Submitted:
05 February 2026
Posted:
06 February 2026
You are already at the latest version
Abstract
Dietary polyphenols such as quercetin, resveratrol, and (-)-epigallocatechin-3-gallate (EGCG) have been shown to be neuroprotective in epidemiologic and experimental studies of Alzheimer’s disease (AD), though clinical evidence remains limited. This re-view highlights the importance of studying glucuronide and sulfate conjugates of these polyphenols, as well as their intestinal microbial metabolites, at bioavailable low na-nomolar concentrations, particularly those that can reach the brain. While many in vitro studies use micromolar concentrations of aglycones, their relevance to understanding neuroprotection is debated. Although polyphenols are redox-sensitive, their direct antioxidant or prooxidant roles may be limited at nanomolar levels, and reactive oxidation products have not been detected in tissues. Instead, their neuroprotective effects appear to be mediated by high-affinity interactions with molecular targets, such as the 67-kDa laminin receptor (67LR). This receptor binds both aglycones and conjugates at low na-nomolar concentrations via a peptide G region containing glycosaminoglycan and palindromic sequences. The same region also binds the prion-amyloid-β complex, sug-gesting that polyphenols could antagonize amyloid-β binding and prevent its neurotoxicity. The peptide G region may also act as a redox sensor. Polyphenol binding to 67LR activates cAMP signaling and downstream neuroprotective pathways involving CREB, SIRT1, and protein phosphatase 2A. Additionally, nanomolar concentrations of resveratrol and quercetin inhibit quinone reductase 2, an enzyme linked to cognitive decline and elevated in AD. Given their low bioavailability in the brain and distinct molecular targets, combining multiple polyphenols at low doses may yield additive and synergistic effects, enhance efficacy, and minimize potential toxicity in preventing AD.
Keywords:
Alzheimer’s disease
; amyloid-β
; cAMP
; epigallocatechin-3-gallate
; glucuronide metabolites
; laminin receptor
; polyphenols
; quercetin
; quinone reductase 2
; resveratrol
1. Introduction
Alzheimer’s disease (AD) is a major global health challenge and the most common neurodegenerative disease of aging. In 2021, an estimated 57 million people worldwide were living with dementia, with AD accounting for ~60–70% of cases; nearly 10 million new dementia cases occur each year [1]. Based on current demographic trends, the global number of people living with dementia is projected to rise to ~152.8 million by 2050 [2]. In the United States, an estimated 7.2 million adults aged ≥65 years are living with Alzheimer’s dementia in 2025, corresponding to about 1 in 9 older adults (≈11%) [3].
Importantly, AD unfolds over a prolonged prodromal period, with biological and cognitive changes emerging years, often well before dementia becomes clinically apparent [4]. Clinically, many individuals pass through amnestic mild cognitive impairment (aMCI), a high-risk state associated with progression to Alzheimer’s dementia at roughly ~10–20% per year in many cohorts [5]. This extended pre-dementia interval represents a crucial window for early intervention, including dietary and other lifestyle-based approaches [6]. To develop effective strategies, it is essential to clarify how dietary agents, particularly polyphenols, can inhibit AD-relevant pathways.
1.1. Unique Focus of the Review
Given the clinical importance of AD, several reviews have addressed the relationship between polyphenols and AD from multiple perspectives [7,8,9,10,11,12,13,14,15,16]. This review focuses on the unique features of dietary polyphenols that limit their bioavailability, rapid conjugation to glucuronide and sulfate metabolites, and the resulting low nanomolar concentrations reached in the brain. We emphasize the functional roles of glucuronide and sulfate conjugates, as well as intestinal microbial metabolites. We also discuss the limitations of direct antioxidant and prooxidant mechanisms and highlight the importance of high-affinity, redox-sensitive molecular targets for polyphenols. Although the human diet contains hundreds of polyphenols, we focus on quercetin, resveratrol, and (−)-epigallocatechin-3-gallate (EGCG) in this review, which have demonstrated consistent, reproducible neuroprotective effects in animal models of AD.
1.2. Multifactorial AD Pathogenesis
The β-amyloid (Aβ) hypothesis has long been a central framework for AD, proposing that abnormal amyloid precursor protein (APP) processing leads to the buildup of pathogenic Aβ species [17,18,19]. In this view, soluble Aβ oligomers are particularly toxic: they impair synaptic plasticity and memory-related signaling and can initiate downstream cascades that include tau dysregulation, neuroinflammation, and ultimately synaptic and neuronal loss [20,21]. Strong genetic support comes from autosomal-dominant (familial) AD, in which pathogenic variants in APP and presenilins alter Aβ generation and promote amyloid pathology [22]. Despite this biological rationale, translating Aβ-lowering into meaningful clinical benefit has been difficult: several approaches have failed in symptomatic disease. More recently, however, anti-amyloid antibodies such as lecanemab and donanemab have produced statistically significant but modest slowing of decline in early symptomatic AD, supporting Aβ as a valid target while also implying that timing (earlier intervention) and/or multi-target strategies may be necessary for larger clinical effects [23].
Currently, there is increasing emphasis on the multifactorial nature of AD. In addition to the important role of Aβ, other frameworks highlight contributions from tau pathology, chronic neuroinflammation, metabolic/mitochondrial dysfunction, dysregulated Ca²⁺ homeostasis, iron dyshomeostasis, and cerebrovascular/ blood-brain barrier (BBB) impairment during disease initiation and progression [24,25,26,27,28,29]. This broader perspective has increased interest in combination and multi-target approaches that engage multiple disease mechanisms rather than focusing on a single pathway [24,30]. Within this context, dietary polyphenols have attracted attention because they can modulate several AD-relevant processes (e.g., Aβ- and tau-related pathways, inflammatory/oxidative signaling, and metabolic stress responses) across experimental systems [7,15].
1.3. Intervention of AD with Dietary Polyphenols
Epidemiological studies suggest that diets rich in plant-based foods are associated with slower cognitive decline [31]. Several plant products enhance memory [32,33]. The Mediterranean-Dash Intervention for Neurodegenerative Delay (MIND) diet, in particular, is consistently linked to a lower risk of AD and related dementias [34,35,36,37,38]. This diet includes foods high in polyphenols, such as fruits, vegetables, olive oil, tea, and moderate amounts of red wine. Several epidemiological studies have reported a lower incidence of dementia and AD associated with green tea consumption [39,40]. Trans-resveratrol has been shown to prevent AD in various animal models [41,42,43,44,45,46]. This protective effect was also reported for quercetin in AD mouse models [47,48,49,50,51,52] (Figure 1). Resveratrol protects isolated neurons from amyloid-β-induced toxicity and other harmful agents [53,54,55,56]. EGCG has demonstrated neuroprotective effects in multiple experimental models of AD [34,57,58].
1.4. Challenges in Understanding the Role of Dietary Polyphenols in AD Prevention
As others have reviewed, quercetin, resveratrol, and EGCG exert neuroprotective effects in vitro against Aβ toxicity and oxidative stress in neuronal cells, but these effects often require 10 to 100 micromolar concentrations [59,60,61,62]. In contrast, they reach only low nanomolar concentrations in the brain. Many dietary polyphenols are absorbed only to a limited extent and undergo rapid phase II metabolism to form glucuronide and sulfate conjugates. Consequently, the parent (aglycone) compounds are present at very low concentrations in plasma and are often undetectable. EGCG is somewhat unique in that a substantial fraction can remain unconjugated. Both the parent compounds and their metabolites are extensively bound to plasma proteins. As a result, only small amounts of aglycones and conjugates, often in the low nanomolar range, reach the brain. This is a critically important consideration when designing experiments to elucidate the mechanisms underlying their neuroprotective effects.
2. Absorption, Distribution, Metabolism, Excretion of Polyphenols
First, we discuss the fraction of administered polyphenols that is absorbed and metabolized, the circulating concentrations of aglycones and conjugated metabolites, the extent of their plasma protein binding, and the low levels that reach the brain. Table 1 summarizes key pharmacokinetic features of quercetin, resveratrol, and EGCG.
2.1. Quercetin
Quercetin is a flavanol-type flavonoid that is present predominantly in vegetables and fruits in the form of glycosides [63]. These glycosides are hydrolyzed by intestinal bacteria to release the quercetin aglycone, which is absorbed in the intestine [64]. Administered quercetin up to 20% absorbed, and its bioavailability is only 2% [65]. In the liver, quercetin is rapidly metabolized by phase II enzymes to form glucuronides, sulfates, mixed glucuronide-sulfate conjugates, and methylated glucuronides [66]. In plasma, only these conjugated metabolites are detected at nanomolar to low micromolar concentrations, depending on the dose of quercetin or quercetin glycosides administered to animals or humans; the quercetin aglycone itself is not detectable [66]. In sheep administered dried onion skin, quercetin was detected in cerebrospinal fluid (CSF) at a concentration of 0.30 nM, whereas quercetin-3-O-glucuronide (Q-3-G) and other conjugated metabolites were not detectable [67]. In contrast, Pasinetti and colleagues quantified Q-3-G in the rat brain at concentrations as low as 1 nM following oral administration of a red wine polyphenol mixture [68].
Pharmacokinetic studies in humans receiving quercetin (500 mg, three times daily) reported an average terminal plasma half-life of ~3.5 h [69]. The elimination half-lives of the major conjugated metabolites were approximately 1.7–5.3 h [70]. Urinary excretion of intact quercetin metabolites accounted for only ~5% of the administered dose [70]. In contrast, a substantial fraction appears to be extensively degraded, with an estimated “biological half-life” of ~20–72 h and substantial recovery as expired CO₂ [71].
2.2. Resveratrol
Resveratrol (3,5,4′-trihydroxy-stilbene) is a non-flavonoid polyphenol found in red wine, grapes, berries, and peanuts [87,88]. The trans isomer of resveratrol (t-RES) is the predominant and more stable form compared to its counterpart, cis-resveratrol [29]. In grapes and other plant sources, resveratrol predominantly exists in glycosylated forms. Upon ingestion, these glycosides are hydrolyzed by microbial glycosidases in the intestine, releasing the aglycone, which is then absorbed [87,88]. Orally administered resveratrol is absorbed in the intestine at nearly 75%, and its oral bioavailability is less than 1% [89]. Following oral administration of red wine containing resveratrol or moderate doses of purified resveratrol (25 mg) in humans, the compound undergoes rapid metabolism. As a result, the aglycone form is typically undetectable in plasma, while its glucuronide and sulfate conjugates are present at low concentrations [89,90,91]. In contrast, administration of high pharmacological doses (100–5000 mg) of resveratrol leads to detectable levels of the resveratrol aglycone in plasma, although only at nanomolar to low micromolar concentrations [89]. The plasma levels of its glucuronide and sulfate conjugates are significantly higher than those of the free aglycone [89]. Resveratrol crosses the BBB in vitro only 2% of the plasma concentration [78]. Therefore, resveratrol and its metabolites reach only low nanomolar concentrations or remain below the detection limit in the brain and CSF [77].
Human studies indicate that the plasma half-life of trans-resveratrol is approximately 1–3 h, whereas its conjugated metabolites have a longer half-life of about 9.2 h [92]. Thus, the parent compound is cleared rapidly, but the pool of conjugates can persist much longer [92]. Most of the orally administered dose is recovered in urine, predominantly as glucuronide and sulfate conjugates, along with microbiota-derived metabolites [89].
2.3. EGCG
EGCG is the most abundant catechin flavonoid in green tea, present along with other catechins such as epicatechin, epigallocatechin, and epicatechin-3-gallate. Unlike polyphenols such as quercetin and resveratrol, these catechins are present predominantly in the unconjugated (aglycone) form in green tea [93]. Following oral administration in humans and rodents, the systemic bioavailability of EGCG is low, estimated to be approximately 0.1–0.3% [94]. When a green tea catechin mixture is administered to humans, EGCG circulates in the plasma both as the aglycone (~45% or higher) and the remaining as conjugated metabolites, primarily EGCG-sulfate and EGCG-glucuronide [80]. Compared with quercetin and resveratrol, EGCG undergoes relatively less extensive conjugation, and a substantial fraction remains in the aglycone form in humans [80].
The extent to which EGCG or its metabolites enter the brain following green tea consumption or oral EGCG administration remains controversial. In one study, oral administration of 3H-labeled EGCG resulted in measurable radioactivity in brain tissue, suggesting CNS uptake; however, the specific chemical form and absolute concentration of EGCG in the brain were not determined [95]. In mouse studies, EGCG was detectable in several peripheral tissues following oral administration, but was undetectable in the brain at standard doses [84]. In rats administered EGCG at 50 mg/kg, brain concentrations of approximately 10 nM were reported [82]; however, this study did not indicate whether residual blood contamination was adequately controlled for. In contrast, another study reported brain EGCG levels of approximately 0.5 nmol/g in rats following very high oral doses (500 mg/kg) of EGCG [83].
In vitro BBB models suggest limited EGCG permeability; one study reported approximately 5.6% translocation across the BBB within one hour, implying that consumption of two cups of green tea could yield brain EGCG concentrations of roughly 10 nM, assuming plasma levels of ~0.2 µM [85]. However, such in vitro BBB models have inherent limitations in predicting in vivo brain uptake. Consistent with this uncertainty, clinical studies have reported that neither EGCG nor its conjugates are detectable in human CSF following oral consumption of green tea [86].
After consumption of green tea or administration of purified EGCG in humans, plasma pharmacokinetics indicate an EGCG half-life of ~3.4 h [96]. Only trace amounts of EGCG are excreted in urine; instead, EGCG is eliminated predominantly via biliary secretion and fecal excretion, with additional biotransformation to microbial metabolites in the gut [97].
2.4. Protein Binding in the Plasma –“Free” Polyphenol/Metabolite Fraction
These polyphenol aglycones are highly protein-bound in plasma: quercetin can be bound as high as 99% [72], resveratrol is nearly 98% protein-bound, and ~50% of its conjugates are bound [74,75], and EGCG approximately 80–90% bound [81]. Studies of many drugs and steroid hormones have shown that extensive protein binding reduces the unbound (free) fraction, thereby limiting tissue availability and narrowing the range of molecular targets accessible to the free compound. However, protein binding is a dynamic equilibrium in which the bound pool can replenish the free fraction as the free compound is distributed, metabolized, or excreted. Protein binding can also slow glomerular filtration, thereby increasing the half-life. In addition, binding to plasma proteins may stabilize polyphenols against auto-oxidation. Despite these potential benefits, extensive protein binding reduces free polyphenol levels in tissues and can further limit their penetration across the BBB. As a result, brain exposure may remain low despite constraints imposed by molecular size and polarity. Moreover, polyphenols and their metabolites are often extracted with organic solvents, which typically measure total (bound + unbound) concentrations rather than the free fraction. Therefore, protein binding should be considered when interpreting exposure measurements and when designing in vitro experiments to elucidate mechanisms of action.
3. Polyphenol Aglycone Actions at Micromolar Concentrations
Collectively, pharmacokinetic studies suggest that polyphenols such as quercetin, resveratrol, and EGCG, or their conjugates, may reach the brain at low nanomolar concentrations, potentially below the limits of detection. Nonetheless, many in vitro studies demonstrating beneficial effects of these polyphenols, including antioxidant and prooxidant activities and inhibition of β-amyloid aggregation, employ micromolar concentrations. While these observations are mechanistically informative and interesting, further studies are required to determine whether such effects are achievable under bioavailable low concentrations and chemical forms, especially in the brain.
3.1. Antioxidants – Limitations
Although dietary polyphenols can, in principle, quench reactive oxygen species (ROS) through direct chemical scavenging, their contribution to antioxidant defense in vivo, particularly in the brain, is thought to be limited because these polyphenol levels are typically in the low-nanomolar range [98,99,100,101]. In addition, polyphenol glucuronides and sulfates conjugates generally exhibit weaker radical-scavenging activity than the corresponding aglycones [63,102]. Instead, quercetin, resveratrol, and EGCG, either as aglycones or as glucuronide/sulfate metabolites, can modulate redox-sensitive signaling pathways that increase the expression of endogenous antioxidant enzymes [103,104,105,106]. Compounds that act mainly through this mechanism have been termed “indirect antioxidants”: rather than directly scavenging ROS, they enhance intrinsic antioxidant defenses, enabling protection that may persist after the compound itself has been cleared from the tissues [107,108,109].
