
Submitted:
07 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Pennycress (Thlaspi arvense L.), a representative and economically valuable cover crop, supports and enhances key ecological processes throughout its life cycle via its root system. It is hypothesized that pennycress selectively modulates its rhizosphere microbial community through root-derived metabolites, which may influence both the crop’s growth and the subsequent crops in rotation. However, systematic investigations comparing the rhizosphere microbiomes and metabolomes among different pennycress lines remain limited. This study employed metagenomic and metabolomic approaches to examine the dynamic changes in the rhizosphere microbial community and metabolite profiles of three pennycress lines with significantly different total alkaloid contents. The goal was to elucidate the interactions between microbes and metabolites. Results indicated significant differences in microbial community structure across the cultivars. JiL67 maintained stable community diversity, while LiN54 (with the lowest alkaloid content) showed reduced diversity. HeL43 (with the highest alkaloid content) exhibited increased diversity but also potential community homogenization, accompanied by the significant enrichment of microbial taxa capable of alkaloid tolerance. Metabolomic analysis identified metabolites such as Portulacaxanthin II, Oleanolic acid, and Soraphen A as significantly enriched in the rhizosphere soil of pennycress. This study reveals the shifts in rhizosphere microbial communities and metabolites linked to different pennycress lines and uncovers their interactive mechanisms, providing a scientific foundation for developing more economically efficient pennycress cultivation strategies.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Measurement Indicators and Methods
2.3. Metagenome Sequencing and Analysis
2.4. Untargeted Metabolomics Assays in the Rhizosphere Soil of the Field Pennycress
2.5. Data Analysis
3. Results
3.1. Agronomic Traits and Physiological Indicators
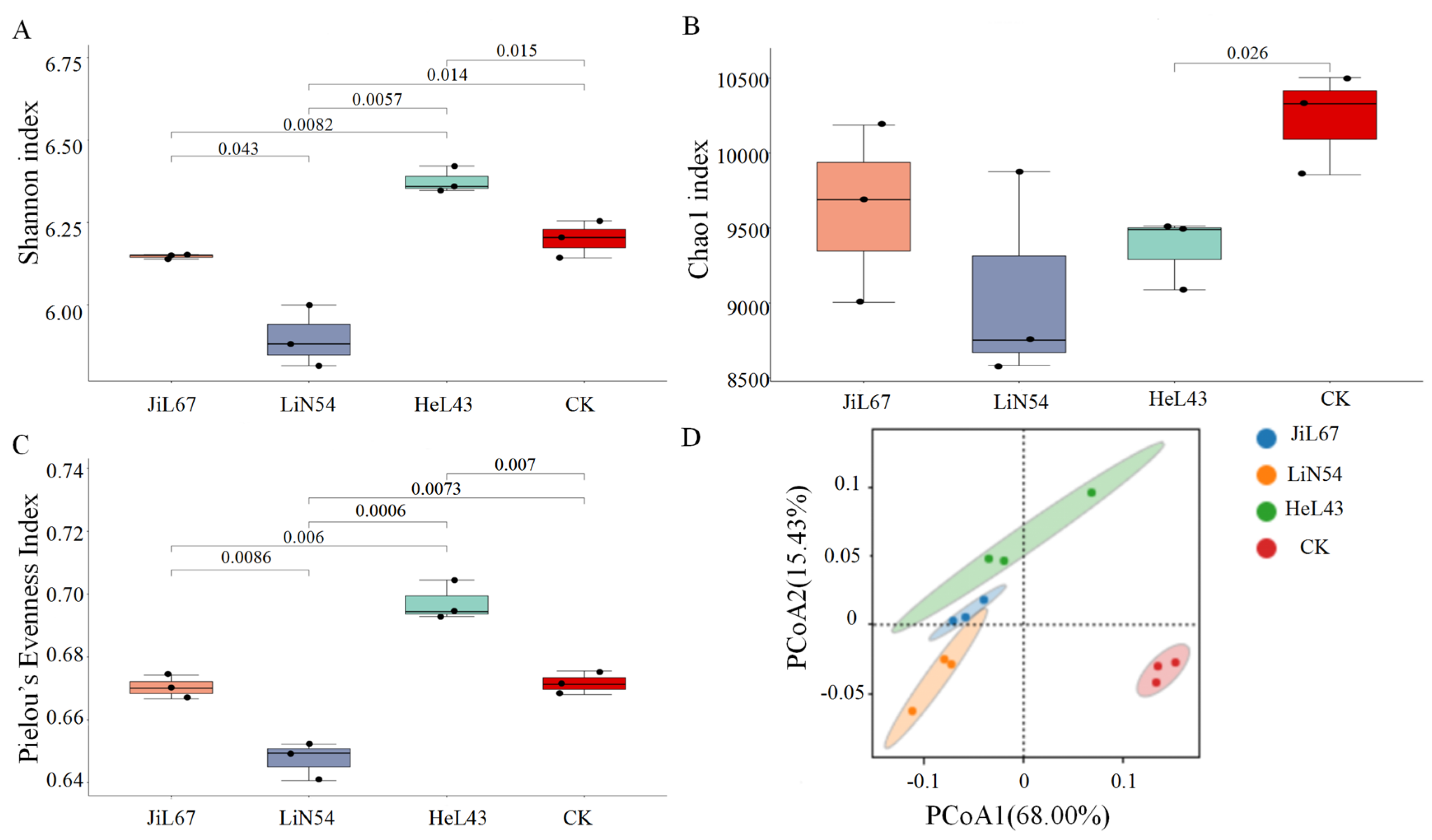
