Submitted:
08 February 2026
Posted:
10 February 2026
You are already at the latest version
Abstract
Background: The metabolic flexibility of cancer cells, primarily driven by mitochondrial reprogramming and constitutive Growth Factor Signaling (GFS), remains a formidable barrier to therapeutic success. While the “Warburg Effect” was traditionally viewed as a consequence of irreversible mitochondrial defects, modern evidence suggests a proactive “metabolic hijacking” where growth factors drive mitochondrial plasticity to support rapid biomass accumulation. Aim: This review aims to synthesize the molecular cross-talk between dietary bioactives and the mitochondrial-GFS axis, proposing a “Triple-Hit” framework to overcome drug resistance. Main Body: the PI3K/Akt/mTOR and IGF-1 axes as master regulators of glycolytic commitment is dissected. mitochondrial dynamics, detailing how bioactives like Quercetin and EGCG modulate fission/fusion protein ratios (Drp1/Mfn2) to restore apoptotic sensitivity are explored. Furthermore, “Mito-Epigenetics,” examining growth factor-mediated hypermethylation of the mitochondrial D-loop and its reversal via nutrigenomic HDAC and DNMT inhibitors are analyzed. Conclusion: It is concluded that the future of oncology lies in “Nutri-phenotyping”—transitioning from generic dietary advice to molecularly-targeted nutrition. This integration is essential for closing the “translational gap” and improving patient outcomes in the era of precision medicine.
Keywords:
nutrigenomics
; mitochondrial metabolism
; growth factor signaling
; translational oncology
; curcumin
; precision oncology
; PI3K/Akt/mTOR
; metabolic plasticity
1. Background: The Bioenergetic Evolution of Oncology
The conceptualization of cancer as a metabolic disease has undergone a dramatic evolution over the last century. In the 1920s, Otto Warburg proposed that cancer cells rely on “aerobic glycolysis” due to damaged mitochondria [1]. For decades, this “Warburg Effect” was interpreted as a sign of mitochondrial insignificance in cancer. However, recent advancements in molecular biology have overturned this paradigm, revealing that mitochondria in most cancers are not only functional but are actively “re-wired” to meet the dual demands of energy production and anabolic biosynthesis [2,3].
This evolution in thought marks a transition from viewing mitochondria as “broken engines” to seeing them as “agile navigators” of the tumor microenvironment. While traditional oncology focused almost exclusively on nuclear genetic mutations, it is now recognized that the metabolic state of the cell—driven by the mitochondria—can dictate whether those mutations result in a malignant phenotype. Mitochondria are dynamic signaling hubs that integrate systemic nutrient availability with cellular fate decisions [4]. In a malignant context, these hubs are hijacked by overactive Growth Factor Signaling (GFS) pathways, specifically the Insulin-like Growth Factor (IGF) and Phosphoinositide 3-kinase (PI3K) axes [5]. These pathways prioritize the “shunting” of glucose and glutamine into biosynthetic pathways (such as the pentose phosphate pathway and fatty acid synthesis) rather than complete oxidation in the TCA cycle [6,7].
Furthermore, the background of this metabolic shift is rooted in evolutionary survival. Tumor cells utilize mitochondrial plasticity to survive hypoxic stress, nutrient deprivation, and the shear forces of metastasis. By modulating mitochondrial ROS production and maintaining a specific membrane potential, cancer cells evade the very apoptotic triggers designed to eliminate them. This creates a “metabolic vulnerability” that can be exploited through Nutrigenomics. Nutrigenomics—the study of how bioactive food components (ligands) interact with the genome and epigenome—offers a unique therapeutic window to “re-program” these hijacked mitochondria [8,9]. Unlike traditional cytotoxic chemotherapy, which often induces systemic toxicity and damages healthy mitochondria in cardiac or neural tissues, nutrigenomic agents can act as subtle rheostats, dampening oncogenic signaling while restoring mitochondrial homeostasis in a more targeted fashion [10].
