
Submitted:
09 February 2026
Posted:
10 February 2026
You are already at the latest version
Abstract
Neonatal diabetes mellitus (NDM) is a rare monogenic disorder characterized by persistent hyperglycemia requiring insulin therapy, typically diagnosed within the first six months of life and less commonly between six and 12 months. NDM may be transient, with frequent relapses during puberty, or permanent and can be associated with extra-pancreatic manifestations or be part of a syndrome. This monogenic form of diabetes is caused by pathogenic variants in genes or chromosomal loci involved in the development and function of pancreatic beta-cells and in insulin synthesis and secretion. Mutations in more than 40 genes have been identified to be implicated in the pathogenesis of NDM. Abnormalities of the 6q24 locus have been recognized as the most common cause of transient NDM, whereas mutations in genes encoding ATP-sensitive potassium (KATP) channels, particularly KCNJ11, are more commonly identified in permanent NDΜ cases. In NDM cases, the clinical course, the presence of extra-pancreatic manifestations, and the optimal treatment depend on the causative gene. Therefore, genetic diagnosis is imperative, as it can facilitate the individualization of management strategies and long-term follow-up, as well as genetic counselling. However, hyperglycemia in the neonatal population, particularly in preterm and/or critically ill neonates, may be observed outside the NDM range due to immaturity and transient beta-cell dysregulation, insulin resistance, epigenetic modifications, or drug administration. The aim of this narrative review is to provide an overview of the genetic basis of NDM and the mechanisms underlying transient hyperglycemic states in neonates.
Keywords:
neonatal hyperglycemia
; neonatal diabetes mellitus
; transient neonatal diabetes mellitus
; permanent neonatal diabetes mellitus
; genetic etiology
; molecular mechanisms
1. Introduction
Hyperglycemia is one of the most common metabolic disturbances in the neonatal intensive care unit (NICU) and is generally transient, reflecting a disturbance in the balance between glucose delivery and insulin activity, rather than a disorder of glucose metabolism [1]. Preterm and low birth weight neonates are more commonly affected, with a prevalence that is inversely related to gestational age and birth weight [2,3]. Furthermore, neonates who are critically ill, those with hypoxic ischemic injury or sepsis, or those receiving medications such as steroids and inotropes are at increased risk of developing hyperglycemia [2].
Neonatal diabetes mellitus (NDM) is a genetic disease presenting during infancy, characterised by severe hyperglycemia requiring treatment [4,5]. It is a rare disease, with a reported prevalence of 1:90,000 to 1:160,000 live births, although significant variation is observed across geographic regions [6]. In countries with a high rate of consanguinity, a prevalence of 1:21,000 has been reported [7,8].
NDM typically presents during the first six months of life, although it may also appear between six and 12 months of age [4,5]. However, occurrence beyond the sixth month is more commonly associated with autoimmune diabetes rather than monogenic forms [9]. Intrauterine growth restriction and low birth weight are frequently observed among neonates with NDM, as insulin, an anabolic hormone, is imperative for normal embryonic growth [10]. The clinical presentation may be insidious, with failure to thrive and classic diabetes symptoms, including polyuria and polydipsia, which are often difficult to recognize in young infants [5,11,12]. In this age group, diabetes may initially present as a sepsis-like illness or acutely as diabetic ketoacidosis [12,13,14]. The diagnosis of NDM should be considered in cases of severe hyperglycemia that persists beyond a week or in cases of extremely high glucose levels (>1000 mg/dL). The diagnosis is further supported by the presence of low or undetectable insulin and C-peptide levels during hyperglycemia [10,12].
Based on the duration of the required treatment, NDM can be distinguished as either transient or permanent and may be isolated or part of a syndrome accompanied by extra-pancreatic manifestations [13]. In permanent NDM (PNDM), lifelong therapy is necessary [5]. In cases of transient NDM (TNDM), remission occurs at various points during the initial postnatal period [6,13]. However, relapse during adolescence or adulthood is common [15]. The diagnosis of TNDM can be challenging due to its similarity to other causes of transient neonatal hyperglycemia, and the high rate of relapse in TNDM cases necessitates a definitive diagnosis in order to establish a follow-up plan.
NDM is a monogenic disease caused by pathogenic variants in genes or chromosomal loci implicated in the development and function of pancreatic beta-cells and in insulin synthesis and secretion. The diagnosis is confirmed through genetic testing [4,11,16,17]. Advances in genetic testing have substantially improved our understanding of the molecular basis of NDM, and pathogenic variants in more than 40 genes have been implicated in its development [17,18]. Gene mutations may occur sporadically or may be inherited in an autosomal or recessive manner [5].
In both TNDM and PNDM, a definitive diagnosis is imperative for predicting clinical progression, detecting extra-pancreatic manifestations, implementing individualized management strategies and long-term follow-up, and providing genetic counselling [13,16]. The current guidelines of the International Society for Pediatric and Adolescent Diabetes (ISPAD) recommend immediate genetic testing for all infants diagnosed with diabetes before the sixth month of age and for those diagnosed between the sixth and the 12th month with negative autoantibodies, extra-pancreatic manifestations, and/or a family history [16].
The primary aim of this narrative review is to provide a comprehensive overview of the genetic basis of NDM, encompassing both transient and permanent forms and the underlying defects in beta-cell development and survival, and insulin synthesis and secretion. A secondary objective, extending beyond monogenic NDM itself, is to summarize the molecular mechanisms underlying transient hyperglycemic states in neonates, with particular emphasis on developmental, epigenetic, and regulatory pathways influencing early life glucose metabolism.
2. Methods
A comprehensive search was conducted to identify relevant articles using the online databases PubMed, Scopus, and Google Scholar from database inception up to November 2025. The search strategy included combinations of the following keywords: “neonatal hyperglycemia”, “neonatal diabetes mellitus”, “transient neonatal diabetes mellitus”, ”permanent neonatal diabetes mellitus”, “genetic etiology”, “molecular mechanisms”. Only full-text, peer-reviewed studies published in English were included. Studies focusing on forms of diabetes other than neonatal diabetes mellitus were excluded. The reference lists of the retrieved articles were manually screened for relevant articles that may have been missed in the initial research. Article screening and data extraction were performed independently by two authors. Any discrepancies were resolved by consensus through discussion with all co-authors.
3. Genetic Causes of NDM
NDM, a rare monogenic disorder, is the clinical manifestation of diverse molecular mechanisms arising from genetically heterogeneous causes. To date, more than 40 genes and genomic loci have been implicated in the pathogenesis of NDM through various molecular pathways. These genetic defects are associated with specific inheritance patterns, variable clinical phenotypes, and extra-pancreatic features.
Although NDM is clinically classified as transient or permanent, based on disease course, research has demonstrated that individual genes may cause either presentation (Figure 1). For the purpose of this narrative review, the genetic causes of NDM have been classified into four categories based on underlying pathogenic mechanisms.
3.1. Imprinting Defects of the 6q24 Locus
It is documented that approximately 70% of TNDM cases are attributable to genetic or epigenetic alterations in an imprinted region of chromosome 6q24 [19]. The 6q24 locus consists of a CpG island whose methylation status depends on parental origin. Normally, it is unmethylated on the paternal and methylated on the maternal allele, leading to expression of the paternal allele, whereas the maternal allele is inactive [4,12]. It has been demonstrated that three distinct mechanisms are implicated in the NDM due to 6q24 alterations: paternal uniparental disomy, unbalanced paternal duplication of 6q24, and hypomethylation of the maternal allele [11].
In paternal uniparental disomy, which accounts for approximately 40% of cases, both alleles are inherited from the father [11,20]. While in most cases it occurs as uniparental isodisomy, it may also be partial [16,19]. Its occurrence is regarded as sporadic, arising from stochastic events during the earliest stages of embryonic development [19,21]. Approximately 30-40% of TNDM cases involving 6q24 alterations are attributable to an unbalanced partial duplication of the long arm of the paternal chromosome 6, resulting in three copies of 6q24, two of which are inherited from the father [5,11,21,22]. Although duplications may occur de novo, they account for the majority of familial cases [16]. Duplications are inherited in an autosomal dominant manner [11]. Affected males have a 50% chance of transmitting the duplication and the disease to their offspring. Affected females have a 50% probability of transmitting the duplication, but their offspring will not be affected by the disease. However, in cases of male offspring who carry the duplication, the next generation may develop diabetes [16,19]. The remaining 20-30% of 6q24-related TNDM is caused by hypomethylation of the maternal allele, and subsequent activation [5,21]. Hypomethylation may result from epigenetic events without an underlying genetic cause or may be part of a generalized hypomethylation involving multiple imprinted loci, such as the recessively inherited mutations of the ZFP57 [16,23]. The ZFP57 gene, located on chromosome 6p22.3, is a transcription factor essential for maintaining methylation of imprinted loci during development [24].
Irrespective of the underlying genetic or epigenetic pathway, the result is the overexpression of the imprinted genes polyadenoma gene-like 1 (PLAGL1) and the hydatidiform mole-associated imprinted transcript (HYMAI) [4]. The PLAGL1 gene encodes a zinc-finger transcription factor that regulates the cell cycle and apoptosis through various molecular mechanisms [25]. It has been demonstrated that the overexpression of PLAGL1 in pancreatic beta-cells results in increased p21 expression and an interaction with the tumor suppressor p53, leading to reduced cell proliferation and enhanced apoptosis [26,27]. Studies in experimental animals have shown that overexpression of PLAGL1 and HYMAI impair insulin excretion and endocrine pancreatic development, leading to a disease resembling TNDM in humans [28,29]. The HYMAI gene encodes a non-coding RNA whose function remains incompletely characterized [13,24,28].
Alterations in the 6q24 locus are exclusively associated with TNDM (Table 1) [30]. A common feature is the early presentation of hyperglycemia, with a median chronological age of onset of less than a week in most cohorts [23,30,31,32,33]. Initial presentation with DKA is rarely reported [34]. The age at which remission most commonly occurs is between three and four months [31,32,33]. Busiah et al. reported that 97% of patients with 6q24 alterations entered remission before the age of one year [30]. However, in a large proportion of patients, diabetes relapses, most commonly during adolescence [4,31,32].
Intrauterine growth restriction is frequently observed in these patients due to insulin's pivotal role as a growth factor during intrauterine development [4]. It has been reported that approximately 80-90% of neonates are born small for gestational age (SGA) [4,30,33]. It is noteworthy that higher birthweight for the gestational age has been associated with earlier remission [23]. Congenital malformations may accompany 6q24 TNDM. Macroglossia and umbilical hernias are the most frequently reported, and less commonly, renal and heart defects, hypothyroidism, and neurological disorders [30]. In the largest cohort to date of 163 patients with 6q24 TNDM, Docherty et al. observed that congenital anomalies were significantly more prevalent in patients with paternal uniparental disomy or hypomethylation at multiple loci, compared with those with paternal duplication of 6q24 or isolated maternal hypomethylation [23]. However, McCullough et al. reported no difference in clinical features, regardless of the genetic etiology [31].
During the early phase, insulin is typically used to achieve euglycemia. However, successful management with oral sulfonylureas has occasionally been reported [11,35]. The management of patients following relapse should be individualized and may include insulin and noninsulin agents [11,36].
As imprinting changes and copy number variations are involved in the 6q24 TNDM, standard sequencing panels are not capable of detection, and methylation-specific assays are required [33]. Alkorta-Aranburu et al. reported that a combination of methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) and next-generation sequencing (NGS) led to genetic diagnosis in 67% of NDM cases, including 6q24 abnormalities [20].
3.2. Defects in Beta-Cell Function and Insulin Secretion (Table 2)
3.2.1. ATP-Sensitive Potassium (KATP) Channels Mutations
Insulin secretion by pancreatic beta-cells in response to glucose is dependent on the function of the ATP-sensitive potassium (KATP) channels [4]. KATP channels are hetero-octameric structures comprised of two subunits. The Kir6.2 subunit, encoded by the KCNJ11 gene, forms a central pore and is surrounded by the regulatory SUR1 subunit, encoded by the ABCC8 gene [5]. The regulation of insulin secretion is achieved through alterations in beta-cell membrane electric activity that are associated with the intracellular metabolic state [37].
In the presence of low glucose concentrations, KATP channels remain open, maintaining hyperpolarization of the cell membrane. As glucose concentrations rise, glucose uptake into pancreatic beta-cells is mediated primarily by facilitative glucose transporters (predominantly GLUT1 in humans, with contributions from GLUT2). An increased intracellular ATP/ADP ratio leads to closure of KATP channels, reducing potassium efflux and causing membrane depolarization. Depolarization subsequently opens voltage-dependent calcium channels, resulting in calcium influx and triggering insulin secretion via exocytosis of insulin secretory granules [4,11,16,38].