3.2. Variation in the Rhizosphere Microbial Community Across Pennycress Lines
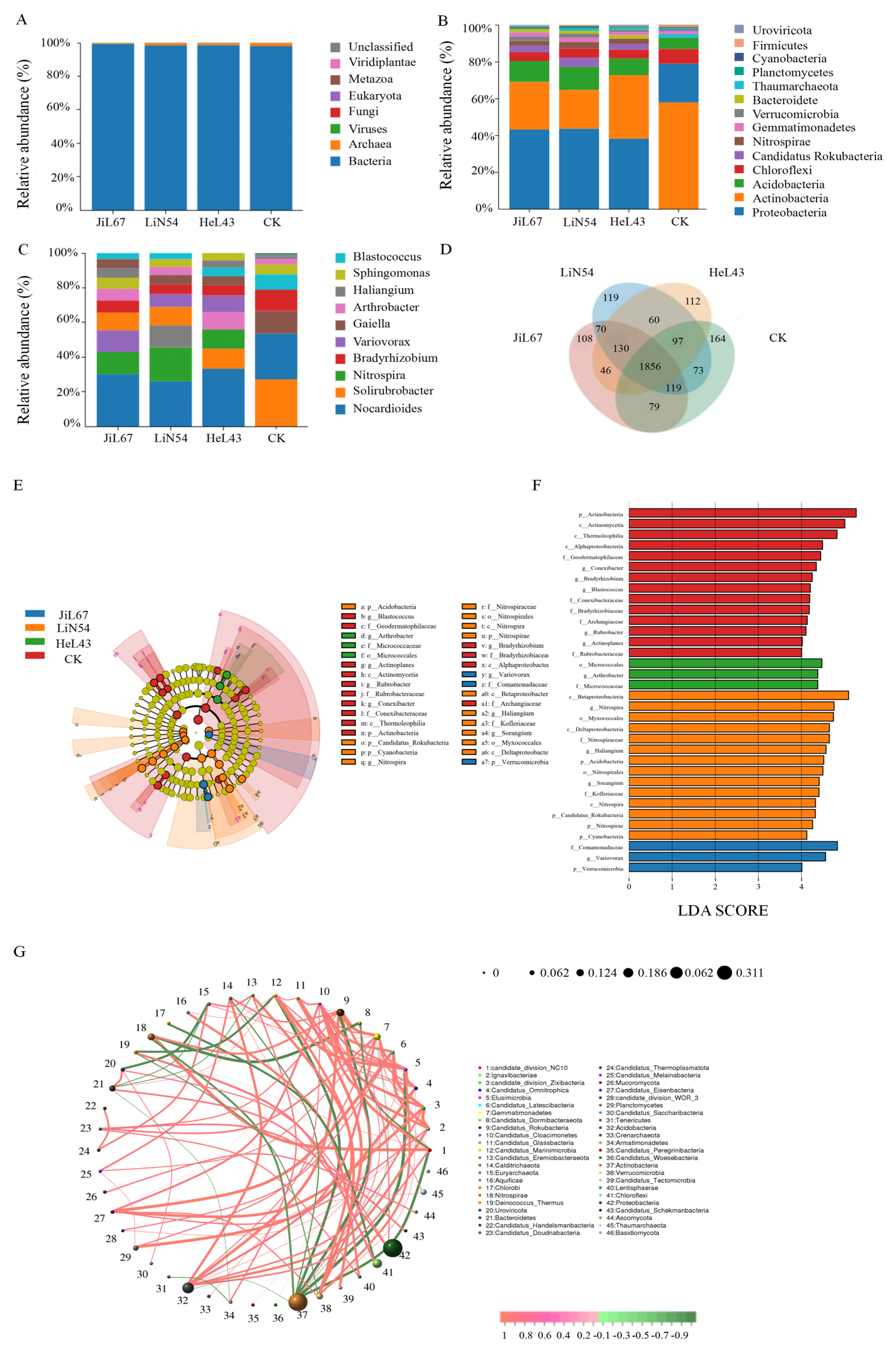
3.3. Composition of the Rhizosphere Microbial Community Across Pennycress Lines
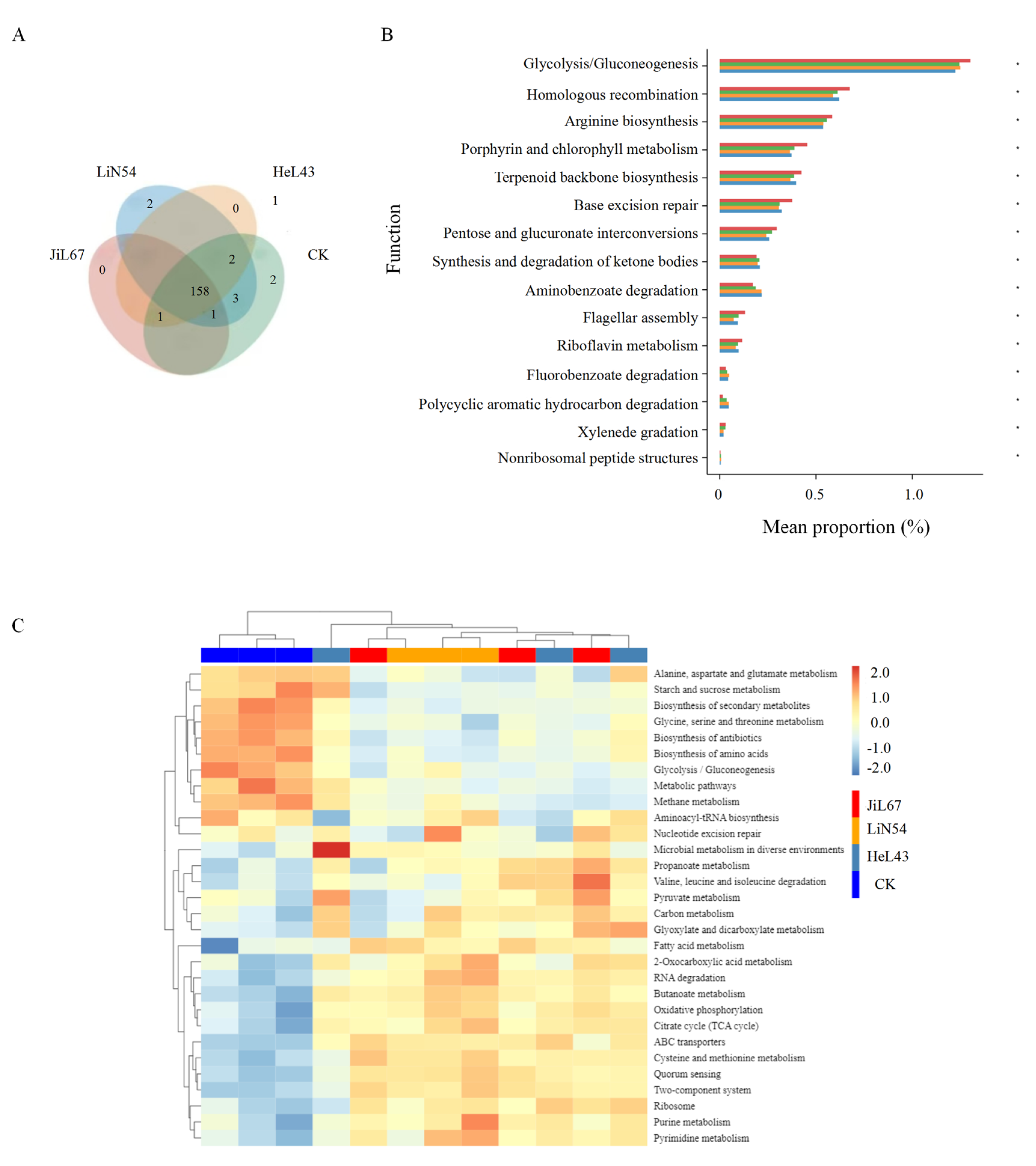
3.4. Potential Functional Pathways of the Rhizosphere Microbiome Across Pennycress Lines
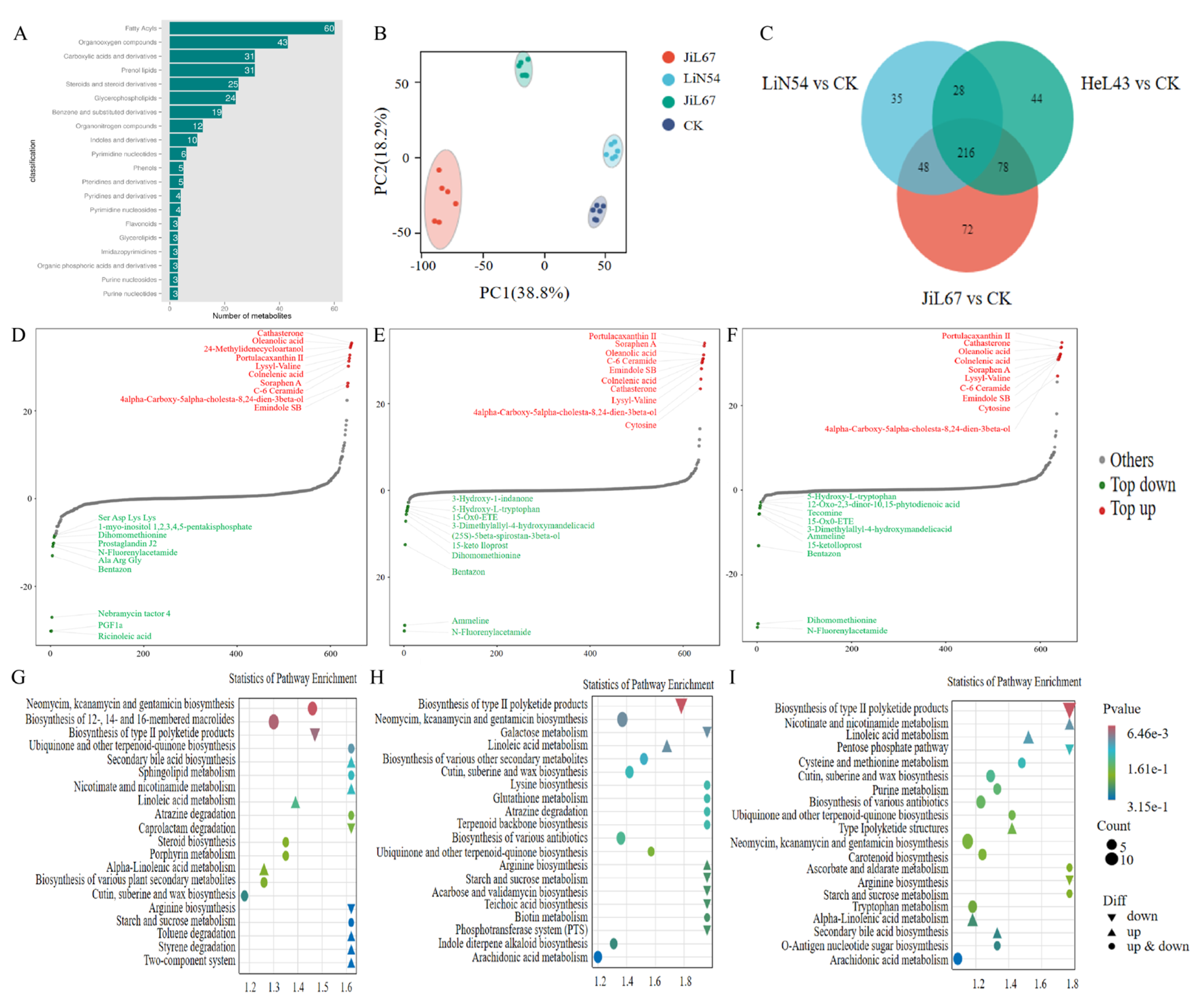
3.5. Non-Targeted Soil Metabolite Analysis
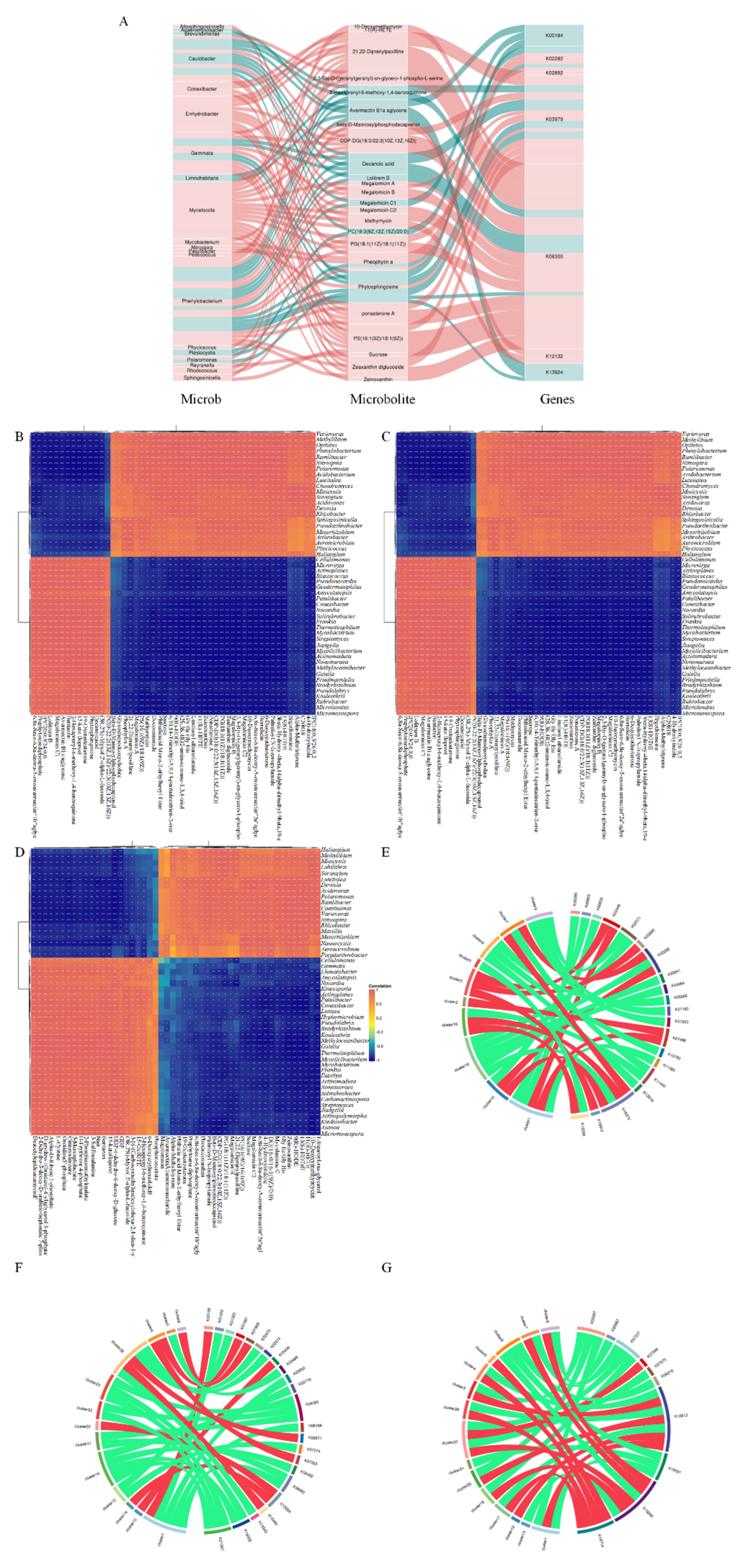
3.6. Joint Analysis of Soil Microorganisms and Metabolites
4. Discussion
4.1. Effects of Pennycress Lines on the Rhizosphere Microbial Community Structure
4.2. Effects of Pennycress Lines on the Rhizosphere Soil Metabolome
4.3. Correlation Between the Soil Metabolome and the Microbial Community
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McGinn, M.; Phippen, W.B.; Chopra, R.; Bansal, S.; Jarvis, B.A.; Phippen, M.E.; Dorn, K.M.; Esfahanian, M.; Nazarenus, T.J.; Cahoon, E.B., Durrett, T.P., Marks, M.D., Sedbrook, J.C. Molecular tools enabling pennycress (Thlaspi arvense L.) as a model plant and oilseed cash cover crop. Plant Biotechnology Journal. 2019, 17(4), 776-788. [CrossRef]