3.2. Prooxidants – Limitations
Others described that quercetin, resveratrol, and EGCG can also act as prooxidants under conditions that favor their oxidation or transition-metal redox cycling, leading to net ROS generation rather than ROS quenching. For example, copper complexes of quercetin and resveratrol can promote ROS formation and oxidative DNA damage [110,111]. At lower, non-toxic exposures, such prooxidant activity may function as a mild oxidative stimulus that engages adaptive defenses (e.g., Nrf2-dependent gene programs), consistent with a hormetic mechanism [112]. Notably, hydrogen peroxide generated by EGCG auto-oxidation has been reported to be protective in human keratinocytes [113]. Prooxidant actions of EGCG have also been discussed as a potential contributor to health benefits, while also possibly mediating adverse effects under certain conditions [114]. Forman and colleagues elegantly discussed how electrophiles derived from dietary polyphenols can increase nucleophilic tone by activating Nrf2 [115]. Shah et al convincingly demonstrated the importance of Nrf2 and heme oxygenase-1 in the flavanol-mediated neuroprotection [116].
Halliwell and colleagues cautioned that cell culture media can generate artefacts because iron-dependent oxidation can produce species such as H₂O₂ and quinones/semiquinones [117]. They also suggested that any prooxidant activities in humans at the low polyphenol concentrations typically achieved after consumption of polyphenol-rich foods remain to be clearly established in vivo [118]. In vitro, quercetin and resveratrol aglycones can form quinone/quinone-methide intermediates and generate glutathione/protein thiol adducts, but such adducts have not been detected in vivo [119,120]. In contrast, EGCG-quinone–cysteine thiol adducts were isolated from the urine of mice given high, toxic doses of EGCG, but not after low doses [121].
3.3. Inhibition of Protein Kinases and Other Enzymes – Limitations
Others have reviewed resveratrol’s inhibition of multiple enzymes, including cyclooxygenase, lipoxygenase, PKCs, ERK1, JNK1, p38, Src, ribonucleotide reductase, DNA polymerases, PKD, and aromatase, typically with IC₅₀ values in the 10–60 μM range [122]. Another review summarized that EGCG inhibits various receptor tyrosine kinases, with effective concentrations generally ranging from 5 to 100 μM [123]. Another review article mentioned EGCG inhibition of cyclooxygenase-1 and -2, with IC₅₀ values of 17–28 μM [124]. Given that parent polyphenol aglycones occur at very low concentrations in plasma and tissues, the physiological relevance of inhibiting these enzymes remains uncertain.
3.4. Inhibition of Aβ Aggregation – Limitations
In vitro, several dietary polyphenols can directly interfere with Aβ self-assembly. Quercetin inhibits Aβ fibril formation and can promote disaggregation of preformed fibrils, at least in peptide/fibril model systems [125]. Resveratrol inhibits Aβ42 fibril formation and reduces Aβ-associated cytotoxicity, although it may not prevent oligomer formation [126]. EGCG can bind natively unfolded Aβ and redirect aggregation away from β-sheet–rich fibrils toward unstructured, off-pathway oligomers [127]. These inhibitory effects were observed at micromolar concentrations of the parent aglycones. Whether comparable Aβ-polyphenol interactions occur at the low nanomolar concentrations of aglycones, or their conjugated metabolites, particularly within the brain, remains to be determined.
4. Polyphenol Metabolites Formed by Gut Microbiota, Gut-Brain Axis
Because parent polyphenol aglycones are present only at very low levels in circulation, attention has shifted to the metabolism of dietary polyphenols by the gut microbiota to explain their health benefits. Because polyphenols are poorly absorbed in the small intestine, a substantial fraction reaches the colon, where the microbiota converts them, often into smaller molecules that are more readily absorbed [128]. Quercetin is degraded via C-ring fission and side-chain modifications into more absorbable phenolic acids, including 3,4-dihydroxyphenylacetic acid [129,130]. Gut microbes reduce resveratrol to dihydroresveratrol and, depending on an individual’s microbiota, further convert it to dehydroxylated products such as lunularin [131,132]. Microbial esterases first degalloylate EGCG, yielding epigallocatechin and gallic acid [133,134,135]. Epigallocatechin is then further metabolized to downstream products, including valerolactone-type metabolites [133,134,135]. Gallic acid itself can be converted to smaller phenolics such as pyrogallol, which is a more potent inducer of Nrf2-associated gene expression than its parent compound, EGCG [136]. The metabolite profile and yields vary substantially among individuals, reflecting differences in microbiota composition and metabolic capacity.
There is growing evidence that these microbial metabolites contribute importantly to some health benefits of dietary polyphenols. However, many of these gut-derived products are phenolic compounds that undergo extensive phase II metabolism in the liver, forming glucuronide and sulfate conjugates. Again, this raises the question of how glucuronide conjugates and intestinal microbial metabolites exert their biological effects. Defining how these microbiota-derived metabolites and their conjugates support neuroprotection through the gut-brain axis in AD remains a critical research priority.
5. Glucuronide and Sulfate Conjugates as Active Metabolites
Given the low circulating concentrations of parent polyphenols and the relatively higher levels of their conjugates, some researchers propose that glucuronide and sulfate metabolites may mediate at least part of the biological actions attributed to the parent compounds [137]. Glucuronide and sulfate conjugates have traditionally been viewed as inactive excretory metabolites; however, this assumption is increasingly being challenged, as a growing number of compounds exhibit biological activity in their conjugated forms. For example, morphine is activated by glucuronidation, and the antihypertensive drug minoxidil is activated by sulfation [138,139]. Morphine-6-glucuronide is actively transported across the BBB and can also be formed in the brain from morphine, where it contributes to analgesia [140,141].
There is direct evidence that glucuronide conjugates of both resveratrol and quercetin can reach CNS compartments, but their net brain exposure is generally low because glucuronides are polar anions and do not readily cross the BBB by passive diffusion. When detected in CSF or brain tissue, their presence is thought to reflect carrier-mediated transport processes at brain barrier sites [142]. A fraction of circulating polyphenol glucuronides may be taken up by BBB-associated cells and locally deconjugated to aglycones; brain tissue also expresses UDP-glucuronosyltransferases and sulfotransferases, enabling local reconjugation [143,144,145,146].
An alternative view is that polyphenol glucuronides and sulfates function primarily as circulating reservoirs. In this model, they can be hydrolyzed locally by β-glucuronidase and sulfatase enzymes to regenerate the corresponding aglycones within target tissues, where the liberated aglycones then exert biological effects [147,148]. This may occur more readily at sites of inflammation, where larger amounts of these hydrolases are released from cells [149,150]. Still, this process would release only small amounts of aglycones, which begs the question of how such low concentrations can produce biological effects.
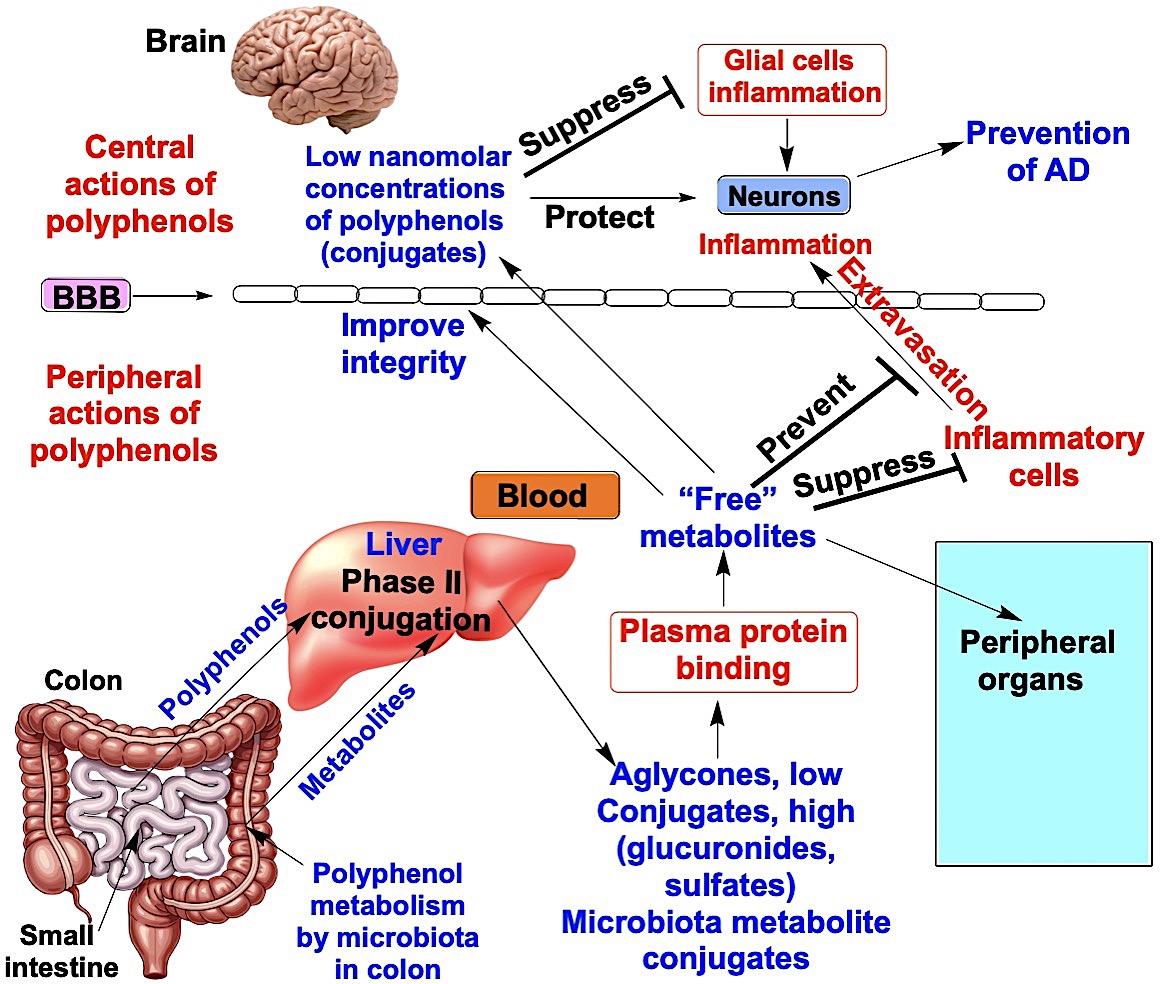
6. Direct Protective Actions of Polyphenols on Neurons – Binding to High-Affinity Receptors and Relevance to the Prevention of AD
Although only low nanomolar concentrations of polyphenols reach the brain after oral administration, they may still exert neuroprotective effects by directly acting on neurons through binding to high-affinity receptors. Two relevant molecular targets are the 67-kDa laminin receptor (67LR) and quinone reductase 2 (QR2); both respond to low nanomolar concentrations of polyphenols and may contribute to AD prevention.
6.1. Neuroprotective Actions of Polyphenols Mediated by 67LR
6.1.1. 67LR as an Evolutionarily Conserved Multifunctional Protein: Friend or Foe
The 67LR is a cell-surface laminin receptor originally implicated in tumor cell adhesion, invasion, and metastasis [151,152,153,154,155,156]. This receptor is evolutionarily conserved and is initially synthesized as a 37-kDa precursor (37LRP), also known as ribosomal protein SA (RPSA), which plays an essential role in ribosomal biogenesis and protein synthesis [155,157]. Through mechanisms that remain incompletely understood, the 37-kDa precursor is converted into the mature 67-kDa cell-surface form that functions as a high-affinity laminin receptor [154,155,156]. In addition, 67LR has been shown to mediate the internalization of a diverse array of pathogenic bacteria and viruses, as well as both cellular and infectious prion proteins [154,155,156,158]. It also serves as a coreceptor for the Aβ-prion protein complex, facilitating Aβ internalization and inducing neuronal death, an important step in the pathogenesis of AD [159,160]. Collectively, these findings indicate that 67LR plays multiple pathological roles.
The widespread tissue distribution of 67LR, including in the brain [161], suggests that it participates in important cellular processes beyond cancer invasion and metastasis, infections, and AD. We and others have demonstrated that 67LR also mediates beneficial functions, including neuritogenesis, neuroprotection, and BBB integrity [162,163,164,165]. Recent studies have further emphasized the importance of 67LR in the axonal growth of dorsal root ganglion neurons [166]. Moreover, 67LR mediates pigment epithelium-derived factor-induced cortical neuronal morphogenesis [167]. Gene knockout studies have demonstrated that homozygous deletion of Rpsa, which encodes 37LRP, results in embryonic lethality, underscoring its critical role in development [154]. Thus, 67LR may exert either pathological or beneficial effects depending on the nature of the ligands that bind to it or are internalized via this receptor.
6.1.2. 67LR as a High-Affinity Target for Dietary Polyphenols and Their Metabolites
Tachibana and colleagues identified high-affinity binding of EGCG (Kd = 40 nM) to 67LR and demonstrated a role for this receptor in inducing cancer cell death at low micromolar concentrations of EGCG [168,169]. This discovery arose from an unbiased screening strategy designed to identify proteins whose expression is upregulated by retinoic acid and that sensitize cancer cells to EGCG-induced apoptosis [168]. In contrast to the cancer cell death observed at micromolar concentrations, EGCG-induced neuroprotection and neuritogenesis occur at low nanomolar concentrations in a 67LR-dependent manner [162,163,170,171].
We and others have demonstrated that EGCG binds to two distinct sites on 67LR, including the peptide G region, a conserved 20-amino-acid sequence critical for ligand interaction [171,172]. Molecular docking studies indicate that the “A” site binds EGCG and other polyphenols with higher affinity and is primarily composed of the second half of the peptide G region, which contains a hydrophobic palindromic sequence encompassing Trp175 and Trp176 (Figure 2, Table 2). Using the isolated peptide G, EGCG was shown to bind this palindrome with high affinity (Kd = 1.95 nM) [171]. In contrast, the “B” site involves amino acid residues from the first half of the peptide G region (outside the palindrome) and binds these polyphenols with lower affinity than the A site. Notably, other dietary polyphenols, including resveratrol and quercetin, also interact with 67LR via the peptide G region [170,171]. In addition, α-tocopherol and α-tocotrienol have been shown to interact with 67LR through Trp176 within the peptide G sequence, further supporting the role of this region as a shared polyphenol-binding motif [173,174].
Interestingly, glucuronide and sulfate conjugates of these polyphenols exhibit higher binding affinity for peptide G than their parent aglycone forms, suggesting that metabolic modification enhances receptor recognitions [170,171,172]. Molecular docking studies further showed that intestinal microbial metabolites of quercetin (3,4-dihydroxyphenylacetic acid), trans-resveratrol (dihydroresveratrol), and EGCG [5-(3′,4′-dihydroxyphenyl)-γ-valerolactone] bind to 67LR, albeit with lower affinity than the corresponding polyphenol aglycones (Table 2). However, glucuronide conjugates of these intestinal microbial metabolites dock with higher affinity than their unconjugated forms (Table 2). Binding studies using peptide G revealed that dihydroresveratrol binds with higher affinity than its parent compound, trans-resveratrol [171]. Dihydroresveratrol also induced elevations in intracellular cAMP, as with trans-resveratrol, via a 67LR-dependent mechanism [171].
Many dietary polyphenols accumulate inside cells only to a limited extent because they are relatively polar and are subject to active efflux by membrane transporters [175]. Glucuronide and sulfate conjugates are even more hydrophilic organic anions; consequently, their cellular entry is often transporter-dependent, while efflux transporters can further limit intracellular exposure [176]. In this context, a cell-surface binding protein such as the 67LR would be well positioned to sense extracellular polyphenols without requiring substantial intracellular accumulation. Collectively, these findings support the concept that 67LR serves as a common receptor for structurally diverse dietary polyphenol aglycones, as well as their glucuronide conjugates, intestinal microbial metabolites, and corresponding glucuronides, thereby potentially mediating their biological and chemopreventive actions.
6.1.3. 67LR Structural Basis for Binding of Polyphenol Glucuronide/Sulfate Conjugates
Glucuronidated and sulfate conjugates of polyphenols are more water-soluble and are generally expected to bind weakly to receptors. Contrary to the long-held view, these polyphenol conjugates bound to 67LR with higher affinity than their aglycones [170,171,172]. The mechanisms by which morphine-6-glucuronide and minoxidil sulfate bind to their receptors with higher affinity and exert greater biological activity remain unclear [138,139]. In contrast, the 67-kDa laminin receptor (67LR) possesses unique structural features that facilitate high-affinity binding of glucuronide- and sulfate-conjugated polyphenols. 67LR interacts with heparan sulfate proteoglycans, a class of glycosaminoglycans (GAGs) characterized by uronic acids (D-glucuronic acid and L-iduronic acid) and sulfate residues [177]. GAG-binding motifs have been identified in other proteins and typically follow the consensus sequences X-B-B-X-B-X and X-B-B-B-X-X-B-X, where B denotes a basic amino acid residue, and X denotes a hydropathic amino acid residue [178,179]. The peptide G region of 67LR contains one such GAG-binding motif, which likely provides additional interaction coordinates necessary for high-affinity binding of polyphenol conjugates at both the A and B binding sites (Figure 3).
