Submitted:
04 February 2026
Posted:
05 February 2026
Read the latest preprint version here
Abstract
Obesity-associated carcinogenesis offers a model to explore the transition from metabolic dysregulation to genomic instability and carcinogenesis. AMPK, the principal cellular energy sensor, coordinates ATP production with metabolic demand; however, in obesity, AMPK activity is impaired, resulting in reduced ATP, elevated AMP, and cellular energy stress. DNA polymerases ε (Pol ε) and δ (Pol δ) maintain replication fidelity via a 3′→5′ exonuclease proofreading activity that removes misincorporated nucleotides. Elevated AMP directly binds and selectively inhibits the exonuclease, conserving energy at the expense of genomic accuracy. As a result, replication errors escape correction and accumulate, some conferring selective advantage and driving carcinogenic evolution. Therapeutic and lifestyle interventions that activate AMPK — including weight loss, exercise, metformin, and aspirin — restore ATP production, lower AMP, and relieve inhibition of exonuclease proofreading, thereby preserving genomic integrity and slowing mutation-driven carcinogenesis. This framework reveals two core biological principles: 1. Energy metabolism and DNA replication fidelity are mechanistically coupled at the DNA polymerase active site. 2. The mutation rate is an adaptive metabolic phenotype, modulated by AMP levels. These concepts redefine the metabolic-genetic interface in carcinogenesis and highlight AMPK activation as a rational target for obesity associated cancer prevention.
Keywords:
AMP
; AMPK
; carcinogenesis
; DNA polymerase
; fidelity
; energy regulation
; metabolism
; mutation
; obesity
; proofreading-exonuclease
1. Introduction
The prevalence of obesity is alarmingly high and continues to increase both in the United States and globally. Concomitantly, the incidences of common cancer types are increasing. The convergence of the obesity epidemic, and the increase in the incidence of various cancers, has provided an opportunity to perform a comparative analysis focused on the link between disordered energy metabolism and mutational carcinogenesis. We sought to understand this relationship because such a link would be a target for preventive intervention. The International Agency for Research on Cancer (IARC) highlight that rising obesity significantly drives future cancer burdens, with projections showing millions more cases and huge economic costs due to treatment and lost productivity, demanding urgent prevention strategies to avert future health crises.
In 2003, Calle et al. demonstrated that higher body weight was correlated with increased mortality rates for many cancer types [1]. A 2014 study from the UK reinforced this finding, showing that a high body mass index (BMI) was linked to 13 different cancer types. Every 5-kg/m² increase in BMI elevated the risk of cancers of the uterus, gallbladder, kidney, liver, colon, cervix, thyroid, ovaries, postmenopausal breast, pancreas, rectum, and esophagus; leukemia; multiple myeloma; and meningioma [2]. Based on more than 1000 epidemiologic studies worldwide, (IARC) reported an association with being overweight, obesity, and weight gain, for at least 13 cancers, primarily epithelial derived adenocarcinomas, including cancer of the esophagus, cancers of the breast (in postmenopausal women), colon, rectum, endometrium (corpus uterus), gallbladder, gastric cardia, kidney (renal cell), liver, ovary, pancreas, and thyroid, as well as meningioma, and multiple myeloma. The strongest association was observed between BMI and endometrial cancer (EC), with a relative risk (RR) of approximately 1.5 for a BMI of 25.0–29.9 kg/m2, 2.5 for a BMI of 30.0–34.9 kg/m2, 4.5, for a BMI of 35.0–39.9 kg/m2, and 7.1 for a >40.0 kg/m2 BMI [3].
The relationship of obesity and cancer begins early in life. A BMI above the normal limit (24.99 kg/m2) in early adulthood, age 20, was found to predict an increased risk of colorectal cancer (CRC) and EC [4,5]. Furthermore, it is a continuous process as the cumulative lifetime excess weight or the weighted years lived overweight or with obesity exhibited an even more robust RR relationship with CRC than a one-time measurement of BMI [6], indicating that the risk factor for obesity associated cancers is an entity which accumulates, e.g., mutations. The risk of developing obesity related cancer has increased in ever younger cohorts in the USA [7].