- Langdale, G.W.; Blevins, R.L.; Karlen, D.L.; McCool, D.K.; Nearing, M.A.; Skidmore, E.L. Cover crop effects on soil erosion by wind and water. In: Cover crops for clean water (Ankeny, Iowa: Soil and Water Conservation Society). 1991, pp. 15–21.
- Gyssels, G.; Poesen, J.; Bochet, E.; Li, Y. Impact of plant roots on the resistance of soils to erosion by water: a review. Progress in Physical Geography. 2005, 29(2), 189–217. [CrossRef]
- Dunn, M.; Ulrich-S, J.D.; Prokopy, L.S.; Myers, R.L.; Watts, C.R.; Scanlon, K. Perceptions and use of cover crops among early adopters: findings from a national survey. Journal of Soil & Water Conserv. 2016, 71(1), 29–40. [CrossRef]
- Han, E.; Li, F.; Perkons, U.; Küpper, P.M.; Bauke, S.L.; Athmann, M.; Thorup-Kristensen, K.; Kautz, T.; Köpke, U. Can precrops uplift subsoil nutrients to topsoil? Plant and Soil. 2021, 463, 329–345. [CrossRef]
- Marcus, G.; E, A.L.; L, S.G.; M, N.M.; Y, E.M.; N, C.T. A temporal analysis and response to nitrate availability of 3D root system architecture in diverse pennycress (Thlaspi arvense L.) accessions. Frontiers in plant science. 2023, 14, 1145389-1145389.
- Winthrop-B, P.; Phippen, M.-E. Soybean seed yield and quality as a response to field pennycress residue. Crop Science. 2012, 52(6), 2767-2773.
- Terry-A, I. US effort in the development of new crops (Lesquerella, PennycressCoriander and Cuphea). Oléagineux, Corps, Lipides. 2009, 16(4-5-6), 205-210.
- Zanetti, T.; Isbell, T.A.; Gesch, R.W.; Evangelista, R.L; Alexopoulou, E.; Moser, B.; Monti, A. Turning a burden into an opportunity: Pennycress (Thlaspi arvense L.) a new oilseed crop for biofuel production. Biomass and Bioenergy. 2019, 130, 105354.
- Li, X.J. Study on anti-inflammayory activity of Thlaspi arvense L. extract and 8 flavonoids. Nanning: Guangxi University, 2020.
- Xue, Y.; Zhang, L.C; Hu, W.; Jia, T.T.; Zhu, J.M.; Zhu, W.K.; Zhu, H.H. Herbal textual research of Thlaspi Herba. Shanghai Journal of Traditional Chinese Medicine. 2023, 57(10), 48-53.