6.1.4. 67LR-Mediated cAMP Signaling and Downstream Events for AD Prevention
We previously reported a rapid, several-fold increase in intracellular cAMP levels in neuronal cells treated with low nanomolar concentrations of resveratrol, EGCG, and quercetin [170,171]. This elevation was prevented by a 67LR-blocking antibody, indicating that 67LR mediates cAMP induction by these polyphenols [171]. Because 67LR is not a G-protein-coupled receptor, it cannot directly initiate adenylyl cyclase (AC) activation [155]. Although resveratrol has been reported to inhibit phosphodiesterases (PDEs), which could elevate intracellular cAMP levels, this effect requires relatively high concentrations (20–40 µM) [180]. Consistent with this, we did not observe inhibition of PDE4B or PDE4D at low nanomolar concentrations of these polyphenols.
Compared with healthy age-matched controls, patients with AD and AD mouse models exhibit markedly reduced levels of cAMP, AC, and PKA in the hippocampus [181]. The cAMP-elevating neuroprotective peptide pituitary adenylyl cyclase-activating polypeptide (PACAP) protects neurons from Aβ oligomer (AβO)-induced cell death [182], yet PACAP levels are reduced in AD patients compared with age-matched controls [183]. Administration of PACAP improves cognitive performance in AD transgenic mice [184]. Phosphodiesterase (PDE) inhibitors are currently under clinical investigation for AD treatment [185]; however, chronic, non-physiological activation of cAMP signaling by these agents can lead to cognitive impairment, hyperexcitability, and hyperalgesia [186]. In contrast, dietary polyphenols induce modest, localized increases in intracellular cAMP and may therefore avoid the adverse effects associated with synthetic PDE inhibitors. However, the mechanisms by which 67LR binding enhances cAMP generation remain unclear.
As shown in Figure 4, the cAMP, elevated in response to dietary polyphenols, activates protein kinase A (PKA), which phosphorylates the transcription factor, cAMP response element-binding protein, CREB at Ser133, leading to increased expression of brain-derived neurotrophic factor (BDNF), a neurotrophin essential for neuronal survival [187,188,189]. Other studies have similarly reported enhanced CREB activation in neurons treated with a low concentration of quercetin-3-glucuronide [68]. In addition, cAMP signaling can enhance the activity of sirtuin 1 (SIRT1), an NAD⁺-dependent deacetylase [190]. The cAMP/CREB pathway increases expression of nicotinamide phosphoribosyltransferase (NAMPT), thereby increasing intracellular NAD⁺ levels required for optimal SIRT1 activity [191]. Activated SIRT1 deacetylates and stimulates transcriptional regulators such as PGC-1α, which is involved in mitochondrial biogenesis and antioxidant defense, and FOXO3 transcription factors, which regulate oxidative stress responses and longevity [192,193]. SIRT1 also deacetylates and inhibits NF-κB, thereby reducing inflammation, an effect that may benefit cognitive function [194]. Resveratrol has been widely shown to increase SIRT1 expression or activity, contributing to neuroprotection against Aβ toxicity in experimental models of AD [195,196]. Quercetin and EGCG have also been reported to activate SIRT1 [197,198]. However, it remains to be determined whether aglycones and conjugates of these polyphenols, at the low concentrations achieved in the brain, are sufficient to activate SIRT1 in neurons. Both BDNF and SIRT1 have been shown to protect neurons from Aβ-induced neurotoxicity and to enhance synaptic plasticity and cognitive function [54,199]. Since cAMP-PKA signaling becomes dysregulated with aging [200], polyphenol-induced increases in cAMP-PKA signaling may be beneficial.
The cAMP/PKA pathway, activated by polyphenol treatment, phosphorylates and activates protein phosphatase 2A (PP2A), a serine/threonine protein phosphatase [201,202]. This is particularly significant because PP2A is a major phosphatase responsible for dephosphorylating the neuronal microtubule-associated protein tau [203]. Activation of PP2A prevents tau hyperphosphorylation, thereby inhibiting the formation of neurofibrillary tangles and subsequent neuronal death. This mechanism is particularly relevant to AD because PP2A activity is reduced in AD patients’ brains [204]. Thus, activation of cAMP signaling by polyphenols via 67LR may represent a central mechanism underlying their neuroprotective effects and their potential to mitigate AD pathology.
6.1.5. 67LR as a Coreceptor for Aβ-Prion Complex – Neuronal Cell Death
Some of the neurotoxicity associated with Aβ is believed to be mediated by high-affinity binding of Aβ oligomers (AβO) to neuronal cell-surface receptors [205,206]. Strittmatter and colleagues identified that the lipid raft-associated cellular prion protein (PrPC) is a major high-affinity receptor for AβO [207,208]. The AβO-PrPC complex can subsequently interact with various cell-surface co-receptors to mediate AβO-induced neuronal toxicity [205,206]. Weiss and his associates found that the 67LR, which is also localized to lipid rafts, is one such co-receptor for the AβO-PrPC complex [159,160]. PrPC binds to 67LR through its peptide G region [177]. The binding of the AβO-PrPC complex to 67LR promotes the internalization of AβO and leads to subsequent neuronal cell death [159,160]. Notably, intranasal administration of a 67LR-blocking antibody reduced neurodegeneration in AD transgenic mice [209].
6.1.6. Potential Antagonism Between AβO-PrPC and Dietary Polyphenols at 67LR
Because both the AβO-PrPC complex and polyphenols and their metabolites bind to the same peptide G region of 67LR [170,171,177], they may compete for this site. Consequently, polyphenols may antagonize AβO-PrPC binding, thereby preventing Aβ internalization and reducing neurotoxicity. Furthermore, polyphenol binding to 67LR induces cAMP/PKA signaling, leading to activation of CREB, SIRT1, and PP2A, and promoting neuroprotection and synaptic plasticity, as illustrated in Figure 4. In addition, cAMP reduces Aβ internalization [210]. Consistent with this mechanism, others have shown that cAMP protects cortical neurons from AβO-induced neuronal cell death [211].
6.1.7. Phenol Red Binds 67LR and Interferes with the Binding of Polyphenols
Phenol red, a commonly used pH-indicator in cell culture media, binds with high affinity to 67LR at its peptide G region. This binding interferes with the interaction of polyphenols and Aβ with 67LR, which occurs at the same site. Commonly used media, including DMEM, RPMI 1640, and Neurobasal, contain phenol red at concentrations as high as 42, 14, and 21 μM, respectively. The high phenol red concentration in the culture medium requires higher polyphenol concentrations to achieve neuroprotective effects. Initially, we were unaware of this interference, and EGCG required high nanomolar concentrations to elicit neuritogenic and neuroprotective effects in phenol red-containing media [162,163]. Subsequently, cells were cultured in phenol red-free medium for two to three passages. Under these conditions, as little as 5 nM EGCG was sufficient to produce neuroprotective effects [171]. These findings underscore the importance of avoiding phenolic compounds such as phenol red in cell culture media when investigating the in vitro effects of polyphenols at low nanomolar concentrations.
6.1.8. 67LR as a Redox Sensor
Physical binding of polyphenols and their metabolites through phenolic groups does not preclude an important role for redox sensitivity of these molecules in redox-mediated cellular regulation. The peptide G region of 67LR, which is involved in the binding of these polyphenols, contains highly redox-sensitive vicinal cysteine residues that can react with hydrogen peroxide [212]. In addition, this region harbors a Met–Trp–Trp–Met motif within its palindromic sequence. Given the established ability of tryptophan residues to function as redox centers and of methionine residues to participate in intramolecular electron transfer, this Met–Trp–Trp–Met motif may support localized charge-transfer or redox interactions, particularly under oxidative conditions. Others have shown sulfhydryl oxidase-like activity for 67LR [213]. At present, it is not known whether transiently formed oxidized polyphenol species, such as semiquinone or quinone intermediates, interact with this redox-active center of 67LR and are subsequently reduced back to their original phenolic forms. Furthermore, binding of EGCG to 67LR has been shown to induce a modest elevation of sublethal levels of H2O2, which are increasingly recognized as important mediators of cellular signaling [214].
6.2. Neuroprotective Actions of Polyphenols by Inhibiting QR2
QR2 is inhibited by resveratrol (Kd = 35 nM) and quercetin (Kd = 50 nM) [215]. Inhibition of this enzyme by these polyphenols leads to the upregulation of antioxidant enzyme expression [215]. QR2 is overexpressed with aging and may contribute to metabolic stress and cognitive deficits [216]. Notably, QR2 is highly expressed in the brains of AD patients, particularly in the hippocampus [217]. Specific QR2 inhibitors have been shown to reduce metabolic burden and reverse AD phenotypes in mouse models [216]. Therefore, inhibition of QR2 by resveratrol and quercetin may improve cognitive function and provide therapeutic benefits for patients with AD.
The exact function of QR2 remains unclear; whether it catalyzes quinone reduction or primarily participates in cell signaling is still under investigation [218]. Unlike typical reductases, QR2 cannot use NAD(P)H as a reducing cofactor, retains a functional FAD cofactor, and can exist in both oxidized and reduced states. These characteristics suggest that QR2 may play a role in redox sensing and regulation rather than acting as a traditional detoxifying enzyme for electrophilic quinones [218]. Given the low brain levels of polyphenol aglycones, it remains to be determined whether their glucuronide and sulfate conjugates or their oxidative metabolites can inhibit QR2 or interact with the enzyme.
7. Indirect Neuroprotective Actions of Polyphenols – AD Prevention
7.1. Actions on Glial Cells in the CNS
Neuroprotective effects of polyphenols may also arise indirectly through their modulation of glial cell function in the brain. Aβ accumulation activates microglia and induces the production of proinflammatory cytokines, leading to a chronic state of neuroinflammation [219]. Resveratrol has been shown to inhibit prostaglandin E2 (PGE2) production and free radical formation by activated microglial cells, primarily by modulating multiple steps in the cyclooxygenase/PGE2 signaling pathway [220]. It also suppresses NF-κB transcriptional activation, thereby reducing cytokine secretion by Aβ-activated astrocytes and microglia [221]. Similarly, EGCG exerts broad anti-inflammatory effects by decreasing cytokine release through transcriptional downregulation [222]. Both resveratrol and EGCG activate SIRT1, which markedly attenuates Aβ-induced NF-κB signaling, contributing to their strong neuroprotective actions [223]. Quercetin, in turn, mitigates inflammatory responses in microglial cells by activating the Nrf2 pathway, which regulates heme oxygenase-1 expression [224]. However, these anti-inflammatory effects have demonstrated in vitro mostly using micromolar concentrations of polyphenol aglycones. Whether similar effects can be elicited by low nanomolar concentrations of their glucuronide and sulfate conjugates remains to be determined. In astrocytes, the PEDF peptide mediates anti-inflammatory actions via the 67LR [225]; however, it remains unclear whether polyphenols also exert their anti-inflammatory effects through this receptor in astrocytes.
7.2. Peripheral Actions
Although brain exposure to dietary quercetin, resveratrol, and EGCG is limited, each may indirectly promote resilience to AD by attenuating systemic drivers of neurodegeneration through peripheral actions. Chronic elevations in circulating cytokines and sustained immune-cell activation can exacerbate cerebrovascular dysfunction and amplify neuroinflammation in AD [226,227]. In vivo, quercetin shows systemic anti-inflammatory activity and can also improve vascular risk factors, including blood pressure, which may translate into protection against cognitive decline [228,229]. Resveratrol can ameliorate peripheral metabolic dysregulation (e.g., insulin resistance) via pathways that include SIRT1.
BBB integrity is compromised in AD, and blood-derived plasma proteins, including fibrinogen, can extravasate into the brain parenchyma and promote neuronal degeneration [230,231,232]. EGCG attenuates serum extravasation by acting via 67LR [164]. Resveratrol also supports endothelial nitric oxide bioavailability and overall vascular function, potentially protecting the brain indirectly by improving cerebral perfusion and preserving BBB integrity [233,234]. In vitro, micromolar concentrations of quercetin have been reported to attenuate Aβ-treated cytotoxicity in human brain microvascular endothelial [235]. Since QR2 is expressed in endothelial cells and can generate ROS [218] that may weaken blood-brain barrier (BBB) integrity, resveratrol and quercetin, by inhibiting QR2, could help preserve BBB function. However, mechanistic evidence remains limited, and it is still unclear whether these protective effects are mediated by the parent aglycones or by bioavailable glucuronide and sulfate conjugates acting through 67LR and QR2.
EGCG also exerts important gut-systemic effects. Although it is extensively metabolized in the gut, EGCG can reshape the gut microbiota and strengthen intestinal barrier function, thereby reducing the translocation of pro-inflammatory microbial products (e.g., lipopolysaccharide) implicated in BBB impairment and neuroinflammatory signaling [236]. Collectively, these peripheral anti-inflammatory, metabolic, gut-barrier, and vasoprotective actions suggest plausible mechanisms by which quercetin, resveratrol, and EGCG could mitigate AD pathobiology despite their low brain concentrations.
8. Combining Low-Dose Polyphenols: A Translational Approach
When polyphenols are administered at low, well-tolerated doses, only very low concentrations of the parent compounds and/or their conjugates typically reach the brain. As a result, receptor occupancy may be suboptimal, leading to limited neuroprotection, for example, insufficient competition with AβO-PrPC complexes for binding to 67LR. One way to increase efficacy for AD prevention would be to raise the dose. However, although higher dosing may increase systemic exposure and CNS delivery, it can also increase the risk of peripheral toxicity. High-dose EGCG/green tea extract has been linked to hepatocellular liver injury in humans and produces marked liver toxicity in preclinical models [237]. High-dose resveratrol has also been associated with renal toxicity in animal studies [238,239]. High-dose quercetin is toxic and can interact with certain drugs [240]. In a randomized, placebo-controlled trial in patients with mild-to-moderate AD, high-dose resveratrol was associated with greater brain volume loss on MRI compared with placebo, an unexpected finding [79].
An alternative strategy is to administer three or four well-characterized dietary polyphenols at low doses in combination. Even if each compound reaches only low concentrations in the brain, agents that converge on the same pathway (e.g., 67LR-mediated signaling) could produce additive effects that approximate the impact of a higher dose of a single compound. Moreover, because these polyphenols differ structurally, EGCG is a galloylated flavanol, quercetin is present largely as glucuronide and sulfate conjugates, and resveratrol as a stilbene (often in conjugated forms), they may also engage complementary mechanisms that enable synergistic interactions. At the same time, such combinations should be evaluated for potential antagonism. Overall, supplementation with a limited set of structurally diverse polyphenols at low doses may be a practical strategy to promote additive or synergistic neuroprotective effects for AD prevention while minimizing toxicity risk.
9. Conclusions and Perspectives
Because AD develops over decades, its extended prodromal phase provides a realistic window for prevention. Yet AD is multifactorial, so meaningful risk reduction will likely require interventions that engage multiple, divergent pathways. Dietary polyphenols are appealing in this context because they can influence several AD-relevant mechanisms.
A central constraint is pharmacokinetics. Most polyphenols are rapidly converted to glucuronide and sulfate conjugates and are extensively protein-bound, leaving only a small unbound fraction available for tissue distribution. Consequently, both aglycones and conjugated metabolites typically reach the brain at only low-nanomolar concentrations after limited passage across the BBB. Microbiota-derived metabolites follow a similar pattern: they are absorbed, quickly conjugated, and circulate predominantly as conjugates. Traditionally, these conjugates have been viewed as inactive excretory products or as reservoirs that can regenerate aglycones. An important unresolved issue, however, is whether conjugated metabolites possess intrinsic biological activity, particularly through engagement of high-affinity targets, and therefore contribute directly to neuroprotection.
Although polyphenols can scavenge ROS, direct antioxidant effects in vivo, especially in the brain, are likely limited because tissue concentrations are low and conjugates generally have weaker radical-scavenging capacity than their parent aglycones. A more plausible mechanism is the “indirect antioxidant” action, in which quercetin, resveratrol, EGCG, and/or their metabolites activate redox-responsive signaling that upregulates endogenous antioxidant and cytoprotective enzymes. Such pathway-level reinforcement can persist even after the compounds themselves are removed, providing a biologically credible route to sustained protection.
Two targets emphasized in this review are particularly relevant to AD pathogenesis and illustrate how low-exposure ligands could still produce meaningful effects. The 67LR binds the AβO-PrPC complex, promotes Aβ internalization, and triggers downstream neurotoxic signaling. In contrast, quercetin, resveratrol, and EGCG, together with their conjugated metabolites, bind the same site on 67LR, competitively antagonize AβO-PrPC binding, and activate neuroprotective signaling. A second target is QR2, an enzyme elevated in AD and associated with cognitive decline; resveratrol and quercetin bind QR2 with high affinity, inhibit its activity, and improve cognition in experimental settings. Notably, both 67LR and QR2 may also function as redox sensors that react with oxidized polyphenol species, suggesting that redox-dependent chemistry can contribute even within a receptor-based framework. Moving forward, it will be important to test whether newly identified polyphenol receptors also contain structural features that enable redox sensing, thereby clarifying when physical binding targets and redox-sensing targets are distinct, and when they may be the same entity.