Gain-of-function mutations in either KCNJ11 or ABCC8 permanently activate KATP channels, rendering them unable to regulate membrane potential. Consequently, beta-cells are incapable to secrete insulin [4,18]. These mutations are the most prevalent genetic cause of NDM, accounting for approximately 50% of cases. They represent the most common genetic cause of PNDM and the second most common in TNDM [11]. KCNJ11 mutations have been demonstrated to be associated with PNDM in 90% of cases, whereas ABCC8 mutations are associated with TNDM in approximately two-thirds of cases [16,39].
Activating heterozygous mutations cause KCNJ11 NDM. In approximately 90% of cases, mutations occur de novo; in the remaining cases, there is autosomal dominant inheritance [16,40]. To date, more than 70 KNCJ11 variants have been reported to be implicated in NDM pathogenesis [38]. The common pathogenic pathway is reduced ATP inhibition of KATP channels. NDM-causing mutations involve both N- and C- terminal domains of KCNJ11 (Kir6.2) and include residues directly involved in ATP binding and residues in close proximity that indirectly affect ATP binding and activity [18,38,41].
Activating ABCC8 mutations causing NDM may be homozygous or heterozygous [4,6]. Moreover, NDM cases caused by compound heterozygosity for both activating and inactivating mutations have been described [42,43]. The SUR1 protein consists of three transmembrane domains and two nucleotide-binding domains (NBD1, NBD2). It has been demonstrated that Mg-nucleotide binding to the NBDs induces structural changes in the NBD dimer, leading to the opening of the Kir6.2 pore, membrane hyperpolarization, and inhibition of insulin secretion [38,44]. NDM is associated with mutations that either enhance Mg-nucleotide binding, stabilize Mg-nucleotide-induced structural alterations, or increase the intrinsic channel open probability [38,41].
The distinction between TNDM and PNDM is not feasible at presentation, and the diagnosis is based on the duration of hyperglycemia [4]. Intrauterine growth restriction is a common feature, affecting approximately half of patients [30,45,46]. In most cases, the diagnosis is made before six months of age. Busiah et al. reported a median time to diagnosis of 45.5 days, significantly longer than in patients with TNDM due to 6q25 (45.5 days vs. 5 days, p < 0.0001) [30]. Presentation with DKA is not uncommon and has been reported in up to 85% of patients [46,47]. In a prospective study involving 71 patients with KATP NDM, 44% were diagnosed with TNDM, with a median time to remission of 39.4 weeks [30].
KATP channels are expressed in neurons across various brain regions, particularly in the hypothalamus, the basal ganglia, and the cortex, providing a rationale for neurological symptoms in approximately 20-30% of patients with KATP NDM [5,30,38,41,48]. A broad spectrum of neurological disorders may be present, ranging from mild psychomotor disorders to severe developmental delay and epilepsy (DEND syndrome), which affects less than 5% of patients [4]. The clinical phenotype has been shown to correlate with the degree to which each mutation reduces ATP sensitivity of the KATP channel [38,49]. DEND phenotype is rare among patients with ABCC8 NDM. However, neurological symptoms may be present, including developmental delay, attention deficit hyperactivity disorder, visual impairments, and spatial dyspraxia [5,6,30]. Malformations of other organ systems are uncommon in patients with KATP [30].
3.2.2. Glucokinase (GCK) Gene Mutations
Glucokinase (GCK), an enzyme considered to be the glucose sensor of beta-cells, catalyzes the first step of glucose metabolism and is essential for maintaining normoglycemia [16]. GCK, a 465-amino acid protein expressed mainly in the liver, pancreas, and brain, is encoded by the GCK gene, which is located on chromosome 7 (7p15.3-p15.1) [50,51,52]. The CKG gene employs tissue-specific promoters to initiate transcription at distinct first exons (1a, 1b, 1c), allowing pancreatic, brain, and liver cells to produce distinct GCK transcripts under distinct regulatory controls [50,51].
GCK catalyzes the first step of glycolysis, the ATP-dependent phosphorylation of glucose to glucose-6-phosphate (G6P). In pancreatic beta-cells, this process promotes insulin secretion, while in the liver, it facilitates glucose metabolism, including glycogen synthesis [51,52]. Through GCK activity, pancreatic beta-cells can regulate glucose phosphorylation and, consequently, glucose metabolism in response to glucose concentration [50]. The low affinity for glucose, the sigmoidal saturation curve, and the lack of GCK inhibition by its product, G6P, enable GCK to function as an effective glucose sensor [50,51].
As GCK plays a pivotal role in regulating insulin secretion, mutations in the GCK gene have been associated with both hyperglycemia and hypoglycemia, and more than 700 mutations have been identified [52]. Heterozygous loss-of-function mutations are associated with a subtype of maturity-onset diabetes of the young (GCK-MODY), which is characterized by mild hyperglycemia [53,54,55]. Homozygous or compound heterozygous loss-of-function mutations are associated with complete loss of GCK activity and PNDM [50,52,55]. Although rare, accounting for 2-3% of PNDM cases, an increased prevalence has been reported in populations with a high rate of consanguinity [56].
Intrauterine growth restriction is a common feature among CKG PNDM patients, with hyperglycemia usually presenting during the first days of life [16,54]. Typically, no associated extra-pancreatic malformations are present [16]. Patients with GCK PNDM require lifelong therapy with insulin. The combination with sulfonylureas has been demonstrated to enhance insulin secretion, thereby improving glycemic control [6,50,55].
3.2.3. SLC2A2 Gene Mutations
The solute carrier family 2 transporter 2 (SLC2A2) gene is located on chromosome 3q26.1-26.3 and encodes GLUT2. GLUT2 is a glucose transporter that contributes to the insulin secretion pathway in pancreatic beta-cells [57]. Slca2 knock-out mice have been demonstrated to present with severe diabetes from birth and early mortality [58]. However, it has been suggested that GLUT1 and GLUT3 can compensate for the absence of GLUT2 in human pancreatic beta-cells, thereby preserving insulin secretion and maintaining normoglycemia [59].
Homozygous or compound heterozygous mutations in the SLC2A2 gene cause Fanconi-Bickel syndrome, which typically presents in late infancy with hepatomegaly, renal Fanconi syndrome, growth retardation, and rickets [59,60]. Neonatal diabetes is rarely reported [61]. However, Sansbury et al., in a cohort of 104 patients with NDM in which common genetic NDM causes had been excluded, identified five patients with homozygous SLC2A2 mutations, four of whom were eventually diagnosed with TNDM. It is noteworthy that only one patient exhibited features of Fanconi-Bickel syndrome at the time of diagnosis, while the remaining four developed them later [60].
3.3. Defects in Insulin Biosynthesis and Beta-Cell Destruction (Table 3)
3.3.1. Insulin (INS) Gene Mutations
Mutations in the insulin gene (INS) represent the second most common cause of PNDM. In the United States and Western Europe, approximately 70% of PNDM cases are attributable to KATP channel and INS mutations [4,42]. INS mutations are a rare cause of TNDM [4].
The insulin biosynthesis is a complex process that occurs in several stages in the endoplasmic reticulum (ER) and the Golgi apparatus [18]. Preproinsulin, the insulin precursor, following its synthesis in ribosomes, is translocated to the ER where it is proteolytically cleaved by a signal peptidase to form proinsulin [18,62]. In the ER, proinsulin undergoes rapid oxidative folding and forms three specific disulfide bonds involving six cysteine residues, which are crucial for its structure and transport. Intracellular trafficking of proinsulin via the Golgi apparatus and packaging into immature secretory granules is followed by proteolytic processing by prohormone convertases to form mature insulin and C-peptide, which are subsequently packaged into secretory granules [18,62,63,64]. The pathway of insulin secretion can be disrupted, leading to NDM through two distinct mechanisms, involving heterozygous gain-of-function mutations and homozygous loss-of-function mutations of the INS gene [17].
Dominant pathogenic variants cause proinsulin misfolding and accumulation within the ER in more than 80% of cases [4,64]. More than 70 dominant INS mutations have been recognized . Proinsulin misfolding may be due to mutations that directly impede disulfide bond formation, such as cysteine mutations, or to non-cysteine mutations that interfere with proper cysteine alignment [63,65,66]. Misfolded proinsulin is trapped and accumulated in the ER, disrupting ER homeostasis and inducing ER stress. This may activate the apoptotic pathway, leading to beta-cell death [17,18,67]. However, mechanisms other than apoptosis have recently been suggested, including impaired beta-cell development due to defects in proliferation or dedifferentiation and loss of beta-cell identity [63,68]. More than 80% of heterozygous INS mutations occur de novo, the remaining are inherited in an autosomal dominant manner [69].
Recessive loss-of-function mutations, homozygous or compound heterozygous, result in markedly reduced or absent insulin biosynthesis. Reduced preproinsulin synthesis may be caused by several mechanisms on the transcriptional or translational level. These include failure of INS transcription due to gene deletion or truncation, mRNA instability, or lack of translation initiation due to loss of the start codon [62,67,70].
Consistent with the presentation of NDM due to other genetic factors, INS NDM patients frequently exhibit IUGR. Most patients are diagnosed during the first six postnatal months, although later presentation is possible [16]. Recessive mutations that result in a lack of insulin biosynthesis from embryonic life are characterised by more severe growth restriction and typically occur earlier, within the first postnatal days [16,18,71]. Typically, no extra-pancreatic features are present (9). Lifelong insulin replacement is necessary [11].
3.3.2. Gene Mutations Associated with ER Stress Response
The most prevalent cause of PNDM in highly consanguineous populations is homozygous mutations in the eukaryotic translation initiation factor 2α kinase 3 (EIF2AK3) gene, which cause Wolcott-Rallison syndrome [72,73]. EIF2AK3, located on chromosome 2p11.2, encodes the PKR-like ER kinase (PERK) [74]. PERK is a transmembrane enzyme that functions as a sensor of misfolded proteins within the ER lumen. It is activated by autophosphorylation and regulates the unfolded protein response [75]. In the absence of functional protein, accumulation of unfolded proteins in the ER leads to apoptosis [72].
Multisystem involvement is characteristic of Wolcott-Rallison syndrome. PNDM is uniformly present, with a median age of presentation of 2.5 months [74]. A recent systematic review, involving 159 patients with Wolcott-Rallison syndrome, reported that the most common manifestations include liver impairment, impaired growth, skeletal abnormalities, neurodevelopmental disorders, renal disorders, and hematopoietic disorders [76]. To date, no genotype-phenotype correlation has been documented [76,77].
The eukaryotic translation initiation factor 2B subunit alpha (EIF2B1) encodes eEIF2Ba, which is part of the eIF2B complex that regulates translation initiation and protein synthesis. Impaired regulation of protein translation leads to severe ER stress in pancreatic beta-cells and apoptosis. De novo heterozygous EIF2B1 variants have been associated with PNDM and transient liver dysfunction. Homozygous EIF2B1 mutations have been associated with a completely distinct phenotype of leukoencephalopathy with vanishing white matter [78].
Mutations in the immediate early response 3 interacting protein 1 (IER3IP1) gene impair ER-to-Golgi proinsulin transport and cause ER accumulation of unfolded proteins, leading to alterations in ER microstructure and ER stress. To date, 10 patients have been described with homozygous IER3IP1 mutations, all presenting with a similar phenotype of microcephaly, epilepsy, and PNDM (MEDS syndrome) [79,80].
Homozygous mutations in the WFS1 gene, which encodes the ER protein wolframin, which plays a pivotal role in maintaining Ca2+ homeostasis and regulating ER stress, have been associated with Wolfram syndrome. This syndrome is characterized by diabetes mellitus, diabetes insipidus, optic atrophy and hearing loss and typically presents during childhood or adolescence [81]. However, De Franco et al. reported five cases of de novo heterozygous missense mutations in WFS1 that presented with a distinct, more severe phenotype. These patients presented with PNDM, congenital sensorineural deafness, and congenital cataract. Functional studies showed that these dominant mutations induced marked ER stress and reduced beta-cell survival, leading to early cell death. The authors suggested that this process may begin during embryonic development [82].