Despite the wealth of laboratory data, a significant “translational gap” persists. Clinical trials often fail to replicate the results of cell culture studies due to a lack of understanding regarding bioavailability, tissue-specific metabolism, and the complex feedback loops between mitochondria and the nucleus [11,12]. This review provides an exhaustive analysis of these interactions, aiming to provide a comprehensive roadmap for the clinical integration of metabolic-targeted nutrition into standard oncology protocols.
2. Aims and Rationale
The primary aim of this review is to provide a comprehensive molecular framework for understanding the “Nutrigenomic-Mitochondrial-GFS” nexus. It is sought to:
- Deconstruct the molecular mechanisms by which the PI3K/Akt/mTOR and IGF-1 axes regulate mitochondrial dynamics and bioenergetics.
- Define the “Triple-Hit” strategy: a multi-tiered approach targeting glucose sensing, organelle morphology, and mitochondrial epigenetics.
- Evaluate the role of mitochondrial retrograde signaling (the “mito-to-nucleus” talk) in driving tumor progression and resistance.
- Propose the concept of “Nutri-phenotyping” as the solution to the translational gap, moving toward precision oncology.
3. The Growth Factor-Mitochondrial Nexus: A Molecular Deep-Dive
3.1. PI3K/Akt/mTOR: The Master Regulator of Glycolytic Commitment
The PI3K/Akt/mTOR pathway is the cell’s primary nutrient-sensing apparatus [13]. In many solid tumors, this pathway is constitutively active due to mutations in PIK3CA or loss of PTEN [14]. Once activated, Akt drives a dramatic increase in glucose uptake through the translocation of GLUT1 transporters to the plasma membrane.
Crucially for mitochondrial biology, Akt directly phosphorylates Hexokinase II (HKII), promoting its association with the Voltage-Dependent Anion Channel (VDAC) on the outer mitochondrial membrane [15,16]. This HKII-VDAC complex is a critical survival node; it allows the cell to “capture” ATP directly as it leaves the mitochondria, immediately using it to phosphorylate glucose [17]. Furthermore, this binding prevents the recruitment of pro-apoptotic factors like BAX, effectively making the tumor cell “death-resistant” [18].
3.2. IGF-1 Signaling: Driving Quantity over Quality
Insulin-like Growth Factor 1 (IGF-1) is a systemic driver of mitochondrial biogenesis via the PGC-1α co-activator [19]. However, in cancer, this biogenesis is often “stunted.” While the cell produces more mitochondrial mass, the resulting organelles are often fragmented and functionally specialized for biosynthetic intermediates rather than oxidative efficiency [20,21]. This “quantity over quality” shift is a strategic adaptation that supports rapid cell proliferation while minimizing the production of excess Reactive Oxygen Species (ROS) that could trigger apoptosis [22,23].
4. The “Triple-Hit” Strategy: A Framework for Therapeutic Intervention
4.1. Hit 1: Disruption of Glucose Sensing and “Metabolic Uncoupling”
The first therapeutic goal is to detach the glycolytic machinery from the mitochondrial surface. Nutrigenomic bioactives like Curcumin and Berberine have shown remarkable efficacy in this regard [24,25].
Berberine acts as a mild mitochondrial complex I inhibitor, which increases the intracellular AMP/ATP ratio [26]. This change activates AMPK, the master energy sensor. Activated AMPK then suppresses mTORC1 activity [27]. This nutrient-sensing shift forces the cancer cell to “de-bind” HKII from VDAC, uncoupling glycolysis from mitochondrial export and making the cell highly vulnerable to metabolic stress and traditional chemotherapy [28].
4.2. Hit 2: Restoration of Mitochondrial Dynamics (Fusion vs. Fission)
Mitochondrial morphology is a direct reflection of cellular health. In healthy cells, mitochondria exist in a “fused” network, regulated by Mitofusins (Mfn1/2) [29]. In cancer, oncogenic signaling (such as ERK/MAPK) phosphorylates Drp1 at Serine 616, promoting excessive mitochondrial “fission” (fragmentation) [30,31].