The ability of steroidal hormones and catecholamines to act at very low concentrations provides a useful analogy: selective binding to a limited set of receptors enables signal amplification through downstream transduction pathways. Polyphenols and their conjugates may similarly exert effects at low concentrations by engaging high-affinity targets that initiate defined signaling cascades. One illustrative mechanism is receptor-mediated elevation of cAMP, a second messenger whose amplification can drive broad transcriptional and functional responses despite minimal ligand exposure.
A major translational hurdle is achieving therapeutically meaningful CNS exposure, as brain levels are typically very low. Simply escalating doses to raise CNS concentrations is not an ideal solution, as higher intake can increase peripheral toxicity risk (e.g., hepatotoxicity reported with high EGCG exposure), raise the likelihood of drug-nutrient interactions, and potentially introduce adverse effects during long-term use (including concerns raised for very high-dose resveratrol or quercetin in some contexts).
A more practical strategy is low-dose combination supplementation using three or four well-characterized polyphenols. Even if each compound reaches only low brain concentrations, agents that converge on the same protective node (for example, 67LR-mediated neuroprotective signaling) could produce additive or synergistic effects approximating those of a higher dose of a single agent while reducing toxicity risk. Overall, defining which circulating conjugates and microbiota-derived metabolites are active, identifying their high-affinity targets, and delineating how redox-dependent processes intersect with receptor signaling will be essential for translating dietary polyphenols into an evidence-based approach for AD prevention in humans.
Author Contributions
Conceptualization, R.S., K.S., W.J.M., and R.G.; methodology, R.S. and R.G.; validation, R.S. and R.G.; formal analysis, R.G.; investigation, R.S. and R.G.; resources, RG. and WJM.; writing—original draft preparation, R.S. and R.G; writing—review and editing, R.S., K.S., W.J.M., and R.G.; supervision, R.G. and W.J.M.; project administration, R.G.; funding acquisition, W.J.M. and R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH grant NINDS/NIA RF1 NS130681 (to WJM and RG).
Informed Consent Statement
Not applicable
Data Availability Statement
Acknowledgments
Given the breadth of the literature, we may have inadvertently missed some relevant studies, and we apologize for any omissions.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| aMCI | Amnestic mild cognitive impairment |
| Aβ | Amyloid-β |
| AβO | Amyloid-β oligomers |
| AD | Alzheimer’s disease |
| BBB | Blood-brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CREB | cAMP response element-binding protein |
| CSF | Cerebrospinal fluid |
| EGCG | (−)-Epigallocatechin-3-gallate |
| GAG | Glycosaminoglycan |
| 67LR | 67-kDa laminin receptor |
| NMNAT | Nicotinamide mononucleotide adenylyltransferase |
| PDE | Phosphodiesterase |
| PKA | Protein kinase A |
| PP2A | Protein phosphatase 2A |
| PrPC | Cellular prion protein |
| Q-3-G | Quercetin-3-O-glucuronide |
| R-3-G | Resveratrol-3-O-glucuronide |
| ROS | Reactive oxygen species |
| SIRT1 | Sirtuin 1 (NAD⁺-dependent deacetylase) |
References
- World Health Organization (WHO). Dementia (Fact sheet). 31 March 2025.
- Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [CrossRef] [PubMed]
- Alzheimer's disease facts and figures. Alzheimer's & Dementia 2025, 21, e70235. [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Tifratene, K.; Robert, P.; Metelkina, A.; Pradier, C.; Dartigues, J.F. Progression of mild cognitive impairment to dementia due to AD in clinical settings. Neurology 2015, 85, 331–338. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer's Disease, and Dementia: From FINGER to World-Wide FINGERS. J Prev Alzheimers Dis 2020, 7, 29–36. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Chen, F.; Ho, L.; Lin, H.Y.; Yuan, C.; Wong, J.; Wang, J. Benefits of dietary polyphenols in Alzheimer's disease. Front Aging Neurosci 2022, 14, 1019942. [Google Scholar] [CrossRef]
- Malar, D.S.; Devi, K.P. Dietary polyphenols for treatment of Alzheimer's disease--future research and development. Curr Pharm Biotechnol 2014, 15, 330–342. [Google Scholar] [CrossRef]
- Bukhari, S.N.A. Dietary Polyphenols as Therapeutic Intervention for Alzheimer's Disease: A Mechanistic Insight. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Kabir, E.R.; Chowdhury, N.M.; Yasmin, H.; Kabir, M.T.; Akter, R.; Perveen, A.; Ashraf, G.M.; Akter, S.; Rahman, M.H.; Sweilam, S.H. Unveiling the Potential of Polyphenols as Anti-Amyloid Molecules in Alzheimer's Disease. Curr Neuropharmacol 2023, 21, 787–807. [Google Scholar] [CrossRef]
- Godos, J.; Micek, A.; Mena, P.; Del Rio, D.; Galvano, F.; Castellano, S.; Grosso, G. Dietary (Poly)phenols and Cognitive Decline: A Systematic Review and Meta-Analysis of Observational Studies. Mol Nutr Food Res 2024, 68, e2300472. [Google Scholar] [CrossRef]
- Colizzi, C. The protective effects of polyphenols on Alzheimer's disease: A systematic review. Alzheimers Dement (N Y) 2019, 5, 184–196. [Google Scholar] [CrossRef]
- Chen, G.; Su, Y.; Chen, S.; Lin, T.; Lin, X. Polyphenols and Alzheimer's Disease: A Review on Molecular and Therapeutic Insights With In Silico Support. Food Sci Nutr 2025, 13, e70496. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Zhang, J.; Yu, G.; Zhu, J.; Nie, Y. Polyphenol consumption and neurodegeneration risk: a systematic meta-analysis of randomized controlled trials bridging nutrition and cognitive health. Food & Function 2026. [Google Scholar] [CrossRef]
- Albadrani, H.M.; Chauhan, P.; Ashique, S.; Babu, M.A.; Iqbal, D.; Almutary, A.G.; Abomughaid, M.M.; Kamal, M.; Paiva-Santos, A.C.; Alsaweed, M.; et al. Mechanistic insights into the potential role of dietary polyphenols and their nanoformulation in the management of Alzheimer's disease. Biomed Pharmacother 2024, 174, 116376. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Khatri, N.; Rahman, Z.N.; Menezes, A.A.; Martini, J.; Shehjar, F.; Mujeeb, N.; Shah, Z.A. Neuroprotective Potential of Flavonoids in Brain Disorders. Brain Sci 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer's Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer's Disease Through Prion Protein and mGluR5. Adv Pharmacol 2018, 82, 293–323. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Aβ profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Molecular Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef]
- Dyck, C.H.v.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. New England Journal of Medicine 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Dai, C.L.; Liu, F.; Iqbal, K. Multi-Targets: An Unconventional Drug Development Strategy for Alzheimer's Disease. Front Aging Neurosci 2022, 14, 837649. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Rong, Z.; Li, J.; Wu, H.; Wei, J. Three-dimensional interactive network: Mitochondrial-metabolic-calcium homeostasis driving Alzheimer’s disease. Genes & Diseases 2025, 101846. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduction and Targeted Therapy 2024, 9, 211. [Google Scholar] [CrossRef]
- Mezzanotte, M.; Stanga, S. Brain Iron Dyshomeostasis and Ferroptosis in Alzheimer's Disease Pathophysiology: Two Faces of the Same Coin. Aging Dis 2024, 16, 2615–2640. [Google Scholar] [CrossRef]
- Klohs, J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegenerative Diseases 2020, 19, 109–127. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Perone, I.; Wang, M.; Yang, S.; Uchendu, S.; Bolat, D.; Datta, D. Dysregulated calcium signaling in the aged primate association cortices: vulnerability to Alzheimer's disease neuropathology. Front Aging Neurosci 2025, 17, 1610350. [Google Scholar] [CrossRef]
- Cummings, J.L.; Burstein, A.H.; Fillit, H. Alzheimer Combination Therapies: Overview and Scenarios. J Prev Alzheimers Dis 2025, 12, 100328. [Google Scholar] [CrossRef]
- Liu, X.; Dhana, K.; Barnes, L.L.; Tangney, C.C.; Agarwal, P.; Aggarwal, N.; Holland, T.M.; Beck, T.; Evans, D.A.; Rajan, K.B. A healthy plant-based diet was associated with slower cognitive decline in African American older adults: a biracial community-based cohort. Am J Clin Nutr 2022, 116, 875–886. [Google Scholar] [CrossRef]
- Mani, V.; Arfeen, M.; Dhaked, D.K.; Mohammed, H.A.; Amirthalingam, P.; Elsisi, H.A. Neuroprotective Effect of Methanolic Ajwa Seed Extract on Lipopolysaccharide-Induced Memory Dysfunction and Neuroinflammation: In Vivo, Molecular Docking and Dynamics Studies. Plants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Mani, V.; Parle, M.; Ramasamy, K.; Abdul Majeed, A.B. Reversal of memory deficits by Coriandrum sativum leaves in mice. J Sci Food Agric 2011, 91, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Liu, W.; Zhou, H.Y.; Gui, Y.R.; Yang, Y.H.; Wu, M.J.; Xiao, Y.F.; Shang, J.T.; Long, G.F.; Shu, X.J. Epigallocatechin-3-gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr Med Sci 2020, 40, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Barnes, L.L.; Bennett, D.A.; Aggarwal, N.T. MIND diet slows cognitive decline with aging. Alzheimers Dement 2015, 11, 1015–1022. [Google Scholar] [CrossRef]
- van den Brink, A.C.; Brouwer-Brolsma, E.M.; Berendsen, A.A.M.; van de Rest, O. The Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) Diets Are Associated with Less Cognitive Decline and a Lower Risk of Alzheimer's Disease-A Review. Adv Nutr 2019, 10, 1040–1065. [Google Scholar] [CrossRef]
- Cherian, L.; Wang, Y.; Fakuda, K.; Leurgans, S.; Aggarwal, N.; Morris, M. Mediterranean-Dash Intervention for Neurodegenerative Delay (MIND) Diet Slows Cognitive Decline After Stroke. J Prev Alzheimers Dis 2019, 6, 267–273. [Google Scholar] [CrossRef]
- Huang, L.; Tao, Y.; Chen, H.; Chen, X.; Shen, J.; Zhao, C.; Xu, X.; He, M.; Zhu, D.; Zhang, R.; et al. Mediterranean-Dietary Approaches to Stop Hypertension Intervention for Neurodegenerative Delay (MIND) Diet and Cognitive Function and its Decline: A Prospective Study and Meta-analysis of Cohort Studies. Am J Clin Nutr 2023, 118, 174–182. [Google Scholar] [CrossRef]
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project 1. Am J Clin Nutr 2006, 83, 355–361. [Google Scholar] [CrossRef]
- Ide, K.; Yamada, H.; Takuma, N.; Park, M.; Wakamiya, N.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Green tea consumption affects cognitive dysfunction in the elderly: a pilot study. Nutrients 2014, 6, 4032–4042. [Google Scholar] [CrossRef]
- Singh, N.; Agrawal, M.; Doré, S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem Neurosci 2013, 4, 1151–1162. [Google Scholar] [CrossRef]
- Rocha-González, H.I.; Ambriz-Tututi, M.; Granados-Soto, V. Resveratrol: a natural compound with pharmacological potential in neurodegenerative diseases. CNS Neurosci Ther 2008, 14, 234–247. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Wang, J.; Ho, L.; Zhao, W.; Dubner, L. Roles of resveratrol and other grape-derived polyphenols in Alzheimer's disease prevention and treatment. Biochim Biophys Acta 2015, 1852, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front Aging Neurosci 2014, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer's and Parkinson's Disease. Front Aging Neurosci 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Dempsey, R.J.; Vemuganti, R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem Int 2015, 89, 75–82. [Google Scholar] [CrossRef]
- Paula, P.C.; Angelica Maria, S.G.; Luis, C.H.; Gloria Patricia, C.G. Preventive Effect of Quercetin in a Triple Transgenic Alzheimer's Disease Mice Model. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Sabogal-Guáqueta, A.M.; Muñoz-Manco, J.I.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer's disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer's disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef]
- Moreno, L.; Puerta, E.; Suárez-Santiago, J.E.; Santos-Magalhães, N.S.; Ramirez, M.J.; Irache, J.M. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer's disease. Int J Pharm 2017, 517, 50–57. [Google Scholar] [CrossRef]
- Lv, M.; Yang, S.; Cai, L.; Qin, L.Q.; Li, B.Y.; Wan, Z. Effects of Quercetin Intervention on Cognition Function in APP/PS1 Mice was Affected by Vitamin D Status. Mol Nutr Food Res 2018, 62, e1800621. [Google Scholar] [CrossRef]
- Dzobo, K.; Hassen, N.; Senthebane, D.A.; Thomford, N.E.; Rowe, A.; Shipanga, H.; Wonkam, A.; Parker, M.I.; Mowla, S.; Dandara, C. Chemoresistance to Cancer Treatment: Benzo-α-Pyrene as Friend or Foe? Molecules 2018, 23. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, J.; Zhong, L.; Wang, N.; Yang, L.; Liu, C.C.; Li, H.; Wang, X.; Zhou, Y.; Zhang, Y.; et al. Quercetin stabilizes apolipoprotein E and reduces brain Aβ levels in amyloid model mice. Neuropharmacology 2016, 108, 179–192. [Google Scholar] [CrossRef]
- Zhu, H.; Huang, J.; Chen, Y.; Li, X.; Wen, J.; Tian, M.; Ren, J.; Zhou, L.; Yang, Q. Resveratrol pretreatment protects neurons from oxygen–glucose deprivation/reoxygenation and ischemic injury through inhibiting ferroptosis. Bioscience, Biotechnology, and Biochemistry 2022, 86, 704–716. [Google Scholar] [CrossRef]