A strong association of obesity with insulin resistance has been recognized since the mid-1990s. Weight loss often correlates with increased insulin sensitivity [8]. Meta-analyses and cohort studies confirm the association of insulin resistance and type two diabetes mellitus (T2DM) with a similar increased cancer risk [9] (as in the obese.)
Increases in the prevalence and severity of a definable disorder related to impaired energy metabolism over 20-30 years correlate with BMI. Similarly, the prevalence of various cancers increases in correlation with BMI. On the basis of this observation, we hypothesized that obesity and cancer are metabolically linked, an idea that warranted further analysis.
2. Methods
This study extends our original biochemical discovery of the first eukaryotic DNA polymerase catalytic core possessing intrinsic proofreading exonuclease activity and its previously unexplained selective inhibition, a phenomenon that permits mutation accumulation in newly synthesized DNA. Here, we advance a mechanistic explanation for this observation by integrating experimental enzymology with metabolic regulation in obesity-associated carcinogenesis. By re-examining and repurposing key experimental findings—both our own and others’—as functional results within a unified mechanistic framework, we identify impaired cellular energy metabolism as a causal regulator of DNA polymerase proofreading fidelity. This work defines a previously unrecognized metabolic control point linking AMP-dependent signaling, DNA replication fidelity, and mutational carcinogenesis, thereby transforming a longstanding biochemical observation into a testable pathogenic mechanism.
Genetic and Metabolic Constraints on Proofreading Fidelity
Pathogenic mutations in the proofreading exonuclease domains of the POLD and POLE genes, encoding DNA polymerase δ and DNA polymerase ε respectively, provide direct genetic evidence that loss of proofreading activity is sufficient to drive mutational carcinogenesis. Germline and somatic exonuclease-deficient variants produce ultra-mutated tumor phenotypes with markedly increased cancer incidence, establishing proofreading fidelity as a dominant barrier to malignant transformation.
In parallel, metabolic disorders characterized by impaired AMPK signaling—most notably Peutz-Jeghers syndrome—demonstrate a strong predisposition to epithelial malignancies, implicating energy dysregulation as an upstream permissive factor in tumorigenesis. When considered together, these genetic and metabolic observations converge on a common vulnerability: the energetic cost of high-fidelity DNA replication. These findings motivated our focused analysis of metabolic modulation of DNA polymerase proofreading as a unifying mechanism of mutation accumulation.
Approach and Rationale
To define a mechanistic link between metabolism and mutational carcinogenesis, we conducted a targeted, hypothesis-driven analysis across two experimentally grounded domains: DNA replication fidelity and energy metabolism in obesity. Rather than cataloging associations, our analysis was explicitly constrained to causal mechanisms capable of modulating mutation formation during DNA synthesis.
Published biochemical, genetic, and metabolic data were interrogated as functional evidence, emphasizing energetic requirements of nucleotide selection, proofreading exonuclease activity, and mismatch correction. Expertise in DNA polymerase enzymology, cancer genetics, and metabolic regulation was applied to integrate these data into a coherent mechanistic model. This synthesis enabled identification of AMP-mediated regulation of proofreading activity as a metabolic checkpoint governing the trade-off between replication fidelity and cellular survival under energetic stress.
Analysis and Synthesis
Through this process, significant associations and correlations between metabolic, mutational, and carcinogenic factors emerged. Features that regulate both energy metabolism in obesity and mutational carcinogenesis were identified, leading to the synthesis of a tenable mechanism underlying the metabolic linkage between them.
3. Results
3.1. The Hallmarks of Cancer
3.1.1. The Hallmarks of Cancer and Genomic Instability
In 2000, Hanahan and Weinberg defined six hallmark biological capabilities acquired during the multistep development of human cancers: sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activation and metastasis [10].
This framework was expanded in 2011 to emphasize genomic instability as a fundamental enabling characteristic that underlies the acquisition of these hallmarks by generating the genetic diversity required for tumor evolution. Genomic instability is now recognized as a defining feature of cancer, contributing directly to intratumoral heterogeneity, disease progression, and therapeutic resistance. At that time, two additional hallmark capabilities were introduced: reprogramming of energy metabolism and evasion of immune destruction [11].