- Tnde, P.; Holb, I.J.; I, S.N.P. Secondary metabolites in fungus-plant interactions. Frontiers in Plant Science. 2015, 6573.
- Li, C.Y.; Zha, W.J.; Li, W.; Wang, J.Y.; You, A.Q. Advances in The Biosynthesis of Terpenoids and Their Ecological Functions in Plant Resistance. International Journal of Molecular Sciences. 2023, 24(14), 11561-11561. [CrossRef]
- Ali, J.; Tonğa, A.; Islam, T.; Mir, S.; Mukarram, M.; Konôpková, A.S.; Chen, R. Defense Strategies and Associated Phytohormonal Regulationin Brassica Plants in Response to Chewing and Sap-Sucking Insects. Frontiers in Plant Science. 2024, 15, 1376917-1376917. [CrossRef]
- Wei, Y.F.; Qin, R.L.; Ding, D.C.; Li, Y.B.; Xie, Y.Y.; Qu, D.C.; Zhao, T.Y.; Yang, S.D. Structural characteristics of soil microbial community in rhizospheres of tomatoes during different growth periods. Journal of Huazhong Agricultural University. 2024, 43(1), 9-21.
- Li, X.N.; Dai, W.Z.; Lin, X.R.; Liao, L.Y.; Lu, W.; Yang, S.D. Differential analysis in the structure of rhizosphere microbialcommunities and identification of functional taxa associated with the content of capsaicin in pepper varieties. Journal of Huazhong Agricultural University. 2016, 1-12.
- Raza, W.; Jiang, G.F.; Eisenhauer, N., Huang, Y.H.; Wei, Z.; Shen, Q.R.; Kowalchuk, A.G.; Jousset, A. Microbe-induced phenotypic variation leads to over yielding in clonal plant populations. Nature Ecology & Evolution. 2024, 8(3), 392-399. [CrossRef]
- Zhou, X.G.; Zhang, J.Y.; Khashi., U.R.M.; Gao, D.M.; Wei Z.; Wu F.Z.; DiniAndreote, F. Interspecific plant interaction via root exudates structures the disease suppressiveness of rhizosphere microbiomes. Molecular plant. 2023,16(5), 849-864.
- Wang, B.; Chen, C.; Xiao, Y.M.; Chen, K.Y.; Wang, J.; Zhao, S.; Liu, N.; Li, J.N.; Zhou, G.Y. Trophic relationships between protists and bacteria and fungi drive the biogeography of rhizosphere soil microbial community and impact plant physiological and ecological functions. Microbiological Research. 2024, 280, 127603. [CrossRef]
- Liu, J.W.; Li, X.Z.; Yao, M.J. Research progress on assembly of plant rhizosphere microbial community. Acta microbiologica Sinica. 2021, 61(2), 231-248.
- Xu, J.; Zhang, Y.Z.; Zhang, P.F.; Trivedi, P.; Riera, N.; Wang, Y.Y.; Liu, X.; Fan, G.Y.; et al. The structure and function of the global citrus rhizosphere microbiome. Nature communications. 2018, 9(1), 4894. [CrossRef]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B.; Dahlberg, J.A.; Coleman-Derr, D.; Lemaux, P.G.; Taylor, J.W. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nature communications. 2020, 11(1), 34. [CrossRef]
- Zhou, Y.; Yi, S.X.; Zang, Y.; Yao, Q.; Zhu, H.H. The predatory myxobacterium Citreicoccus inhibens gen. nov. sp. nov. showed Antifungal Activity and Bacteriolytic Property against Phytopathogens. Microorganisms. 2021, 9(10), 2137. [CrossRef]
- Li, L.; Rodriguez-Concepcion, M.; Al-Babili, S. Recent advances in understanding carotenoid-derived signaling molecules in regulating plant growth and development. Frontiers in Plant Science. 2015, 6790.
- Pang, Z.Q.; Chen, J.; Wang, T.H.; Gao, C.S.; Li, Z.M.; Guo, L.T.; Xu, J.P.; Cheng, Y. Linking plant secondary metabolites and plant microbiomes: a review. Frontiers in Plant Science. 2021, 12, 621276. [CrossRef]
- Liu, C.C.; Yu, J.S.; Ying, J.Z.; Zhang, K.; Hu, Z.G.; Liu, Z.X.; Chen, S.L. Integrated metagenomics and metabolomics analysis reveals changes in the microbiome and metabolites in the rhizosphere soil of Fritillaria unibracteata. Frontiers in Plant Science. 2023, 14, 1223720. [CrossRef]
- Zhang, L.; Xu, H.M.; Zhu, B.L. Association of rhizosphere soil microbiome with the occurrence and development of replant disease - A review. Acta Microbiologica Sinica. 2016, 56(8), 1234-1241.