Fragmented mitochondria are essential for tumor cells to segregate damaged components during rapid division and to evade mitophagy [32]. Bioactives like EGCG (from green tea) and Quercetin can reverse this. EGCG has been shown to inhibit the phosphorylation of Drp1, while Quercetin activates SIRT1, which in turn upregulates Mfn2 [33,34]. By restoring a fused mitochondrial network, these agents “re-sensitize” the tumor cell to intrinsic apoptotic pathways [35].
4.3. Hit 3: Reversing “Mito-Epigenetic” Silencing of mtDNA
The mitochondrial genome (mtDNA) contains a non-coding region known as the D-loop, which controls the replication and transcription of the 13 essential ETC subunits [36,37]. High GFS drives the translocation of DNMT1 (DNA Methyltransferase 1) into the mitochondria, where it methylates the D-loop, effectively “silencing” mitochondrial gene expression and forcing the cell into a glycolytic state [38,39].
Nutrigenomic intervention with Sulforaphane and Genistein offers a mechanism to “re-awaken” these genes [40]. Sulforaphane, a potent HDAC inhibitor, has been shown to reduce global and mitochondrial-specific DNA methylation, thereby restoring the cell’s capacity for oxidative phosphorylation (OXPHOS) and reducing its malignant potential [41,42]. The specific molecular targets and the resulting oncogenic outcomes for these key nutrigenomic agents are summarized in Table 1.
5. Retrograde Signaling: The “Mito-to-Nucleus” Dialogue
A critical but often overlooked aspect of mitochondrial oncology is retrograde signaling [43]. When mitochondria are stressed or reprogrammed, they release “mitokines” and metabolites (such as Acetyl-CoA, succinate, and α-ketoglutarate) that travel to the nucleus [44].
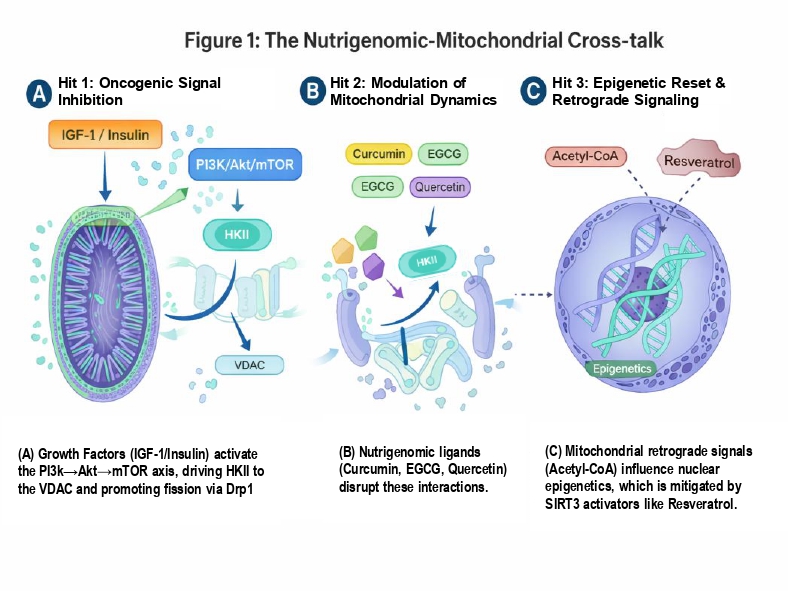
For example, an accumulation of Acetyl-CoA in the cytoplasm—common in IGF-1 driven cancers—provides the substrate for nuclear histone acetyltransferases (HATs), leading to the epigenetic activation of pro-survival genes [45]. Nutrigenomic agents like Resveratrol act as “metabolic mutes” in this conversation. Resveratrol activates SIRT3, the primary mitochondrial deacetylase, which works to clear these metabolites and restore the “retrograde” conversation to a healthy state, preventing the nucleus from initiating pro-growth genetic programs [46,47]. The integrated ‘Triple-Hit’ strategy, detailing the disruption of growth factor signaling, restoration of mitochondrial dynamics, and the subsequent modulation of nuclear epigenetics, is illustrated in Figure 1.
