Submitted:
06 February 2026
Posted:
11 February 2026
You are already at the latest version
Abstract
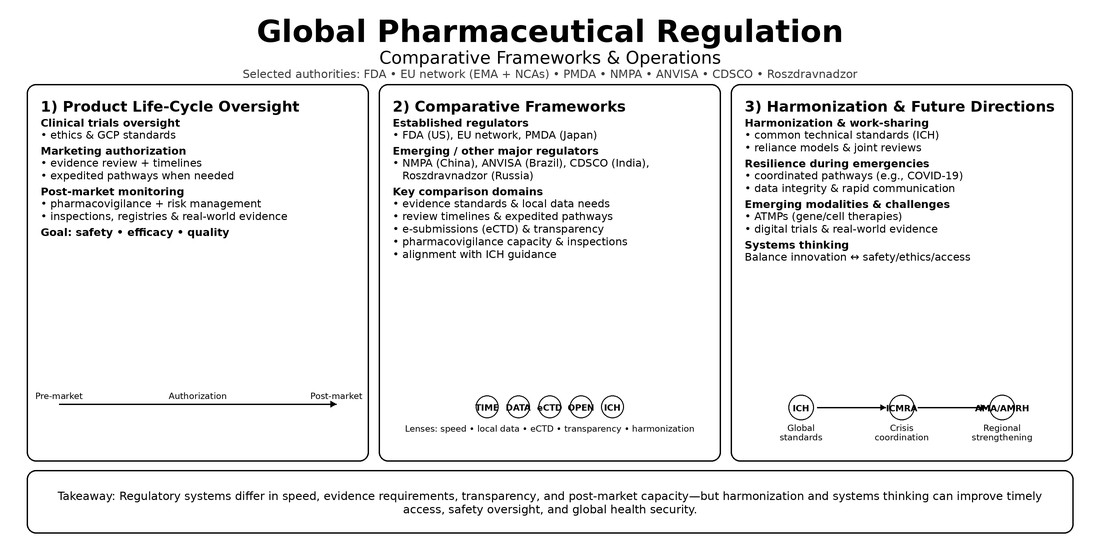
Pharmaceutical regulation and healthcare governmental agencies are central to protecting public health by governing clinical trials, market authorization, and post-market safety monitoring of medicinal products worldwide. Although substantial literature describes major established systems, particularly the United States Food and Drug Administration (FDA), Japan's Pharmaceuticals and Medical Devices Agency (PMDA), and the European Union regulatory network coordinated by the European Medicines Agency (EMA) together with national competent authorities, comparative analysis that integrate both established and emerging regulatory authorities remain limited. This review examines the core functions of regulatory affairs across the product life cycle and compares key features of global regulatory frameworks, including approval pathways, evidentiary expectations, data transparency, and pharmacovigilance approaches. It emphasizes the need for systems thinking to balance innovation with safety, efficacy, and quality, while anticipating unintended consequences of new therapies. This evaluation also highlights how region‑specific constraints and enabling infrastructures, such as national drug‑utilization registries in parts of Europe, can shape regulatory decision‑making and post‑market evaluation. Finally, this paper discusses opportunities for stronger international alliances and greater harmonization to improve efficiency, support timely patient access to essential therapies, strengthen risk management, and reinforce global health security in an increasingly interconnected healthcare environment.
Keywords:
pharmacovigilance
; drug development
; post-marketing surveillance
; global pharmaceutical regulation
; regulatory frameworks
1. Introduction
The pharmaceutical industry operates within an increasingly complex and interconnected regulatory environment designed to ensure the safety, efficacy, and quality of medicinal products worldwide. Regulatory affairs (RA) is a key function of the pharmaceutical industry, overseeing the development, approval, manufacturing, and ongoing monitoring or post-marketing surveillance of pharmaceutical agents to ensure they meet ever-evolving regulatory standards. As healthcare challenges become more global in nature and medical innovations advance at an unprecedented pace, regulatory affairs play an essential role in ensuring consistency, stability, and compliance with international best practices and standards. Regulatory frameworks must therefore continue to evolve while maintaining their fundamental role in protecting public health [1].
The last decade has witnessed significant transformations in regulatory approaches, driven by technological advancements, emerging health threats, and the growing need for international collaboration [2]. These changes include the adoption of expedited review pathways, reliance models, the integration of real-world evidence, and increased emphasis on post-marketing surveillance systems capable of proactive risk detection. The COVID-19 pandemic and the emergence of the COVID-19 vaccines revealed the importance of cross-border regulatory collaboration, catalyzing joint reviews, emergency use authorizations, and digital innovation in regulatory operations.
Beyond compliance, regulatory affairs professionals facilitate drug approvals by navigating complex submission processes, addressing regulatory requirements across different regions, and ensuring timely market entry. They also serve to align global dossiers across regions to meet the essential needs of medical humanity. Furthermore, post-marketing surveillance mechanisms overseen by Regulatory Affairs (RA) help identify safety signals, enabling proactive risk management and ongoing product life cycle oversight. International collaboration amongst regulatory authorities across countries brings several advantages, including better access to affordable medications, the exchange of expertise, alignment of regulatory standards, and initiatives to strengthen regulatory capabilities [3].
While the literature is replete with of the United States Food and Drug Administration (FDA) [4,5,6,7], the European Medicines Agency (EMA) [6,7,8], and Japan's Pharmaceuticals and Medical Devices Agency (PMDA) [3,9], this paper further examines the critical roles of regulatory affairs in pharmaceutical development and marketing, emphasizing the importance of international collaboration and harmonization in an increasingly interconnected global medical ecosystem. It also compares both established and emerging regulatory authorities across regions exploring how different regulatory authorities approach these challenges while considering regional requirements and populational heterogeneity.
2. Global Regulatory Landscape
2.1. Global Regulatory Authorities
Global markets have undergone significant evolution in recent decades, characterized by an unprecedented expansion of cross-border trade and investment activities, including healthcare. This heightened interdependence among nations underscores the necessity for robust regulatory frameworks to manage risks and promote international relations effectively. Regulatory authorities play a central role in these systems, ensuring safety, uniformity and stability, and compliance across jurisdictions.
2.1.1. Established Major Regulatory Authorities
Amongst several other agencies, the FDA, PMDA, and the European medicines regulatory network – coordinated through the EMA in partnership with national competent authorities (NCAs) in EU/EEA Member States - constitute the principal regulatory bodies governing clinical trials, market authorization, and post-marketting surveillance worldwide. These systems, encompassing the United States, Europe, and Japan, maintain distinctive regulatory frameworks that shape the landscape of pharmaceutical drug development and approval processes. Together, they represent some of the most comprehensive pharmaceutical regulatory approaches, with highly structured review processes and international impact [10].
- The US Food and Drug Administration (FDA)
The Food and Drug Administration (FDA), one of the oldest regulatory bodies, was established to address safety concerns surrounding medical supplies and patent medicines, leading to the enforcement of the Pure Food and Drug Act of 1906 [11]. Initially, its authority was limited to preventing the adulteration and misbranding of food and drugs, as outlined before the Federal Food, Drug, and Cosmetic (FD&C Act) of 1938 [12]. As an example, the thalidomide-related birth defects prompted the introduction of the Kefauver-Harris Amendments, which mandated premarket approval and required evidence of both safety and effectiveness [13]. These reforms shaped the pre-market regulatory framework still in use today. The FDA enforces laws and guidelines that form the basis of its regulatory authority, primarily outlined in the FD&C Act [14]. The FDA is responsible for protecting public health by ensuring the safety and efficacy of drugs, medical devices, food, cosmetics, and tobacco products, advancing medical innovations, providing science-based information to the public, and supporting national counterterrorism efforts [15,16].
- 2.
- Europe’s medicines regulatory network: the European Medicines Agency (EMA) and national competent authorities (NCAs).