3.4. Defects in Pancreatic Morphogenesis
3.4.1. PDX1 Gene Mutations
The pancreatic and duodenal homeobox 1 (PDX1) gene plays a key role in pancreatic organogenesis and in the maturation of beta-cells and their ability to secrete insulin [83]. Recessive loss-of-function mutations are a known cause of pancreatic agenesis or hypoplasia and PNDM [84,85,86]. However, more recently, cases of biallelic PDX1 variants presenting with PNDM and no clinical manifestations of exocrine pancreatic deficiency or pancreatic agenesis have been documented [87,88,89,90]. It has been suggested that a correlation between the mutation type and phenotype exists. Pancreatic agenesis has been observed to result from null mutations that disrupt protein function, whereas hypomorphic mutations, which partially diminish protein function, have been associated with PNDM and occasionally exocrine insufficiency [88,89].
3.4.2. PTF1A Gene Mutations
The pancreas transcription factor 1 alpha (PTF1A) gene encodes PTF1A, a basic helix-loop-helix protein crucial for pancreas and cerebellum development [91]. Homozygous loss-of-function truncation mutations have been associated with a severe phenotype characterized by NDM, pancreatic agenesis, and cerebellar hypoplasia or agenesis, with early mortality [92,93,94]. A distinct, milder phenotype has been documented in patients with recessive inactivating mutations in an enhancer located 25kb downstream of the PTF1A. This enhancer is pancreatic-specific and critical for pancreatic progenitor differentiation [95]. The phenotype comprises pancreatic agenesis, leading to PNDM and exocrine pancreatic insufficiency, with no cerebral involvement [96,97,98,99]. In a recent multicenter study of 30 patients with NDM caused by PTF1A enhancer mutations, Demirbilek et al. reported a median age of diabetes presentation of five days and exocrine pancreatic insufficiency in all patients. Growth retardation, anemia, and cholestasis were the most commonly reported additional features [91].
3.4.3. GATA6 Gene Mutations
The transcription factor GATA6, encoded by the GATA6 gene, contains two zinc-finger domains and is critical for pancreatic organogenesis and for the generation and function of both endocrine and exocrine cells [100]. De novo heterozygous inactivating GATA6 mutations have been recognized as the most common cause of pancreatic agenesis [101]. Other commonly reported features include cardiac, hepatobiliary, and intestinal malformations [100,101,102]. However, considerable heterogeneity in phenotype is evident [100]. A wide spectrum of diabetes presentation in patients with GATA6 mutations has been documented, including mainly NDM, as well as childhood-onset and adult-onset diabetes [100]. A genotype-phenotype correlation cannot be established due to the observed variability of phenotype in patients carrying the same mutation. It has been suggested that genetic, environmental and stochastic factors may contribute to the clinical heterogeneity [103,104]. However, pancreatic involvement has been documented to be more common in the presence of inactivating mutations within the second zinc-finger domain of GATA6 [104,105].
3.4.4. HNF1B Gene Mutations
The hepatocyte nuclear factor 1b (HNF1B) gene is located on chromosome 17q12 and encodes the hepatocyte nuclear factor 1 beta (HNF1b), a transcription factor crucial for the development of many organs, including the kidneys, pancreas, liver, and intestines [106,107]. Heterozygous loss-of-function mutations in the HNF1B gene are associated with a multisystem disorder, most commonly presenting as diabetes (maturity-onset diabetes of the young 5 [MODY5]) and renal cysts (RCAD syndrome) [108]. The role of HNF1B in early kidney and pancreatic development has been demonstrated in studies with experimental animals [109,110]. However, heterozygous HNF1B mutations have been rarely reported as a cause of NDM. A few cases of both TNDM and PNDM have been described, typically accompanied by pancreatic hypoplasia and renal malformations [106,107,111,112].
3.5. Defects in Beta-Cell Development (Table 4)
3.5.1. NEUROG3 Gene Mutations
Neurogenin-3 (NEUROG3) is a basic helix-loop-helix transcription factor essential for the endocrine cell development in the pancreas, intestine, and hypothalamus. Neurog3 null mice are characterized by the absence of endocrine cells in the pancreas and intestine and by lethality during the early postnatal period [113].
In humans, autosomal recessive NEUROG3 mutations have been associated with variable phenotypes, including enteric anendocrinosis, which presents as severe malabsorptive congenital diarrhea, diabetes, and, less frequently, hypothyroidism, hypogonadotrophic hypogonadism, and short stature [114,115,116,117]. Diabetes is present in more than half of the cases either as NDM or childhood-onset diabetes [114]. It has been suggested that null mutations lead to NDM, whereas hypomorphic mutations lead to childhood-onset diabetes [118]. However, the occurrence during adolescence has been reported among patients carrying null mutations, suggesting a potential role of epigenetic modifications or environmental factors in the occurrence of different phenotypes [119,120].
3.5.2. NEUROD1 Gene Mutations
NEUROD1, encoded by the NEUROD1 gene, is a basic helix-loop-helix transcription factor with an important role in the development and functional maturation of the endocrine pancreas [121]. Experimental studies have demonstrated that Neurod1 is essential for beta-cell maturation and that Neurod1 deficiency compromises the proliferation, differentiation, and functional properties of endocrine cells, resulting in impaired insulin production. Neurod1-knockout mice exhibit profound beta-cell dysfunction, resulting in severe diabetes and death in the perinatal or early postnatal period [121,122].
In humans, heterozygous loss-of-function mutations have been associated with MODY, particularly MODY6 and adult-onset diabetes [121]. However, homozygous inactivating NEUROD1 mutations have been reported as a rare cause of PNDM [123]. Rubio-Cabezas et al. identified two patients with homozygous NEUROD1 mutations who presented with PNDM, including a morphologically normal pancreas, cerebellar hypoplasia, sensorineural deafness, and visual impairments . Moreover, Demirbilek et al. reported another case of homozygous NEUROD1 mutation presenting with a similar phenotype [123]. Therefore, biallelic loss-of-function NEUROD1 mutations are regarded as a rare cause of syndromic PNDM.
4. Transient Hyperglycemic States in Neonates
Transient hyperglycemia is a common metabolic disturbance, particularly among preterm and critically ill neonates. It reflects reversible disturbances in glucose homeostasis, rather than the genetic susceptibility underlying NDM. In contrast to NDM, which is characterized by persistent defects in beta-cell development and survival, insulin biosynthesis or secretion due to monogenic mutations, transient hyperglycemia is attributable to developmental immaturity and stress responses that impair glucose regulation during early postnatal life [1].
Hyperglycemia is very common in preterm neonates, with a reported prevalence varying from 20-80% [3]. The risk of hyperglycemia is inversely correlated with gestational age and birthweight [3]. It has been demonstrated that neonates with a birthweight of less than 1000g have 18 times higher risk of hyperglycemia compared to neonates with a birthweight of 1000-2000g [124]. Predisposition to hyperglycemia in preterm neonates is multifactorial, reflecting beta-cell functional immaturity, decreased insulin sensitivity, increased counter-regulatory hormone activity, and exogenous factors such as high glucose infusion rates and administration of vasoactive drugs or glucocorticoids [1].
Reduced beta-cell mass and functional immaturity of beta-cells in preterm neonates may result in insufficient insulin secretion and poor insulin secretory response to glucose [2,125,126,127]. Impaired insulin secretion in neonates, and particularly in very preterm neonates, may reflect delayed maturation of glucose-sensing mechanisms. Key elements of glucose-sensing, such as GLUT2 and GCK, have been shown to have reduced expression and functional activity in the immature beta-cell [126]. The response of beta-cells in preterm neonates to hyperglycemia is characterized by increased secretion of proinsulin, a non-processed precursor with approximately one-tenth the biological activity of mature insulin [128,129,130]. Proinsulin is normally processed by proconvertases in the immature secretory granules of the beta-cell to form mature insulin; however, in preterm beta-cells, this processing is inefficient. Consequently, an increased proinsulin/insulin ratio is observed in preterm neonates, which underlies a defective processing system in the immature beta-cell [3,131].
It has been demonstrated that gluconeogenesis continues in the preterm neonate, despite glucose infusion or total parenteral nutrition, and that this may persist up to the chronological age of five weeks [132,133]. This has been at least partly attributed to the immaturity of hepatic glucose sensing and regulation. Low levels of the glucose transporters GLUT2 and GLUT4, associated with immaturity, may contribute to this phenomenon [2]. GLUT2 facilitates the bidirectional transportation of glucose within the liver, in accordance with alterations in plasma glucose levels. Consequently, low levels of GLUT2 are associated with impaired glucose sensing and incomplete suppression of endogenous glucose production [3,131]. Lower pancreatic GLUT2 is associated with impaired insulin secretion in response to glucose [3]. In addition to functional beta-cell immaturity, delayed introduction of enteral feeding, which is common in very preterm neonates, is associated with reduced incretin levels, which are known to promote insulin secretion [2].
However, persistent glucose production despite high glucose levels may not be solely attributed to sustained gluconeogenesis. Alternative mechanisms, including impaired hepatic glucose sensing and altered intracellular glucose metabolism, may contribute [128]. Reduced GCK activity in preterm neonates may result in reduced phosphorylation of glucose to G6P, and ineffective glucose metabolism may lead to failure of hepatic glucose output suppression [128,131]. Moreover, the less abundant insulin-sensitive tissues in preterm neonates, such as skeletal muscle and adipose tissue, and reduced GLUT4 levels may contribute to reduced insulin-stimulated glucose uptake, thereby contributing to peripheral insulin resistance [2,3]. An additional potential contributor to reduced peripheral glucose uptake and decreased glycogen synthesis may be zinc deficiency, which is common in very preterm neonates. Zinc increases intracellular signaling by promoting PI3K/Akt pathway activation, thereby promoting GLUT4 translocation to the plasma membrane, enhancing peripheral glucose uptake. Zinc also inhibits glycogen synthase kinase 3-beta, thereby promoting glycogen synthesis. Therefore, zinc deficiency may impair glucose uptake and glycogen storage, contributing to insulin resistance and hyperglycemia [3].
Additional factors contributing to insulin resistance are increased levels of pro-inflammatory cytokines, such as interleukin-1, interleukin-6, and tumor necrosis factor-alpha, which are elevated in sepsis, necrotizing enterocolitis, and other morbidities associated with prematurity. Moreover, exposure to vasoactive agents and corticosteroids exacerbates insulin resistance by increasing counter-regulatory hormones and impairing insulin receptor signaling [3].
Impaired beta-cell development has also been documented in neonates with intrauterine growth-restriction (IUGR) [134]. Pancreatic islets of severely affected fetuses have been demonstrated to be smaller, less vascularized, with fewer beta-cells, resulting in lower insulin secretion [134,135]. The severity of these alterations correlates with the degree of placental insufficiency [134,136,137]. Intrauterine hypoxemia and the increased catecholamine secretion secondary to placental insufficiency induce adaptive metabolic responses that impact pancreatic islets development and inhibit insulin secretion [138,139]. In addition, increased hepatic glucose production in utero due to increased hepatic phosphoenolpyruvate carboxykinase (PEPCK) and G6P expression may persist postnatally and contribute to the development of insulin resistance [138]. Moreover, it has been suggested that epigenetic modifications in key developmental genes may contribute to the association of IUGR and long-term disturbances of glucose homeostasis [140]. Experimental studies in mice have shown that Pdx1 expression, a key regulator of pancreatic organogenesis and beta-cell maturation, is persistently reduced in beta-cells from growth restricted (IUGR) offspring, supporting an epigenetic mechanism of developmental programming [140,141].
Alterations in glucose homeostasis are commonly observed in neonates following hypoxic ischemic injury, and hyperglycemia has been reported in up to 50% of affected neonates. It has been suggested that early hyperglycemia probably reflects reduced cerebral glucose uptake and severe cellular energy failure, characterized by increased adenosine triphosphate consumption and enhanced anaerobic glycolysis with hydrogen ion accumulation [142]. Inflammation, mitochondrial dysfunction, and oxidative stress are the main molecular cascades underlying brain injury in hypoxic ischemic encephalopathy and are associated with increased insulin resistance [143]. Neonates with hypoxic-ischemic injury have elevated circulating proinflammatory cytokines, including interleukin-1, interleukin-6, and tumor necrosis factor-alpha, which have been shown to impair insulin receptor signaling and promote insulin resistance [2,144]. Moreover, activation of the hypothalamic-pituitary-adrenal axis leads to increased levels of corticosteroids and counter-regulating hormones, which enhance gluconeogenesis and glycogenolysis, and increase insulin resistance, thereby contributing to hyperglycemia [142,145].
