: Performance, Result Interpretation, and Cost-Effectiveness. J. Clin. Microbiol. 2015, 53, 3723–3728. [Google Scholar] [CrossRef]
- Caurio, C.F.B.; Allende, O.S.; Kist, R.; Santos, K.L.; Vasconcellos, I.C.S.; Rozales, F.P.; Lana, D.F.D.; Praetzel, B.M.; Alegretti, A.P.; Pasqualotto, A.C. Clinical Validation of an In-House Quantitative Real-Time PCR Assay for Cytomegalovirus Infection Using the 1st WHO International Standard in Kidney Transplant Patients. J. Bras. Nefrol. 2021, 43, 530–538. [Google Scholar] [CrossRef]
- Sanghavi, S.K.; Abu-Elmagd, K.; Keightley, M.C.; St George, K.; Lewandowski, K.; Boes, S.S.; Bullotta, A.; Dare, R.; Lassak, M.; Husain, S.; et al. Relationship of Cytomegalovirus Load Assessed by Real-Time PCR to pp65 Antigenemia in Organ Transplant Recipients. J. Clin. Virol. 2008, 42, 335–342. [Google Scholar] [CrossRef]
- Haynes, R.J.; Holden, M.J.; Kline, M.C.; Butler, J.M. Cytomegalovirus DNA Candidate Standard Reference Material (SRM): Sequencing of qPCR Target Regions and Digital PCR–Based Quantification. In Proceedings of the Association for Molecular Pathology (AMP) Annual Meeting, San Jose, CA, USA, 18–20 November 2010; National Institute of Standards and Technology (NIST): Gaithersburg, MD, USA, 2010. [Google Scholar]
Figure 1.
Agreement patterns between the laboratory-developed qualitative CMV PCR assay and the reference method across different specimen types. BALF, bronchoalveolar lavage fluid; CB, colonoscopic biopsy; AF, amniotic fluid.
Figure 1.
Agreement patterns between the laboratory-developed qualitative CMV PCR assay and the reference method across different specimen types. BALF, bronchoalveolar lavage fluid; CB, colonoscopic biopsy; AF, amniotic fluid.
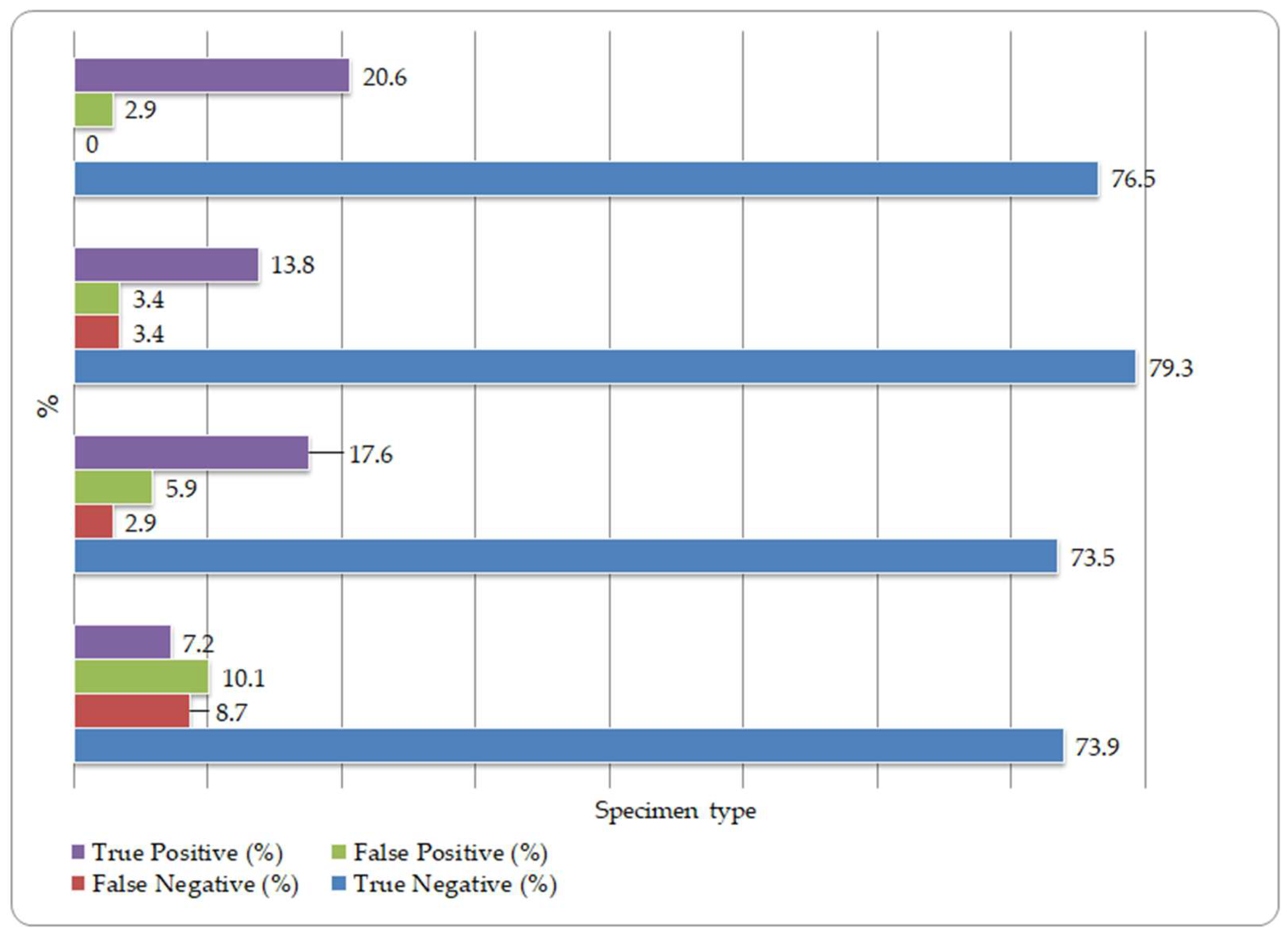

Table 1.