Medicines regulation in Europe is delivered through a coordinated network of the European Medicines Agency (EMA), the European Commission, and national competent authorities (NCAs) in EU/EEA Member States. Established in 1995, EMA fosters scientific excellence in the evaluation and supervision of medicines, and supports timely patient access to innovative therapies, through its adaptive pathways’ initiative [17,18]. In the centralized procedure, the EMA is responsible for the scientific evaluation of marketing authorization applications; once granted by the European Commission, the centralized marketing authorization is valid in all EU Member States as well as Iceland, Norway, and Liechtenstein [19]. NCAs are primarily responsible for authorizing medicines that do not fall under the scope of the centralized procedure and may also be responsible for other activities related to medicinal products, including authorization of clinical trials [20,21]. In pharmacovigilance, EU law requires marketing authorization holders, NCAs, and EMA to operate pharmacovigilance systems, with overall safety monitoring operating through cooperation between Member States, EMA, and the European Commission [22].
Finland’s Finnish Medicines Agency (Fimea), provides an example of a national authority within the EU network, combining regulation, medicines information, and pharmacovigilance. Fimea regulates and develops the pharmaceutical sector and promotes the rational use of medicines by producing and providing drug information, while also overseeing pharmacovigilance nationally as part of the EU’s agency network [17,23]. Finland’s Kanta Prescription Centre stores all electronic prescriptions and associated dispensing notes in a central service, supporting medication safety and enabling comprehensive analyses of national prescription drug utilization [24,25].
Similarly, Sweden’s Medical Products Agency (Läkemedelsverket) is the national authority responsible for the regulation and surveillance of the development, manufacturing, and sale of medicinal products [26]. Sweden also maintains a National Prescribed Drug Register, established in 2005, which contains all prescribed drugs dispensed in pharmacies and provides a strong foundation for official statistics and real-world drug utilization research [27].
- 3.
- Japan's Pharmaceuticals and Medical Devices Agency (PMDA)
The Pharmaceutical and Medical Device Agency (PMDA) is a Japanese regulatory agency governed by the Ministry of Health, Labour, and Welfare, recognized for its role in protecting public health by assuring the safety, efficacy and quality of pharmaceuticals and medical devices [28]. This agency was established and became functional on April 1, 2004, under the Law for the Pharmaceuticals and Medical Devices Agency, as a consolidation of the services of the Pharmaceuticals and Medical Devices Evaluation Center of the National Institute of Health Sciences (PMDEC), the Organization for Pharmaceutical Safety and Research (OPSR/KIKO), and parts of the Japan Association for the Advancement of Medical Equipment (JAAME) [29].
2.1.2. Emerging Major Regulatory Authorities
The BRIC countries (Brazil, Russia, India, and China) featured below constitute major emerging pharmaceutical markets distinguished by their large, fast-growing economies and significant regional influence, positioning them as increasingly important regulatory powers in the global pharmaceutical landscape [30]. In general, each agency of the BRIC countries regulates pharmaceutical and healthcare products in broad accordance to the framework of the FDA, PMDA, and EMA.
- China's National Medical Products Administration (NMPA)
The National Medical Products Administration (NMPA), previously known as the China Food and Drug Administration (CFDA), has its origins in the State Food and Drug Administration (SFDA). In March 2013, the SFDA was renamed and restructured as the CFDA, achieving the status of a ministerial-level agency [31]. Subsequently, as part of China's governmental reorganization, the NMPA experienced another transformation in March 2018, which involved its consolidation into the newly established State Administration for Market Regulation (SAMR) [31]. The National Medical Products Administration (NMPA) is responsible for regulating the safety, quality, registration, and post-marketing management of drugs, medical devices, and cosmetics, while also formulating policies, standards, and laws, overseeing professional qualifications, and engaging in international cooperation in these areas [31].
- 2.
- Brazil's National Health Surveillance Agency (ANVISA)
The Brazilian Health Surveillance Agency (Agencia Nacional de Vigilancia Sanitaria – ANVISA) was established in January 1999 [32,33]. Its primary objective is to protect public health by managing risks associated with health-related goods and services. ANVISA’s main focus includes maintaining scientific standards, regulating, and inspecting products that may endanger health. Operating under the Brazilian National Health System (SUS), Anvisa is responsible for overseeing the production of health goods and healthcare services in both the public and private sectors [32,33]. Its duties include enforcing industry regulations, inspecting manufacturing practices, and registering and monitoring health products and procedures. Furthermore, ANVISA plays a significant role in the implementation of healthcare policies, such as pharmaceutical assistance, and in directing scientific research related to product development [32,33].
- 3.
- India's Central Drugs Standard Control Organization (CDSCO)
The Central Drugs Standard Control Organization (CDSCO) serves as the central authority under the Drugs and Cosmetics Act, overseeing the approval of new drugs, clinical trials, drug standards, and quality control of imported drugs, and coordinating state drug control activities [34,35]. The Drug Controller General of India (DCGI) is responsible for approving licenses for specified categories of drugs such as blood and blood products, intravenous (IV) fluids, vaccines, and sera [34]. CDSCO’S mission is to safeguard public health in India by ensuring the safety, efficacy, and quality of drugs, cosmetics, and medical devices [34].
- 4.
- Russia's Federal Service for Surveillance in Healthcare (Roszdravnadzor)
The Federal Service for Surveillance in Healthcare (Roszdravnadzor) was established by the President of the Russian Federation through Decree No. 314 dated March 9, 2004, "On the System and Structure of the Federal Executive Bodies," and serves as a federal executive body responsible for overseeing and regulating the healthcare system [36]. It also protects public health by ensuring safety and quality of medicines and medical devices manufactured in or imported into the Russian Federation.
2.1.3. Other Regulatory Authorities
While the FDA, the European medicines regulatory network (EMA and national competent authorities), PMDA, and emerging BRIC authorities command significant influence on the pharmaceutical landscape, other regulatory bodies also contribute essential regional perspectives and represent important pharmaceutical markets in their respective regions. Apart from Brazil's ANVISA, Argentina's ANMAT provides complementary South American representation as it follows a distinct regulatory approach and serves the second-largest pharmaceutical market in the region. This dual representation from South America mirrors our approach to Europe and North America, where the EU medicines regulatory network and the FDA provide established reference frameworks for large regional markets. The following regulatory authorities are therefore from regions not previously addressed, ensuring that all inhabited continents are represented in this global regulatory review:
- Argentina's National Administration of Drugs, Food and Medical Technology (ANMAT)
ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica) of Argentina represents a key regulatory authority in South America, having been awarded the status of "Regulatory Authority of Regional Reference" by the Pan American Health Organization and gaining recognition as an ICH observer member in 2019 [37]. ANMAT's responsibilities extend beyond the scope of pharmaceutics, to include food safety, medical technology evaluation, and managing registration requirements for imported medical products, operating within a comprehensive regulatory framework that serves as a benchmark for neighboring countries [38].
- 2.
- Australia's Therapeutic Goods Administration (TGA)
Australia's Therapeutic Goods Administration (TGA) stands as the preeminent regulatory authority for Oceania, with its influence extending throughout the Asia-Pacific region [39]. It also served as the 2022 chair of the International Medical Device Regulators Forum (IMDRF) and participated in international work-sharing initiatives with other major global regulatory authorities [39]. The TGA's core activities include the regulation of medicines, medical devices, blood and blood products, and vaping products, advertising; however, it does not regulate food, dietary supplements or veterinary drugs. The agency also oversees import and export controls to ensure consistent quality standards across Australia and neighboring Pacific countries [39].
- 3.