5. Conclusions
Monogenic NDM is a rare genetic disorder caused by diverse single-gene mutations that impair pancreatic beta-cell development or survival, insulin biosynthesis and secretion, or pancreatic morphogenesis. Rapid advances in molecular techniques have led to a continuously expanding understanding of the genetic basis of NDM. More than 40 genes and chromosomal loci have been identified to be implicated in the pathogenesis of NDM. A precise genetic diagnosis can be established in more than 80% of affected individuals. The clinical course of diabetes, the presence of extra-pancreatic malformations, and long-term prognosis vary according to the underlying genetic defect. However, phenotype cannot always be accurately predicted, and further studies are needed to refine genotype-phenotype correlations.
TNDM is characterized by early remission, although relapse occurs in more than half of the patients. TNDM is most commonly associated with abnormalities at the imprinted 6q24 locus. However, other genes, such as ABCC8 and, less frequently, KCNJ11 and INS, may be implicated. In contrast, PNDM requires lifelong antidiabetic treatment and most commonly results from activating variants in genes encoding KATP channel subunits, particularly KCNJ11. However, a wide variety of other genes may also be implicated, including mutations in INS, GCK, and in genes encoding transcription factors essential for pancreatic development, such as PDX1 and PTF1A.
In contrast to NDM, transient hyperglycemic states frequently observed in preterm and critically ill neonates, such as those with sepsis or HIE, are not driven by monogenic defects but reflect developmental immaturity of glucose homeostasis or stress-related responses. These conditions typically resolve with maturation or recovery from critical illness and do not confer long-term implications as monogenic NDM. Distinguishing NDM from transient hyperglycemic states is essential to guide appropriate genetic testing, management, and long-term follow-up. Differentiating TNDM and transient hyperglycemia is particularly challenging, as both conditions remit. Therefore, clinical judgement and molecular testing are required for accurate diagnosis, which is crucial to ensure long-term follow-up in TNDM, given the high rate of relapse.
Molecular diagnosis is therefore central to NDM diagnosis, enabling accurate classification and optimization of care. As molecular diagnostic technologies continue to advance, additional genes involved in NDM pathogenesis are expected to be identified, further expanding current knowledge. Moreover, further genetic and functional studies are essential to more accurately define genotype-phenotype correlations, improve prognosis prediction, and ensure optimal follow-up and management, and thus improve outcomes for patients with NDM.
Author Contributions
Conceptualization, V.G. and A.S..; methodology, F.B., M.B. and C.M.T.; formal analysis, F.B. and C.M.T.; investigation, N.D. and C.K.; writing—original draft preparation, N.D. and C.K.; writing—review and editing, M.B., V.G and A.S; supervision, V.G. and A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hay, W. W.; Rozance, P. J. Neonatal Hyperglycemia—Causes, Treatments, and Cautions. J. Pediatr. 2018, 200, 6–8. [Google Scholar] [CrossRef]
- Beardsall, K. Hyperglycaemia in the Newborn Infant. Physiology Verses Pathology. Front. Pediatr. 2021, 9. [Google Scholar] [CrossRef]
- Angelis, D.; Jaleel, M. A.; Brion, L. P. Hyperglycemia and Prematurity: A Narrative Review. Pediatr. Res. 2023, 94, 892–903. [Google Scholar] [CrossRef]
- Beltrand, J.; Busiah, K.; Vaivre-Douret, L.; Fauret, A. L.; Berdugo, M.; Cavé, H.; Polak, M. Neonatal Diabetes Mellitus. Front. Pediatr. 2020, 8. [Google Scholar] [CrossRef]
- Hammoud, B.; Greeley, S. A. W. Growth and Development in Monogenic Forms of Neonatal Diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2022, 29, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Golshan-Tafti, M.; Dastgheib, S. A.; Bahrami, R.; Yeganegi, M.; Aghasipour, M.; Marzbanrad, Z.; Saeida-Ardekani, M.; Shahbazi, A.; Omidi, A.; Lookzadeh, M. H.; et al. Genetic Underpinnings of Neonatal Diabetes: A Review of Current Research. Egyptian Journal of Medical Human Genetics 2024, 25, 119. [Google Scholar] [CrossRef]
- Al-Khawaga, S.; Mohammed, I.; Saraswathi, S.; Haris, B.; Hasnah, R.; Saeed, A.; Almabrazi, H.; Syed, N.; Jithesh, P.; El Awwa, A.; et al. The Clinical and Genetic Characteristics of Permanent Neonatal Diabetes (PNDM) in the State of Qatar. Mol. Genet. Genomic Med. 2019, 7(10). [Google Scholar] [CrossRef]
- Habeb, A. M.; Al-Magamsi, M. S.; Eid, I. M.; Ali, M. I.; Hattersley, A. T.; Hussain, K.; Ellard, S. Incidence, Genetics, and Clinical Phenotype of Permanent Neonatal Diabetes Mellitus in Northwest Saudi Arabia. Pediatr. Diabetes 2012, 13, 499–505. [Google Scholar] [CrossRef] [PubMed]
- RubioCabezas, O.; Flanagan, S. E.; Damhuis, A.; Hattersley, A. T.; Ellard, S. KATP Channel Mutations in Infants with Permanent Diabetes Diagnosed after 6 Months of Life. Pediatr. Diabetes 2012, 13, 322–325. [Google Scholar] [CrossRef]
- Kabeer, A. S. VH. Genetics of Neonatal Diabetes: An Update. Genetic Clinics 2024, 17(4), 17–23. [Google Scholar]
- Lemelman, M. B.; Letourneau, L.; Greeley, S. A. W. Neonatal Diabetes Mellitus. Clin. Perinatol. 2018, 45, 41–59. [Google Scholar] [CrossRef]
- Shah, B.; Kataria, A.; Palliyil Gopi, R.; Mally, P. Neonatal Diabetes Mellitus: Current Perspective. Res. Rep. Neonatol. 2014, 55. [Google Scholar] [CrossRef]
- Dahl, A.; Kumar, S. Recent Advances in Neonatal Diabetes. Diabetes Metab. Syndr. Obes. 2020, Volume 13, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, L. R.; Carmody, D.; Wroblewski, K.; Denson, A. M.; Sanyoura, M.; Naylor, R. N.; Philipson, L. H.; Greeley, S. A. W. Diabetes Presentation in Infancy: High Risk of Diabetic Ketoacidosis. Diabetes Care 2017, 40, e147–e148. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Bowman, P.; Kraljevic, I.; Tripolski, M.; Houghton, J. A. L.; De Franco, E.; Shepherd, M. H.; Skrabic, V.; Patel, K. A. Transient Neonatal Diabetes: An Etiologic Clue for the Adult Diabetologist. Can. J. Diabetes 2020, 44, 128–130. [Google Scholar] [CrossRef]
- Greeley, S. A. W.; Polak, M.; Njølstad, P. R.; Barbetti, F.; Williams, R.; Castano, L.; Raile, K.; Chi, D. V.; Habeb, A.; Hattersley, A. T.; et al. ISPAD Clinical Practice Consensus Guidelines 2022: The Diagnosis and Management of Monogenic Diabetes in Children and Adolescents. Pediatr. Diabetes 2022, 23, 1188–1211. [Google Scholar] [CrossRef]
- Barbetti, F.; Deeb, A.; Suzuki, S. Neonatal Diabetes Mellitus around the World: Update 2024. J. Diabetes Investig. 2024, 15, 1711–1724. [Google Scholar] [CrossRef]
- Gobble, M. R. S.; Stone, S. I. Neonatal and Syndromic Forms of Diabetes. Curr. Diab. Rep. 2025, 25, 26. [Google Scholar] [CrossRef]
- Temple, I. K.; Shield, J. P. H. 6q24 Transient Neonatal Diabetes. Rev. Endocr. Metab. Disord. 2010, 11, 199–204. [Google Scholar] [CrossRef]
- Alkorta-Aranburu, G.; Sukhanova, M.; Carmody, D.; Hoffman, T.; Wysinger, L.; Keller-Ramey, J.; Li, Z.; Johnson, A. K.; Kobiernicki, F.; Botes, S.; et al. Improved Molecular Diagnosis of Patients with Neonatal Diabetes Using a Combined Next-Generation Sequencing and MS-MLPA Approach. Journal of Pediatric Endocrinology and Metabolism 2016, 29. [Google Scholar] [CrossRef]
- Mackay, D. J. G.; Temple, I. K. Transient Neonatal Diabetes Mellitus Type 1. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 335–342. [Google Scholar] [CrossRef]
- Polak, M.; Cavé, H. Neonatal Diabetes Mellitus: A Disease Linked to Multiple Mechanisms. Orphanet J. Rare Dis. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Docherty, L. E.; Kabwama, S.; Lehmann, A.; Hawke, E.; Harrison, L.; Flanagan, S. E.; Ellard, S.; Hattersley, A. T.; Shield, J. P. H.; Ennis, S.; et al. Clinical Presentation of 6q24 Transient Neonatal Diabetes Mellitus (6q24 TNDM) and Genotype–Phenotype Correlation in an International Cohort of Patients. Diabetologia 2013, 56, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Touati, A.; Errea-Dorronsoro, J.; Nouri, S.; Halleb, Y.; Pereda, A.; Mahdhaoui, N.; Ghith, A.; Saad, A.; Perez de Nanclares, G.; H’mida ben brahim, D. Transient Neonatal Diabetes Mellitus and Hypomethylation at Additional Imprinted Loci: Novel ZFP57 Mutation and Review on the Literature. Acta Diabetol. 2019, 56, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Vega-Benedetti, A. F.; Saucedo, C.; Zavattari, P.; Vanni, R.; Zugaza, J. L.; Parada, L. A. PLAGL1: An Important Player in Diverse Pathological Processes. J. Appl. Genet. 2017, 58, 71–78. [Google Scholar] [CrossRef]
- Liu, P.-Y.; Hsieh, T.-Y.; Liu, S.-T.; Chang, Y.-L.; Lin, W.-S.; Wang, W.-M.; Huang, S.-M. Zac1, an Sp1-like Protein, Regulates Human P21 Gene Expression in HeLa Cells. Exp. Cell Res. 2011, 317, 2925–2937. [Google Scholar] [CrossRef]
- Huang, S.-M.; Schönthal, A. H.; Stallcup, M. R. Enhancement of P53-Dependent Gene Activation by the Transcriptional Coactivator Zac1. Oncogene 2001, 20, 2134–2143. [Google Scholar] [CrossRef]
- Ma, D.; Shield, J. P. H.; Dean, W.; Leclerc, I.; Knauf, C.; Burcelin, R.; Rutter, G. A.; Kelsey, G. Impaired Glucose Homeostasis in Transgenic Mice Expressing the Human Transient Neonatal Diabetes Mellitus Locus, TNDM. Journal of Clinical Investigation 2004, 114, 339–348. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. Transient Neonatal Diabetes Mellitus Gene Zac1 Impairs Insulin Secretion in Mice through Rasgrf1. Mol. Cell. Biol. 2012, 32, 2549–2560. [Google Scholar] [CrossRef]
- Busiah, K.; Drunat, S.; Vaivre-Douret, L.; Bonnefond, A.; Simon, A.; Flechtner, I.; Gérard, B.; Pouvreau, N.; Elie, C.; Nimri, R.; et al. Neuropsychological Dysfunction and Developmental Defects Associated with Genetic Changes in Infants with Neonatal Diabetes Mellitus: A Prospective Cohort Study. Lancet Diabetes Endocrinol. 2013, 1, 199–207. [Google Scholar] [CrossRef]