In 2022, Hanahan, further refined the model by identifying phenotypic plasticity and disrupted differentiation as discrete hallmark capability, and by recognizing non-mutational epigenetic reprogramming and polymorphic microbiomes as it drives the accumulation of oncogenic mutations that permit the emergence and maintenance of hallmark traits [12].
Accordingly, understanding carcinogenesis requires understanding relevant genomic instability, including an examination of the mechanisms that normally preserve genomic stability, starting with the fundamentals of eukaryotic DNA replication and fidelity preservation.
3.1.2. DNA Replication Errors and Genomic Instability in Carcinogenesis
Two principal models have been proposed to explain the origin of carcinogenic mutations not of obvious extraneous cause and responsible for genomic instability: the mutator DNA polymerase hypothesis and the stochastic DNA replication error theory. Loeb and colleagues posited that errors introduced during DNA synthesis by cellular polymerases are causally related to malignant transformation. They also emphasized the integral relationship between DNA replication fidelity, genomic stability, and energy cost, noting that higher fidelity demands progressively more energy (Figure 1) [13].
The mutator phenotype hypothesis of oncogenesis was expanded to include defective base excision and mismatch repair, spontaneous deamination of cytosine to uracil, alterations in deoxynucleotide triphosphate pools, and incorporation of ribonucleotides into DNA as sources of mutations [14]. Evidence supporting the mutator phenotype hypothesis came from next-generation sequencing demonstrating that each tumor is unique, containing tens to hundreds of thousands of mutations, which primarily are misincorporated base substitutions and small base insertions or deletions causing frameshift changes [15]. However, the way most of these mutations occur and the identity of the mutator DNA polymerase has remained elusive.
Vogelstein and collaborators likewise underscored the central role of DNA replication errors in carcinogenesis proposing a model of malignant transformation being driven by a series of mutations that confer a selective growth advantage to cells [16]. Next-generation sequencing revealed that while numerous somatic mutations are found in the majority tumors, most do not enhance the selective growth advantage of cells. The terms “driver” and “passenger” were coined to distinguish between mutations that actively promote carcinogenesis and the numerous others that do not. A mathematical analysis of the evolution of somatic mutations in cancers revealed that the number of mutations in self-renewing tissues correlated with the patient’s age at diagnosis and indicated that half or more of the somatic mutations are “passenger” mutations occurring before the onset of neoplasia. Furthermore, many mutations in human cancer were attributable to random errors occurring during somatic cell DNA replication, accumulating as the individual ages. In fact, most human cancers were caused by two to eight sequential driver gene mutations that develop stochastically accumulating over 20 to 30 years [17]. Further study indicated that the lifetime risk of different cancer types strongly correlates (0.81) with the number of divisions of normal self-renewing cells. Only one-third of the variation in cancer risk among tissues was attributable to environmental or inherited predisposition. Most cases were due to random mutations arising during DNA replication in noncancerous stem cells [18].
Cancer genome sequencing and epidemiological data also indicated that DNA replication errors account for two-thirds of the mutations in human cancers. Approximately 66% of cancer mutations result from DNA replication copying errors, 29% can be attributed to lifestyle or environmental factors, while the remaining 5% are inherited [19]. In common solid tumors, such as those of the colon, breast, brain, pancreas, or endometrium, an average of 33 to 66 genes harbored subtle somatic mutations. Approximately 95% of these mutations were single-base substitutions, with the remainder being deletions or insertions of one or a few bases [20]. Accordingly, an examination of the process of DNA replication and the mechanisms by which the high fidelity of base incorporation is accomplished was incorporated into the analysis.
3.1.3. DNA Replication and Fidelity in Eukaryotes
Eukaryotic genomic DNA replication is primarily carried out by DNA polymerase δ (pol δ) and DNA polymerase ε (pol ε). Both enzymes possess intrinsic 3ʹ→5ʹ exonuclease proofreading activity, which enables removal of misincorporated nucleotides allowing DNA polymerase to insert the correct nucleotide and thereby increases replication fidelity by preventing the propagation of replication errors.