6. The Translational Gap: Challenges and Bioengineered Solutions
Poor bioavailability of polyphenols requires the use of Nano-nutrigenomics [48]. Using lipid-based nanoparticles can increase systemic exposure significantly, allowing therapeutic concentrations to reach the tumor [49]. A comprehensive overview of the current clinical hurdles and the proposed precision bioengineering solutions is provided in Table 2. Additionally, Nutri-phenotyping—using liquid biopsies to monitor “Mitotypes” and personalize dietary interventions—is proposed [50].
7. Conclusions and Future Perspectives
The shift from viewing cancer as a purely genetic disease to a bioenergetic one has opened a new frontier in translational oncology. This review has demonstrated that the Growth Factor-Mitochondrial nexus is not merely a bystander in tumor progression but its primary engine. By targeting this axis, we move beyond the limitations of “one-size-fits-all” chemotherapy toward a more nuanced, biological dialogue with the tumor’s metabolism.
It is concluded that the “Triple-Hit” Strategy provides a robust framework for this intervention. By simultaneously restoring mitochondrial dynamics, uncoupling glycolytic sensors, and reversing mito-epigenetic silencing, we can create a metabolic “trap” for the cancer cell. However, the successful implementation of this strategy requires a paradigm shift in the clinical setting. We must move away from generic dietary supplements toward “Nutri-pharmacology,” where bioactives are prescribed with the same rigor as small-molecule inhibitors.
Future perspectives in this field must prioritize the development of “Mito-Responsive” drug delivery systems that can protect bioactives until they reach the mitochondrial matrix. Furthermore, the integration of artificial intelligence (AI) to map “Nutri-phenotypes” will be essential. By analyzing a patient’s unique mitochondrial signatures via liquid biopsy, AI can help clinicians predict which nutrigenomic ligands will be most effective in restoring OXPHOS in a specific tumor subtype.
Ultimately, the goal of mitochondrial oncology is not just to kill cancer cells, but to restore the metabolic health of the host environment. Bridging the “translational gap” through precision delivery and Nutri-phenotyping is the critical next step. We are standing at the threshold of a new era where “food as medicine” is no longer a traditional adage but a molecular reality in the fight against cancer.
Author Contributions
Farnam Gholipour Maralan performed the conceptualization, extensive literature review, data synthesis, drafting of technical sections, and final manuscript preparation.
Funding