- McCullough, M. E.; Letourneau-Freiberg, L. R.; Bowden, T. L.; Kandasamy, B.; Ray, A.; Wroblewski, K.; del Gaudio, D.; Mackay, D. J. G.; Philipson, L. H.; Greeley, S. A. W. Clinical Characteristics and Remission Monitoring of 6q24-Related Transient Neonatal Diabetes. Pediatr. Diabetes 2024. [Google Scholar] [CrossRef]
- Bonfanti, R.; Iafusco, D.; Rabbone, I.; Diedenhofen, G.; Bizzarri, C.; Patera, P. I.; Reinstadler, P.; Costantino, F.; Calcaterra, V.; Iughetti, L.; et al. Differences between Transient Neonatal Diabetes Mellitus Subtypes Can Guide Diagnosis and Therapy. Eur. J. Endocrinol. 2021, 184, 575–585. [Google Scholar] [CrossRef]
- Pietrusiński, M.; Grzybowska-Adamowicz, J.; Płoszaj, T.; Skoczylas, S.; Borowiec, M.; Piekarska, K.; Skowrońska, B.; Wajda-Cuszlag, M.; Mazur, A.; Zmysłowska, A. The Clinical and Diagnostic Characterization of 6q24-Related Transient Neonatal Diabetes Mellitus: A Polish Pediatric Cohort Study. Biomedicines 2025, 13, 2492. [Google Scholar] [CrossRef]
- Shoucri, B. M.; Thambundit, A.; Chia, D. J. 6q24-Related Transient Neonatal Diabetes Mellitus Presenting With Severe Diabetic Ketoacidosis and Multiorgan Failure. JCEM Case Reports 2025, 3. [Google Scholar] [CrossRef]
- Garcin, L.; Kariyawasam, D.; Busiah, K.; Fauret-Amsellem, A.-L.; Le Bourgeois, F.; Vaivre-Douret, L.; Cavé, H.; Polak, M.; Beltrand, J. Successful Off-Label Sulfonylurea Treatment of Neonatal Diabetes Mellitus Due to Chromosome 6 Abnormalities. Pediatr. Diabetes 2018, 19, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Carmody, D.; Beca, F. A.; Bell, C. D.; Hwang, J. L.; Dickens, J. T.; Devine, N. A.; Mackay, D. J. G.; Temple, I. K.; Hays, L. R.; Naylor, R. N.; et al. Role of Noninsulin Therapies Alone or in Combination in Chromosome 6q24-Related Transient Neonatal Diabetes: Sulfonylurea Improves but Does Not Always Normalize Insulin Secretion. Diabetes Care 2015, 38, e86–e87. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F. M. ATP-Sensitive Potassium Channelopathies: Focus on Insulin Secretion. Journal of Clinical Investigation 2005, 115, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P. J.; Ashcroft, F. M. New Insights into KATP Channel Gene Mutations and Neonatal Diabetes Mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef]
- Babenko, A. P.; Polak, M.; Cavé, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus. New England Journal of Medicine 2006, 355, 456–466. [Google Scholar] [CrossRef]
- Edghill, E. L.; Gloyn, A. L.; Goriely, A.; Harries, L. W.; Flanagan, S. E.; Rankin, J.; Hattersley, A. T.; Ellard, S. Origin of de Novo KCNJ11 Mutations and Risk of Neonatal Diabetes for Subsequent Siblings. J. Clin. Endocrinol. Metab. 2007, 92, 1773–1777. [Google Scholar] [CrossRef]
- Shimomura, K.; Maejima, Y. KATP Channel Mutations and Neonatal Diabetes. Internal Medicine 2017, 56, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Matsutani, N.; Furuta, H.; Matsuno, S.; Oku, Y.; Morita, S.; Uraki, S.; Doi, A.; Furuta, M.; Iwakura, H.; Ariyasu, H.; et al. Identification of a Compound Heterozygous Inactivating ABCC 8 Gene Mutation Responsible for Young-onset Diabetes with Exome Sequencing. J. Diabetes Investig. 2020, 11, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Flanagan, S. E.; Girard, C. A.; Patch, A.-M.; Harries, L. W.; Parrish, A.; Edghill, E. L.; Mackay, D. J. G.; Proks, P.; Shimomura, K.; et al. Permanent Neonatal Diabetes Caused by Dominant, Recessive, or Compound Heterozygous SUR1 Mutations with Opposite Functional Effects. The American Journal of Human Genetics 2007, 81, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E. L.; Flanagan, S. E.; Ellard, S. Permanent Neonatal Diabetes Due to Activating Mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef]
- Besser, R. E. J.; Flanagan, S. E.; Mackay, D. G. J.; Temple, I. K.; Shepherd, M. H.; Shields, B. M.; Ellard, S.; Hattersley, A. T. Prematurity and Genetic Testing for Neonatal Diabetes. Pediatrics 2016, 138. [Google Scholar] [CrossRef]
- Ngoc, C. T. B.; Dien, T. M.; De Franco, E.; Ellard, S.; Houghton, J. A. L.; Lan, N. N.; Thao, B. P.; Khanh, N. N.; Flanagan, S. E.; Craig, M. E.; et al. Molecular Genetics, Clinical Characteristics, and Treatment Outcomes of KATP-Channel Neonatal Diabetes Mellitus in Vietnam National Children’s Hospital. Front. Endocrinol. (Lausanne) 2021, 12. [Google Scholar] [CrossRef]
- Mujahed, R. H.; Shawabka, A. M.; Daghlis, N. M.; Thweib, A. A.; Rabba, H. A.; Makhlouf, T. Z. De Novo KCNJ11 Mutation in an Infant With Neonatal Diabetes Mellitus Presenting as Diabetic Ketoacidosis: A Case Report and Literature Review. J. Investig. Med. High Impact Case Rep. 2025, 13. [Google Scholar] [CrossRef]
- Li, B.; Xi, X.; Roane, D. S.; Ryan, D. H.; Martin, R. J. Distribution of Glucokinase, Glucose Transporter GLUT2, Sulfonylurea Receptor-1, Glucagon-like Peptide-1 Receptor and Neuropeptide Y Messenger RNAs in Rat Brain by Quantitative Real Time RT-PCR. Molecular Brain Research 2003, 113, 139–142. [Google Scholar] [CrossRef]
- Ashcroft, F. M. ATP-Sensitive K + Channels and Disease: From Molecule to Malady. American Journal of Physiology-Endocrinology and Metabolism 2007, 293, E880–E889. [Google Scholar] [CrossRef]
- Esquiaveto-Aun, A. M.; De Mello, M. P.; Paulino, M. F. V. M.; Minicucci, W. J.; Guerra-Júnior, G.; De Lemos-Marini, S. H. V. A New Compound Heterozygosis for Inactivating Mutations in the Glucokinase Gene as Cause of Permanent Neonatal Diabetes Mellitus (PNDM) in Double-First Cousins. Diabetol. Metab. Syndr. 2015, 7, 101. [Google Scholar] [CrossRef]
- Osbak, K. K.; Colclough, K.; Saint-Martin, C.; Beer, N. L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A. L. Update on Mutations in Glucokinase ( GCK ), Which Cause Maturity-Onset Diabetes of the Young, Permanent Neonatal Diabetes, and Hyperinsulinemic Hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Abu Aqel, Y.; Alnesf, A.; Aigha, I. I.; Islam, Z.; Kolatkar, P. R.; Teo, A.; Abdelalim, E. M. Glucokinase (GCK) in Diabetes: From Molecular Mechanisms to Disease Pathogenesis. Cell. Mol. Biol. Lett. 2024, 29, 120. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, A.; Chakera, A. J.; Thomsen, S. K.; Colclough, K.; Barrett, A.; De Franco, E.; Chatelas, A.; Demirbilek, H.; Akcay, T.; Alawneh, H.; et al. Phenotypic Severity of Homozygous GCK Mutations Causing Neonatal or Childhood-Onset Diabetes Is Primarily Mediated through Effects on Protein Stability. Hum. Mol. Genet. 2014, 23, 6432–6440. [Google Scholar] [CrossRef] [PubMed]
- Bolu, S. A Family With Novel Homozygous Deletion Mutation (c.1255delT; p.Phe419Serfs*12) in GCK Gene Which Is A Rare Cause of Permanent Neonatal Diabetes Mellitus. Turk Pediatri Ars. 2019. [Google Scholar] [CrossRef] [PubMed]
- Oza, C. M.; Karguppikar, M. B.; Khadilkar, V.; Khadilkar, A. Variable Presentations of GCK Gene Mutation in a Family. BMJ Case Rep. 2022, 15, e246699. [Google Scholar] [CrossRef]
- Al Senani, A.; Hamza, N.; Al Azkawi, H.; Al Kharusi, M.; Al Sukaiti, N.; Al Badi, M.; Al Yahyai, M.; Johnson, M.; De Franco, E.; Flanagan, S.; et al. Genetic Mutations Associated with Neonatal Diabetes Mellitus in Omani Patients. Journal of Pediatric Endocrinology and Metabolism 2018, 31, 195–204. [Google Scholar] [CrossRef]
- Sun, B.; Chen, H.; Xue, J.; Li, P.; Fu, X. The Role of GLUT2 in Glucose Metabolism in Multiple Organs and Tissues. Mol. Biol. Rep. 2023, 50, 6963–6974. [Google Scholar] [CrossRef]
- Guillam, M.-T.; Hümmler, E.; Schaerer, E.; Wu, J.-Y.; Birnbaum, M. J.; Beermann, F.; Schmidt, A.; Dériaz, N.; Thorens, B. Early Diabetes and Abnormal Postnatal Pancreatic Islet Development in Mice Lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Khandelwal, P.; Sinha, A.; Jain, V.; Houghton, J.; Hari, P.; Bagga, A. Fanconi Syndrome and Neonatal Diabetes: Phenotypic Heterogeneity in Patients with GLUT2 Defects. CEN Case Rep. 2018, 7, 1–4. [Google Scholar] [CrossRef]
- Sansbury, F. H.; Flanagan, S. E.; Houghton, J. A. L.; Shuixian Shen, F. L.; Al-Senani, A. M. S.; Habeb, A. M.; Abdullah, M.; Kariminejad, A.; Ellard, S.; Hattersley, A. T. SLC2A2 Mutations Can Cause Neonatal Diabetes, Suggesting GLUT2 May Have a Role in Human Insulin Secretion. Diabetologia 2012, 55, 2381–2385. [Google Scholar] [CrossRef]
- Yoo, H.-W.; Shin, Y.-L.; Seo, E.-J.; Kim, G.-H. Identification of a Novel Mutation in the GLUT2 Gene in a Patient with Fanconi-Bickel Syndrome Presenting with Neonatal Diabetes Mellitus and Galactosaemia. Eur. J. Pediatr. 2002, 161, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, J.; Cui, J.; Chen, W.; Guo, H.; Barbetti, F.; Arvan, P. INS-Gene Mutations: From Genetics and Beta Cell Biology to Clinical Disease. Mol. Aspects Med. 2015, 42, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sui, L.; Du, Q.; Haataja, L.; Yin, Y.; Viola, R.; Xu, S.; Nielsson, C. U.; Leibel, R. L.; Barbetti, F.; et al. Permanent Neonatal Diabetes-Causing Insulin Mutations Have Dominant Negative Effects on Beta Cell Identity. Mol. Metab. 2024, 80, 101879. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Saint-Martin, C.; Xu, J.; Ding, L.; Wang, R.; Feng, W.; Liu, M.; Shu, H.; Fan, Z.; Haataja, L.; et al. Biological Behaviors of Mutant Proinsulin Contribute to the Phenotypic Spectrum of Diabetes Associated with Insulin Gene Mutations. Mol. Cell. Endocrinol. 2020, 518, 111025. [Google Scholar] [CrossRef]
- Haataja, L.; Manickam, N.; Soliman, A.; Tsai, B.; Liu, M.; Arvan, P. Disulfide Mispairing During Proinsulin Folding in the Endoplasmic Reticulum. Diabetes 2016, 65, 1050–1060. [Google Scholar] [CrossRef]
- Haataja, L.; Arunagiri, A.; Hassan, A.; Regan, K.; Tsai, B.; Dhayalan, B.; Weiss, M. A.; Liu, M.; Arvan, P. Distinct States of Proinsulin Misfolding in MIDY. Cellular and Molecular Life Sciences 2021, 78, 6017–6031. [Google Scholar] [CrossRef]
- Yang, Y.; Shu, H.; Hu, J.; Li, L.; Wang, J.; Chen, T.; Zhen, J.; Sun, J.; Feng, W.; Xiong, Y.; et al. A Novel Nonsense INS Mutation Causes Inefficient Preproinsulin Translocation Into the Endoplasmic Reticulum. Front. Endocrinol. (Lausanne). 2022, 12. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin Mutations Impair Beta-Cell Development in a Patient-Derived IPSC Model of Neonatal Diabetes. Elife 2018, 7. [Google Scholar] [CrossRef]