- Selected African Regulatory Agencies (NAFDAC, SAHPRA, TMDA, EDA, Ghana’s FDA, MCAZ, ARP, Rwanda’s FDA)
The African Medicines Regulatory Harmonization (AMRH) initiative, launched in 2009, represents a landmark effort to address the continent’s challenges in ensuring access to quality, safe, and affordable medical products by promoting collaborative regional regulatory oversight and harmonized standards across the continent's eight Regional Economic Communities (RECs) [40]. Alongside this, the WHO benchmark, conducted with its Global Benchmarking Tool, evaluates regulatory systems against more than 250 indicators. Maturity Level 4, the highest level, signifies an advanced regulatory system committed to ongoing improvement. Maturity Level 3 indicates a stable, well-functioning, and integrated regulatory system [41]. Currently, out of 54 African countries, only 8 have achieved WHO Maturity Level 3 certification for their national medicines regulatory authorities [41] including:
- Nigeria (NAFDAC) – National Agency for Food and Drug Administration and Control
- South Africa (SAHPRA) - South African Health Products Regulatory Authority
- Tanzania (TMDA) - Tanzania Medicines and Medical Devices Authority
- Egypt (EDA) - Egyptian Drug Authority
- Ghana (FDA) – Food and Drugs Authority
- Zimbabwe (MCAZ) - Medicines Control Authority of Zimbabwe
- Senegal (ARP) - Agence Sénégalaise de Réglementation Pharmaceutique
- Rwanda (FDA) - Food and Drugs Authority
Nigeria's NAFDAC
NAFDAC merits focus as Nigeria's pharmaceutical market represents the largest in West Africa, accounting for 60% of the region’s market [42]. NAFDAC stands out among the eight WHO Maturity Level 3 certified regulatory authorities in Africa for its leadership in regional pharmaceutical regulation, and for overcoming severe challenges with counterfeit medications [43]. Another notable success was the pioneering of the Mobile Authentication Service (MAS), which allows consumers to verify product authenticity via short message service, otherwise known as SMS [44,45]. NAFDAC has also appointed accredited laboratories in China and India, to effect pre-shipment analysis and certification of drugs and pharmaceutical supplies coming into Nigeria [43]. Overall, the agency regulates and controls importation, exportation, manufacture, advertisement, distribution, sale and use of food, drugs, cosmetics, medical devices, bottled water, Chemicals and detergents [46].
2.2. Harmonization Initiatives and Comparative Regulatory Frameworks
2.2.1. Global Harmonization Efforts
Despite the local advances made in the regulatory landscape, there is an ongoing demand for global harmonization of science-based standards to streamline drug development and evaluation to enhance product quality, safety, and efficacy globally. In recent years, steps have been taken to harmonize and streamline application requirements across different countries through the establishment of several initiatives aimed at minimize the need for additional (and sometimes redundant) clinical trials, reduce costs, address disparities in safety monitoring systems and access to healthcare, and facilitating distribution of drugs while ensuring their safety and efficacy of the pharmaceutical agents.
The International Council for Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH) was established in 1990 [47]. It was designed to achieve greater harmonisation worldwide to ensure that safe, effective, and high-quality medicines are developed, and registered and maintained in the most resource-efficient manner whilst meeting high standards [47]. ICH has six founding members: US, EU, Japan, Canada, Swissmedic, and WHO [48]. The ICH includes standing regulatory members Health Canada, Canada; Swissmedic, Switzerland along with standing observers, including the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) and the WHO [49]. This harmonization has reduced duplication of clinical trials and procedures, lowering bureaucracy and shortening the time to market for new drugs [50].
2.2.2. Regional Harmonization Networks
Nations around the world often participate in a variety of regional harmonization networks that support and reinforce the global harmonization efforts. Regional harmonization can increase the efficiency of regulatory authorities by drawing from a shared pool of resources and expertise. Less-resourced National Regulatory Authorities (NRAs) see harmonization as a way to enhance collaboration, strengthen decision-making, share resources, reduce duplication, and improve public health outcomes [50].
2.3. Comparative Analysis of Regulatory Authorities
Regulatory systems vary considerably in their legal mandates, operational frameworks, review models, and degree of alignment with international standards. To characterize these differences systematically, Table 1 provides a comparative overview of core regulatory features across a set of established (FDA, EMA, PMDA) and emerging (NMPA, ANVISA, CDSCO, Roszdravnadzor) agencies. For a detailed version including twenty feature comparisons and explanatory descriptions, see Table S1. The features assessed span pre- and post-market regulatory requirements, scientific and procedural standards, and engagement with global harmonization initiatives, thereby offering a multidimensional perspective on the evolving global regulatory landscape.
3. Drug Development and Approval Process
3.1. Clinical Trial Oversight
3.1.1. Global Clinical Trial Standards
As a crucial process in drug development, the role of clinical trials cannot be overstated. Clinical trials ensure novel drugs are rigorously tested for efficacy and safety while adhering to ethical standards. Global frameworks such as the Belmont Report have become foundational practices held by clinical researchers and pharmaceutical industries when conducting studies involving human subjects to protect study participants [62]. Other important global standards include the International Ethical Guidelines for Biomedical Research Involving Human Subjects issued by the Council for International Organizations of Medical Sciences [63] and International Council for Harmonisation (ICH) and Guideline for Good Clinical Practice (GCP). These guidelines are globally recognized and are upheld as a tool for guiding clinical trial processes for new medicines [64,65].
3.1.2. Innovation in Clinical Trial Regulation
Innovative approaches are reshaping clinical trial regulations to enhance efficiency and adaptability. A recent study by Rosa et al. suggested leveraging real-world data collected through digital technologies for improving clinical trial processes [66]. The study also highlighted the absence of sufficient regulatory frameworks to guide the use of digital tools in clinical trials [66]. The authors argued for the data quality and representativeness, of data that could potentially be sourced to electronic records of study participants in clinical trials thereby simplifying design and collection methods. The advancement and growing need for innovative approaches in clinical trial design calls for clear and well-defined regulatory guidelines in ensuring simpler evidence generation and study implementation. The US Food and Drug Authority (FDA) has responded to this need by establishing the Center for Clinical Trials Innovation (C3TI) primarily to guide and foster integration of innovative approaches to enhance efficiency of drug development in clinical trials [67]. Such proactive measures by regulatory agencies exemplify the potential for improving efficiency in drug development, ultimately making life-saving treatments accessible more quickly.
Furthermore, a study by Scheppler et al. identified the role regulatory authorities play in accelerating global access to life-saving vaccines and advocated for harmonization and collaboration of National regulatory agencies in clinical trial processes [68]. Collaboration among the regulatory agencies is key to helping solve the lengthy drug approval challenge and ultimately help ensure patients receive the safe and effective treatments they deserve.
In Sub-Saharan Africa, a recently ratified collaboration shows the interest of African regulatory agencies in working toward well-regulated clinical trials through the African Medicines Agency (AMA) [69]. Hwenda et al explored the paucity of clinical trial data in Africa - which accounts for less than 3% of global clinical trial genomics data and the readiness of the AMA to accelerate drug development and improve clinical trials in Africa [69]. Africa has untapped potential to contribute to clinical trial data given its 1.3 billion population, diverse population and disease burden [70]. As a regulatory initiative, the AMA is well positioned to improve the continent's contribution to clinical research and innovation by strengthening existing regulatory systems, harmonizing protocols and standards to simplify trial set-up, streamlining trial approvals, and leveraging partnerships such as the European and Development Country Clinical Trial Partnership (EDCTP) to ensure that clinical trials are set up with the participation of the right local partners to maximize successful trials [69]. By addressing the challenges of clinical trial readiness, the AMA could significantly bolster Africa's role in drug development.
3.2. Review and Approval Mechanisms
Drug review and approval mechanisms vary considerably between countries. In the US, FDA relies on preclinical and clinical trial data from drug development to make an approval decision for a New Drug Application (NDA). Each NDA undergoes a thorough review typically lasting for 6 to 10 months to verify new drug efficacy, safety and the adequacy of drug development processes. A decision for approval is passed when the review committee has determined the new drug is safe and effective for intended use. FDA review processes can also be expedited through accelerated approval and priority review pathways which allows faster evaluations of therapies addressing serious or life-threatening conditions such as COVID-19 vaccines [11,71]. The European Commission utilizes a centralized drug review and approval mechanism through the European Medicines Agency (EMA) Committee for Medicinal products for Human Use (CHMP). CHMP provides recommendations after a new drug review and the EC authorizes based on this recommendation [21]. Such a harmonized strategy makes approved medicines accessible to patients in 27 European Union member states and illustrates how centralized strategic approach can accelerate access to new therapies in a timely fashion.
3.3. Challenges in Multinational Clinical Trial Regulations and Potential Solutions
Conducting multinational clinical trials presents significant challenges, as these trials must comply with diverse local and international laws, regulations, and guidelines that differ from one country to another. The incompatibility of these regulatory frameworks, combined with a lack of adequately trained Good Clinical Practice (GCP) staff at clinical sites and difficulties in ensuring effective oversight across different jurisdictions, can result in delays in studying timelines and increased costs.