- Zhao, Y.X.; Zhou, J.Y.; Chen, L.L.; Li, S.; Yin, Y.; Jeyaraj, A.; Liu, S.J.; Zhuang, J.; Wang, Y.H.; Chen, X.; Li, X.H. Allelopathic Effect of Osmanthus fragrans Changes the Soil Microbial Community and Increases the Soil Nutrients and the Aroma Quality of Tea Leaves. Journal of agricultural and food chemistry. 2025, 73(22), 13818-13831. [CrossRef]
- Xu, Y.; Tan, Z.R.; Yin, Y.; Hua, J.M.; Han, Y.X.; Lin, J.X.; Wang, A.; Wang, J.H. Research progress on the effects of invasive plants on soil characteristics. Chinese Journal of Plant Ecology. 2025,49(11), 1767-1777.
- Zhang, H.; Ma, R.J.; Wang, N.L.; Li, G. Allelopathic Effects of Different Plants on the Major Weed Ligularia sagitata in Alpine and Cold Grassland. Acta Botanica Boreali-Occidentalia Sinica. 2006, (11), 2307-2311.
- Bressan, M.; Roncato, M-A.; Bellvert, F.; Comte, G.; Haichar, F.E.; Achouak, W.; Berge, O. Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. The ISME Journal. 2009, 3(11), 1243–1257. [CrossRef]
- Aleklett, K.; Leff, J.; Fierer, N.; Hart, M. Wild plant species growing closely connected in a subalpine meadow host distinct root-associated bacterial communities. PeerJ. 2015, 3(8), e804. [CrossRef]
- Zeng, W.A.; Yang, Z.Y.; Huang, Y.; Gu, Y.B.; Tao, J.M.; Liu, Y.J.; Xie, P.F.; Cai, H.L.; Yin, H.Q. Response of soil bacterial community structure and co-occurrence network topology properties to soil physicochemical properties in long-term continuous cropping farmland. Acta Microbiological Sinica. 2022, 62(06), 2403-2416.
- Pham, H.N.; Pham, P.A.; Nguyen, T.T.H.; Meiffren, G.; Brothier, E.; Lamy, I.; Michalet, S.; Dijoux-Franca, M.G.; Nazaret, S. Influence of metal contamination in soil on metabolic profiles of Miscanthus × giganteus belowground parts and associated bacterial communities. Applied Soil Ecology. 2018, 125, 240-249. [CrossRef]
- Xiao, D.; Huang, Y.; Feng, S.Z.; Ge, Y.H.; Zhang, W.; He, X.Y.; Wang, K.L. Soil organic carbon mineralization with fresh organic substrate and inorganic carbon additions in a red soil is controlled by fungal diversity along a pH gradient. Geoderma. 2018, 321, 79-89. [CrossRef]
- Chen, L.F.; He, Z.B.; Zhao, W.Z.; Kong, J.Q.; Gao, Y. Empirical evidence for microbial regulation of soil respiration in alpine forests. Ecological Indicators. 2021, 126, 107710. [CrossRef]
- Zhang, Z.X.; Li, J.H.; Zhang, L.N.; Su, M.; Chen, J.; Yang, Z.P.; Gao, Z.Q.; Qiao, Y.J.; Zhang, X.Y.; Xue, Z.Q.; Zhang, C.L. Effects of combined application of selenium and nitrogen fertilizers on selenium and nitrogen accumulation in oat plants, rhizospheric soil microbial community, and metabolites. Chinese Journal of Applied and Environmental Biology. 2023, 29(05), 1241-1252.
- Yang, J.; Lin, Y.; Yang, W.H.; Zhou, B.Q.; Mao, Y.L.; Xing, S.H. Soluble organic nitrogen variation rate and its response to bacteria under different fertilization treatments in paddy soil. Journal of Agro-Environment Science. 2021, 40(07), 1509-1518.
- Montel, M.C.; Reitz, J.; Talon, R.; Berdague, J.; Rousset-Akrim, S. Biochemical activities of Micrococcaceae and theireffects on the aromatic profiles and odours of a dry sausage model. Food Microbiology. 1996, 13(6), 489-499. [CrossRef]
- Fan, Q.S.; Wanapat, M.; Hou, F.J. Chemical composition of milk and rumen microbiome diversity of yak, Impacting by herbage grown at Different Phenological Periods on the Qinghai-Tibet Plateau. Animals. 2020, 10(6), 1030. [CrossRef]
- Vandera, E.; Samiotaki, M.; Parapouli, M.; Panayotou, G.; Koukkou, A.I. Comparative proteomic analysis of Arthrobacter phenanthrenivorans Sphe3 on phenanthrene, phthalate and glucose. Journal of Proteomics. 2015, 113, 73-89. [CrossRef]
- Yin, L.; Liu, J.X.; Tao, J.P.; Xing, G.M.; Tan, G.F.; Li, S.; Duan, A.Q.; Ding, X.; Xu, Z.S.; Xiong, A.S. The gene encoding lycopene epsilon cyclase of celery enhanced lutein and β-carotene contents and confers increased salt tolerance in Arabidopsis. Plant Physiology and Biochemistry. 2020, 157, 339-347. [CrossRef]
- Tian, L. Recent advances in understanding carotenoid-derived signaling molecules in regulating plant growth and development. Frontiers in Plant Science. 2015, 6, 790. [CrossRef]
- Priyanka, M.; Asit, R.; Sudipta, J.; Sanghamitra, N.; Sujata, M. Influence of extraction methods and solvent system on the chemical composition and antioxidant activity of Centella asiatica L. leaves. Biocatalysis and Agricultural Biotechnology. 2021, 33, 101971.