- Edghill, E. L.; Flanagan, S. E.; Patch, A.-M.; Boustred, C.; Parrish, A.; Shields, B.; Shepherd, M. H.; Hussain, K.; Kapoor, R. R.; Malecki, M.; et al. Insulin Mutation Screening in 1,044 Patients With Diabetes. Diabetes 2008, 57, 1034–1042. [Google Scholar] [CrossRef]
- Garin, I.; Edghill, E. L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J. M.; Maestro, M. A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive Mutations in the INS Gene Result in Neonatal Diabetes through Reduced Insulin Biosynthesis. Proceedings of the National Academy of Sciences 2010, 107, 3105–3110. [Google Scholar] [CrossRef]
- Støy, J.; Steiner, D. F.; Park, S.-Y.; Ye, H.; Philipson, L. H.; Bell, G. I. Clinical and Molecular Genetics of Neonatal Diabetes Due to Mutations in the Insulin Gene. Rev. Endocr. Metab. Disord. 2010, 11, 205–215. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Patch, A.-M.; Minton, J. A. L.; Flanagan, S. E.; Edghill, E. L.; Hussain, K.; Balafrej, A.; Deeb, A.; Buchanan, C. R.; Jefferson, I. G.; et al. Wolcott-Rallison Syndrome Is the Most Common Genetic Cause of Permanent Neonatal Diabetes in Consanguineous Families. J. Clin. Endocrinol. Metab. 2009, 94, 4162–4170. [Google Scholar] [CrossRef]
- Habeb, A. M.; Flanagan, S. E.; Deeb, A.; Al-Alwan, I.; Alawneh, H.; Balafrej, A. A. L.; Mutair, A.; Hattersley, A. T.; Hussain, K.; Ellard, S. Permanent Neonatal Diabetes: Different Aetiology in Arabs Compared to Europeans. Arch. Dis. Child. 2012, 97, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Asl, S. N.; Vakili, R.; Vakili, S.; Soheilipour, F.; Hashemipour, M.; Ghahramani, S.; De Franco, E.; Yaghootkar, H. Wolcott-Rallison Syndrome in Iran: A Common Cause of Neonatal Diabetes. Journal of Pediatric Endocrinology and Metabolism 2019, 32, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein Kinase R-like ER Kinase and Its Role in Endoplasmic Reticulum Stress-Decided Cell Fate. Cell Death Dis. 2015, 6, e1822–e1822. [Google Scholar] [CrossRef] [PubMed]
- Aldrian, D.; Bochdansky, C.; Kavallar, A. M.; Mayerhofer, C.; Deeb, A.; Habeb, A.; Romera Rabasa, A.; Khadilkar, A.; Uçar, A.; Knoppke, B.; et al. Natural History of Wolcott-Rallison Syndrome: A Systematic Review and Follow-up Study. Liver International 2024, 44, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, F.; Habibi, M.; Enayati, S.; Bitarafan, F.; Razzaghy-Azar, M.; Sotodeh, A.; Omran, S. P.; Maroofian, R.; Amoli, M. M. A Genotype-First Approach for Clinical and Genetic Evaluation of Wolcott-Rallison Syndrome in a Large Cohort of Iranian Children With Neonatal Diabetes. Can. J. Diabetes 2018, 42, 272–275. [Google Scholar] [CrossRef] [PubMed]
- De Franco, E.; Caswell, R.; Johnson, M. B.; Wakeling, M. N.; Zung, A.; Dũng, V. C.; Bích Ngọc, C. T.; Goonetilleke, R.; Vivanco Jury, M.; El-Khateeb, M.; et al. De Novo Mutations in EIF2B1 Affecting EIF2 Signaling Cause Neonatal/Early-Onset Diabetes and Transient Hepatic Dysfunction. Diabetes 2020, 69, 477–483. [Google Scholar] [CrossRef]
- Yang, J.; Zhen, J.; Feng, W.; Fan, Z.; Ding, L.; Yang, X.; Huang, Y.; Shu, H.; Xie, J.; Li, X.; et al. IER3IP1 Is Critical for Maintaining Glucose Homeostasis through Regulating the Endoplasmic Reticulum Function and Survival of β Cells. Proceedings of the National Academy of Sciences 2022, 119. [Google Scholar] [CrossRef]
- Montaser, H.; Leppänen, S.; Vähäkangas, E.; Bäck, N.; Grace, A.; Eurola, S.; Ibrahim, H.; Lithovius, V.; Stephens, S. B.; Barsby, T.; et al. IER3IP1 Mutations Cause Neonatal Diabetes Due to Impaired Proinsulin Trafficking. Diabetes 2025, 74, 514–527. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J.; Li, D.; Zhang, Z.; Ni, Q.; Han, R.; Ye, L.; Zhang, Y.; Hong, J.; Wang, W.; et al. Novel WFS1 Variants Are Associated with Different Diabetes Phenotypes. Front. Genet. 2024, 15. [Google Scholar] [CrossRef]
- De Franco, E.; Flanagan, S. E.; Yagi, T.; Abreu, D.; Mahadevan, J.; Johnson, M. B.; Jones, G.; Acosta, F.; Mulaudzi, M.; Lek, N.; et al. Dominant ER Stress–Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes 2017, 66, 2044–2053. [Google Scholar] [CrossRef]
- Ebrahim, N.; Shakirova, K.; Dashinimaev, E. PDX1 Is the Cornerstone of Pancreatic β-Cell Functions and Identity. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zanfardino, A.; Schiaffini, R.; Ishibashi, J.; Daniel, B.; Haemmerle, M. W.; Rapini, N.; Piscopo, A.; Miraglia del Giudice, E.; Digilio, M. C.; et al. Neonatal Diabetes–Associated Missense PDX1 Variant Disrupts Chromatin Association and Protein-Protein Interaction. JCI Insight 2025, 10. [Google Scholar] [CrossRef] [PubMed]
- Schwitzgebel, V. M.; Mamin, A.; Brun, T.; Ritz-Laser, B.; Zaiko, M.; Maret, A.; Jornayvaz, F. R.; Theintz, G. E.; Michielin, O.; Melloul, D.; et al. Agenesis of Human Pancreas Due to Decreased Half-Life of Insulin Promoter Factor 1. J. Clin. Endocrinol. Metab. 2003, 88, 4398–4406. [Google Scholar] [CrossRef] [PubMed]
- Stoffers, D. A.; Zinkin, N. T.; Stanojevic, V.; Clarke, W. L.; Habener, J. F. Pancreatic Agenesis Attributable to a Single Nucleotide Deletion in the Human IPF1 Gene Coding Sequence. Nat. Genet. 1997, 15, 106–110. [Google Scholar] [CrossRef]
- Jeffery, N.; Al Nimri, O.; Houghton, J. A. L.; Globa, E.; Wakeling, M. N.; Flanagan, S. E.; Hattersley, A. T.; Patel, K. A.; De Franco, E. Widening the Phenotypic Spectrum Caused by Pathogenic PDX1 Variants in Individuals with Neonatal Diabetes. BMJ Open Diabetes Res. Care 2024, 12, e004439. [Google Scholar] [CrossRef]
- De Franco, E.; Shaw-Smith, C.; Flanagan, S. E.; Edghill, E. L.; Wolf, J.; Otte, V.; Ebinger, F.; Varthakavi, P.; Vasanthi, T.; Edvardsson, S.; et al. Biallelic PDX1 (Insulin Promoter Factor 1) Mutations Causing Neonatal Diabetes without Exocrine Pancreatic Insufficiency. Diabetic Medicine 2013, 30. [Google Scholar] [CrossRef]
- Nicolino, M.; Claiborn, K. C.; Senée, V.; Boland, A.; Stoffers, D. A.; Julier, C. A Novel Hypomorphic PDX1 Mutation Responsible for Permanent Neonatal Diabetes With Subclinical Exocrine Deficiency. Diabetes 2010, 59, 733–740. [Google Scholar] [CrossRef]
- Sahebi, L.; Niknafs, N.; Dalili, H.; Amini, E.; Esmaeilnia, T.; Amoli, M.; Farrokhzad, N. Iranian Neonatal Diabetes Mellitus Due to Mutation in PDX1 Gene: A Case Report. J. Med. Case Rep. 2019, 13, 258. [Google Scholar] [CrossRef]
- Demirbilek, H.; Cayir, A.; Flanagan, S. E.; Yıldırım, R.; Kor, Y.; Gurbuz, F.; Haliloğlu, B.; Yıldız, M.; Baran, R. T.; Akbas, E. D.; et al. Clinical Characteristics and Long-Term Follow-up of Patients with Diabetes Due To PTF1A Enhancer Mutations. J. Clin. Endocrinol. Metab. 2020, 105, e4351–e4359. [Google Scholar] [CrossRef]
- Sellick, G. S.; Barker, K. T.; Stolte-Dijkstra, I.; Fleischmann, C.; J Coleman, R.; Garrett, C.; Gloyn, A. L.; Edghill, E. L.; Hattersley, A. T.; Wellauer, P. K.; et al. Mutations in PTF1A Cause Pancreatic and Cerebellar Agenesis. Nat. Genet. 2004, 36, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Tutak, E.; Satar, M.; Yapicioğlu, H.; Altintaş, A.; Narli, N.; Hergüner, O.; Bayram, Y. A Turkish Newborn Infant with Cerebellar Agenesis/Neonatal Diabetes Mellitus and PTF1A Mutation. Genet. Couns. 2009, 20, 147–152. [Google Scholar] [PubMed]
- Al-Shammari, M.; Al-Husain, M.; Al-Kharfy, T.; Alkuraya, F. A Novel PTF1A Mutation in a Patient with Severe Pancreatic and Cerebellar Involvement. Clin. Genet. 2011, 80, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Escalada, I.; Maestro, M. Á.; Balboa, D.; Elek, A.; Bernal, A.; Bernardo, E.; Grau, V.; García-Hurtado, J.; Sebé-Pedrós, A.; Ferrer, J. Pancreas Agenesis Mutations Disrupt a Lead Enhancer Controlling a Developmental Enhancer Cluster. Dev. Cell 2022, 57, 1922–1936.e9. [Google Scholar] [CrossRef]
- Paksaz, M.; Saneifard, H.; Mirdehghan, A.; Mosallanejad, A.; Shakiba, M.; Saberi, M. A Rare PTF1A Enhancer Mutation Causing Neonatal Diabetes Mellitus with Pancreatic Agenesis: Case Report and Considerations for Genetic Evaluation. Int. J. Endocrinol. Metab. 2025, 23. [Google Scholar] [CrossRef]
- Gonc, E. N.; Ozon, A.; Alikasifoglu, A.; Haliloğlu, M.; Ellard, S.; Shaw-Smith, C.; Kandemir, N. Variable Phenotype of Diabetes Mellitus in Siblings with a Homozygous PTF1A Enhancer Mutation. Horm. Res. Paediatr. 2015, 84, 206–211. [Google Scholar] [CrossRef]
- Gabbay, M.; Ellard, S.; De Franco, E.; Moisés, R. S. Pancreatic Agenesis Due to Compound Heterozygosity for a Novel Enhancer and Truncating Mutation in the PTF1A Gene. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 274–277. [Google Scholar] [CrossRef]
- Evliyaoğlu, O.; Ercan, O.; Ataloğlu, E.; Zübarioğlu, Ü.; Özcabı, B.; Dağdeviren, A.; Erdoğan, H.; De Franco, E.; Ellard, S. Neonatal Diabetes: Two Cases with Isolated Pancreas Agenesis Due to Homozygous PTF1A Enhancer Mutations and One with Developmental Delay, Epilepsy, and Neonatal Diabetes Syndrome Due to KCNJ11 Mutation. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 168–174. [Google Scholar] [CrossRef]
- Yue, X.; Luo, Y.; Wang, J.; Huang, D. Monogenic Diabetes with GATA6 Mutations: Characterization of a Novel Family and a Comprehensive Analysis of the GATA6 Clinical and Genetics Traits. Mol. Biotechnol. 2024, 66, 467–474. [Google Scholar] [CrossRef]
- Allen, H. L.; Flanagan, S. E.; Shaw-Smith, C.; De Franco, E.; Akerman, I.; Caswell, R.; Ferrer, J.; Hattersley, A. T.; Ellard, S. GATA6 Haploinsufficiency Causes Pancreatic Agenesis in Humans. Nat. Genet. 2012, 44, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Chao, C. S.; McKnight, K. D.; Cox, K. L.; Chang, A. L.; Kim, S. K.; Feldman, B. J. Novel GATA6 Mutations in Patients with Pancreatic Agenesis and Congenital Heart Malformations. PLoS One 2015, 10, e0118449. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; De Franco, E.; Cardenas-Diaz, F. L.; Letourneau-Freiberg, L. R.; Sanyoura, M.; Osorio-Quintero, C.; French, D. L.; Greeley, S. A. W.; Hattersley, A. T.; Gadue, P. A Non-Coding Disease Modifier of Pancreatic Agenesis Identified by Genetic Correction in a Patient-Derived IPSC Line. Cell Stem Cell 2020, 27, 137-146.e6.
