
Submitted:
11 February 2026
Posted:
13 February 2026
You are already at the latest version
Abstract
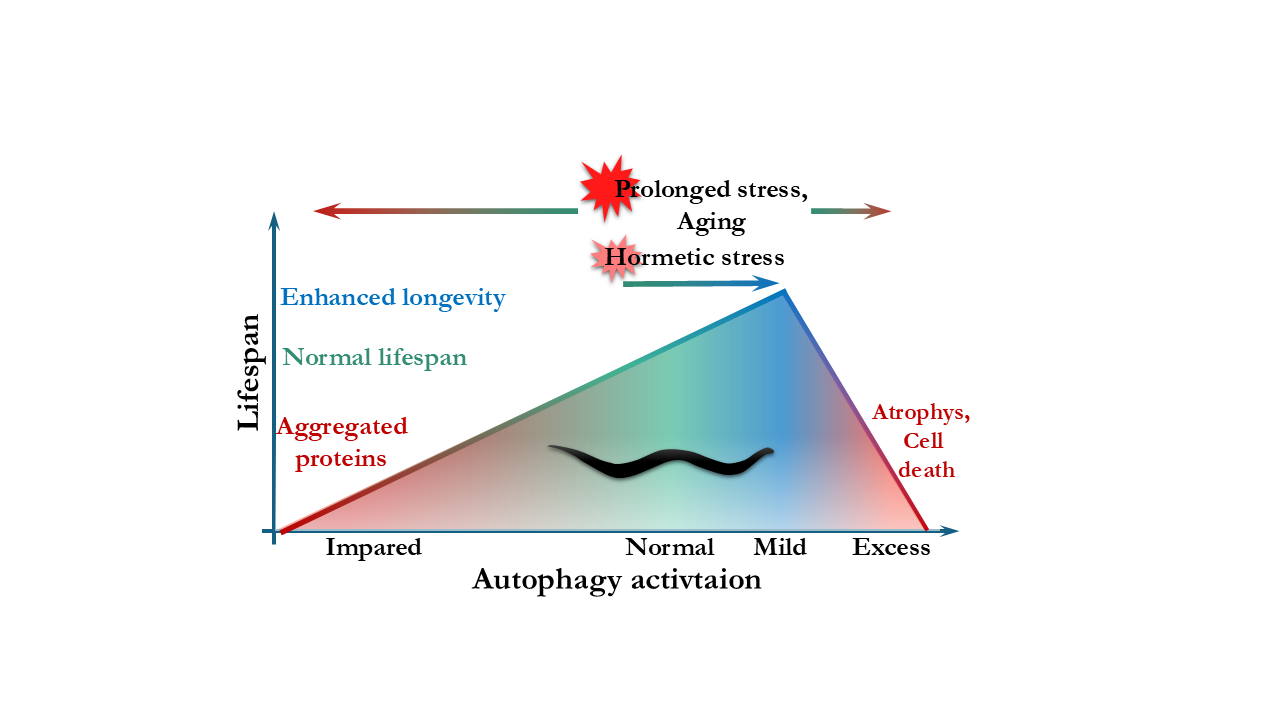
Autophagy is a tightly regulated catabolic process essential for cellular homeostasis, stress adaptation, and regeneration. In the nematode Caenorhabditis elegans, with its short lifespan, transparent body, and well-defined genetics, the process can be investigated at tissue- and age-specific manner, making it an excellent model to study the connection between autophagy and longevity. While autophagy is indispensable for development and homeostasis, recent studies have revealed that its role in aging is more complex than previously thought. During post-reproductive life, autophagic flux and the degradative capacity of lysosomes decline, resulting in the accumulation of undegraded material and cellular stress. Several studies have demonstrated that the experimental modulation of core autophagy in aged or post-reproductive C. elegans, particularly in neurons, can improve proteostasis, preserve tissue integrity, and extend lifespan. Here we review the current results obtained using the genetic model system Caenorhabditis elegans that link autophagy to lifespan regulation. We focus on studies that investigate unexpected, context-dependent, or deleterious effects of inhibiting autophagy-related genes during aging. We also discuss how age- and tissue-specific modulation of autophagy could define the most effective strategies for promoting healthy aging. This could provide relevant insights for the therapeutic targeting of autophagy in humans.