- Renju, K; John, J.A.; Sabulal, B. Centella asiatica: secondary metabolites, biological activities and biomass sources. Phytomedicine Plus. 2022, 2(1), 100176.
- Dipesh, D.; Thi, L.; Prasad, R.; Kumar, J.; RitBahadur, G.; Prakash, P.; Raj, P.A.; Cheol, Y.J.; Kyung, S.J. Enhanced production of nargenicin A (1) and generation of novel glycosylated derivatives. Applied biochemistry and biotechnology. 2015, 175(6), 2934-2949.
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene Biosynthesis in Plants. Annual Review of Plant Biology. 2014, 65(1), 225-257.
- Han, L.X; Zhang, H.P.; Long, Z.N.; Ge, Q.Q.; Mei, J.J.; Yu, Y.L.; Fang, H. Exploring microbial community structure and biological function in manured soil during ten repeated treatments with chlortetracycline and ciprofloxacin. Chemosphere. 2019, 228, 469-4777. [CrossRef]
- Zhang, F.S.; Wang, J.Q.; Zhang, W.F.; Cui, Z.L.; Ma, W.Q.; Chen, X.P.; Jiang, R.F. Nutrient Use Efficiencies of Major Cereal Crops in China and Measures for Improvement. Acta Pedologica Sinica. 2008, 05, 915-924.
- Kazuhiro, T.; Kunihiko, F.; Ken-ichi, Y.; Naoki, H.; Akio, I. Biodegradation of melamine and its hydroxy derivatives by a bacterial consortium containing a novel Nocardioides species. Applied microbiology and biotechnology. 2012, 94(6), 1647-1656.
- Khan, M.; Iqbal, N.; Masood, A.; Mobin, M.; Anjum, N.A.; Khan, N.A. Modulation and significance of nitrogen and sulfur metabolism in cadmium challenged plants. Plant Growth Regulation. 2016, 78(1), 1-11.. [CrossRef]
- Ullah, K.I.; Neeli, H.; Xiao, M.; Huang, X.; Ullah, K.N.; Wan-Taek, I.; Iftikhar, A.; Zhi, X.Y.; Li, W.J. Phenylobacterium terrae sp. nov., isolated from a soil sample of Khyber-Pakhtun-Khwa, Pakistan. Antonie van Leeuwenhoek. 2018, 111(10), 1767-1775.
- Hoa, N.X.; Wai, N.K.; Seong, L.Y.; Hwan, K.Y.; Hak, M.J.; Yong, K.K. Antagonism of antifungal metabolites from Streptomyces griseus H7602 against Phytophthora capsici. Journal of basic microbiology. 2015, 55(1), 45-53.
- Xiong, Z.Q.; Zhang, Z.P.; Li, J.H.; Wei, S.J.; Tu, G.Q. Characterization of Streptomyces padanus JAU4234, a producer of actinomycin X2, fungichromin, and a new polyene macrolide antibiotic. Applied and Environ Microbiology. 2012, 78(2), 589-592. [CrossRef]
- Tong, X.M, Chen, F, Yu, J, A, S.C.R.; L, S.J.B.; Wang, W.; Liang, Y.; Zheng, X.G.; Wang, J. Analysis of bacterial flora structure and diversity in snow on zhuoyou peak(8201m). Chinese Science Bulletin. 2008, 53(18), 2216-2222.
- Qian, J.; Wu, Z.Y.; Zhu, Y.Z.; Zhang, Y.; Guo, X.K.; Liu, C. Correlation between microecology and antibiotic resistance atthe pig-soil interface in Chongming District, Shanghai, China. Chinese Journal of Microecology. 2024, 36(02), 135-146.
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. Journal of neuro-oncology. 2013, 113(1), 1-11. [CrossRef]

| Varieties | Plant Height (cm) |
Fresh weight (g) | Dry weight (g) | D/F Ratio (%) | Protein content (mg/g) | Total alkaloids (mg/g) |
|---|---|---|---|---|---|---|
| JiL67 | 64.00 ± 9.64a | 53 | 15.25 | 0.30 ± 0.05a | 150.68 ± 19.73a | 1.50 ± 0.40b |
| LiN54 | 74.67 ± 20.01a | 40.35 | 13.55 | 0.35 ± 0.05a | 161.15 ± 5.10a | 1.40 ± 0.49b |
| HeL43 | 67.33 ± 3.06a | 41.1 | 15.17 | 0.37 ± 0.05a | 173.42 ± 1.44a | 2.65 ± 0.06a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