The first eukaryotic DNA polymerase exhibiting proofreading activity was identified in 1976 and designated DNA polymerase δ (pol δ) [21]. This polymerase was recognized as the founding member of a previously uncharacterized class of eukaryotic DNA polymerases, and its 3ʹ→5ʹ exonuclease function was determined to reside within its 122-kDa catalytic subunit [22]. In 1981, a second proofreading-competent eukaryotic DNA polymerase was described [23]. Initially considered related to pol δ, this enzyme was subsequently shown to interact with proliferating cell nuclear antigen (PCNA) [24], a sliding-clamp protein essential for highly processive DNA synthesis across extended genomic regions [25].
Further biochemical studies revealed two functional forms of pol δ: a PCNA-dependent and a PCNA-independent form. In 1989, a highly processive DNA polymerase with 3ʹ→5ʹ exonuclease activity and independence from PCNA was purified and characterized. This enzyme, which also contains a 122-kDa catalytic core, [26]was designated DNA polymerase ε (pol ε) [27]. As nomenclature evolved, PCNA-dependent polymerases were classified as pol δ, whereas PCNA-independent polymerases were reclassified as pol ε[28]. This reorganization subsequently created terminological confusion: the originally described pol δ, along with the PCNA-dependent form of pol δ were concurrently isolated and demonstrated to be structurally and immunologically distinct. Importantly, the originally described enzyme was confirmed to be highly processive and PCNA-independent; nevertheless, in accordance with the revised nomenclature, it was retroactively reassigned as pol ε, an important distinction for subsequent discussion [29].
Both pol δ and pol ε were cloned in the early 1990s, firmly establishing them as the principal replicative polymerases responsible for the bulk of nuclear DNA synthesis in eukaryotic cells [30,31]. A prevalent model has pol ε synthesizing the leading strand in a continuous manner and pol δ the lagging strand in a discontinuous manner.
DNA Polymerase Replication and Stepwise Fidelity Improvement
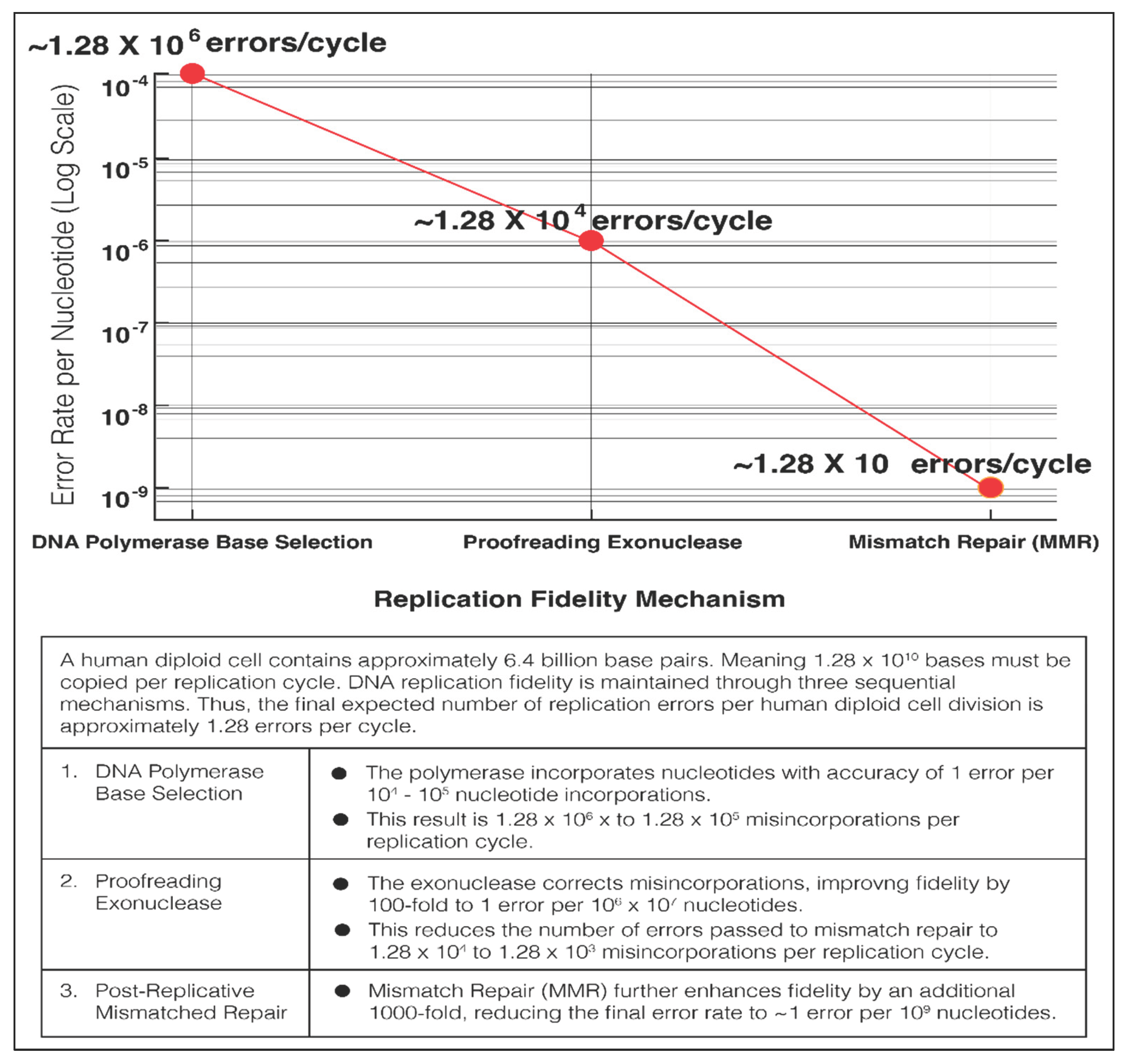