Keywords:
1. Introduction
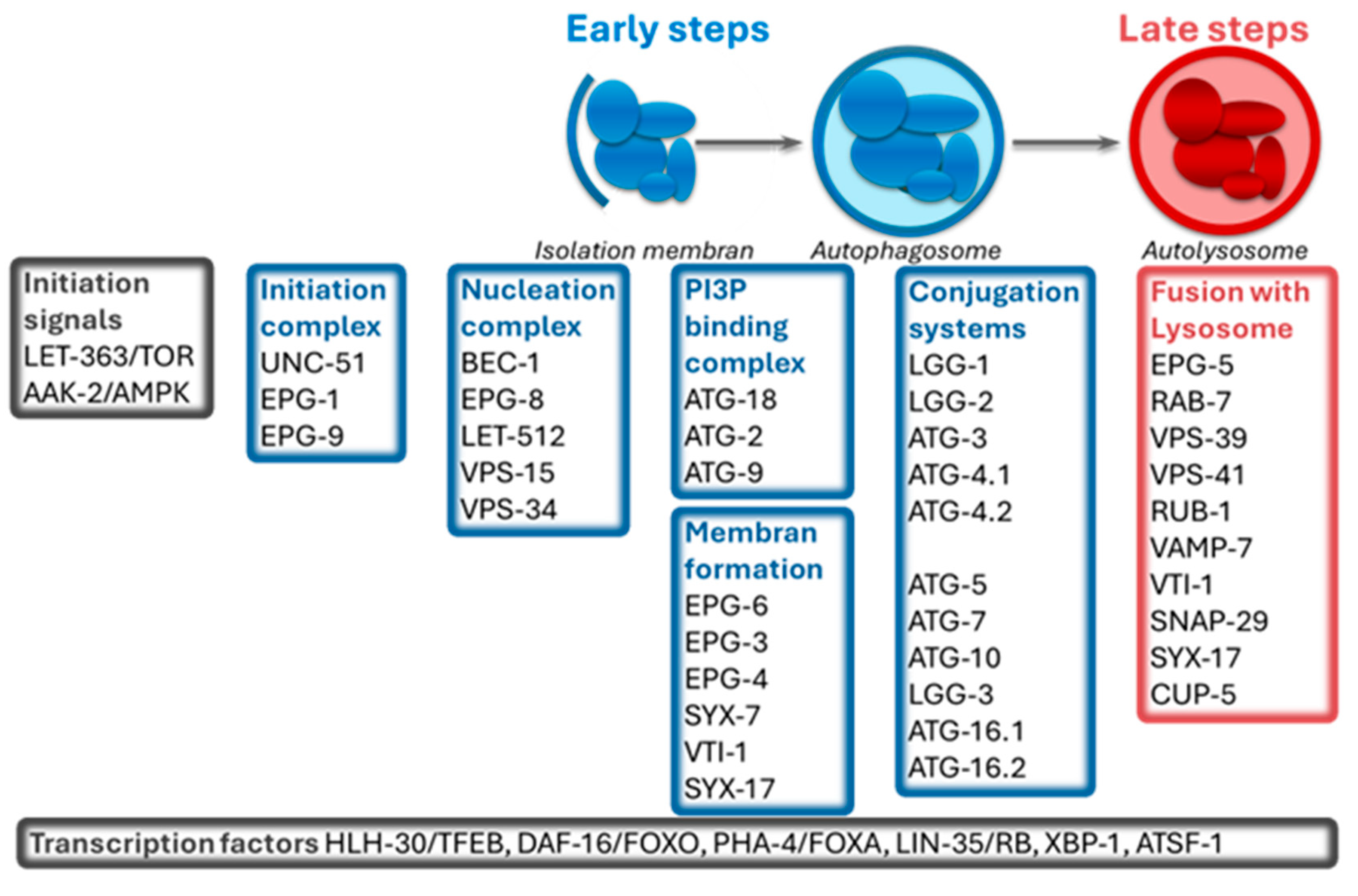
2. C. elegans as a Model Organism to Study Autophagy
3. Autophagy as a Conserved Driver of Lifespan Extension in C. elegans
4. Dysregulation of Autophagy Can Lead to Disadvantageous Effects in C. elegans
5. Antagonistic Pleiotropy and Age-Dependent Reversal of Autophagy Function
6. Spatiotemporal and Compensatory Regulation of Autophagy During Aging
7. Relevance to Human Disease
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AID | Auxin-inducible degradation |
| atg | Autophagy-related genes |
| C. elegans | Caenorhabditis elegans |
| DR | Dietary restriction |
| FOXA | Forkhead box A |
| HLH-30 | Helix-loop-helix protein 30 |
| IGF-1 | Insulin growth factor 1 |
| PHA-4 | Pharynx Defective protein 4 |
| PLPs | Pseudocoelomic lipoprotein pools |
| RNAi | RNA interference |
| SID | Systemic RNA interference defective |
| TFEB | Transcription factor EB |
| TOR | Target of Rapamycin |
| TLs | Tubular lysosomes |
References
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: renovation of cells and tissues. Cell. 2011, 147(4), 728–41. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Hollenstein, D.M.; Kraft, C. Autophagosomes are formed at a distinct cellular structure. Curr. Opin. Cell Biol. 2020, 65, 50–57. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; et al. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Palmisano, N.J.; Meléndez, A. Autophagy in C. elegans development. Dev. Biol. 2019, 447, 103–125. [Google Scholar] [CrossRef]
- Tian, Y.; et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef]
- Tóth, M.L.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Hars, E.S.; et al. Autophagy regulates ageing in C. elegans. Autophagy 2007, 3, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; et al. Neuronal inhibition of autophagy extends lifespan. Genes Dev. 2017, 31, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Ezcurra, M.; et al. Intestinal autophagy in aging. Curr. Biol. 2018, 28, 2544–2556.e5. [Google Scholar] [CrossRef]
- Alberti, A.; Michelet, X.; Djeddi, A.; Legouis, R. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 2010, 6, 622–633. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. In vivo mitophagy monitoring in Caenorhabditis elegans to determine mitochondrial homeostasis. Bio Protoc. 2017, 7, e2215. [Google Scholar] [CrossRef]
- Kumsta, C.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans. Nat. Commun. 2019, 10, 5648. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR signaling in Caenorhabditis elegans development, metabolism, and aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef]
- Vellai, T.; et al. Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef]
- Kenyon, C.; et al. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; et al. Translational regulation of non-autonomous mitochondrial stress response promotes longevity. Cell Rep. 2019, 28, 1050–1062.e6. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.H.; et al. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 2007, 447, 550–555. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Ruvkun, G. MXL-3 and HLH-30 link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 2013, 15, 668–676. [Google Scholar] [CrossRef]
- Visvikis, O.; et al. Innate host defense requires TFEB-mediated transcription. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef]