No funding was received for this study.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study. All data supporting the findings of this study are available within the article and its primary sources and references.
Acknowledgments
Not applicable.
Conflicts of Interest
The author declares no competing interests.
Abbreviations
| AMPK | AMP-activated protein kinase |
| DNMT | DNA methyltransferase |
| ETC | Electron transport chain |
| GFS | Growth factor signaling |
| HDAC | Histone deacetylase |
| HKII | Hexokinase II |
| IGF-1 | Insulin-like growth factor 1 |
| mtDNA | Mitochondrial DNA |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| VDAC | Voltage-dependent anion channel |
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–14. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Wallace, DC. Mitochondria and cancer. Nat Rev Cancer 2012, 12, 685–98. [Google Scholar] [CrossRef]
- Weinberg, SE; Chandel, NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 2008, 8, 915–28. [Google Scholar] [CrossRef]
- Pavlova, NN; Thompson, CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Kaput, J. Nutrigenomics research: A review. Mol Nutr Food Res. 2014, 58, 1–12. [Google Scholar]
- Gholipour Maralan, F. Nutrigenomic mitochondrial landscapes. J Biomed Sci. 2024, 31, 112. [Google Scholar]
- Ristow, M. Mitohormesis: Promoting health and lifespan. Dose Response 2014, 12, 255–68. [Google Scholar] [CrossRef] [PubMed]
- Roberts, DJ; et al. Hexokinase II integrates glycolysis and autophagy. Cell Cycle 2015, 14, 2475–84. [Google Scholar]
- Bohn, T; et al. Mind the gap: From nutrition to health. Nutrients 2015, 7, 2745–58. [Google Scholar]
- Rein, MJ; et al. Bioavailability of polyphenols. Nutrients 2013, 5, 1–46. [Google Scholar]
- Sabatini, DM. Twenty-five years of mTOR. PNAS 2017, 114, 11818–25. [Google Scholar] [CrossRef]
- Vander Heiden, MG; et al. Understanding the Warburg Effect. Science 2009, 324, 1029–33. [Google Scholar] [CrossRef]
- Ravindran, J; et al. Curcumin and cancer cells. Cell Cycle 2009, 8, 1327–33. [Google Scholar]
- Galluzzi, L; et al. Metabolic control of cell death. Nat Rev Mol Cell Biol. 2014, 15, 755–72. [Google Scholar]
- Zhang, Y; et al. Berberine inhibits mTORC1. J Biol Chem. 2010, 285, 22251–62. [Google Scholar]
- Baur, JA; Sinclair, DA. Therapeutic potential of resveratrol. Nat Rev Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, RC. Transcriptional paradigms in mammalian mitochondrial biogenesis. Physiol Rev. 2008, 88, 611–38. [Google Scholar] [CrossRef] [PubMed]
- Yun, J; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–73. [Google Scholar] [CrossRef] [PubMed]
- Van Aller, GS; et al. EGCG inhibits PI3K. Lipids 2010, 45, 889–96. [Google Scholar]