One potential solution to enhance global collaboration in clinical trials is the creation of a more unified regulatory framework that offers all trial sites a common reference structure for regulatory review and informed consent processes. Such a system would enable national regulators to collaborate on approvals, despite differences in political, social, and economic contexts, while maintaining decentralized oversight within each country. Additionally, establishing a centralized support network could provide mentorship to less experienced researchers and facilitate the exchange of expertise and technology throughout the trial process, addressing challenges related to limited professional expertise and infrastructure.
4. Post-Market Safety Monitoring
4.1. Pharmacovigilance Systems
Pharmacovigilance (PV) systems are an extension of the original adverse drug reaction (ADR) monitoring and reporting systems and are also internationally recognized systems that must align with the entire drug life cycle and post marketing supervision of drugs.
Internationally-recognized Pharmacovigilance (PV) Systems
Various internationally-recognized drug safety surveillance frameworks exist worldwide: the European Union (EU) monitoring network, the World Health Organization (WHO)-Uppsala Monitoring Centre approach, and the International Conference on Harmonization (ICH) system. Despite differences in operational structures and implementations across these three frameworks, they all aim to promote safe medication use [72]. The ten pioneering members of the Uppsala Monitoring Centre include Australia, Canada, the Federal Republic of Germany, Ireland, the Netherlands, New Zealand, Sweden, the United Kingdom, the USA and former Czechoslovakia. These nations were the first to respond to the thalidomide tragedy by establishing medication evaluation committees, creating standardized Adverse Drug Reaction (ADR) documentation forms, and implementing safety protocols [71].
The WHO's global initiative for medication safety surveillance, known as the Programme for International Drug Monitoring (WHO-PIDM), comprises approximately 80 high-income countries. While PV systems require significant financial resources to operate, low- and middle-income countries (LMICs) often struggle with budgetary constraints, leading to limited or non-functional PV systems. In contrast, high-income nations can invest in robust and effective PV activities, demonstrating a direct relationship between a country's income level and its medicine safety measures [71].
4.2. Risk Management and Mitigation
Across the globe, regulatory authorities have prioritized risk assessment as well as risk mitigation strategies [73]. These approaches help ensure continued product safety while maintaining market access.
4.2.1. Risk Management Plan (RMP)
One key guide that applies to risk management involves the development of a Risk Management Plan (RMP) that summarizes product knowledge and target population data at the time of application, encompassing clinical development findings, population epidemiology, disease natural history, and identifying unstudied populations who may receive the drug post-authorization. This information is crucial for assessing post-marketing adverse events and potential risks. While not all identified and potential risks need to be addressed, companies must prioritize significant risks affecting benefit-risk balance through early collaboration between clinical pharmacology, drug development, epidemiology, pharmacovigilance teams, and regulatory agencies [74].
4.2.2. Risk Evaluation and Mitigation Strategy (REMS)
While the EMA mandates a product’s RMP for all applications, the U.S. Food and Drug Administration (FDA) may mandate a Risk Evaluation and Mitigation Strategy (REMS) for certain medications to ensure that benefits outweigh potential risks.
REMS is a drug safety program that the FDA requires for certain medications with serious safety concerns. While all medications have labeling that describes risks, REMS specifically focuses on preventing, monitoring and/or managing serious risks through education and reinforcement of safe medication use behaviors, rather than mitigating all adverse events [75].
REMS focuses on specific strategies that may be outlined in Part V of the RMP, which may include the following: a Medication guide educating patients regarding safety risks and proper product usage; a Communication Plan for healthcare providers regarding safety concerns; and Elements to Assure Safe Use (ETASU), which may restrict product distribution through mechanisms such as requiring prescriber and pharmacist certification, limiting use to inpatient settings, patient monitoring, restricting dispensing to patients with documentation of safe-use conditions, or mandating enrollment in a patient registry [74]. Although the implementation of an RMP and REMS occurs in the post-approval stage, planning and study-design, including development of effective post-approval tools and risk reduction approaches should be established during the pre-approval phase [74].
5. Emerging Challenges and Future Directions
5.1. Advanced Therapy Medicinal Products (ATMPs)
As new and emerging drug–resistant or even untreatable diseases increase, so does the need for innovative therapies, many of which are more advanced in nature compared with existing therapies. These therapies, collectively termed Advanced Therapy Medicinal Products (ATMPs) in the European regulatory context, include gene therapies, cell therapies, and tissue-engineered products, irrespective of their cellular targets, often present novel and complex mechanisms of action [8,76]. Their longer-term effects may be delayed, uncertain, or unknown due to limited data, unlike conventional medications, making it challenging for regulatory authorities especially when considering their entirety of effects, including safety, efficacy, and generational transfection. Ethical concerns surrounding these therapies, especially gene therapies, in terms of their long-term effects have sparked debate and skepticism regarding their acceptance in recent years [77,78].
Recognizing the distinct challenges posed by ATMPs, global regulatory authorities have developed tailored classification systems and approval pathways [63,79]. However, differences in terminology, regulatory structures, and post-marketing requirements persist across regions, potentially delaying patient access and complicating global harmonization efforts [79,80]. International regulatory bodies such as the International Council for Harmonisation (ICH) and the World Health Organization (WHO) have sought to address these gaps by promoting standardization and convergence in regulatory practices [73,81].
Major regulatory agencies have adopted distinct frameworks to govern ATMPs and similar innovative therapies, shaped by differences in regulatory maturity, healthcare infrastructure, and ethical considerations (Table 2).
5.2. Global Health Emergencies
The International Coalition of Medicines Regulatory Authorities (ICMRA) is a voluntary coalition of leaders from medicines regulatory authorities that provides strategic direction to improve communication and enhance effective global crisis response mechanisms. Across the globe, agencies responsible for medicine regulation have established collaborative efforts through ICMRA to accelerate and strengthen processes for developing and approving COVID-19 therapeutics, including preventive vaccines and treatment options; and since October 1st, 2019, leadership of ICMRA has been provided by the EMA’s Executive Director [92]. The coalition acknowledges the World Health Organization’s (WHO) international leadership in managing global health emergencies. Together, ICMRA and WHO encourage pharmaceutical manufacturers to ensure comprehensive access to clinical trial data for novel therapeutics and immunizations - regardless of whether these products receive full authorization, or limited approval, emergency authorization, or are ultimately denied market access [93].
5.2.1. Public Health Emergency of International Concern (PHEIC)
A Public Health Emergency of International Concern (PHEIC) is defined as an extraordinary event that constitutes a public health risk to other states through the international spread of disease and potentially requires a coordinated international response. This phenomenon implies a situation that is serious, sudden, unusual or unexpected; that carries implications for public health beyond the affected State’s national borders; and which may require immediate international action [94].
5.2.2. Examples of Major Scenarios of Global Health Emergencies
- Case Type 1: Quality or safety issues with medicines on the market that affect public health across multiple countries [31]. For example, investigating widespread problems with medication that could be causing harm.
- Case Type 2: Shortage of approved medicines during crises, where previously available products become scarce or unavailable due to supply problems [31]. A clear example is a viral outbreak where antiviral treatments or vaccines are in short supply.
- Case Type 3: Urgent need for new treatments or vaccines during emerging health threats such as the COVID-19 pandemic. These situations typically arise when the WHO declares a Public Health Emergency, prompting national health authorities and WHO to act using existing international health guidelines [31].
To improve coordination between ICMRA members and the WHO, the coalition encourages the WHO to contact ICMRA leadership when regulatory action is needed during global health emergencies [31].
5.3. Case Studies in Regulatory Innovation
COVID-19 Vaccine Development
The recent COVID-19 pandemic was an event that challenged the regulatory authorities worldwide to achieve unprecedented levels of innovation. As the world grappled with the reality of increased deaths from the novel infectious disease, there was a pressing need for treatment options. Table 3 summarizes regulatory processes through which selected agencies approve COVID-19 vaccines in accordance with the WHO Emergency Use Listing (EUL) Procedure for vaccines. In 2021, the FDA approved the first COVID-19 vaccine after adjudging that it met its rigorous scientific standards for emergency use authorization [95,96]. The EMA did the same [97,98]. The successful emergence of the COVID-19 vaccine in record time was based on the utilization of highly adaptable vaccine platforms such as messenger RNA (mRNA) and the adaptation of structural biology tools to design agents (immunogens) that powerfully stimulate the immune system [99]. This decision follows the principle of beneficence, which minimizes risk and maximizes benefits for the sake of promoting health and wellbeing [100].