- Hansen, M.; et al. Autophagy in lifespan extension by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef]
- Guo, B.; et al. Genome-wide screen identifies regulators of autophagy in C. elegans. EMBO Rep. 2014, 15, 705–713. [Google Scholar] [CrossRef]
- Bördén, K.; Vellai, T.; Sigmond, T. Developing endogenous autophagy reporters in C. elegans. Int. J. Mol. Sci. 2025, 26, 10178. [Google Scholar] [CrossRef]
- Conte, D.; et al. RNA interference in C. elegans. Curr. Protoc. Mol. Biol. 2015, 109, 26.3.1–26.3.30. [Google Scholar] [CrossRef]
- Maher, K.N.; et al. Large-scale gene knockdown in C. elegans. J. Vis. Exp. 2013, 79, e50693. [Google Scholar] [CrossRef]
- Calixto, A.; et al. Enhanced neuronal RNAi in C. elegans. Nat. Methods 2010, 7, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Whangbo, J.S.; et al. SID-1 domains important for dsRNA import. G3 2017, 7, 3887–3899. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.; et al. Engineering the auxin-inducible degron system. Nat. Commun. 2025, 16, 10848. [Google Scholar] [CrossRef] [PubMed]
- Son, H.G.; et al. Age-dependent biomarkers in C. elegans. Aging Cell 2019, 18, e12853. [Google Scholar] [CrossRef]
- Meléndez, A.; et al. Autophagy genes are essential for dauer development. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef]
- Long, X.; et al. TOR deficiency causes developmental arrest. Curr. Biol. 2002, 12, 1448–1461. [Google Scholar] [CrossRef]
- Gelino, S.; et al. Intestinal autophagy improves healthspan. PLoS Genet. 2016, 12, e1006135. [Google Scholar] [CrossRef]
- Hsieh, P.N.; et al. Conserved KLF-autophagy pathway. Nat. Commun. 2017, 8, 914. [Google Scholar] [CrossRef]
- Vérièpe-Salerno, J.; et al. MALT-1 shortens lifespan. Autophagy Rep. 2023, 2, 2277584. [Google Scholar] [CrossRef]
- Villalobos, T.V.; et al. Tubular lysosome induction. Nat. Aging 2023, 3, 1091–1106. [Google Scholar] [CrossRef]
- Sigmond, T.; Vellai, T. Lysosomal alteration links food limitation to longevity. Nat. Aging 2023, 3, 1048–1050. [Google Scholar] [CrossRef]
- Dolese, D.A.; et al. Tubular lysosomes link pexophagy and aging. Autophagy 2021, 1–12. [Google Scholar] [CrossRef]
- Kumsta, C.; et al. Hormetic heat stress induces autophagy. Nat. Commun. 2017, 8, 14337. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, LR; De Magalhaes Filho, CD; McQuary, PR; Chu, CC; Visvikis, O; Chang, JT; Gelino, S; Ong, B; Davis, AE; Irazoqui, JE; Dillin, A; Hansen, M. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; et al. Amino acid-mediated lifespan extension. BMC Genet. 2015, 16, 8. [Google Scholar] [CrossRef]
- Yang, J.; et al. miR-34 regulates lifespan via atg9. Age 2013, 35, 11–22. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy and longevity. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Madeo, F.; et al. Essential role for autophagy in lifespan extension. J. Clin. Invest. 2015, 125, 85–93. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; et al. Genetics of longevity. Adv. Genet. 2015, 90, 1–101. [Google Scholar] [CrossRef]