- Feng, X.; Liang, N.; Zhu, D.; Gao, Q.; Peng, L.; Dong, H.; Yue, Q.; Liu, H.; Bao, L.; Zhang, J.; et al. Resveratrol inhibits β-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS One 2013, 8, e59888. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Dave, K.R.; Pérez-Pinzón, M.A. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab 2006, 26, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci 2005, 25, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res 2008, 1214, 177–187. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, N.; Liu, X. Resveratrol and Amyloid-Beta: Mechanistic Insights. Nutrients 2017, 9, 1122. [Google Scholar] [CrossRef]
- Sousa, J.C.e.; Santana, A.C.F.; MagalhÃEs, G.J.P. Resveratrol in Alzheimer's disease: a review of pathophysiology and therapeutic potential. Arq Neuropsiquiatr 2020, 78, 501–511. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Chen, J.-Y.; Ouyang, D.; Lu, J.-H. Quercetin in Animal Models of Alzheimer’s Disease: A Systematic Review of Preclinical Studies. International Journal of Molecular Sciences 2020, 21, 493. [Google Scholar] [CrossRef]
- Cascella, M.; Bimonte, S.; Muzio, M.R.; Schiavone, V.; Cuomo, A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer's disease: an overview of pre-clinical studies and translational perspectives in clinical practice. Infect Agent Cancer 2017, 12, 36. [Google Scholar] [CrossRef]
- Moon, J.-H.; Tsushida, T.; Nakahara, K.; Terao, J. Identification of quercetin 3-O-β-D-glucuronide as an antioxidative metabolite in rat plasma after oral administration of quercetin. Free Radical Biology and Medicine 2001, 30, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Tamura, G.; Gold, C.; Ferro-Luzzi, A.; Ames, B.N. Fecalase: a model for activation of dietary glycosides to mutagens by intestinal flora. Proceedings of the National Academy of Sciences 1980, 77, 4961–4965. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Zhang, Y.; Li, T.; Tang, Y.; Song, S.-Y.; Zhou, Q.; Wang, Y. A detailed overview of quercetin: implications for cell death and liver fibrosis mechanisms. Frontiers in Pharmacology 2024, 15–2024. [Google Scholar] [CrossRef] [PubMed]
- Sudaka, Y.; Mitsui, T.; Kida, H.; Sultana, M.J.; Nishikawa, M.; Ikushiro, S.; Yamaguchi, N. Validation of a quantitation method for conjugated quercetin in human plasma. Journal of Pharmaceutical and Biomedical Analysis 2025, 258, 116738. [Google Scholar] [CrossRef]
- Wiczkowski, W.; Skipor, J.; Misztal, T.; Szawara-Nowak, D.; Topolska, J.; Piskula, M.K. Quercetin and isorhamnetin aglycones are the main metabolites of dietary quercetin in cerebrospinal fluid. Molecular Nutrition & Food Research 2015, 59, 1088–1094. [Google Scholar] [CrossRef]
- Ho, L.; Ferruzzi, M.G.; Janle, E.M.; Wang, J.; Gong, B.; Chen, T.Y.; Lobo, J.; Cooper, B.; Wu, Q.L.; Talcott, S.T.; et al. Identification of brain-targeted bioactive dietary quercetin-3-O-glucuronide as a novel intervention for Alzheimer's disease. Faseb j 2013, 27, 769–781. [Google Scholar] [CrossRef]
- Moon, Y.J.; Wang, L.; DiCenzo, R.; Morris, M.E. Quercetin pharmacokinetics in humans. Biopharm Drug Dispos 2008, 29, 205–217. [Google Scholar] [CrossRef]
- Mullen, W.; Edwards, C.A.; Crozier, A. Absorption, excretion and metabolite profiling of methyl-, glucuronyl-, glucosyl- and sulpho-conjugates of quercetin in human plasma and urine after ingestion of onions. Br J Nutr 2006, 96, 107–116. [Google Scholar] [CrossRef]
- Walle, T.; Walle, U.K.; Halushka, P.V. Carbon dioxide is the major metabolite of quercetin in humans. J Nutr 2001, 131, 2648–2652. [Google Scholar] [CrossRef]
- Boulton, D.W.; Walle, U.K.; Walle, T. Extensive binding of the bioflavonoid quercetin to human plasma proteins. J Pharm Pharmacol 1998, 50, 243–249. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Robinson, K.; Mock, C.; Liang, D. Pre-formulation studies of resveratrol. Drug Dev Ind Pharm 2015, 41, 1464–1469. [Google Scholar] [CrossRef]
- Burkon, A.; Somoza, V. Quantification of free and protein-bound trans-resveratrol metabolites and identification of trans-resveratrol-C/O-conjugated diglucuronides - two novel resveratrol metabolites in human plasma. Mol Nutr Food Res 2008, 52, 549–557. [Google Scholar] [CrossRef]
- Juan, M.E.; Maijó, M.; Planas, J.M. Quantification of trans-resveratrol and its metabolites in rat plasma and tissues by HPLC. J Pharm Biomed Anal 2010, 51, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Menet, M.C.; Baron, S.; Taghi, M.; Diestra, R.; Dargère, D.; Laprévote, O.; Nivet-Antoine, V.; Beaudeux, J.L.; Bédarida, T.; Cottart, C.H. Distribution of trans-resveratrol and its metabolites after acute or sustained administration in mouse heart, brain, and liver. Mol Nutr Food Res 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Alquisiras-Burgos, I.; González-Herrera, I.G.; Alcalá-Alcalá, S.; Aguilera, P. Nose-to Brain Delivery of Resveratrol, a Non-Invasive Method for the Treatment of Cerebral Ischemia. Drugs and Drug Candidates 2024, 3, 102–125. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Hayashi, A.; Terasaka, S.; Nukada, Y.; Kameyama, A.; Yamane, M.; Shioi, R.; Iwashita, M.; Hashizume, K.; Morita, O. 4″-Sulfation Is the Major Metabolic Pathway of Epigallocatechin-3-gallate in Humans: Characterization of Metabolites, Enzymatic Analysis, and Pharmacokinetic Profiling. J Agric Food Chem 2022, 70, 8264–8273. [Google Scholar] [CrossRef]
- Eaton, J.D.; Williamson, M.P. Multi-site binding of epigallocatechin gallate to human serum albumin measured by NMR and isothermal titration calorimetry. Biosci Rep 2017, 37. [Google Scholar] [CrossRef]
- Lin, L.C.; Wang, M.N.; Tseng, T.Y.; Sung, J.S.; Tsai, T.H. Pharmacokinetics of (-)-epigallocatechin-3-gallate in conscious and freely moving rats and its brain regional distribution. J Agric Food Chem 2007, 55, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Miyazawa, T. Absorption and distribution of tea catechin, (-)-epigallocatechin-3-gallate, in the rat. J Nutr Sci Vitaminol 1997, 43, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Lee, M.-J.; Lu, H.; Meng, X.; Hong, J.J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-Gallate Is Absorbed but Extensively Glucuronidated Following Oral Administration to Mice. The Journal of Nutrition 2003, 133, 4172–4177. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (-)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem Biophys Rep 2017, 9, 180–186. [Google Scholar] [CrossRef]
- Zini, A.; Del Rio, D.; Stewart, A.J.; Mandrioli, J.; Merelli, E.; Sola, P.; Nichelli, P.; Serafini, M.; Brighenti, F.; Edwards, C.A.; et al. Do flavan-3-ols from green tea reach the human brain? Nutr Neurosci 2006, 9, 57–61. [Google Scholar] [CrossRef]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6. [Google Scholar] [CrossRef]
- Walle, T. Bioavailability of resveratrol. Ann N Y Acad Sci 2011, 1215, 9–15. [Google Scholar] [CrossRef]
- Vitaglione, P.; Sforza, S.; Galaverna, G.; Ghidini, C.; Caporaso, N.; Vescovi, P.P.; Fogliano, V.; Marchelli, R. Bioavailability of trans-resveratrol from red wine in humans. Mol Nutr Food Res 2005, 49, 495–504. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Almeida, L.; Vaz-da-Silva, M.; Falcão, A.; Soares, E.; Costa, R.; Loureiro, A.I.; Fernandes-Lopes, C.; Rocha, J.F.; Nunes, T.; Wright, L.; et al. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol Nutr Food Res 2009, 53 Suppl 1, S7–15. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial effects of green tea--a review. J Am Coll Nutr 2006, 25, 79–99. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Suganuma, M.; Okabe, S.; Oniyama, M.; Tada, Y.; Ito, H.; Fujiki, H. Wide distribution of [3H](-)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 1998, 19, 1771–1776. [Google Scholar] [CrossRef]
- Lee, M.J.; Maliakal, P.; Chen, L.; Meng, X.; Bondoc, F.Y.; Prabhu, S.; Lambert, G.; Mohr, S.; Yang, C.S. Pharmacokinetics of tea catechins after ingestion of green tea and (-)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev 2002, 11, 1025–1032. [Google Scholar] [PubMed]
- Capasso, L.; De Masi, L.; Sirignano, C.; Maresca, V.; Basile, A.; Nebbioso, A.; Rigano, D.; Bontempo, P. Epigallocatechin Gallate (EGCG): Pharmacological Properties, Biological Activities and Therapeutic Potential. Molecules 2025, 30. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Rafter, J.; Jenner, A. Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: direct or indirect effects? Antioxidant or not? American Journal of Clinical Nutrition 2005, 81, 268S–276S. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Halliwell, B. Do polyphenols enter the brain and does it matter? Some theoretical and practical considerations. Genes Nutr 2012, 7, 99–109. [Google Scholar] [CrossRef]
- Sies, H. Polyphenols and health: update and perspectives. Arch Biochem Biophys 2010, 501, 2–5. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.; Rice-Evans, C. Flavonoids: antioxidants or signalling molecules? Free Radic Biol Med 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Justino, G.C.; Santos, M.R.; Canário, S.; Borges, C.; Florêncio, M.H.; Mira, L. Plasma quercetin metabolites: structure-antioxidant activity relationships. Arch Biochem Biophys 2004, 432, 109–121. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, S.Z.; Chiou, A.L.; Williams, L.R.; Hoffer, B.J. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. Journal of Neuroscience 1997, 17, 4341–4348. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2008, 294, L478–488. [Google Scholar] [CrossRef] [PubMed]
- Pullikotil, P.; Chen, H.; Muniyappa, R.; Greenberg, C.C.; Yang, S.; Reiter, C.E.; Lee, J.W.; Chung, J.H.; Quon, M.J. Epigallocatechin gallate induces expression of heme oxygenase-1 in endothelial cells via p38 MAPK and Nrf-2 that suppresses proinflammatory actions of TNF-α. J Nutr Biochem 2012, 23, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Fujii, M.; Hou, D.X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med 2007, 42, 1690–1703. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res 2008, 52 Suppl 1, S128–138. [Google Scholar] [CrossRef]
- Jung, K.A.; Kwak, M.K. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef]
- Ahmad, A.; Syed, F.A.; Singh, S.; Hadi, S.M. Prooxidant activity of resveratrol in the presence of copper ions: mutagenicity in plasmid DNA. Toxicol Lett 2005, 159, 1–12. [Google Scholar] [CrossRef]
- Tan, J.; Wang, B.; Zhu, L. DNA binding and oxidative DNA damage induced by a quercetin copper(II) complex: potential mechanism of its antitumor properties. J Biol Inorg Chem 2009, 14, 727–739. [Google Scholar] [CrossRef]
- Plauth, A.; Geikowski, A.; Cichon, S.; Wowro, S.J.; Liedgens, L.; Rousseau, M.; Weidner, C.; Fuhr, L.; Kliem, M.; Jenkins, G.; et al. Hormetic shifting of redox environment by pro-oxidative resveratrol protects cells against stress. Free Radic Biol Med 2016, 99, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Elbling, L.; Herbacek, I.; Weiss, R.M.; Jantschitsch, C.; Micksche, M.; Gerner, C.; Pangratz, H.; Grusch, M.; Knasmuller, S.; Berger, W. Hydrogen peroxide mediates EGCG-induced antioxidant protection in human keratinocytes. Free Radic Biol Med 2010, 49, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, K.; Liu, Z.; Huang, J. Prooxidant Effects of Epigallocatechin-3-Gallate in Health Benefits and Potential Adverse Effect. Oxid Med Cell Longev 2020, 2020, 9723686. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic Biol Med 2014, 66, 24–35. [Google Scholar] [CrossRef]
- Shah, Z.A.; Li, R.C.; Ahmad, A.S.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Doré, S. The flavanol (-)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab 2010, 30, 1951–1961. [Google Scholar] [CrossRef]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (-)-epigallocatechin, (-)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochemical & Biophysical Research Communications 2000, 273, 50–53. [Google Scholar]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch Biochem Biophys 2008, 476, 107–112. [Google Scholar] [CrossRef]
- Awad, H.M.; Boersma, M.G.; Vervoort, J.; Rietjens, I.M. Peroxidase-catalyzed formation of quercetin quinone methide-glutathione adducts. Arch Biochem Biophys 2000, 378, 224–233. [Google Scholar] [CrossRef]
- Steenwyk, R.C.; Tan, B. In vitro evidence for the formation of reactive intermediates of resveratrol in human liver microsomes. Xenobiotica 2010, 40, 62–71. [Google Scholar] [CrossRef]
- Sang, S.; Yang, I.; Buckley, B.; Ho, C.T.; Yang, C.S. Autoxidative quinone formation in vitro and metabolite formation in vivo from tea polyphenol (-)-epigallocatechin-3-gallate: studied by real-time mass spectrometry combined with tandem mass ion mapping. Free Radic Biol Med 2007, 43, 362–371. [Google Scholar] [CrossRef]
- Pervaiz, S.; Holme, A.L. Resveratrol: its biologic targets and functional activity. Antioxid Redox Signal 2009, 11, 2851–2897. [Google Scholar] [CrossRef]
- Kim, H.S.; Quon, M.J.; Kim, J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol 2014, 2, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, F.; Sun, Y.; Li, B.; Oi, N.; Chen, H.; Lubet, R.A.; Bode, A.M.; Dong, Z. Select dietary phytochemicals function as inhibitors of COX-1 but not COX-2. PLoS One 2013, 8, e76452. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Aliaga, K.; Bermejo-Bescós, P.; Benedí, J.; Martín-Aragón, S. Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci 2011, 89, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X.P.; Yang, S.G.; Wang, Y.J.; Zhang, X.; Du, X.T.; Sun, X.X.; Zhao, M.; Huang, L.; Liu, R.T. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, Z.; Zhang, N.; Liu, L.; Li, S.; Wei, H. In vitro catabolism of quercetin by human fecal bacteria and the antioxidant capacity of its catabolites. Food Nutr Res 2014, 58. [Google Scholar] [CrossRef]