6. Conclusions
Looking ahead, regulatory authorities worldwide should, more than ever, adopt systems thinking, balancing the need for innovations in disease management with the responsibility of ensuring safety, efficacy, and ethical integrity, while also considering the unintended consequences of novel therapies. These agencies must continue to proactively update and revise regulatory guidelines to meet the demands of an ever-evolving healthcare landscape, including scientific advances and emerging health threats. The distinct regulatory approaches to advanced therapies, as seen across jurisdictions, underscore the urgency of strengthening international dialogue and convergence. As the global population grows and pharmaceutical innovation accelerates, the need for stronger international alliances and harmonization of regulatory authorities becomes even more critical. Such collaboration will be instrumental in supporting effective risk management, promoting equitable access, and enhancing global health security. In this rapidly changing environment, regulatory frameworks must remain agile and forward-looking, capable of addressing new therapeutic models while still fulfilling their core role of protecting public health. International cooperation and harmonization will not just facilitate innovation but will increasingly define the responsiveness of health systems worldwide.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1, Table S1: Comparing Regulatory Framework and Harmonization Efforts Across Key Regulatory Authorities.
Author Contributions
Conceptualization, T.O.A.; methodology, T.O.A., and O.T.U.; validation, T.O.A.; formal analysis, T.O.A., O.T.U., and A.S.A.; investigation, T.O.A., O.T.U., A.S.A., and O.A.; resources, T.O.A.; data curation, T.O.A.; writing—original draft preparation, T.O.A., O.T.U., A.S.A., and O.A.; writing—review and editing, O.T.U., A.S.A., O.A., B.S.B, H.S.D, H.L., and T.O.A.; supervision, T.O.A.; project administration, T.O.A.; funding acquisition, T.O.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADR | Adverse Drug Reaction |
| AMA | African Medicines Agency |
| BLA | Biologics License Application |
| CHMP | Committee for Medicinal Products for Human Use |
| CIRS | Center for Innovation in Regulatory Science |
| EUL | Emergency Use Listing |
| IND | Investigational New Drug |
| HCT/Ps | Cellular and Tissue-Based Products |
| CFR | Code of Federal Regulations |
| IND | Investigational New Drug |
| DCGI | Drugs Controller General of India |
| GMP | Good Manufacturing Practice |
| NDA | New Drug Application |
| PV | Pharmacovigilance |
| RA | Regulatory Affairs |
| REMS | Risk Evaluation and Mitigation Strategy |
| RMP | Risk Management Plan |
| RMAT | Regenerative Medicine Advanced Therapy |
| RWE | Real-World Evidence |
| WHO | World Health Organization |
References
- Rupnawar, M., and P. Wani. 2024. “Pharmaceutical Regulatory Affairs: An Overview of Global Regulatory Frameworks and Emerging Trends.” International Journal of Scientific Research and Technology 2 (12): 1–1. [CrossRef]
- Hussaarts, L., S. Mühlebach, V. P. Shah, et al. 2017. “Equivalence of Complex Drug Products: Advances in and Challenges for Current Regulatory Frameworks.” Annals of the New York Academy of Sciences 1407 (1): 39–49. [CrossRef]
- Raut, N., K. Bajaj, P. Matte, et al. 2023. “Building Bridges: Harmonization Efforts for Enhanced Collaboration between Developed and Developing Countries.” International Journal of Drug Regulatory Affairs 11 (3): 68–79. [CrossRef]
- Darrow, J. J., J. Avorn, and A. S. Kesselheim. 2020. “FDA Approval and Regulation of Pharmaceuticals, 1983–2018.” JAMA 323 (2): 164–176. [CrossRef]
- Tobin, J. J., and G. Walsh. 2023. Medical Product Regulatory Affairs: Pharmaceuticals, Diagnostics, Medical Devices. Hoboken, NJ: John Wiley & Sons.
- Gherghescu, I., and M. B. Delgado-Charro. 2021. “The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA.” Pharmaceutics 13 (1): 48. [CrossRef]
- Muselík, J., A. Komersová, K. Kubová, K. Matzick, and B. Skalická. 2021. “A Critical Overview of FDA and EMA Statistical Methods to Compare In Vitro Drug Dissolution Profiles of Pharmaceutical Products.” Pharmaceutics 13 (10): 1703. [CrossRef]
- Chen, R., R. Manochakian, L. James, et al. 2020. “Emerging Therapeutic Agents for Advanced Non-Small Cell Lung Cancer.” Journal of Hematology & Oncology 13 (1): 58. [CrossRef]
- Ali, F., A. Ilyas, and S. Ahmadeen. 2024. “Regulations in Japan.” In Global Regulations of Medicinal, Pharmaceutical, and Food Products. Boca Raton, FL: CRC Press.
- Clinical Stage. n.d. “Regulatory Differences across Regions.” Accessed March 20, 2025. https://www.clinicalstage.net/regulatory-differences-across-regions.
- US Food and Drug Administration (FDA). 2019. “Step 4: FDA Drug Review.” April 18, 2019. Accessed January 17, 2025. https://www.fda.gov/patients/drug-development-process/step-4-fda-drug-review.
- Van Norman, G. A. 2016. “Drugs and Devices: Comparison of European and U.S. Approval Processes.” JACC: Basic to Translational Science 1 (5): 399–412. [CrossRef]
- Hussain, F. 2013. “Kefauver-Harris Drug Amendments 1962.” Pharma Mirror Magazine, December 7. Accessed January 20, 2025. https://www.pharmamirror.com/knowledge-base/pharmaceutical-dictionary/kefauver-harris-drug-amendments-1962/.
- US Food and Drug Administration (FDA). 2018. Federal Food, Drug, and Cosmetic Act (FD&C Act). November 3, 2018. Accessed January 20, 2025. https://www.fda.gov/regulatory-information/laws-enforced-fda/federal-food-drug-and-cosmetic-act-fdc-act.
- US Food and Drug Administration (FDA). 2024a. “What We Do.” August 9, 2024. Accessed January 20, 2025. https://www.fda.gov/about-fda/what-we-do.
- Wang, W., and A. I. Wertheimer. 2022. “History, Status, and Politicization of the FDA.” Research in Social and Administrative Pharmacy 18 (5): 2811–2816. [CrossRef]
- Finnish Medicines Agency (Fimea). n.d.-a. “Pharmaceutical safety and information.” Accessed January 12, 2026. https://fimea.fi/en/pharmaceutical_safety_and_information.
- European Medicines Agency (EMA). 2015. “Adaptive Pathways.” November 20, 2015. Accessed January 21, 2025. https://www.ema.europa.eu/en/human-regulatory-overview/research-development/adaptive-pathways.
- European Medicines Agency (EMA). n.d.-b. “Marketing authorisation.” Accessed January 12, 2026. https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation.
- European Medicines Agency (EMA). n.d.-c. “National competent authorities (human).” Accessed January 12, 2026. https://www.ema.europa.eu/en/partners-networks/eu-partners/eu-member-states/national-competent-authorities-human.
- European Medicines Agency (EMA). 2019b. “Authorization of Medicines.” March 4, 2019. Accessed January 17, 2025. https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines.
- European Medicines Agency (EMA). n.d.-d. “Pharmacovigilance: Overview.” Accessed January 12, 2026. https://www.ema.europa.eu/en/human-regulatory-overview/pharmacovigilance-overview.
- Finnish Medicines Agency (Fimea). n.d.-b. “Pharmacovigilance.” Accessed January 12, 2026. https://fimea.fi/en/supervision/pharmacovigilance.
- Kanta Services. 2025a. “Prescriptions and medicines.” February 6, 2025. Accessed January 12, 2026. https://www.kanta.fi/en/prescriptions-and-medicines.
- Kanta Services. 2025b. “Prescription.” February 18, 2025. Accessed January 12, 2026. https://www.kanta.fi/en/professionals/prescription.
- Swedish Medical Products Agency (Läkemedelsverket). n.d. “About the Swedish Medical Products Agency.” Accessed January 12, 2026. https://www.lakemedelsverket.se/en/about-the-swedish-mpa.