- Gelino, S.; Hansen, M. Autophagy and aging. J. Clin. Exp. Pathol. 2012, Suppl. 4. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Kroemer, G. Autophagy in stress and disease. Cell Death Differ. 2015, 22, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in aging and disease. Cell Death Differ. 2009, 16, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.L.; et al. Autophagy genes and neurodegeneration. J. Cell Sci. 2007, 120, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; You, Y.; Avery, L. Dual roles of autophagy in starvation. Genes Dev. 2007, 21, 2161–2171. [Google Scholar] [CrossRef]
- Zhou, B.; et al. Mitochondrial permeability and lifespan. Cell 2019, 177, 299–314.e16. [Google Scholar] [CrossRef]
- Sakai, N.; et al. TORC2 signaling in learning. PLoS ONE 2017, 12, e0177900. [Google Scholar] [CrossRef]
- Franco-Juárez, B.; et al. High glucose induces autophagy. Aging 2018, 10, 2657–2667. [Google Scholar] [CrossRef]
- Takacs, Z.; et al. ATG-18 and EPG-6 in lifespan control. Cells 2019, 8, 236. [Google Scholar] [CrossRef]
- Lu, Q.; et al. EPG-6 regulates autophagosome formation. Dev. Cell 2011, 21, 343–357. [Google Scholar] [CrossRef]
- Polson, H.E.J.; et al. WIPI2 regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Hashimoto, Y.; et al. Suppression of autophagy extends lifespan. Genes Cells 2009, 14, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.; Wilhelm, T.; Richly, H. Neuronal autophagy and longevity. Aging 2017, 9, 1953–1954. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.F.; Lehner, B. Vitellogenins in C. elegans. Front. Physiol. 2019, 10, 1067. [Google Scholar] [CrossRef]
- Mizunuma, M.; et al. mTORC2-SGK-1 in longevity. Aging Cell 2014, 13, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; et al. Monitoring autophagy in aging. Methods Enzymol. 2008, 451, 493–520. [Google Scholar] [CrossRef]
- Palmisano, N.J.; et al. RAB-10 promotes autophagy. Autophagy 2017, 13, 1742–1753. [Google Scholar] [CrossRef]
- Jenzer, C.; et al. Autophagy in apoptosis. Autophagy 2019, 15, 228–241. [Google Scholar] [CrossRef]
- Jenzer, C.; et al. Human GABARAP rescues autophagy. Autophagy 2014, 10, 1868–1872. [Google Scholar] [CrossRef]
- McGhee, J.D. The C. elegans intestine. WormBook 2007, 1–36. [Google Scholar] [CrossRef]
- Yang, Y.; et al. ATG-16.2 mediates neuronal autophagy. Nat. Aging 2024, 4, 198–212. [Google Scholar] [CrossRef]
- Jung, R.; et al. Tissue-specific resistance to aggregation. PLoS Biol. 2023, 21, e3002284. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Campos, M.; et al. KFERQ-selective autophagy. PLoS ONE 2025, 20, e0330339. [Google Scholar] [CrossRef]
- Mukherjee, A.; et al. Selective microautophagy in Drosophila. Autophagy 2016, 12, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy in neurodegeneration. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Murley, A.; et al. Macroautophagy and lysosomal damage. Cell 2025, 188, 2670–2686.e14. [Google Scholar] [CrossRef]
- Zhong, R.; Richardson, C.E. Lysosomal expansion protects neurons. PLoS Biol. 2025, 23, e3002957. [Google Scholar] [CrossRef]
- Bansal, A.; et al. Lifespan vs. healthspan. Proc. Natl. Acad. Sci. USA 2015, 112, E277–E286. [Google Scholar] [CrossRef]
- Huang, C.; et al. Physiological predictors of lifespan. Proc. Natl. Acad. Sci. USA 2004, 101, 8084–8089. [Google Scholar] [CrossRef]
- Podshivalova, K.; et al. Aging and late-life decline. Cell Rep. 2017, 19, 441–450. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; et al. Targeting autophagy in cancer. JCMT 2019. [Google Scholar] [CrossRef]