- De Franco, E. Neonatal Diabetes Caused by Disrupted Pancreatic and Β-cell Development. Diabetic Medicine 2021, 38. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Wasson, L. K.; AL Willcox, J.; Morton, S. U.; Gorham, J. M.; DeLaughter, D. M.; Neyazi, M.; Schmid, M.; Agarwal, R.; Jang, M. Y.; et al. GATA6 Mutations in HiPSCs Inform Mechanisms for Maldevelopment of the Heart, Pancreas, and Diaphragm. Elife 2020, 9. [Google Scholar] [CrossRef]
- Edghill, E. L.; Bingham, C.; Slingerland, A. S.; Minton, J. A. L.; Noordam, C.; Ellard, S.; Hattersley, A. T. Hepatocyte Nuclear Factor-1 Beta Mutations Cause Neonatal Diabetes and Intrauterine Growth Retardation: Support for a Critical Role of HNF-1β in Human Pancreatic Development. Diabetic Medicine 2006, 23, 1301–1306. [Google Scholar] [CrossRef]
- Yorifuji, T.; Kurokawa, K.; Mamada, M.; Imai, T.; Kawai, M.; Nishi, Y.; Shishido, S.; Hasegawa, Y.; Nakahata, T. Neonatal Diabetes Mellitus and Neonatal Polycystic, Dysplastic Kidneys: Phenotypically Discordant Recurrence of a Mutation in the Hepatocyte Nuclear Factor-1β Gene Due to Germline Mosaicism. J. Clin. Endocrinol. Metab. 2004, 89, 2905–2908. [Google Scholar] [CrossRef]
- Li, Y.; Han, X. The HNF1B Mutations and Deletion Associated with Diabetes and Their Resulting Diabetic Phenotypes: A Systematic Review. Int. J. Diabetes Dev. Ctries. 2025, 45, 24–32. [Google Scholar] [CrossRef]
- Poll, A. V; Pierreux, C. E.; Lokmane, L.; Haumaitre, C.; Achouri, Y.; Jacquemin, P.; Rousseau, G. G.; Cereghini, S.; Lemaigre, F. P. A VHNF1/TCF2-HNF6 Cascade Regulates the Transcription Factor Network That Controls Generation of Pancreatic Precursor Cells. Diabetes 2006, 55(1), 61–69. [Google Scholar] [CrossRef]
- Maestro, M. A.; Boj, S. F.; Luco, R. F.; Pierreux, C. E.; Cabedo, J.; Servitja, J. M.; German, M. S.; Rousseau, G. G.; Lemaigre, F. P.; Ferrer, J. Hnf6 and Tcf2 (MODY5) Are Linked in a Gene Network Operating in a Precursor Cell Domain of the Embryonic Pancreas. Hum. Mol. Genet. 2003, 12, 3307–3314. [Google Scholar] [CrossRef]
- Pezzino, G.; Ruta, R.; Rapini, N.; Chiodo, D. C.; Mucciolo, M.; Tomaselli, L.; Cianfarani, S.; Barbetti, F. A Rare Cause of Transient Neonatal Diabetes Mellitus: Spontaneous HNF1B Splice Variant. Diabetic Medicine 2024, 41. [Google Scholar] [CrossRef]
- Iafusco, F.; Meola, S.; Pecoraro, C.; Mazzaccara, C.; Iafusco, D.; Tinto, N. Prenatal Diagnosis of HNF1b Mutation Allows Recognition of Neonatal Dysglycemia. Acta Diabetol. 2021, 58(3), 393–395. [Google Scholar] [CrossRef]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. Neurogenin3 Is Required for the Development of the Four Endocrine Cell Lineages of the Pancreas. Proceedings of the National Academy of Sciences 2000, 97(4), 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Wejaphikul, K.; Srilanchakon, K.; Kamolvisit, W.; Jantasuwan, S.; Santawong, K.; Tongkobpetch, S.; Theerapanon, T.; Damrongmanee, A.; Hongsawong, N.; Ukarapol, N.; et al. Novel Variants and Phenotypes in NEUROG3 -Associated Syndrome. J. Clin. Endocrinol. Metab. 2022, 108, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S. E.; Oliver-Krasinski, J.; Ernst, L.; Hughes, N.; Patel, P.; Stoffers, D. A.; Russo, P.; De León, D. D. Neonatal Diabetes and Congenital Malabsorptive Diarrhea Attributable to a Novel Mutation in the Human Neurogenin-3 Gene Coding Sequence. J. Clin. Endocrinol. Metab. 2011, 96, 1960–1965. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Jensen, J. N.; Hodgson, M. I.; Codner, E.; Ellard, S.; Serup, P.; Hattersley, A. T. Permanent Neonatal Diabetes and Enteric Anendocrinosis Associated With Biallelic Mutations in NEUROG3. Diabetes 2011, 60, 1349–1353. [Google Scholar] [CrossRef]
- Hancili, S.; Bonnefond, A.; Philippe, J.; Vaillant, E.; De Graeve, F.; Sand, O.; Busiah, K.; Robert, J.; Polak, M.; Froguel, P.; et al. A Novel NEUROG3 Mutation in Neonatal Diabetes Associated with a Neuro-intestinal Syndrome. Pediatr. Diabetes 2018, 19, 381–387. [Google Scholar] [CrossRef]
- Zhang, X.; McGrath, P. S.; Salomone, J.; Rahal, M.; McCauley, H. A.; Schweitzer, J.; Kovall, R.; Gebelein, B.; Wells, J. M. A Comprehensive Structure-Function Study of Neurogenin3 Disease-Causing Alleles during Human Pancreas and Intestinal Organoid Development. Dev. Cell 2019, 50, 367–380.e7. [Google Scholar] [CrossRef]
- Solorzano-Vargas, R. S.; Bjerknes, M.; Wang, J.; Wu, S. V.; Garcia-Careaga, M. G.; Pitukcheewanont, P.; Cheng, H.; German, M. S.; Georgia, S.; Martín, M. G. Null Mutations of NEUROG3 Are Associated with Delayed-Onset Diabetes Mellitus. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Gómez, J. L.; Gleisner, A.; Hattersley, A. T.; Codner, E. Hypogonadotropic Hypogonadism and Short Stature in Patients with Diabetes Due to Neurogenin 3 Deficiency. J. Clin. Endocrinol. Metab. 2016, 101, 3555–3558. [Google Scholar] [CrossRef]
- Bohuslavova, R.; Smolik, O.; Malfatti, J.; Berkova, Z.; Novakova, Z.; Saudek, F.; Pavlinkova, G. NEUROD1 Is Required for the Early α and β Endocrine Differentiation in the Pancreas. Int. J. Mol. Sci. 2021, 22, 6713. [Google Scholar] [CrossRef]
- Bohuslavova, R.; Fabriciova, V.; Smolik, O.; Lebrón-Mora, L.; Abaffy, P.; Benesova, S.; Zucha, D.; Valihrach, L.; Berkova, Z.; Saudek, F.; et al. NEUROD1 Reinforces Endocrine Cell Fate Acquisition in Pancreatic Development. Nat. Commun. 2023, 14(1), 5554. [Google Scholar] [CrossRef]
- Demirbilek, H.; Hatipoglu, N.; Gul, U.; Tatli, Z. U.; Ellard, S.; Flanagan, S. E.; De Franco, E.; Kurtoglu, S. Permanent Neonatal Diabetes Mellitus and Neurological Abnormalities Due to a Novel Homozygous Missense Mutation in NEUROD1. Pediatr. Diabetes 2018, 19, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Morley, R.; Cole, T. J. Adverse Neurodevelopmental Outcome of Moderate Neonatal Hypoglycaemia. BMJ 1988, 297, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Jermendy, A.; Toschi, E.; Aye, T.; Koh, A.; Aguayo-Mazzucato, C.; Sharma, A.; Weir, G. C.; Sgroi, D.; Bonner-Weir, S. Rat Neonatal Beta Cells Lack the Specialised Metabolic Phenotype of Mature Beta Cells. Diabetologia 2011, 54, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C. C.; Hussain, K.; Jones, P. M.; Persaud, S.; Löbner, K.; Boehm, A.; Clark, A.; Christie, M. R. Low Levels of Glucose Transporters and Kþ ATP Channels in Human Pancreatic Beta Cells Early in Development. Diabetologia 2007, 50, 1000–1005. [Google Scholar] [CrossRef]
- Parappil, H.; Gaffari, M.; Paramban, R.; Rijims, M.; Skaria, S.; Ahmed, S. N. Management of Hyperglycemia in the Neonatal Unit. J. Clin. Neonatol. 2022, 11, 38–44. [Google Scholar] [CrossRef]
- Mitanchez, D. Glucose Regulation in Preterm Newborn Infants. Horm. Res. Paediatr. 2007, 68, 265–271. [Google Scholar] [CrossRef]
- Hey, E. Hyperglycaemia and the Very Preterm Baby. Semin. Fetal Neonatal Med. 2005, 10, 377–387. [Google Scholar] [CrossRef]
- Decaro, M. H.; Vain, N. E. Hyperglycaemia in Preterm Neonates: What to Know, What to Do. Early Hum. Dev. 2011, 87, S19–S22. [Google Scholar] [CrossRef]
- Mitanchez-Mokhtari, D.; Lahlou, N.; Kieffer, F.; Magny, J.-F.; Roger, M.; Voyer, M. Both Relative Insulin Resistance and Defective Islet β-Cell Processing of Proinsulin Are Responsible for Transient Hyperglycemia in Extremely Preterm Infants. Pediatrics 2004, 113, 537–541. [Google Scholar] [CrossRef]
- Chacko, S. K.; Ordonez, J.; Sauer, P. J. J.; Sunehag, A. L. Gluconeogenesis Is Not Regulated by Either Glucose or Insulin in Extremely Low Birth Weight Infants Receiving Total Parenteral Nutrition. J. Pediatr. 2011, 158, 891–896. [Google Scholar] [CrossRef]
- Cowett, R. M.; Andersen, G. E.; Maguire, C. A.; Oh, W. Ontogeny of Glucose Homeostasis in Low Birth Weight Infants. J. Pediatr. 1988, 112, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, B. H.; Limesand, S. W.; Rozance, P. J. The Impact of IUGR on Pancreatic Islet Development and β-Cell Function. Journal of Endocrinology 2017, 235, R63–R76. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, F. A.; De Prins, F.; Aerts, L.; Verjans, M. The endocrine pancreas in small-for-dates infants. Bjog 1977, 84, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Green, A. S.; Rozance, P. J.; Limesand, S. W. Consequences of a Compromised Intrauterine Environment on Islet Function. Journal of Endocrinology 2010, 205, 211–224. [Google Scholar] [CrossRef]
- Limesand, S. W.; Rozance, P. J.; Zerbe, G. O.; Hutton, J. C.; Hay, W. W. Attenuated Insulin Release and Storage in Fetal Sheep Pancreatic Islets with Intrauterine Growth Restriction. Endocrinology 2006, 147, 1488–1497. [Google Scholar] [CrossRef]
- Thorn, S.; Rozance, P.; Brown, L.; Hay, W. The Intrauterine Growth Restriction Phenotype: Fetal Adaptations and Potential Implications for Later Life Insulin Resistance and Diabetes. Semin. Reprod. Med. 2011, 29, 225–236. [Google Scholar] [CrossRef]
- Yates, D. T.; Macko, A. R.; Chen, X.; Green, A. S.; Kelly, A. C.; Anderson, M. J.; Fowden, A. L.; Limesand, S. W. Hypoxaemia-induced Catecholamine Secretion from Adrenal Chromaffin Cells Inhibits Glucose-stimulated Hyperinsulinaemia in Fetal Sheep. J. Physiol. 2012, 590, 5439–5447. [Google Scholar] [CrossRef]
- Park, J. H.; Stoffers, D. A.; Nicholls, R. D.; Simmons, R. A. Development of Type 2 Diabetes Following Intrauterine Growth Retardation in Rats Is Associated with Progressive Epigenetic Silencing of Pdx1. Journal of Clinical Investigation 2008. [Google Scholar] [CrossRef]
- Stoffers, D. A.; Desai, B. M.; DeLeon, D. D.; Simmons, R. A. Neonatal Exendin-4 Prevents the Development of Diabetes in the Intrauterine Growth Retarded Rat. Diabetes 2003, 52, 734–740. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, H.; Wu, J.; Yu, Z.; Li, Q.; Jiang, C.-M. Association between Glycemia and Outcomes of Neonates with Hypoxic-Ischemic Encephalopathy: A Systematic Review and Meta-Analysis. BMC Pediatr. 2024, 24, 699. [Google Scholar] [CrossRef]
- Pedroza-García, K. A.; Calderón-Vallejo, D.; Quintanar, J. L. Neonatal Hypoxic–Ischemic Encephalopathy: Perspectives of Neuroprotective and Neuroregenerative Treatments. Neuropediatrics 2022, 53, 402–417. [Google Scholar] [CrossRef]
- Šumanović-Glamuzina, D.; Čulo, F.; Čulo, M. I.; Konjevoda, P.; Jerković-Raguž, M. A Comparison of Blood and Cerebrospinal Fluid Cytokines (IL-1β, IL-6, IL-18, TNF-α) in Neonates with Perinatal Hypoxia. Bosn. J. Basic Med. Sci. 2017. [Google Scholar] [CrossRef]
- Sheng, J. A.; Bales, N. J.; Myers, S. A.; Bautista, A. I.; Roueinfar, M.; Hale, T. M.; Handa, R. J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front. Behav. Neurosci. 2021, 14. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the association between specific genetic causes and transient or permanent neonatal diabetes mellitus (NDM), as well as treatment implications for selected genetic forms of permanent NDM (e.g., KATP-related NDM). The oval sizes reflect approximate frequencies among genetically confirmed NDM cases.