- Socialstyrelsen (National Board of Health and Welfare). 2020. “National Prescribed Drug Register.” August 21, 2020. Accessed January 12, 2026. https://www.socialstyrelsen.se/en/statistics-and-data/registers/national-prescribed-drug-register/.
- Pharmaceuticals and Medical Devices Agency (PMDA). n.d.-a. “Outline of PMDA.” Accessed January 21, 2025. https://www.pmda.go.jp/english/about-pmda/outline/0005.html.
- Pharmaceuticals and Medical Devices Agency (PMDA). n.d.-b. “History.” Accessed January 21, 2025. https://www.pmda.go.jp/english/about-pmda/outline/0002.html.
- Wileman, H., and A. Mishra. 2010. “Drug Lag and Key Regulatory Barriers in the Emerging Markets.” Perspectives in Clinical Research 1 (2): 51–56. Accessed March 20, 2025. https://journals.lww.com/picp/fulltext/2010/01020/Drug_Lag_and_Key_Regulatory_Barriers_in_the.2.aspx.
- International Coalition of Medicines Regulatory Authorities (ICMRA). n.d. “Framework for the Involvement of Health Regulatory Authorities in the Management of Global Health Crises.” Accessed January 17, 2025. https://www.icmra.info/drupal/sites/default/files/2019-04/ICMRA%20SOP%20for%20Crisis%20Management.pdf.
- Ferreira, C. G. 2019. “ES10.05 Brazilian Health Regulatory Agency – Agência Nacional de Vigilância Sanitária.” Journal of Thoracic Oncology 14 (10): S39. [CrossRef]
- Huynh-Ba, K., and A. Beumer Sassi. 2018. “ANVISA: An Introduction to a New Regulatory Agency with Many Challenges.” AAPS Open 4 (1): 9. [CrossRef]
- Dhole, S. D., and R. B. Darade. 2024. “Indian Pharmaceutical Regulatory Authority: Review.” International Journal in Pharmaceutical Sciences 2 (5): 1109–1119. [CrossRef]
- Central Drugs Standard Control Organization (CDSCO). 2025. “Introduction.” Accessed January 21, 2025. https://cdsco.gov.in/opencms/opencms/hi/About-us/Introduction/.
- Federal Service for Surveillance in Healthcare. n.d. “Regulatory Framework for Federal Service for Surveillance in Healthcare.” Accessed January 21, 2025. https://roszdravnadzor.gov.ru/en.
- Vargas-Pelaez, C. M., M. T. Drago, A. Acosta, and M. R. Farias. 2017. “Pharmaceutical Policy in Argentina.” In Pharmaceutical Policy in Countries with Developing Healthcare Systems, 97–121. [CrossRef]
- Argentina.gob.ar. 2025. “What Is ANMAT?” Accessed March 20, 2025. https://www.argentina.gob.ar/anmat/anmat-en/what-anmat.
- Skerritt, J. 2022. “Australia’s Robust Strategy for Regional and Global Medical Product Engagement.” U.S. Food and Drug Administration. September 20, 2022. Accessed March 20, 2025. https://www.fda.gov/international-programs/australias-robust-strategy-regional-and-global-medical-product-engagement.
- FHI Clinical. n.d. “Increasing Regulatory Harmonization in Africa Is Streamlining Marketing Authorization of Your Products.” Accessed March 20, 2025. https://fhiclinical.com/increasing-regulatory-harmonization-in-africa/.
- World Health Organization (WHO). 2024. “Senegal and Rwanda Achieve WHO Maturity Level 3 in Medicines Regulation.” December 5, 2024. Accessed March 19, 2025. https://www.who.int/news/item/05-12-2024-senegal-and-rwanda-achieve-who-maturity-level-3-in-medicines-regulation.
- Ozili, P. K. 2021. “Covid-19 Pandemic and Economic Crisis: The Nigerian Experience and Structural Causes.” Journal of Economic and Administrative Sciences 37 (4): 401–418. [CrossRef]
- Olatunji, O. E. 2014. “Leadership Effectiveness and Regulatory Performance in the Public Sector: The Experience of the National Agency for Food and Drug Administration and Control (NAFDAC).” International Journal of Innovative Research & Development 3 (3): 217–225.
- Omijeh, B. O., and O. Raheem. 2021. “An Improved Drug Verification System for NAFDAC.” Accessed March 19, 2025. https://www.africanscholarpublications.com/wp-content/uploads/2021/06/AJASD_Vol20_No2_March_2021-2.pdf.
- Spink, J., D. C. Moyer, and M. R. Rip. 2016. “Addressing the Risk of Product Fraud: A Case Study of the Nigerian Combating Counterfeiting and Sub-standard Medicines Initiatives.” Journal of Forensic Science & Criminology 4 (2): 201.
- National Agency for Food and Drug Administration and Control (NAFDAC). n.d. “About NAFDAC.” Accessed March 20, 2025. https://nafdac.gov.ng/about-nafdac/.
- International Council for Harmonisation (ICH). n.d.-a “Welcome to the ICH Official Website.” Accessed January 21, 2025. https://www.ich.org/.
- Adleberg, J. 2024. “ICH Overview.” U.S. Food & Drug Administration. FDA/HC ICH Regional Public Meeting, February 22, 2024. Accessed January 21, 2025. https://www.fda.gov/media/177713/download.
- Vbnm,International Council for Harmonisation (ICH). n.d.-b. “Management Committee.” Accessed January 21, 2025. https://www.ich.org/page/management-committee.
- Reggi, V. 2017. “Medicines Regulatory Harmonization: International Collaboration as a Key to Improve Public Health.” Medicine Access @ Point of Care 1: maapoc.0000001. [CrossRef]
- Eurofins Scientific. n.d. “Three Key Regulations in the Pharmaceutical Industry: EMA, FDA and ANVISA.” Accessed March 21, 2025. https://www.eurofins.com/assurance/resources/articles/three-key-regulations-in-the-pharmaceutical-industry-ema-fda-and-anvisa/.
- Center for Innovation in Regulatory Science (CIRS). 2021. New Drug Approvals in Six Major Authorities 2011–2020: Focus on Facilitated Regulatory Pathways and Worksharing. https://www.cirsci.org/wp-content/uploads/dlm_uploads/2021/06/CIRS-RD-Briefing-81-6-agencies-v5.pdf.
- Vashisth, S., G. Singh, and A. Nanda. 2012. “A Comparative Study of Regulatory Trends of Pharmaceuticals in Brazil, Russia, India and China (BRIC) Countries.” Journal of Generic Medicines 9 (3): 128–143. [CrossRef]
- Clini India. 2024. “Understanding US FDA vs. EMA vs. CDSCO: How Do They Differ?” Accessed March 21, 2024. https://www.cliniindia.com/understanding-us-fda-vs-ema-vs-cdsco-how-do-they-differ/.
- National Medical Products Administration (NMPA). 2025. “Measures Facilitate Approval of 48 First-in-Class Innovative Drugs.” March 21, 2025. Accessed May 6, 2025. https://english.nmpa.gov.cn/2025-03/21/c_1080286.htm.
- Agência Nacional de Vigilância Sanitária (Anvisa). n.d. “Drugs.” Accessed May 6, 2025. https://www.gov.br/anvisa/pt-br/english/regulation-of-products/drugs.
- Deepika, M., and J. Karthi. 2025. “A Comparative Study of USFDA, EMA, and CDSCO Regulatory Guidelines.” International Journal of Research Publication and Reviews 6 (3): 1429–1431. https://ijrpr.com/uploads/V6ISSUE3/IJRPR39725.pdf.
- “Exploring CDSCO’s Approval Process for New Drugs.” n.d. Issuu. Accessed May 6, 2025. https://issuu.com/corpseedgroup/docs/exploring_cdsco_s_approval_process_for_new_drugs.
- US Food and Drug Administration (FDA). 2025. “FDA Acceptance of Foreign Clinical Studies Not Conducted under an IND: Frequently Asked Questions.” February 3, 2025. Accessed May 6, 2025. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-acceptance-foreign-clinical-studies-not-conducted-under-ind-frequently-asked-questions.
- Naito, C. 1998. “Ethnic Factors in the Acceptability of Foreign Clinical Data.” Drug Information Journal 32 (1, suppl): 1283S–1292S. [CrossRef]
- National Medical Products Administration (NMPA). 2020. “Clinical Technical Requirements for Drugs Marketed Overseas but Not Marketed in China.” November 18, 2020. Accessed May 7, 2025. https://english.nmpa.gov.cn/2020-11/18/c_568155.htm.