- Kasahara, K.; Kerby, R.L.; Aquino-Martinez, R.; Evered, A.H.; Cross, T.-W.L.; Everhart, J.; Ulland, T.K.; Kay, C.D.; Bolling, B.W.; Bäckhed, F.; et al. Gut microbes modulate the effects of the flavonoid quercetin on atherosclerosis. npj Biofilms and Microbiomes 2025, 11, 12. [Google Scholar] [CrossRef]
- Bode, L.M.; Bunzel, D.; Huch, M.; Cho, G.S.; Ruhland, D.; Bunzel, M.; Bub, A.; Franz, C.M.; Kulling, S.E. In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am J Clin Nutr 2013, 97, 295–309. [Google Scholar] [CrossRef]
- Iglesias-Aguirre, C.E.; Vallejo, F.; Beltrán, D.; Aguilar-Aguilar, E.; Puigcerver, J.; Alajarín, M.; Berná, J.; Selma, M.V.; Espín, J.C. Lunularin Producers versus Non-producers: Novel Human Metabotypes Associated with the Metabolism of Resveratrol by the Gut Microbiota. J Agric Food Chem 2022, 70, 10521–10531. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, A.; Nanjo, F. Metabolism of (-)-epigallocatechin gallate by rat intestinal flora. J Agric Food Chem 2010, 58, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Roowi, S.; Stalmach, A.; Mullen, W.; Lean, M.E.; Edwards, C.A.; Crozier, A. Green tea flavan-3-ols: colonic degradation and urinary excretion of catabolites by humans. J Agric Food Chem 2010, 58, 1296–1304. [Google Scholar] [CrossRef]
- Liu, Z.; de Bruijn, W.J.C.; Bruins, M.E.; Vincken, J.P. Reciprocal Interactions between Epigallocatechin-3-gallate (EGCG) and Human Gut Microbiota In Vitro. J Agric Food Chem 2020, 68, 9804–9815. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Boeren, S.; Miro Estruch, I.; Rietjens, I. The Gut Microbial Metabolite Pyrogallol Is a More Potent Inducer of Nrf2-Associated Gene Expression Than Its Parent Compound Green Tea (-)-Epigallocatechin Gallate. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Lasa, A.; Churruca, I.; Eseberri, I.; Andrés-Lacueva, C.; Portillo, M.P. Delipidating effect of resveratrol metabolites in 3T3-L1 adipocytes. Mol Nutr Food Res 2012, 56, 1559–1568. [Google Scholar] [CrossRef]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: a quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br J Anaesth 2014, 113, 935–944. [Google Scholar] [CrossRef]
- Buhl, A.E.; Waldon, D.J.; Baker, C.A.; Johnson, G.A. Minoxidil sulfate is the active metabolite that stimulates hair follicles. J Invest Dermatol 1990, 95, 553–557. [Google Scholar] [CrossRef]
- Bourasset, F.; Cisternino, S.; Temsamani, J.; Scherrmann, J.M. Evidence for an active transport of morphine-6-beta-d-glucuronide but not P-glycoprotein-mediated at the blood-brain barrier. J Neurochem 2003, 86, 1564–1567. [Google Scholar] [CrossRef]
- Yamada, H.; Ishii, K.; Ishii, Y.; Ieiri, I.; Nishio, S.; Morioka, T.; Oguri, K. Formation of highly analgesic morphine-6-glucuronide following physiologic concentration of morphine in human brain. J Toxicol Sci 2003, 28, 395–401. [Google Scholar] [CrossRef]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? Aaps j 2017, 19, 931–939. [Google Scholar] [CrossRef]
- Ishisaka, A.; Mukai, R.; Terao, J.; Shibata, N.; Kawai, Y. Specific localization of quercetin-3-O-glucuronide in human brain. Arch Biochem Biophys 2014, 557, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Castillejo, S.; Macià, A.; Motilva, M.J.; Catalán, Ú.; Solà, R. Endothelial Cells Deconjugate Resveratrol Metabolites to Free Resveratrol: A Possible Role in Tissue Factor Modulation. Mol Nutr Food Res 2019, 63, e1800715. [Google Scholar] [CrossRef] [PubMed]
- Ouzzine, M.; Gulberti, S.; Ramalanjaona, N.; Magdalou, J.; Fournel-Gigleux, S.f. The UDP-glucuronosyltransferases of the blood-brain barrier: their role in drug metabolism and detoxication. Front Cell Neurosci 2014, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Sabolovic, N.; Heurtaux, T.; Humbert, A.C.; Krisa, S.; Magdalou, J. cis- and trans-Resveratrol are glucuronidated in rat brain, olfactory mucosa and cultured astrocytes. Pharmacology 2007, 80, 185–192. [Google Scholar] [CrossRef]
- Patel, K.R.; Andreadi, C.; Britton, R.G.; Horner-Glister, E.; Karmokar, A.; Sale, S.; Brown, V.A.; Brenner, D.E.; Singh, R.; Steward, W.P.; et al. Sulfate metabolites provide an intracellular pool for resveratrol generation and induce autophagy with senescence. Sci Transl Med 2013, 5, 205ra133. [Google Scholar] [CrossRef]
- Menendez, C.; Dueñas, M.; Galindo, P.; González-Manzano, S.; Jimenez, R.; Moreno, L.; Zarzuelo, M.J.; Rodríguez-Gómez, I.; Duarte, J.; Santos-Buelga, C.; et al. Vascular deconjugation of quercetin glucuronide: the flavonoid paradox revealed? Mol Nutr Food Res 2011, 55, 1780–1790. [Google Scholar] [CrossRef]
- Shi, Q.; Haenen, G.R.; Maas, L.; Arlt, V.M.; Spina, D.; Vasquez, Y.R.; Moonen, E.; Veith, C.; Van Schooten, F.J.; Godschalk, R.W.L. Inflammation-associated extracellular β-glucuronidase alters cellular responses to the chemical carcinogen benzo[a]pyrene. Arch Toxicol 2016, 90, 2261–2273. [Google Scholar] [CrossRef]
- El Masri, R.; Crétinon, Y.; Gout, E.; Vivès, R.R. HS and Inflammation: A Potential Playground for the Sulfs? Frontiers in Immunology 2020, 11–2020. [Google Scholar] [CrossRef]
- Rao, N.C.; Barsky, S.H.; Terranova, V.P.; Liotta, L.A. Isolation of a tumor cell laminin receptor. Biochem Biophys Res Commun 1983, 111, 804–808. [Google Scholar] [CrossRef]
- Lesot, H.; Kühl, U.; Mark, K. Isolation of a laminin-binding protein from muscle cell membranes. Embo j 1983, 2, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Malinoff, H.L.; Wicha, M.S. Isolation of a cell surface receptor protein for laminin from murine fibrosarcoma cells. J Cell Biol 1983, 96, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- DiGiacomo, V.; Meruelo, D. Looking into laminin receptor: critical discussion regarding the non-integrin 37/67-kDa laminin receptor/RPSA protein. Biol Rev Camb Philos Soc 2016, 91, 288–310. [Google Scholar] [CrossRef]
- Nelson, J.; McFerran, N.V.; Pivato, G.; Chambers, E.; Doherty, C.; Steele, D.; Timson, D.J. The 67 kDa laminin receptor: structure, function and role in disease. Bioscience Reports 2008, 28, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Rea, V.E.; Rossi, F.W.; De Paulis, A.; Ragno, P.; Selleri, C.; Montuori, N. 67 kDa laminin receptor: structure, function and role in cancer and infection. Infez Med 2012, 20 Suppl 2, 8–12. [Google Scholar]
- Ardini, E.; Pesole, G.; Tagliabue, E.; Magnifico, A.; Castronovo, V.; Sobel, M.E.; Colnaghi, M.I.; Ménard, S. The 67-kDa laminin receptor originated from a ribosomal protein that acquired a dual function during evolution. Mol Biol Evol 1998, 15, 1017–1025. [Google Scholar] [CrossRef]
- Rieger, R.; Edenhofer, F.; Lasmézas, C.I.; Weiss, S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med 1997, 3, 1383–1388. [Google Scholar] [CrossRef]
- Pinnock, E.C.; Jovanovic, K.; Pinto, M.G.; Ferreira, E.; Dias, B.D.C.; Penny, C.; Knackmuss, S.; Reusch, U.; Little, M.; Schatzl, H.M.; et al. LRP/LR Antibody Mediated Rescuing of Amyloid-β-Induced Cytotoxicity is Dependent on PrPc in Alzheimer’s Disease. Journal of Alzheimer’s Disease 2016, 49, 645–657. [Google Scholar] [CrossRef]
- Da Costa Dias, B.; Jovanovic, K.; Gonsalves, D.; Moodley, K.; Reusch, U.; Knackmuss, S.; Weinberg, M.S.; Little, M.; Weiss, S.F. The 37kDa/67kDa laminin receptor acts as a receptor for Abeta42 internalization. Sci Rep 2014, 4, 5556. [Google Scholar] [CrossRef]
- Liu, M.; Li, N.; Guo, W.; Jia, L.; Jiang, H.; Li, Z.; Wang, J.; Zhang, X.; Zhu, R.; Bao, C.; et al. RPSA distribution and expression in tissues and immune cells of pathogen-infected mice. Microb Pathog 2021, 152, 104609. [Google Scholar] [CrossRef]
- Gundimeda, U.; McNeill, T.H.; Schiffman, J.E.; Hinton, D.R.; Gopalakrishna, R. Green tea polyphenols potentiate the action of nerve growth factor to induce neuritogenesis: possible role of reactive oxygen species. Journal of Neuroscience Research 2010, 88, 3644–3655. [Google Scholar] [CrossRef]
- Gundimeda, U.; McNeill, T.H.; Fan, T.K.; Deng, R.; Rayudu, D.; Chen, Z.; Cadenas, E.; Gopalakrishna, R. Green tea catechins potentiate the neuritogenic action of brain-derived neurotrophic factor: role of 67-kDa laminin receptor and hydrogen peroxide. Biochem Biophys Res Commun 2014, 445, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Jeong, M.J.; Kang, T.C. Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Luo, Q.; Shen, N.; Qin, X.; Jia, C.; Chao, Z.; Zhang, L.; Qin, H.; Liu, X.; Quan, X.; et al. PEDF Protects Endothelial Barrier Integrity during Acute Myocardial Infarction via 67LR. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Mrówczyńska, E.; Machalica, K.; Mazur, A.J. Non-integrin laminin receptor (LamR) plays a role in axonal outgrowth from chicken DRG via modulating the Akt and Erk signaling. Frontiers in Cell and Developmental Biology 2024, 12–2024. [Google Scholar] [CrossRef]
- Blazejewski, S.M.; Bennison, S.A.; Ha, N.T.; Liu, X.; Smith, T.H.; Dougherty, K.J.; Toyo-Oka, K. Rpsa Signaling Regulates Cortical Neuronal Morphogenesis via Its Ligand, PEDF, and Plasma Membrane Interaction Partner, Itga6. Cereb Cortex 2022, 32, 770–795. [Google Scholar] [CrossRef]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nature Structural & Molecular Biology 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Fujimura, Y.; Sumida, M.; Sugihara, K.; Tsukamoto, S.; Yamada, K.; Tachibana, H. Green Tea Polyphenol EGCG Sensing Motif on the 67-kDa Laminin Receptor. PLoS ONE [Electronic Resource] 2012, 7. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Oh, A.; Hou, L.; Lee, E.; Aguilar, J.; Li, A.; Mack, W.J. Flavonoid quercetin and its glucuronide and sulfate conjugates bind to 67-kDa laminin receptor and prevent neuronal cell death induced by serum starvation. Biochem Biophys Res Commun 2023, 671, 116–123. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Aguilar, J.; Oh, A.; Lee, E.; Hou, L.; Lee, T.; Xu, E.; Nguyen, J.; Mack, W.J. Resveratrol and its metabolites elicit neuroprotection via high-affinity binding to the laminin receptor at low nanomolar concentrations. FEBS Lett 2024, 598, 995–1007. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Aguilar, J.; Lee, E.; Mack, W.J. Neuroprotection by resveratrol-glucuronide and quercetin-glucuronide via binding to polyphenol- and glycosaminoglycan-binding sites in the laminin receptor. Neural Regeneration Research 2025, 20. [Google Scholar]
- Hayashi, D.; Mouchlis, V.D.; Okamoto, S.; Namba, T.; Wang, L.; Li, S.; Ueda, S.; Yamanoue, M.; Tachibana, H.; Arai, H.; et al. Vitamin E functions by association with a novel binding site on the 67 kDa laminin receptor activating diacylglycerol kinase. J Nutr Biochem 2022, 110, 109129. [Google Scholar] [CrossRef] [PubMed]
- Namba T., H.D., Fukuda I., Ueda S., and Shirai Y. Tocotrienols activate diacylglycerol kinase a via 67 KDa laminin receptor. Functional Food Science 2025, 5, 160–169. [CrossRef]
- Liu, Z.; Hu, M. Natural polyphenol disposition via coupled metabolic pathways. Expert Opin Drug Metab Toxicol 2007, 3, 389–406. [Google Scholar] [CrossRef]
- Järvinen, E.; Deng, F.; Kiander, W.; Sinokki, A.; Kidron, H.; Sjöstedt, N. The Role of Uptake and Efflux Transporters in the Disposition of Glucuronide and Sulfate Conjugates. Frontiers in Pharmacology 2022, 12–2021. [Google Scholar] [CrossRef]
- Hundt, C.; Peyrin, J.M.; Haïk, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.P.; Dormont, D.; Lasmézas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. Embo j 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis: An Official Journal of the American Heart Association, Inc. 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Crijns, H.; Adyns, L.; Ganseman, E.; Cambier, S.; Vandekerckhove, E.; Pörtner, N.; Vanbrabant, L.; Struyf, S.; Gerlza, T.; Kungl, A.; et al. Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence. Int J Mol Sci 2021, 23. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal 2018, 42, 281–291. [Google Scholar] [CrossRef]
- Onoue, S.; Endo, K.; Ohshima, K.; Yajima, T.; Kashimoto, K. The neuropeptide PACAP attenuates beta-amyloid (1-42)-induced toxicity in PC12 cells. Peptides 2002, 23, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Caselli, R.J.; Baxter, L.; Serrano, G.; Yin, J.; Beach, T.G.; Reiman, E.M.; Shi, J. Association of pituitary adenylate cyclase-activating polypeptide with cognitive decline in mild cognitive impairment due to Alzheimer disease. JAMA Neurol 2015, 72, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Rat, D.; Schmitt, U.; Tippmann, F.; Dewachter, I.; Theunis, C.; Wieczerzak, E.; Postina, R.; van Leuven, F.; Fahrenholz, F.; Kojro, E. Neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) slows down Alzheimer's disease-like pathology in amyloid precursor protein-transgenic mice. Faseb j 2011, 25, 3208–3218. [Google Scholar] [CrossRef]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer's Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J Alzheimers Dis Rep 2020, 4, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Song, X.J.; Wang, Z.B.; Gan, Q.; Walters, E.T. cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J Neurophysiol 2006, 95, 479–492. [Google Scholar] [CrossRef]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.-K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer's disease. Life Sciences 2020, 257, 118020. [Google Scholar] [CrossRef]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem 2011, 116, 1–9. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer's disease and other cognitive disorders. Rev Neurosci 2011, 22, 153–169. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell 2011, 44, 851–863. [Google Scholar] [CrossRef]