Distribution of clinical specimens and CMV positivity.
| Specimen Type | Total Samples (n) | CMV Positive n (%) | CMV Negative n (%) |
| BALF | 69 | 11 (15.9) | 58 (84.1) |
| Stool | 34 | 7 (20.6) | 27 (79.4) |
| Urine | 29 | 5 (17.2) | 24 (82.8) |
| CB / AF / Ocular | 34 | 7 (20.6) | 27 (79.4) |
| Total | 166 | 30 (18.1) | 136 (81.9) |
BALF, bronchoalveolar lavage fluid; CB, colonoscopic biopsy; AF, amniotic fluid; Ocular, intraocular fluid; CMV, cytomegalovirus. Percentages are calculated based on the total number of specimens within each specimen type.
Table 2.
Diagnostic agreement and performance by specimen type.
| Specimen type | TP | FP | FN | TN | PPA (%) | NPA (%) | OPA (%) | Cohen’s κ (95% Cl) |
| BALF | 5 | 7 | 6 | 51 | 45.5 | 87.9 | 81.2 | 0.36 (0.03–0.61) |
| Stool | 6 | 2 | 1 | 25 | 85.7 | 92.6 | 91.2 | 0.79 (0.47–1.00) |
| Urine | 4 | 1 | 1 | 23 | 80.0 | 95.8 | 93.1 | 0.74 (0.44–1.00) |
| CB/ AF / Ocular | 7 | 1 | 0 | 26 | 100 | 96.3 | 97.1 | 0.93 (0.75–1.00) |
| All specimens | 22 | 11 | 8 | 125 | 73.3 | 91.9 | 88.6 | 0.66 (0.48–0.78) |
BALF, bronchoalveolar lavage fluid; CB, colonoscopic biopsy specimens; AF, amniotic fluid; Ocular, ocular fluid; TP, true positive; FP, false positive; FN, false negative; TN, true negative; PPA, positive percent agreement; NPA, negative percent agreement; OPA, overall percent agreement; CI, confidence interval. TP, FP, FN, and TN are presented as absolute numbers.
Table 3.
Evaluation of the analytical sensitivity (LoD95) of the CMV qPCR test using probit.
| regression analysis | ||||
| Parameter | Estimate | Standard error | z-value | p-value |
| Fixed (β₀) | −5.35 | 0.57 | −9.43 | <0.001 |
| Log10 (copy/µL) (β₁) | 3.88 | 0.38 | 10.20 | <0.001 |
The relationship between target concentration and detection probability was evaluated using a probit regression model, and all model parameters were found to be statistically significant (p < 0.001). Accordingly, the LoD95 corresponding to a 95% detection probability of the test was determined to be 63.8 copies/µL.
Table 4.
Diagnostic performance by specimen type.
| Specimen type | TP | FP | FN | TN | Sensitivity | Specificity | PPV | NPV |
| BALF | 5 (7.2) | 7 (10.1) | 6 (8.7) | 51 (73.9) | 45.5 (26.9–65.3) | 87.9 (78.5–96.0) | 41.7 (24.5–61.2) | 89.5 (78.9–95.1) |
| Stool | 6 (17.6) | 2 (5.9) | 1 (2.9) | 25 (73.5) | 85.7 (48.7–97.4) | 92.6 (76.6–97.9) | 75.0 (40.9–92.9) | 96.2 (81.1–99.3) |
| Urine | 4 (13.8) | 1 (3.4) | 1 (3.4) | 23 (79.3) | 80.0 (37.6–96.4) | 95.8 (79.8–99.3) | 80.0 (37.6–96.4) | 95.8 (79.8–99.3) |
| CB / AF/ Ocular | 7 (20.6) | 1 (2.9) | 0 (0.0) | 26 (76.5) | 100 (64.6–100) | 96.3 (81.7–99.3) | 87.5 (52.9–97.8) | 100 (87.1–100) |
| All specimens | 22 (13.3) | 11 (6.6) | 8 (4.8) | 125 (75.3) | 73.3 (57.0–84.6) | 91.9 (88.5–97.3) | 66.7 (49.6–80.2) | 94.0 (88.6–96.9) |
BALF, bronchoalveolar lavage fluid; CB, colonoscopic biopsy specimens; AF, amniotic fluid; Ocular, ocular fluid; TP, true positive; FP, false positive; FN, false negative; TN, true negative; PPV, positive predictive value; NPV, negative predictive value. TP, FP, FN, and TN are presented as number and percentage (%). Sensitivity, specificity, PPV, and NPV are presented as percentages with 95% confidence intervals. Ninety-five percent confidence intervals were calculated using the Wilson score method.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