Figure 1.
Schematic representation of the association between specific genetic causes and transient or permanent neonatal diabetes mellitus (NDM), as well as treatment implications for selected genetic forms of permanent NDM (e.g., KATP-related NDM). The oval sizes reflect approximate frequencies among genetically confirmed NDM cases.
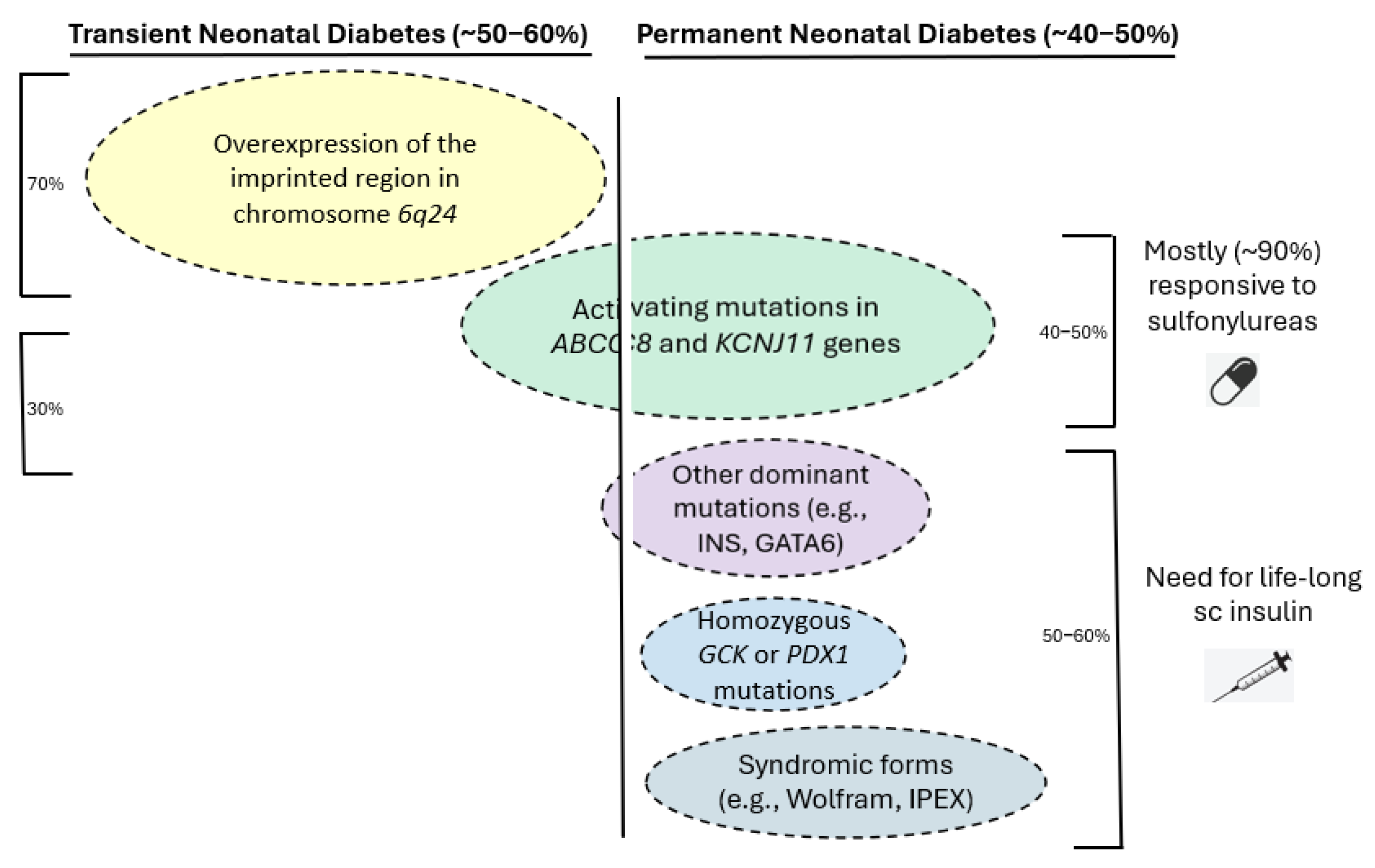

Table 1.
Clinical and genetic characteristics of patients with 6q24 neonatal diabetes mellitus.
| Author | Population | Genetic etiology | Median age of diagnosis, days | Median age of remission, weeks | SGA | Congenital anomalies/disorders |
|---|---|---|---|---|---|---|
| Busiah, 2013[30] | 40 | 45% UPD6pat 33% 6q24 duplication 23% maternal hypomethylation |
5.0 (1.0–14.5) | 14.4 (7.8–28.3) | 92% | Macroglossia 32% Cardiac 8% Urinary tract 17% Neurological disorders 18% Umbilical hernia 8% |
| Docherty, 2013 [23] | 163 | 41% UPD6pat 33% paternal 6q24 duplication 26% maternal hypomethylation |
4.0 | 12.0 | ND | Macroglossia 44% Umbilical hernia 21% Dysmorphic facial features 18% Urinary tract 9% Cardiac 9% Hypothyroidism 4% |
| Bonfanti, 2021 [32] | 12 | 50% UPD6pat 17% paternal 6q24 duplication 33% maternal hypomethylation |
7.0 | 12.0 | ND | Macroglossia/Umbilical hernia 33% |
| McCullough, 2024 [31] | 33 | 52% UPD6pat 21% paternal 6q24 duplication 24% maternal hypomethylation |
2.0 (1.0–7.0) | 12.0 (8.0-24.0) | 71% | Macroglossia 56% Umbilical hernia 22% Speech pathologies 36% |
| Pietrusinski, 2025 [33] | 5 | 60% UPD6pat/maternal hypomethylation | 4.4 | 16.0 | 80% | Macroglossia 0% Umbilical hernia 40% Neurodevelopmental disorders 40% |
UPD6pat: paternal uniparental disomy of chromosome 6; ND: No data.
Table 2.
Overview of the main genetic causes of neonatal diabetes mellitus associated with beta-cell dysfunction and impaired insulin secretion.
Table 2.
Overview of the main genetic causes of neonatal diabetes mellitus associated with beta-cell dysfunction and impaired insulin secretion.
| Gene | Location | Mode of inheritance | Gene product | Type of NDM | Associated malformations |
|---|---|---|---|---|---|
| KCNJ11 | 11p15.1 | de novo/ AD | Kir6.2 | PNDM (more often)/TNDM | Neurodevelopmental impairment, DEND syndrome |
| ABCC8 | 11p15.1 | de novo/ AD | SUR1 | PNDM/TNDM (more often) | Neurodevelopmental impairment, DEND syndrome (very rare) |
| GCK | 7p15.3-p15.1 | AR | GCK | PNDM | Not present |
| SLC2A2 | 3q26.1-26.3 | AR | GLUT2 | PNDM/TNDM | Fanconi-Bickel syndrome (renal Fanconi syndrome, rickets, growth retardation, hepatomegaly, impaired galactose metabolism, diabetes) |
| SLC19A | 1q23.3 | AR | THTR1 | PNDM | Rogers syndrome (Thiamine-responsive megaloblastic anemia, sensorineural deafness, diabetes) |
AD: autosomal dominant; AR: autosomal recessive; NDM: Neonatal diabetes mellitus; PNDM: Permanent neonatal diabetes mellitus; TNDM: Transient neonatal diabetes mellitus; DEND: Developmental delay, epilepsy, and neonatal diabetes.
Table 3.
Overview of the main genetic causes of neonatal diabetes mellitus associated with defects in insulin biosynthesis and beta-cell survival.
Table 3.
Overview of the main genetic causes of neonatal diabetes mellitus associated with defects in insulin biosynthesis and beta-cell survival.
| Gene | Location | Mode of inheritance | Gene product | Type of NDM | Associated malformations |
|---|---|---|---|---|---|
| INS | 11p15.5 | de novo, AD (more often)/AR | Insulin | PNDM (more often)/TNDM | Not present |
| EIF2AK3 | 2p11.2 | AR | PERK | PNDM | Wolcott-Rallison syndrome (neurodevelopmental disorders, skeletal dysplasias, renal disorders, liver impairment, diabetes) |
| EIF2B1 | 12q24.31 | de novo, AD | eEIF2Ba | PNDM | Liver impairment |
| IER3IP1 | 18q12 | AR | IER3IP1 | PNDM | MEDS syndrome (diabetes, microcephaly, epilepsy) |
| WFS1 | 4p16.1 | AR/ de novo, AD | wolframin | PNDM | Homozygous: Wolfram syndrome (diabetes mellitus, diabetes insipidus, optic atrophy, deafness) Heterozygous: diabetes, congenital sensorineural deafness, congenital cataract |
AD: autosomal dominant; AR: autosomal recessive; NDM: Neonatal diabetes mellitus; PNDM: Permanent neonatal diabetes mellitus; TNDM: Transient neonatal diabetes mellitus.
Table 4.
Overview of the main genetic causes of neonatal diabetes mellitus associated with defects in beta-cell development and pancreatic morphogenesis.
Table 4.
Overview of the main genetic causes of neonatal diabetes mellitus associated with defects in beta-cell development and pancreatic morphogenesis.
| Gene | Location | Mode of inheritance | Gene product | Type of NDM | Associated malformations |
|---|---|---|---|---|---|
| PDX1 | 13q12.1 | AD | PDX1 | PNDM | Nonsense mutations: pancreatic agenesis/hypoplasia Hypomorphic mutations: +/- exocrine pancreatic insufficiency |
| PTF1A | 10p12.2 | AR/AD | PTF1A | PNDM | Homozygous: pancreatic agenesis, cerebellar hypoplasia/agenesis Heterozygous: pancreatic agenesis Growth retardation, anemia, cholestasis |
| HNF1B | 17q12 | AD | HNF1B | PNDM/TNDM | Pancreatic hypoplasia, renal cysts |
| GATA6 | 18q11.2 | AD | GATA6 | PNDM | Pancreatic agenesis, cardiac, hepatobiliary, intestinal malformations |
| GATA4 | 8p23.1 | AD | GATA4 | PNDM | Pancreatic agenesis, congenital heart defects |
| CNOT1 | 16q21 | de novo, AD | CNOT1 | PNDM | Pancreatic agenesis, holoprosencephaly |
| RFX6 | 6q22.1 | AR | RFX6 | PNDM | Mitchell–Riley syndrome (diabetes, bowel atresia, gallbladder agenesis/hypoplasia) |
| NEUROG3 | 10q21.3 | AR | NEUROG3 | PNDM | Malabsorptive congenital diarrhea (enteric anendocrinosis), hypogonadotrophic hypogonadism, short stature |
| NEUROD1 | 2q31.3 | AR | NEUROD1 | PNDM | Cerebellar hypoplasia, sensorineural deafness, visual impairments |
| NKX2-2 | 20p11.22 | AR | NKX2-2 | PNDM | Neurodevelopmental impairment, corpus callosum hypoplasia |
| GLIS3 | 9p24.2 | AR | GLIS3 | PNDM | congenital hypothyroidism, polycystic kidneys, hepatomegaly, cholestasis, characteristic facial features |
AD: autosomal dominant; AR: autosomal recessive; NDM: Neonatal diabetes mellitus; PNDM: Permanent neonatal diabetes mellitus; TNDM: Transient neonatal diabetes mellitus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