- Mitani, T.; Watanabe, S.; Wada, K.; Fujii, H.; Nakamura, S.; Katayama, S. Intracellular cAMP contents regulate NAMPT expression via induction of C/EBPβ in adipocytes. Biochemical and Biophysical Research Communications 2020, 522, 770–775. [Google Scholar] [CrossRef]
- Wu, S.-k.; Wang, L.; Wang, F.; Zhang, J. Resveratrol improved mitochondrial biogenesis by activating SIRT1/PGC-1α signal pathway in SAP. Scientific Reports 2024, 14, 26216. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Chen, Y.; Dong, Y. Unraveling the AMPK-SIRT1-FOXO Pathway: The In-Depth Analysis and Breakthrough Prospects of Oxidative Stress-Induced Diseases. Antioxidants (Basel) 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxid Med Cell Longev 2020, 2020, 6782872. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sun, Z.; Han, X.; Li, S.; Jiang, X.; Chen, S.; Zhang, J.; Lu, H. Neuroprotective Effect of Resveratrol via Activation of Sirt1 Signaling in a Rat Model of Combined Diabetes and Alzheimer’s Disease. Frontiers in Neuroscience 2020, 13–2019. [Google Scholar] [CrossRef]
- Raghavan, A.; Shah, Z.A. Sirtuins in neurodegenerative diseases: a biological-chemical perspective. Neurodegener Dis 2012, 9, 1–10. [Google Scholar] [CrossRef]
- Jin, T.; Zhang, Y.; Botchway, B.O.A.; Huang, M.; Lu, Q.; Liu, X. Quercetin activates the Sestrin2/AMPK/SIRT1 axis to improve amyotrophic lateral sclerosis. Biomed Pharmacother 2023, 161, 114515. [Google Scholar] [CrossRef]
- Dong, X.-W.; Fang, W.-L.; Li, Y.-H.; Chai, Y.-R. Epigallocatechin-Gallate: Unraveling Its Protective Mechanisms and Therapeutic Potential. Cell Biochemistry and Function 2025, 43, e70056. [Google Scholar] [CrossRef]
- Deng, H.; Mi, M.T. Resveratrol Attenuates Aβ25-35 Caused Neurotoxicity by Inducing Autophagy Through the TyrRS-PARP1-SIRT1 Signaling Pathway. Neurochem Res 2016, 41, 2367–2379. [Google Scholar] [CrossRef]
- Leslie, S.N.; Datta, D.; Wang, M.; van Dyck, C.H.; Arnsten, A.F.T.; Nairn, A.C. Biochemical characterization of age-related calcium-cAMP-PKA signaling dysregulation and its effect on tau pathology in rhesus monkey cortex. Alzheimer's & Dementia 2020, 16, e042017. [Google Scholar] [CrossRef]
- Ahn, J.H.; McAvoy, T.; Rakhilin, S.V.; Nishi, A.; Greengard, P.; Nairn, A.C. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci U S A 2007, 104, 2979–2984. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Huang, Y.; Umeda, D.; Yamada, S.; Yamashita, S.; Kumazoe, M.; Kim, Y.; Murata, M.; Yamada, K.; Tachibana, H. 67-kDa laminin receptor-dependent protein phosphatase 2A (PP2A) activation elicits melanoma-specific antitumor activity overcoming drug resistance. J Biol Chem 2014, 289, 32671–32681. [Google Scholar] [CrossRef]
- Planel, E.; Yasutake, K.; Fujita, S.C.; Ishiguro, K. Inhibition of Protein Phosphatase 2A Overrides Tau Protein Kinase I/Glycogen Synthase Kinase 3β and Cyclin-dependent Kinase 5 Inhibition and Results in Tau Hyperphosphorylation in the Hippocampus of Starved Mouse *. Journal of Biological Chemistry 2001, 276, 34298–34306. [Google Scholar] [CrossRef]
- Sontag, J.-M.; Sontag, E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Frontiers in Molecular Neuroscience 2014, e 7 - 2014. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-beta Receptors: The Good, the Bad, and the Prion Protein. J Biol Chem 2016, 291, 3174–3183. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer's Disease. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Smith, L.M.; Kostylev, M.A.; Lee, S.; Strittmatter, S.M. Systematic and standardized comparison of reported amyloid-β receptors for sufficiency, affinity, and Alzheimer's disease relevance. J Biol Chem 2019, 294, 6042–6053. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.; Bignoux, M.J.; Otgaar, T.C.; Tagliatti, N.; Jovanovic, K.; Letsolo, B.T.; Weiss, S.F.T. LRP/LR specific antibody IgG1-iS18 impedes neurodegeneration in Alzheimer's disease mice. Oncotarget 2018, 9, 27059–27073. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, R.; Lin, C.Y.; Oh, A.; Le, C.; Yang, S.; Hicks, A.; Kindy, M.S.; Mack, W.J.; Bhat, N.R. cAMP-induced decrease in cell-surface laminin receptor and cellular prion protein attenuates amyloid-β uptake and amyloid-β-induced neuronal cell death. FEBS Lett 2022, 596, 2914–2927. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Calandra, V.; Roberts, S.B.; Posmantur, R. cAMP delays beta-amyloid (25-35) induced cell death in rat cortical neurons. Neuroreport 2000, 11, 2293–2297. [Google Scholar] [CrossRef]
- Vilas-Boas, F.; Bagulho, A.; Tenente, R.; Teixeira, V.H.; Martins, G.; da Costa, G.; Jerónimo, A.; Cordeiro, C.; Machuqueiro, M.; Real, C. Hydrogen peroxide regulates cell adhesion through the redox sensor RPSA. Free Radic Biol Med 2016, 90, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.L.; Taubner, L.; Sample, Y.K.; Kazmin, D.A.; Copié, V.; Starkey, J.R. Tumor shedding of laminin binding protein modulates angiostatin production in vitro and interferes with plasmin-derived inhibition of angiogenesis in aortic ring cultures. International Journal of Cancer 2006, 118, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Gundimeda, U.; McNeill, T.H.; Elhiani, A.A.; Schiffman, J.E.; Hinton, D.R.; Gopalakrishna, R. Green tea polyphenols precondition against cell death induced by oxygen-glucose deprivation via stimulation of laminin receptor, generation of reactive oxygen species, and activation of protein kinase Cepsilon. Journal of Biological Chemistry 2012, 287, 34694–34708. [Google Scholar] [CrossRef] [PubMed]
- Buryanovskyy, L.; Fu, Y.; Boyd, M.; Ma, Y.; Hsieh, T.C.; Wu, J.M.; Zhang, Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004, 43, 11417–11426. [Google Scholar] [CrossRef]
- Gould, N.L.; Scherer, G.R.; Carvalho, S.; Shurrush, K.; Kayyal, H.; Edry, E.; Elkobi, A.; David, O.; Foqara, M.; Thakar, D.; et al. Specific quinone reductase 2 inhibitors reduce metabolic burden and reverse Alzheimer's disease phenotype in mice. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Hashimoto, T.; Nakai, M. Increased hippocampal quinone reductase 2 in Alzheimer's disease. Neurosci Lett 2011, 502, 10–12. [Google Scholar] [CrossRef]
- Islam, F.; Shilton, B. Insights into the cellular function and mechanism of action of quinone reductase 2 (NQO2). Biochem J 2025, 482, 309–324. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer's disease. J Cell Biol 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Kim, Y.A.; Lim, S.Y.; Rhee, S.H.; Park, K.Y.; Kim, C.H.; Choi, B.T.; Lee, S.J.; Park, Y.M.; Choi, Y.H. Resveratrol inhibits inducible nitric oxide synthase and cyclooxygenase-2 expression in beta-amyloid-treated C6 glioma cells. Int J Mol Med 2006, 17, 1069–1075. [Google Scholar]
- Ma, T.; Tan, M.S.; Yu, J.T.; Tan, L. Resveratrol as a therapeutic agent for Alzheimer's disease. Biomed Res Int 2014, 2014, 350516. [Google Scholar] [CrossRef]
- Regan, P.; Hole, K.L.; Sero, J.; Williams, R.J. Epigallocatechin Gallate Modulates Microglia Phenotype to Suppress Pro-inflammatory Signalling Cues and Inhibit Phagocytosis. Mol Neurobiol 2024, 61, 4441–4453. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Chen, Z.; Jasmer, K.J.; Chuang, D.Y.; Gu, Z.; Hannink, M.; Simonyi, A. Quercetin Attenuates Inflammatory Responses in BV-2 Microglial Cells: Role of MAPKs on the Nrf2 Pathway and Induction of Heme Oxygenase-1. PLoS One 2015, 10, e0141509. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.; He, Q.; Ao, G.; Xu, N.; He, W.; Liu, X.; Huang, L.; Yu, Q.; Kanamaru, H.; et al. PEDF-34 attenuates neurological deficit and suppresses astrocyte-dependent neuroinflammation by modulating astrocyte polarization via 67LR/JNK/STAT1 signaling pathway after subarachnoid hemorrhage in rats. J Neuroinflammation 2024, 21, 178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduction and Targeted Therapy 2023, 8, 267. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, G.; Luo, Y.; Jiang, L.; Chi, H.; Tian, G. Neuroinflammation in Alzheimer's disease: insights from peripheral immune cells. Immun Ageing 2024, 21, 38. [Google Scholar] [CrossRef]
- Dos Santos, M.G.; Arbo, B.D.; Hort, M.A. Effects of quercetin and its derivatives in in vivo models of neuroinflammation: A systematic review and meta-analysis. Neural Regen Res 2026, 21, 1783–1792. [Google Scholar] [CrossRef]
- Serban, M.C.; Sahebkar, A.; Zanchetti, A.; Mikhailidis, D.P.; Howard, G.; Antal, D.; Andrica, F.; Ahmed, A.; Aronow, W.S.; Muntner, P.; et al. Effects of Quercetin on Blood Pressure: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J Am Heart Assoc 2016, 5. [Google Scholar] [CrossRef]
- Chen, Y.; He, Y.; Han, J.; Wei, W.; Chen, F. Blood-brain barrier dysfunction and Alzheimer's disease: associations, pathogenic mechanisms, and therapeutic potential. Front Aging Neurosci 2023, 15, 1258640. [Google Scholar] [CrossRef]
- Nehra, G.; Bauer, B.; Hartz, A.M.S. Blood-brain barrier leakage in Alzheimer’s disease: From discovery to clinical relevance. Pharmacology & Therapeutics 2022, 234, 108119. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging 2015, 36, 608–617. [Google Scholar] [CrossRef]
- Yang, A.J.T.; Bagit, A.; MacPherson, R.E.K. Resveratrol, Metabolic Dysregulation, and Alzheimer's Disease: Considerations for Neurogenerative Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Förstermann, U.; Li, H. Resveratrol and endothelial nitric oxide. Molecules 2014, 19, 16102–16121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, S.; Li, J.; Sun, Y.; Hasimu, H.; Liu, R.; Zhang, T. Quercetin protects human brain microvascular endothelial cells from fibrillar β-amyloid1–40-induced toxicity. Acta Pharmaceutica Sinica B 2015, 5, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, W.; Chen, J.; Xie, D.; Wang, Y.; Zhou, J. Tea Polyphenol Epigallocatechin Gallate and the Gut–Health Axis: Unraveling Structural Characteristics, Metabolic Pathways, and Systemic Benefits. Advances in Nutrition 2025, 16, 100545. [Google Scholar] [CrossRef]
- Andersen, J.K.; Davies, K.J.; Forman, H.J. Reactive oxygen and nitrogen species in neurodegeneration . Free Radic Biol Med 2013, 62, 1–3. [Google Scholar] [CrossRef]
- Crowell, J.A.; Korytko, P.J.; Morrissey, R.L.; Booth, T.D.; Levine, B.S. Resveratrol-associated renal toxicity. Toxicol Sci 2004, 82, 614–619. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Andres, S.; Pevny, S.; Ziegenhagen, R.; Bakhiya, N.; Schäfer, B.; Hirsch-Ernst, K.I.; Lampen, A. Safety Aspects of the Use of Quercetin as a Dietary Supplement. Mol Nutr Food Res 2018, 62. [Google Scholar] [CrossRef]
Figure 1.
Structures of polyphenols aglycones (left three), glucuronide conjugates of polyphenols (middle three), glucuronide conjugates of intestinal microbial metabolites (right three).
Figure 1.
Structures of polyphenols aglycones (left three), glucuronide conjugates of polyphenols (middle three), glucuronide conjugates of intestinal microbial metabolites (right three).
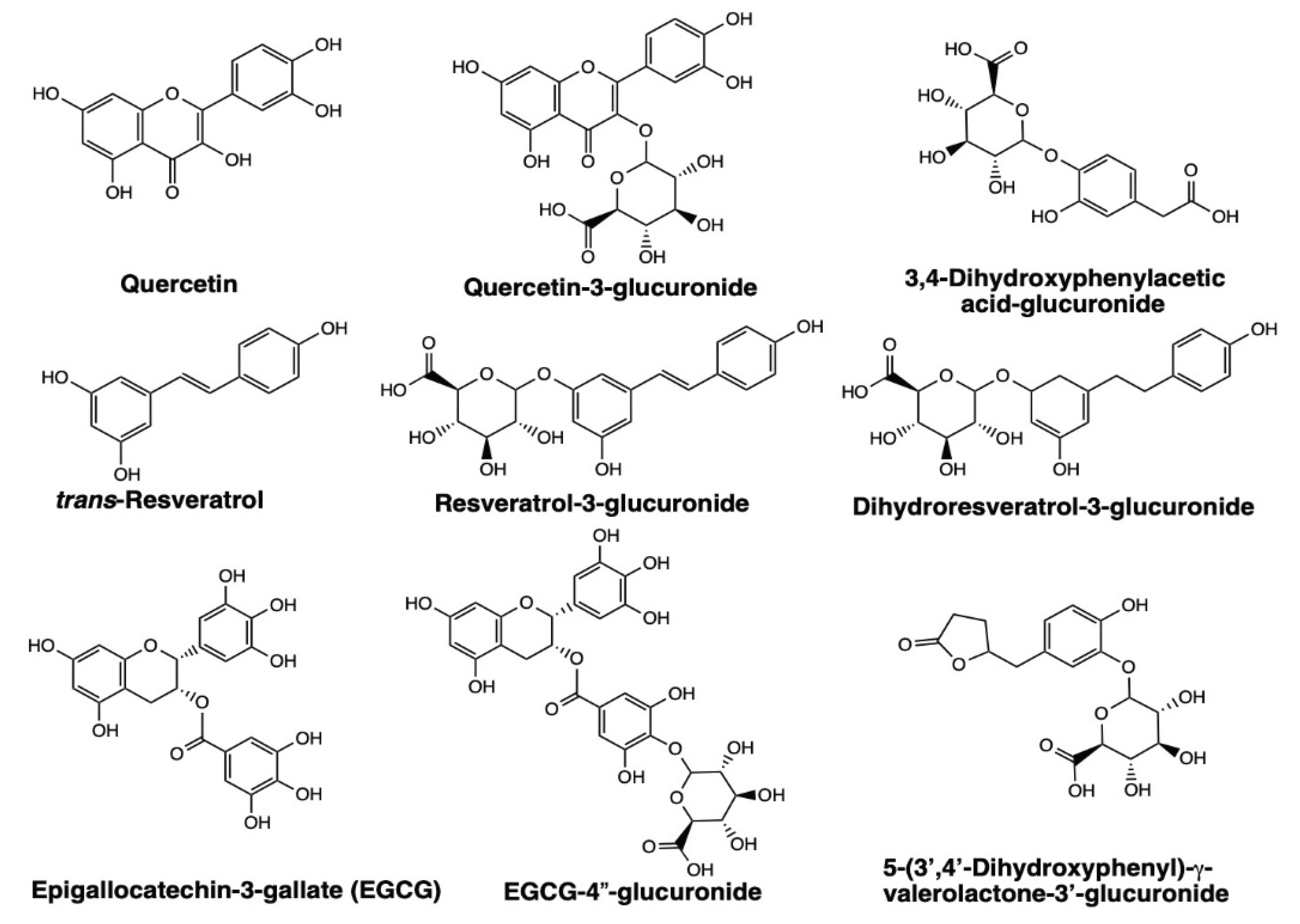
Figure 2.
PyMOL-generated images showing the crystal structure of truncated 37LRP (cyan) after docking of polyphenols and their glucuronide metabolites. The peptide G region is shown in red, the palindromic sequence in yellow, and the bound ligands in blue. Docked ligands are shown as follows: polyphenol aglycones (left three), glucuronide conjugates of polyphenols (middle three), and glucuronide conjugates of intestinal microbial metabolites (right three).
Figure 2.
PyMOL-generated images showing the crystal structure of truncated 37LRP (cyan) after docking of polyphenols and their glucuronide metabolites. The peptide G region is shown in red, the palindromic sequence in yellow, and the bound ligands in blue. Docked ligands are shown as follows: polyphenol aglycones (left three), glucuronide conjugates of polyphenols (middle three), and glucuronide conjugates of intestinal microbial metabolites (right three).
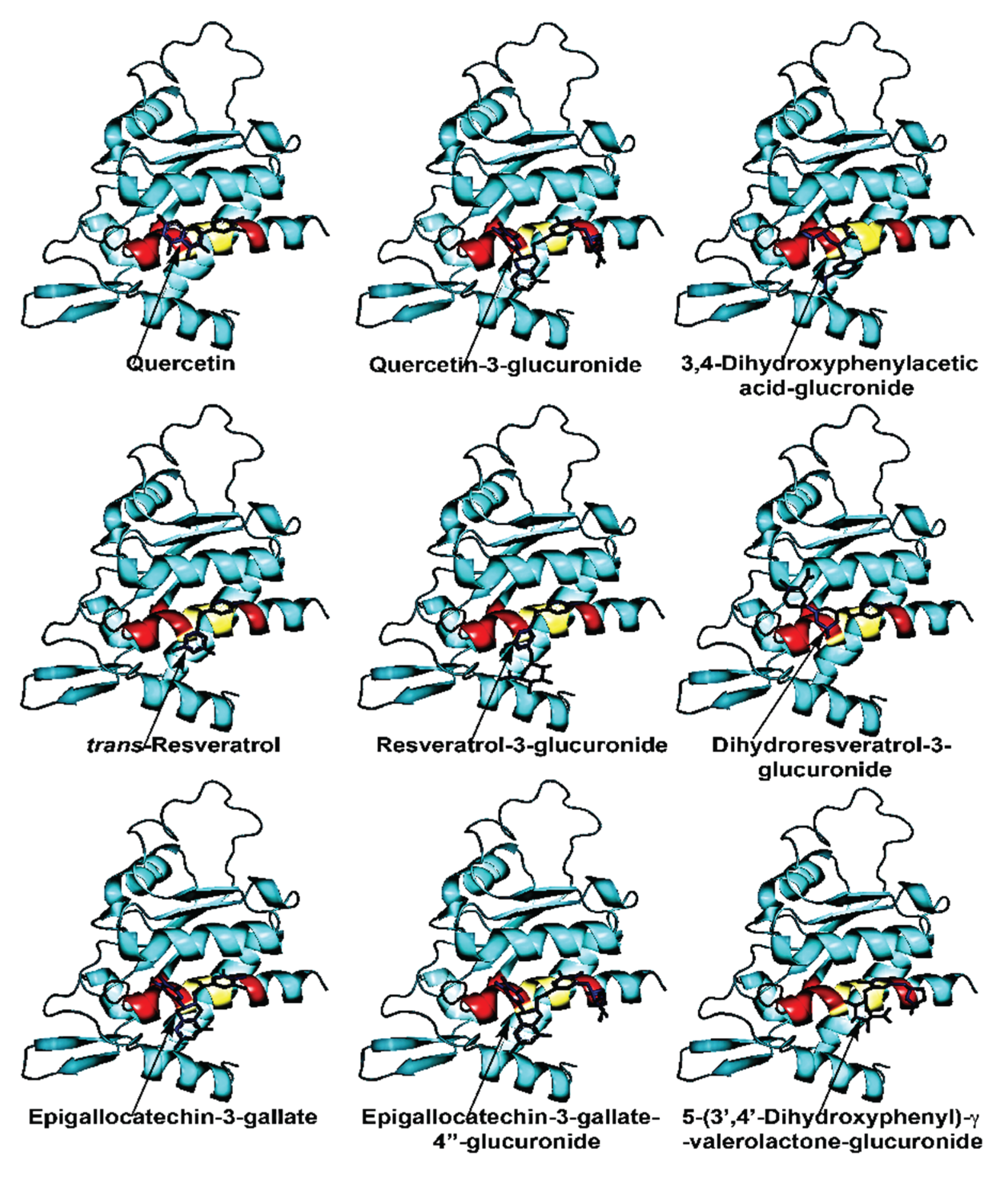
Figure 3.
Binding sites “A” and “B” for polyphenol and their glucuronides in the peptide G region of 67LR. The peptide G region contains a palindromic sequence (blue) and a GAG-binding motif (red). The “A” site includes Leu173, Trp175, and Trp176 within the palindromic sequence, along with other amino acids, and mediates high-affinity binding of polyphenols and their glucuronides. His169, located in the GAG-binding motif, also contributes to the high-affinity binding of polyphenols at the A site. The “B” site comprises Asn165 and Lys166 within the GAG-binding motif, along with additional residues, and is involved in the binding of polyphenols and their glucuronides.
Figure 3.
Binding sites “A” and “B” for polyphenol and their glucuronides in the peptide G region of 67LR. The peptide G region contains a palindromic sequence (blue) and a GAG-binding motif (red). The “A” site includes Leu173, Trp175, and Trp176 within the palindromic sequence, along with other amino acids, and mediates high-affinity binding of polyphenols and their glucuronides. His169, located in the GAG-binding motif, also contributes to the high-affinity binding of polyphenols at the A site. The “B” site comprises Asn165 and Lys166 within the GAG-binding motif, along with additional residues, and is involved in the binding of polyphenols and their glucuronides.