- US Department of Health and Human Services. 2018. “Read the Belmont Report.” January 15, 2018. Accessed January 17, 2025. https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html.
- Abou-El-Enein, M., A. Elsanhoury, and P. Reinke. 2016. “Overcoming Challenges Facing Advanced Therapies in the EU Market.” Cell Stem Cell 19 (3): 293–297. [CrossRef]
- International Council for Harmonisation (ICH). 2016. Guideline for Good Clinical Practice E6(R2). Accessed January 16, 2025. https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf.
- Council for International Organizations of Medical Sciences, and World Health Organization, eds. 2002. International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva: CIOMS.
- Rosa, C., L. A. Marsch, E. L. Winstanley, M. Brunner, and A. N. C. Campbell. 2021. “Using Digital Technologies in Clinical Trials: Current and Future Applications.” Contemporary Clinical Trials 100: 106219. [CrossRef]
- US Food and Drug Administration (FDA). 2024b. “FDA Promotes Clinical Trial Innovation.” FDA Voices. August 9, 2024. Accessed January 17, 2025. https://www.fda.gov/news-events/fda-voices/fda-promotes-clinical-trial-innovation.
- Scheppler, L., N. De Clercq, M. McGoldrick, and J. Dias. 2021. “Regulatory Harmonization and Streamlining of Clinical Trial Applications Globally Should Lead to Faster Clinical Development and Earlier Access to Life-Saving Vaccines.” Vaccine 39 (5): 790–796. [CrossRef]
- Hwenda, L., M. Sidibe, and M. Makanga. 2022. “The African Medicines Agency: The Key to Unlocking Clinical Research in Africa.” Lancet Global Health 10 (8): e1088–e1089. [CrossRef]
- Jackson, C. 2020. “Africa’s Missing Genomic Data and Its Impact on Health Care.” GEN – Genetic Engineering and Biotechnology News, September 8, 2020. Accessed January 17, 2025. https://www.genengnews.com/topics/omics/africas-missing-genomic-data-and-its-impact-on-health-care/.
- Khan, M. A. A., S. Hamid, and Z. U. D. Babar. 2023. “Pharmacovigilance in High-Income Countries: Current Developments and a Review of Literature.” Pharmacy 11 (1): 10. [CrossRef]
- Cui, X., L. X. Wang, G. Y. Liu, and Y. M. Xie. 2021. “Enlightenment of International Pharmacovigilance System on Establishment of Pharmacovigilance System of Chinese Medicine.” China Journal of Chinese Materia Medica 46: 5450–5455. [CrossRef]
- World Health Organization (WHO). 2020. 72nd and 73rd Report: WHO Technical Report Series No. 1030. Accessed May 7, 2025. https://www.who.int/publications/i/item/9789240024373.
- Dieck, G. S., and R. G. Sharrar. 2013. “Preparing for Safety Issues Following Drug Approval: Pre-Approval Risk Management Considerations.” Therapeutic Advances in Drug Safety 4 (5): 220–228. [CrossRef]
- US Food and Drug Administration (FDA). 2023b. “Risk Evaluation and Mitigation Strategies (REMS).” May 16, 2023. Accessed January 17, 2025. https://www.fda.gov/drugs/drug-safety-and-availability/risk-evaluation-and-mitigation-strategies-rems.
- Allison, S. J. 2022. “Novel Anti-Cancer Agents and Cellular Targets and Their Mechanism(s) of Action.” Biomedicines 10 (8): 1767. [CrossRef]
- Rothschild, J. 2020. “Ethical Considerations of Gene Editing and Genetic Selection.” Journal of General and Family Medicine 21 (3): 37–47. [CrossRef]
- “China Grants Conditional Approval for First COVID Vaccine.” 2020. National Health Commission of the People’s Republic of China. December 31, 2020. Accessed May 7, 2025. https://en.nhc.gov.cn/2020-12/31/c_82609.htm.
- Hanna, E., C. Rémuzat, P. Auquier, and M. Toumi. 2016. “Advanced Therapy Medicinal Products: Current and Future Perspectives.” Journal of Market Access & Health Policy 4: 31036. [CrossRef]
- Cuende, N., J. E. J. Rasko, M. B. C. Koh, M. Dominici, and L. Ikonomou. 2018. “Cell, Tissue and Gene Products with Marketing Authorization in 2018 Worldwide.” Cytotherapy 20 (11): 1401–1413. [CrossRef]
- International Council for Harmonisation (ICH). n.d.-c. “Mission.” Accessed May 7, 2025. https://www.ich.org/page/mission.
- US Food and Drug Administration (FDA). n.d.-a. “Regulation of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps).” Accessed May 7, 2025. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products.
- US Food and Drug Administration (FDA). n.d.-b. “Regenerative Medicine Advanced Therapy (RMAT) Designation.” Accessed May 7, 2025. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/regenerative-medicine-advanced-therapy-designation.
- European Medicines Agency (EMA). 2024. “Advanced Therapy Medicinal Products: Overview.” Updated March 2024. Accessed May 7, 2025. https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview.
- Pharmaceuticals and Medical Devices Agency (PMDA). n.d.-d. “Review of Regenerative Medical Products.” Accessed May 7, 2025. https://www.pmda.go.jp/english/review-services/reviews/0003.html.
- National Health Surveillance Agency – Anvisa. 2020. “10154json-file-1.pdf.” Accessed May 7, 2025. https://www.gov.br/anvisa/pt-br/assuntos/reunioesdadicol/processos-deliberados/5de2020/arquivos/10154json-file-1/view.
- Brazilian Health Regulatory Agency (Anvisa). 2023. “Anvisa Approves Specific Good Manufacturing Practice Regulation for Advanced Therapy Products.” 2023. Accessed May 7, 2025. https://www.gov.br/anvisa/pt-br/assuntos/noticias-anvisa/2023/anvisa-aprova-norma-especifica-de-boas-praticas-de-fabricacao-para-produtos-de-terapias-avancadas.
- Roszdravnadzor. n.d. “Federal Law No. 180-FZ on Biomedical Cell Products.” Accessed May 7, 2025. https://www.consultant.ru/document/cons_doc_LAW_200349/.
- Jotwani, G. 2017. National Guidelines for Stem Cell Research. Indian Council of Medical Research and Department of Biotechnology. https://dbtindia.gov.in/sites/default/files/National_Guidelines_StemCellResearch-2017.pdf.
- Yin, C., J. Gao, G. Li, et al. 2022. “Gene and Cell Therapies in China: Booming Landscape under Dual-Track Regulation.” Journal of Hematology & Oncology 15 (1): 139. [CrossRef]
- Li, Y., F. Verter, B. Wang, and N. Gu. 2019. “Regulations on Cell Therapy Products in China: A Brief History and Current Status.” Regenerative Medicine 14 (8): 791–803. [CrossRef]
- International Coalition of Medicines Regulatory Authorities (ICMRA). 2016. “International Coalition of Medicines Regulatory Authorities (ICMRA).” European Medicines Agency. October 13, 2016. Accessed January 17, 2025. https://www.ema.europa.eu/en/partners-networks/international-activities/multilateral-coalitions-initiatives/international-coalition-medicines-regulatory-authorities-icmra.
- World Health Organization (WHO). 2021. “Joint Statement on Transparency and Data Integrity: International Coalition of Medicines Regulatory Authorities (ICMRA) and WHO.” May 7, 2021. Accessed January 17, 2025. https://www.who.int/news/item/07-05-2021-joint-statement-on-transparency-and-data-integrityinternational-coalition-of-medicines-regulatory-authorities-(icmra)-and-who.
- World Health Organization (WHO). 2019. “Emergencies: International Health Regulations and Emergency Committees.” December 19, 2019. Accessed January 22, 2025. https://www.who.int/news-room/questions-and-answers/item/emergencies-international-health-regulations-and-emergency-committees.