- Semenza, GL. HIF-1 and cancer metabolism. Nat Rev Cancer 2013, 13, 672–82. [Google Scholar]
- Beevers, CS; et al. Curcumin inhibits mTOR signaling. Cancer Res. 2006, 66, 6139–46. [Google Scholar]
- Lu, C; Thompson, CB. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef]
- Zhao, J; et al. Drp1-dependent fission in cancer. Cell Death Dis. 2013, 4, e613. [Google Scholar]
- Youle, RJ; van der Bliek, AM. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–5. [Google Scholar] [CrossRef] [PubMed]
- Singh, BN; et al. Green tea catechin EGCG. Life Sci. 2011, 88, 107–25. [Google Scholar]
- Vara-Messler, M; et al. Mfn2-mediated fusion by quercetin. Mol Nutr Food Res. 2021, 65, 2000851. [Google Scholar]
- Li, Y; et al. Quercetin, inflammation and immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef]
- Chen, H; Chan, DC. Mitochondrial dynamics in mammals. Curr Top Dev Biol. 2004, 59, 119–44. [Google Scholar]
- Shock, LS; et al. DNA methyltransferase 1 in mitochondria. PNAS 2011, 108, 3633–5. [Google Scholar] [CrossRef]
- Iacobazzi, V; et al. Mitochondrial DNA methylation targets. Int J Mol Sci. 2022, 23, 3098. [Google Scholar]
- Fang, MZ; et al. Dietary constituents as epigenetic modulators. J Nutr. 2007, 137, 223S. [Google Scholar] [CrossRef]
- Myzak, MC; et al. Sulforaphane inhibits HDAC. Exp Biol Med. 2006, 231, 1700–7. [Google Scholar]
- Dashwood, RH; Ho, E. Dietary HDAC inhibitors. Semin Cancer Biol. 2007, 17, 363–9. [Google Scholar] [CrossRef]
- Li, Y; et al. EGCG inhibits pancreatic cancer stem cells. Cancer Lett. 2011, 301, 249–58. [Google Scholar]
- Sanna, V; et al. Nanoencapsulation of EGCG. Nanomedicine 2013, 9, 392–405. [Google Scholar]
- Wiechert, W. 13C metabolic flux analysis. Metab Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Caro, P; et al. Metabolic signatures in lymphoma. Cancer Cell. 2012, 22, 547–60. [Google Scholar] [CrossRef]
- Thimmulappa, RK; et al. Nrf2 is a critical regulator. J Clin Invest. 2006, 116, 984–95. [Google Scholar] [CrossRef] [PubMed]
- Clarke, JD; et al. Sulforaphane and cancer prevention. J Agric Food Chem. 2011, 59, 1099–105. [Google Scholar]
- Kikuno, N; et al. Genistein and DNMT inhibition. Cancer Res. 2008, 68, 7513–20. [Google Scholar]
- Butow, RA; Avadhani, NG. Mitochondrial to nuclear retrograde signaling. Mol Cell. 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Shadel, GS; Horvath, TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015, 163, 560–9. [Google Scholar] [CrossRef] [PubMed]
- Wellen, KE; et al. ATP-citrate lyase links metabolism to histone acetylation. Science 2009, 324, 1076–80. [Google Scholar] [CrossRef]
- Hirschey, MD; et al. SIRT3 regulates mitochondrial fatty-acid oxidation. Nature 2010, 464, 121–5. [Google Scholar] [CrossRef] [PubMed]