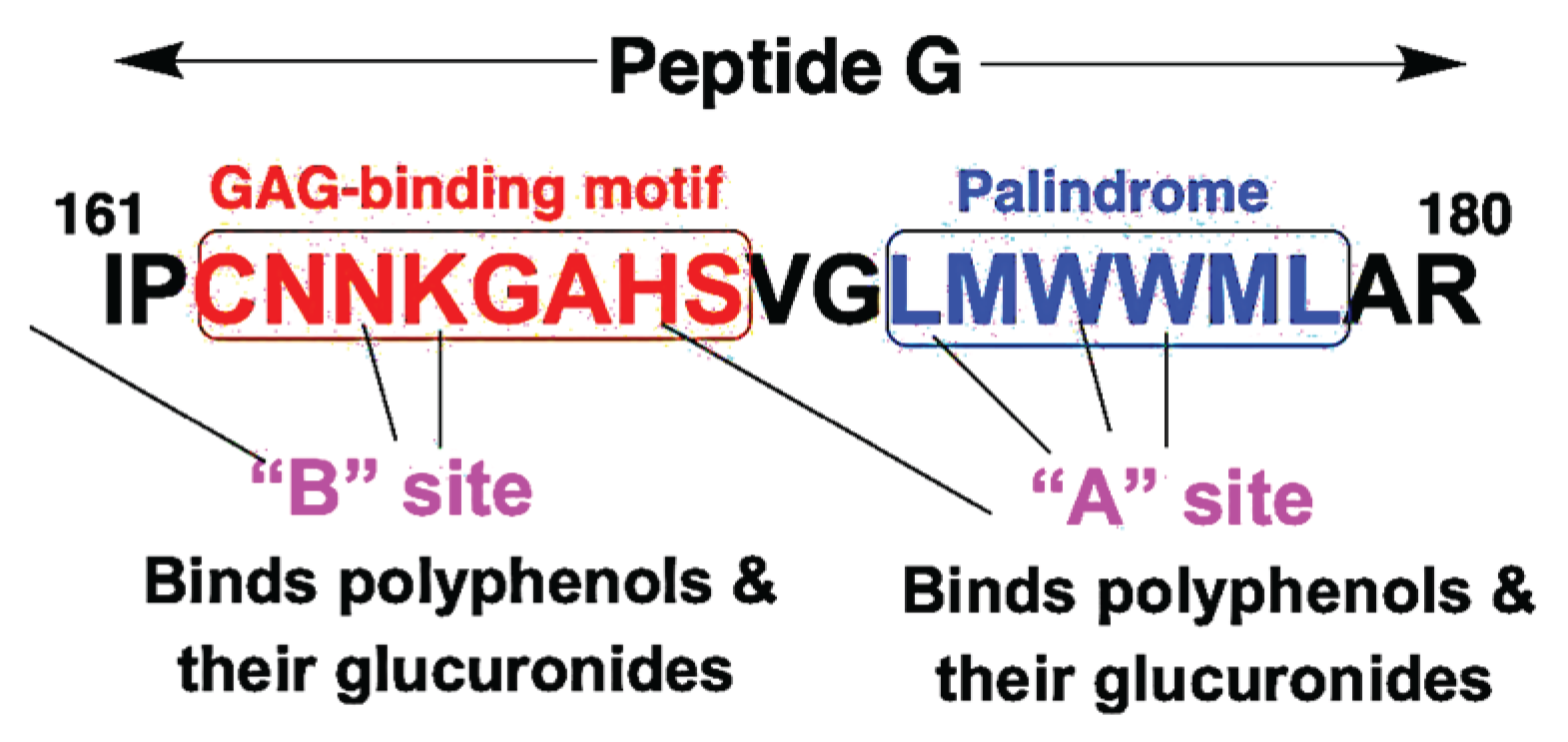
Figure 4.
The Aβ-PrPᶜ complex binds to 67LR, triggering signaling that promotes neuronal death, whereas polyphenols and their glucuronide metabolites compete for the same receptor and activate neuroprotective pathways. Binding of the Aβ-PrPᶜ complex to 67LR promotes Aβ internalization, elevates ROS, induces synaptic toxicity, and ultimately drives neuronal death and AD progression (events shown in red). In contrast, polyphenols and their conjugates bind 67LR and increase intracellular cAMP, thereby activating PKA and leading to the subsequent phosphorylation of CREB. Activated CREB promotes transcription of BDNF and NMNAT (nicotinamide mononucleotide adenylyl transferase). NMNAT supports SIRT1 activation, which deacetylates transcription factors such as FOXO3a and Nrf2, inducing antioxidant enzymes that reduce ROS. PKA also activates protein phosphatase 2A (PP2A), which dephosphorylates tau and limits its hyperphosphorylation and neurofibrillary tangle formation. Together, these events (shown in blue) promote neuroprotection and neuroplasticity, improve cognition, and may slow AD progression. Polyphenols such as resveratrol and quercetin also inhibit QR2 (quinone reductase 2), an enzyme linked to cognitive decline and elevated in AD.
Figure 4.
The Aβ-PrPᶜ complex binds to 67LR, triggering signaling that promotes neuronal death, whereas polyphenols and their glucuronide metabolites compete for the same receptor and activate neuroprotective pathways. Binding of the Aβ-PrPᶜ complex to 67LR promotes Aβ internalization, elevates ROS, induces synaptic toxicity, and ultimately drives neuronal death and AD progression (events shown in red). In contrast, polyphenols and their conjugates bind 67LR and increase intracellular cAMP, thereby activating PKA and leading to the subsequent phosphorylation of CREB. Activated CREB promotes transcription of BDNF and NMNAT (nicotinamide mononucleotide adenylyl transferase). NMNAT supports SIRT1 activation, which deacetylates transcription factors such as FOXO3a and Nrf2, inducing antioxidant enzymes that reduce ROS. PKA also activates protein phosphatase 2A (PP2A), which dephosphorylates tau and limits its hyperphosphorylation and neurofibrillary tangle formation. Together, these events (shown in blue) promote neuroprotection and neuroplasticity, improve cognition, and may slow AD progression. Polyphenols such as resveratrol and quercetin also inhibit QR2 (quinone reductase 2), an enzyme linked to cognitive decline and elevated in AD.
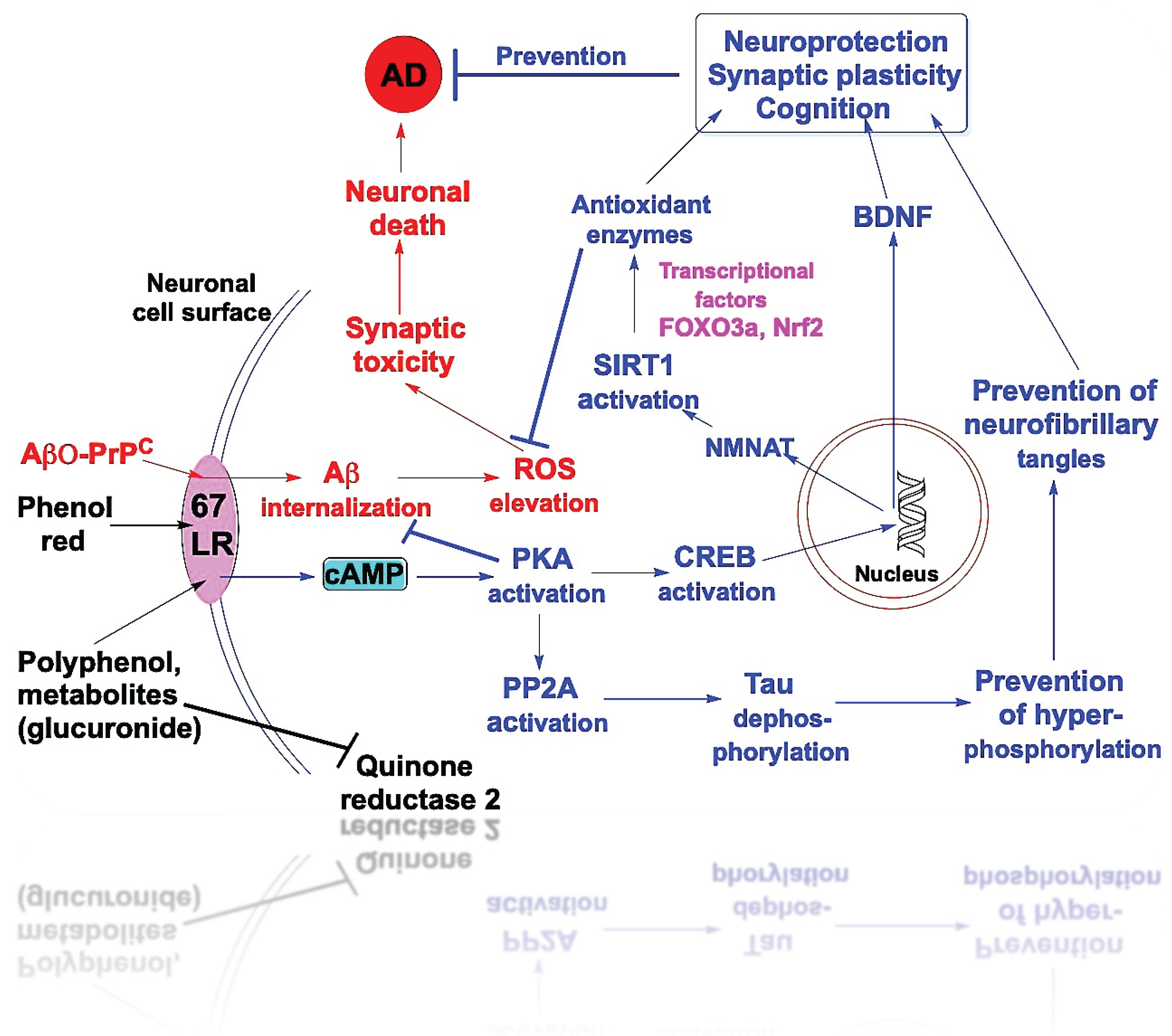

Table 1.
Distribution of quercetin, resveratrol, and EGCG and their metabolites in plasma and brain.
Table 1.
Distribution of quercetin, resveratrol, and EGCG and their metabolites in plasma and brain.
| Polyphenol | Plants | Plasma | Brain | CSF |
| Quercetin |
Glycoside form | Aglycone, N.D.a Q-3-G, 500 nM Q-3-S, 300 nM Q-7G/4′S, 300 nM Methylated-Q, 75 nM Protein binding: Aglycone: 99%b |
Aglycone, N.D.c Q-3-G, 1 nM |
Aglycone, 0.3 nMd Conjugates, N.D. |
| Resveratrol |
Glycoside form | Aglycone, <0.02 μMe Conjugates all, ~2 μM Protein bindingf: Aglycone: 98% Conjugates: 50% |
Aglycone, 0.17 nmol/g R-3-G, 1.06 nmol/gh R-3-S, 0.44 nmol/gh BBB permeability: 2%i |
Aglycone, 2 nMj R-3-G, 20 nM R-4’-G, 27 nM R-3-S, 39 nM |
| EGCG |
Aglycone | Aglycone, 230 nMk EGCG-4″-S, 170 nM EGCG-4″-G, 75 nM Protein binding: 90%l |
EGCG, 10 nMm EGCG, 0.5 nmol/gn EGCG, N.D.o BBB permeability: 2.8%p |
EGCG, N.D.q |
a. Quercetin glycoside (equivalent to 100 mg quercetin) was administered orally to humans [66]. b. Quercetin plasma protein binding in humans is approximately 99% [72]. c. A red wine polyphenol mixture (150 mg) was administered orally to rats [68]. d. Powdered dry onion skin was administered to sheep by intraruminal intubation [67]. e. Humans received an oral dose of trans-resveratrol (25 mg) [73]. f. Resveratrol binding to human plasma proteins [74,75]. g. Rats were administered trans-resveratrol (15 mg/kg) intravenously, and resveratrol levels were measured after 90 min [76]. h. Mice were administered trans-resveratrol (150 mg/kg) by gavage, and resveratrol levels were measured after 30 min [77]. i. Resveratrol crosses the BBB with a permeability of approximately 2% of the plasma concentration [78]. j. CSF levels were measured in humans after oral administration of resveratrol for 52 weeks [79]. k. Humans were orally administered a green tea catechin mixture containing 135 mg EGCG [80]. l. EGCG plasma protein binding [81]. m. In rats administered EGCG at 50 mg/kg [82]. n. In rats, following very high oral doses of EGCG (500 mg/kg) [83]. o. Intragastric administration of EGCG to mice [84]. p. EGCG crosses the BBB with a permeability of 2.8% at 30 min and 5.6% at 1 h [85]. q. CSF levels were measured in six multiple sclerosis patients 2 h after the administration of 250 ml of green tea. The limit of detection was 10 nM [86]. N.D., not detectable; Q-3-G, quercetin-3-glucuronide; Q-3-S, quercetin-3-sulfate; Q-7G/4′-S, quercetin-7-glucuronide-4′-sulfate; R-3-G, resveratrol-3-glucuronide; R-3-S, resveratrol-3-sulfate.
Table 2.
Binding energies of polyphenol aglycones and their intestinal microbial metabolites to 37LRP, along with the predicted amino acids involved in ligand binding. Three-dimensional structures of all compounds were generated using ChemDraw and converted to PDB format with Avogadro. Molecular docking was performed using the crystal structure of truncated 37LRP (PDB ID: 3BCH; amino acid residues 9–183) with AutoDock Vina (version 1.2.5) and AutoDockTools (version 1.5.7). Ligand-binding amino acid residues on 37LRP were identified using Discovery Studio (version 24.1). All glucuronide conjugates are primarily formed in the liver.
Table 2.
Binding energies of polyphenol aglycones and their intestinal microbial metabolites to 37LRP, along with the predicted amino acids involved in ligand binding. Three-dimensional structures of all compounds were generated using ChemDraw and converted to PDB format with Avogadro. Molecular docking was performed using the crystal structure of truncated 37LRP (PDB ID: 3BCH; amino acid residues 9–183) with AutoDock Vina (version 1.2.5) and AutoDockTools (version 1.5.7). Ligand-binding amino acid residues on 37LRP were identified using Discovery Studio (version 24.1). All glucuronide conjugates are primarily formed in the liver.
| Polyphenol aglycones or their metabolites |
Binding energy (kcal/mol) |
Amino acids involved in the ligand binding to “A” site in 37LRP (a precursor protein for 67LR) |
| Quercetin | -8.34 | Thr82, Arg85, Ala86, Lys89, Trp175, Trp176, Ala179 # (Phe90, Ala93, Ala168, His169, Gly172, Leu173) |
| Quercetin-3-glucuronide | -8.43 | Arg85, Ala86, Lys89, Gly172, Trp175, Trp176 (Asn81, Thr82, Phe90, Ala93, His169, Leu173, Ala179) |
| *3,4-Dihydroxyphenylacetic acid |
-5.06 | Ala86, Lys89, Gly172 (Arg85, Phe90, Leu173, Trp175, Trp176, Ala179) |
| 3,4-Dihydroxyphenylacetic acid-glucuronide |
-7.11 | Ala86, Ala168, His169, Leu173, Trp176 (Lys17, Ala20, Ala21, Arg85, Lys89, Gly172) |
| trans-Resveratrol | -6.65 | Ala86, Lys89, His169, Gly172, Trp175, Trp176, Ala179 (Phe90, Ala93, Leu173) |
| Resveratrol-3-glucuronide | -7.78 | Ala86, Lys89, His169, Trp175, Trp176, Ala 179 (Lys17, Ala20, Ala21, Phe90, Ala93, Gly172, Leu173) |
| *Dihydroresveratrol | -6.38 | Ala86, Lys89, His169, Trp175, Trp176, Ala179 (Phe90, Ala93, Gly172, Leu173) |
| Dihydroresveratrol-3- glucuronide |
-8.18 | Thr82, Arg85, Ala86, Lys89, Trp175, Trp176, Ala179 (Asn81, Gln84, Phe90, Ala93, Ala168, Gly172) |
| Epigallocatechin-3-gallate | -9.31 | Thr82, Arg85, Lys89, Gly172, Leu173, Trp176 (Lys17, Ala20, Ala21, Ala86, Phe90, Ala93, Ala168, His169, Trp175, Ala179) |
| Epigallocatechin-3-gallate- 4″-glucuronide |
-9.96 | Ala21, Thr82, Arg85, Lys89, Ala168, Leu173, Trp176 (Lys17, Ala20, Ala86, Phe90, Ala93, His169, Gly172, Trp175, Ala179, Arg180, Leu183) |
| *5-(3′,4′-Dihydroxyphenyl)- γ-valerolactone |
-5.99 | Ala86, Lys89, Trp175 (Arg85, Phe90, Gly172, Trp176, Ala179) |
| 5-(3′,4′-Dihydroxyphenyl)-γ- valerolactone-3′-glucuronide |
-6.84 | Lys 89, Gly172, Trp176, Ala179 (Ala86, Phe90, Ala93, Leu173, Trp175, Leu183) |
* Denotes intestinal microbial metabolites. # Amino acid residues contributing to van der Waals interactions are indicated in parentheses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