- Tanne, J. H. 2021. “Covid-19: FDA Approves Pfizer-BioNTech Vaccine in Record Time.” BMJ 374: n2096. [CrossRef]
- US Food and Drug Administration (FDA). 2020. “FDA Takes Key Action in Fight Against COVID-19 by Issuing Emergency Use Authorization for First COVID-19 Vaccine.” December 14, 2020. Accessed January 17, 2025. https://www.fda.gov/news-events/press-announcements/fda-takes-key-action-fight-against-covid-19-issuing-emergency-use-authorization-first-covid-19.
- European Medicines Agency (EMA). 2020. “EMA Recommends First COVID-19 Vaccine for Authorisation in the EU.” December 21, 2020. Accessed January 17, 2025. https://www.ema.europa.eu/en/news/ema-recommends-first-covid-19-vaccine-authorisation-eu.
- European Centre for Disease Prevention and Control (ECDC). 2020. “First COVID-19 Vaccine Authorised for Use in the European Union.” December 21, 2020. Accessed January 17, 2025. https://www.ecdc.europa.eu/en/news-events/first-covid-19-vaccine-authorised-use-european-union.
- Fauci, A. S. 2021. “The Story Behind COVID-19 Vaccines.” Science 372 (6538): 109. [CrossRef]
- Brear, M. R., and R. Gordon. 2021. “Translating the Principle of Beneficence into Ethical Participatory Development Research Practice.” Journal of International Development 33 (1): 109–126. [CrossRef]
- European Medicines Agency (EMA). 2016. “Conditional Marketing Authorisation.” February 15, 2016. Accessed May 7, 2025. https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/conditional-marketing-authorisation.
- Pharmaceuticals and Medical Devices Agency (PMDA). 2021. “Special Approval for Emergency on First COVID-19 Vaccine in Japan.” February 16, 2021. Accessed May 7, 2025. https://www.pmda.go.jp/english/about-pmda/0003.pdf.
- Mattos Filho. 2020. “Anvisa Issues Emergency Use Authorization Rules to Accelerate Access to Covid-19 Vaccines.” December 8, 2020. Accessed May 7, 2025. https://www.mattosfilho.com.br/en/unico/anvisa-autorizacao-uso-emergencial-vacinas/.
- Mikule, E., T. Reissaar, J. Villers, A. S. Takoupo Penka, A. Temerev, and L. Rozanova. 2021. “The Fast Approval and Slow Rollout of Sputnik V: Why Is Russia’s Vaccine Rollout Slower than That of Other Nations?” Epidemiologia 2 (3): 360–376. [CrossRef]
- Ministry of Health and Family Welfare, Government of India. n.d. “General FAQ.” Accessed May 7, 2025. https://dshm.delhi.gov.in/(S(anjsnxdbajexzs2iqruzkarz))/COWIN/English/FAQHWFrontline.pdf.

Table 1.
Comparing Regulatory Framework and Harmonization Efforts Across Key Regulatory Authorities.
Table 1.
Comparing Regulatory Framework and Harmonization Efforts Across Key Regulatory Authorities.
|
Feature Category |
Feature | FDA (USA)a | EU regulatory network (EMA + NCAs)a | PMDA (Japan)b | NMPA (China)c | ANVISA (Brazil)d | CDSCO (India)e | Roszdravnadzor (Russia)c |
| General Framework | Public Health Mission | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Legal Basis | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Organizational Model | Team | Rapporteur | Team | Provincial | Centralized | Centralized | Federal | |
| Pre-Market Requirements | Review Timeline (months) | 6–10f | 7–11g | 12h | ~10i | ~12j | 12–24k | 18–26 |
| Expedited Pathways | Multiple | PRIME | Sakigake | Yes | Yes | Limited | Limited | |
| Clinical Data: Local Required | Nol | Partialm | Yes | Yesn | Partial | Yes | Yes | |
| Electronic Submissions (eCTD) | Yes | Yes | Yes | In progress | In progress | Partial | Paper/electronic hybrid | |
| Post-Market Oversight | Safety Monitoring | REMS | RMP | Re-exam | Yes | Yes | Limited | Basic |
| Transparency | High | Moderate | Moderate | Low | Moderate | Low | Low | |
| Inspection Capacity | High | High | High | Medium | Medium | Limited | Periodic | |
| Global Alignment | ICH Participation | Full | Full | Full | Member | Observer | Limited | None |
| Accept RWE (Real Word Evidence) | Growing | Developing | Developing | Limited | Limited | Minimal | Minimal |
✓ = Feature Present "Partial" = Conditional or limited requirement "High/Moderate/Low" = Degree of implementation/maturitya Sources: [10,51].b Source: [52].c Source: [53].d Sources: [51,53].e Sources: [53,54].f Source: [11].g Source: [21].h Source: [28].i Source: [55].j Source: [56].k Sources: [57,58].l Source: [59].m Source: [60].n Source: [61]."EU" reflects the EU medicines regulatory network coordinated by EMA and implemented with national competent authorities (e.g., Finland’s Fimea), which perform substantial regulatory, inspection, and pharmacovigilance activities at Member State level.
Table 2.
Comparative Overview of ATMP Regulatory Frameworks Across Major Jurisdictions.
| Agency/Region | Terminology | Legal Framework | Approval Pathway | Special Features | Post-Marketing Oversight |
| FDA (USA)a | HCT/Ps, Cell/Gene Therapies | 21 CFR Part 1271 | IND + Biologics License Application (BLA) | RMAT, Breakthrough Therapy, Fast Track options | REMS and post-marketing surveillance |
| EU (EMA + Member States/NCAs)b | Advanced Therapy Medicinal Products (ATMPs) | Regulation (EC) No. 1394/2007 | Centralized authorization via EMA | Hospital exemption (implemented at Member State level via NCAs, e.g., Fimea); optional classification procedure | Risk Management Plans (RMPs) required |
| PMDA (Japan)c | Regenerative Medical Products | PMD Act (2014), Act on the Safety of Regenerative Medicine | Conditional/time-limited approval | Accelerated approval with post-market conditions | Mandatory follow-up studies |
|
ANVISA (Brazil)d |
Produtos de Terapias Avançadas | RDC 338/2020 | Clinical trials + GMP + regulatory submission | Special pathways for rare disease therapies | Traceability and pharmacovigilance |
| Roszdravnadzor (Russia)e | Biomedical Cell Products (BCPs) | Federal Law No. 180-FZ (2016), Law No. 61-FZ | State registration post-expert review | Centralized registry and annual reporting | Biosecurity, ethics board, and monitoring |
| CDSCO (India)f | Stem Cell and Cell-Based Products (SCCPs) | Guidelines for Stem Cell Research (2017) | Ethics + regulatory clearance from DCGI | Oversight by Institutional Ethics Committees | Ethical and informed consent focus |
| NMPA (China)g | Cell and Gene Therapy Products | Technical Guidelines (2021), ICH-aligned | IND-equivalent process + GMP | Fast-track review pathways | Enhanced post-marketing safety guidance |
Table 3.
Regulatory Processes of COVID-19 Vaccine Emergency Approval Across Selected Agencies.
| Agency/Region | Expedited Pathway | Clinical Data | Post Authorization Monitoring |
| FDA (USA)a | Issues Emergency Use Authorization | Phase III data from clinical trial | RWE and active safety monitoring required |
| EU (EMA + European Commission)b | Issues Conditional Marketing Authorization e.g. Comirnaty Approval | Phase III data except rare diseases. | Continued data submission required to maintain status |
| PMDA (Japan)c | Issues a Special Approval for Emergency e.g. Pfizer-BioNTech vaccine | Rely on Japan or foreign Clinical trial Phase III data | Post approval monitoring activities required. |
|
ANVISA (Brazil)d |
Issues Emergency Use Authorization | Require Phase III data | Follow-up data on vaccine quality, safety and efficacy required. |
| Roszdravnadzor (Russia)e | Issues Emergence Approval e.g. Sputnik V | Phase III Clinical data not required. | Post approval monitoring of data is required. |
| CDSCO (India)f | Issues Emergency Use Authorization e.g. Covishield |
Phase II Clinical data required. Bridging trials may be required. | Post Approval follow-up is required. |
| NMPA (China)g | Issues a conditional market approval e.g. Sinovac's CoronaVac |
Phase III data required. | Required to continue relevant research, fulfill conditional requirements and submit the results of subsequent research in a timely manner. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