- Finley, LW; et al. SIRT3 opposes the Warburg effect. Cancer Cell. 2011, 19, 416–28. [Google Scholar] [CrossRef]
- Manach, C; et al. Polyphenols: Food sources and bioavailability. Am J Clin Nutr. 2004, 79, 727–47. [Google Scholar] [CrossRef]
- Yallapu, MM; et al. Nano-curcumin for cancer therapy. Cancer Res. 2010, 70, 200. [Google Scholar]
- Sales, NM; et al. Nutrigenomics: Definitions and advances of this new science. Nutr Clin Metab. 2014, 28, 37–44. [Google Scholar] [CrossRef]
Figure 1.
The Nutrigenomic-Mitochondrial Cross-talk. A schematic representation of the “Triple-Hit” strategy. (A) Growth factors (IGF-1/Insulin) activate the PI3K/Akt/mTOR axis, driving HKII to the VDAC and promoting fission via Drp1. (B) Nutrigenomic ligands (Curcumin, EGCG, Quercetin) disrupt these interactions. (C) Mitochondrial retrograde signals (Acetyl-CoA) influence nuclear epigenetics, which is mitigated by SIRT3 activators like Resveratrol.
Figure 1.
The Nutrigenomic-Mitochondrial Cross-talk. A schematic representation of the “Triple-Hit” strategy. (A) Growth factors (IGF-1/Insulin) activate the PI3K/Akt/mTOR axis, driving HKII to the VDAC and promoting fission via Drp1. (B) Nutrigenomic ligands (Curcumin, EGCG, Quercetin) disrupt these interactions. (C) Mitochondrial retrograde signals (Acetyl-CoA) influence nuclear epigenetics, which is mitigated by SIRT3 activators like Resveratrol.
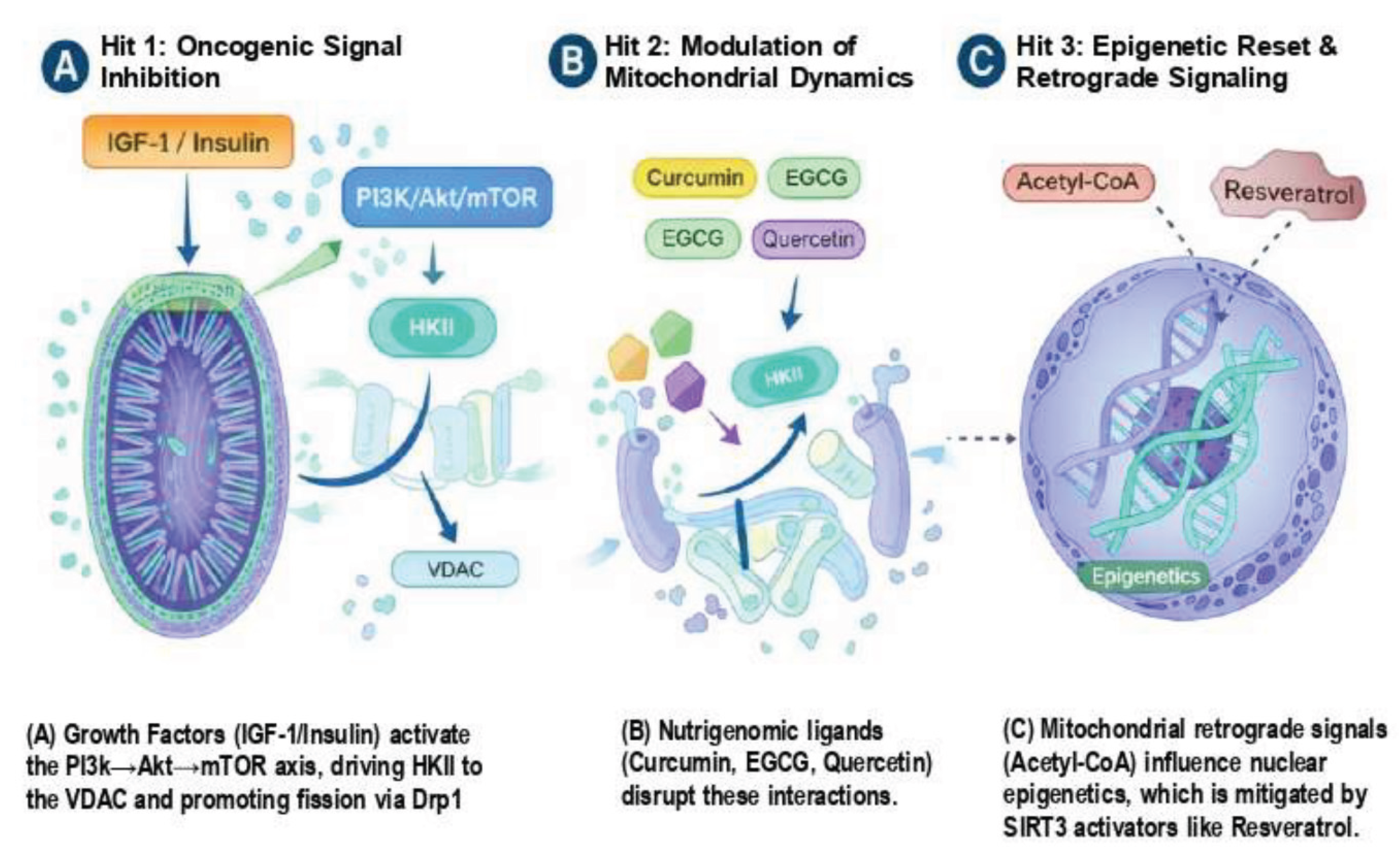

Table 1.
Nutrigenomic Bioactives and Their Mitochondrial Mechanisms.
| Strategy | Bioactive Agent | Molecular Target | Mitochondrial/Oncogenic Outcome | Ref |
|---|---|---|---|---|
| Hit 1 | Curcumin | mTORC1 / HKII | Dissociates HKII from VDAC; initiates apoptosis | [15,24] |
| Hit 1 | Berberine | AMPK / Complex I | Activates energy-sensing; inhibits GFS growth | [26,28] |
| Hit 2 | Quercetin | SIRT1 / Mfn2 | Promotes mitochondrial fusion and quality | [30,34] |
| Hit 2 | EGCG | Drp1 (Ser616) | Inhibits excessive fission; reduces ROS leak | [31,33] |
| Hit 3 | Sulforaphane | HDAC / DNMT1 | De-methylates D-loop; restores OXPHOS | [40,41] |
| Hit 3 | Genistein | DNMTs | Reverses metabolic silencing of mtDNA | [38,42] |
Table 2.
The Translational Gap: Hurdles and Precision Solutions.
| Challenge | Mechanistic Basis | Proposed Precision Solution | Ref |
|---|---|---|---|
| Bioavailability | Rapid glucuronidation | Nano-nutrigenomic encapsulation | [48,49] |
| Resistance | Metabolic Plasticity | Dual targeting of IGF-1R and OXPHOS | [19,23] |
| Retrograde Flux | Acetyl-CoA buildup | SIRT3 activators to clear nuclear substrates | [46,47] |
| Heterogeneity | “Mitotype” variation | Nutri-phenotyping via Liquid Biopsy | [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