- Thorburn, A.; et al. Autophagy and cancer therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; et al. Autophagy in aging. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Djajadikerta, A.; et al. Autophagy induction as therapy. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rhim, H. Autophagy in neurodegeneration. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef]
- Kovács, T.; et al. AUTEN-99 protects against neurodegeneration. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef]
- Papp, D.; et al. AUTEN-67 and neuroprotection. Autophagy 2016, 12, 273–286. [Google Scholar] [CrossRef]
- Billes, V.; et al. AUTEN-67 in Huntington’s disease. J. Huntingtons Dis. 2016, 5, 133–147. [Google Scholar] [CrossRef]
- Madonna, R.; et al. Empagliflozin and autophagy. Cardiovasc. Res. 2023, 119, 1175–1189. [Google Scholar] [CrossRef]
- Wei, S.; et al. Arsenic-induced autophagy. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Hartmann, J.; et al. Secretory autophagy and neurodegeneration. Nat. Commun. 2024, 15, 2635. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]

| Conditions | Activation trigger category | Animal age | Autophagy gene studied | Inactivation/ overexpression |
Tissue | Ref. |
|---|---|---|---|---|---|---|
| Benefical effets of autophagy | ||||||
| TOR pathway inhibition | Endogene activation | Early adulthood | bec-1, unc-51, atg-18 | Mutants/RNAi1 | Multiple tissues | [11,20,54] |
| Reduced insulin/IGF-1 signaling | Endogene activation | Early adulthood | bec-1, atg-7, lgg-1 | Mutants/RNAi1 | Multiple tissues | [11,26,35] |
| Mitochondrial dysfunction | Mitochondrial stress | Early adulthood | unc-51, bec-1, atg-18, dct-1 | Mutants/RNAi1 | Multiple tissues | [11,18] |
| Dietary restriction | Mild stress activation | Early adulthood | Multiple autophagy genes | Mutants/RNAi1 | Intestine | [11,37] |
| MALT-1 inhibition | Direct autophagy activation | Early adulthood | hlh-30 | Mutants | Intestine | [39] |
| TFEB/HLH-30 overexpression | Direct transcriptional activation | Early adulthood | hlh-30 | Overexpression | Intestine | [44] |
| SQST-1/p62 overexpression | Activation of selective autophagy | Early adulthood | sqst-1 | Overexpression | Intestin, neurons | [24] |
| Normal homeostasis | Neutral / homeostatic | Lifelong | Core autophagy genes | RNAi1 | Multiple tissues | [11,52] |
| Prolonged stress induced overexpression of autophagy | ||||||
| Proteotoxicity in mec-4(d) mutants | Proteotoxic stress | Adulthood | bec-1, atg-7 | RNAi1 | Neurons | [53] |
| Constitutive MPK-1 activation | Starvation | Early adulthood | bec-1 | RNAi1 | Pharynx | [54] |
| sgk-1 inhition | mPTP activation | Adulthood | bec-1, atg-16 | RNAi1 | Intestine | [55] |
| High glucose diet | Metabolic stress | Adulthood | hlh-30 | RNAi1 | Intestine | [58] |
| Age-dependent negative effects of autophagy | ||||||
| Aging-related dysregulation | Neutral / homeostatic | Postreproductive | unc-51, bec-1, atg-7, atg-9 | RNAi1 | Multiple tissues | [62] |
| Aging-related dysregulation | Proteotoxic stress with aging | Postreproductive | atg-7, bec-1 | Tissue-specific RNAi | Neurons | [13] |
| Aging-related hyperfunction | Eccessive autophagy with aging | Postreproductive | bec-1, atg-13, atg-18 | RNAi1 | Intestine | [14] |
| Cellular toxicity | Progressive lysosomal overload | Late adulthood | Multiple autophagy genes | Mutants | Multiple tissues | [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