During each cell replication cycle, a human diploid cell genome of 6.4 billion base pairs, or 1.28 x 1010 nucleotides must be copied. The high intrinsic fidelity of the DNA polymerases results in an estimated error rate of more than 1 in 105 nucleotide incorporations. However, this error rate results in roughly 100,000 base misincorporations when copying 6.4 billion base pair. These misincorporations by the DNA polymerase would be the source of the carcinogenic mutations of the Mutator Hypothesis of Dr. Loeb and the Stochastic Error Theory of Dr. Vogelstein. Eventually less than 10 errors per cell is arrived at by stepwise reduction of the errors (Figure 2) [32].
3.1.4. Inhibition of Proofreading Activity Is Mutagenic
Selective inhibition of the exonuclease activity of DNA polymerases by nucleotide 5’monophosphates, especially adenosine 5’-monophosphate (AMP) was reported in 1977 as an extension of the description of the first eukaryotic DNA polymerase with proofreading exonuclease activity (pol δ, reclassified as pol ε). The addition of AMP to the DNA polymerase reaction selectively inhibited exonuclease activity and decreased fidelity but left polymerase activity unaffected [33]. The half maximum inhibiting concentration (IC50) of AMP to selectively inhibit the proofreading exonuclease was approximately 150 uM [33].
It is noteworthy that essentially identical findings that 5’ AMP and other 5’ nucleoside monophosphates also selectively inhibited the proofreading exonuclease of E. coli DNA polymerase 1 were obtained/demonstrated [34]. The selective inhibition is caused by AMP binding to the product site of the exonuclease [34]. A reason for the functional disassociation of the exonuclease proofreading activity from the DNA polymerase activity induced by AMP, a prevalent metabolite, has remained curiously unexplained.
The effect of selective exonuclease inhibition on eukaryotic DNA polymerase fidelity was examined via the reversion of bacteriophage M13mp2 mutants. The accuracy DNA polymerase referred to as δ II was high, as it produced fewer than one single-base substitution error for every 106 nucleotides polymerized. Upon AMP-mediated inhibition of this exonuclease activity, overall fidelity decreased, indicating that the exonuclease improved fidelity by as much as 100-fold [35]. The polymerase was later identified as Pol ε. A subsequent study using a forward mutation assay combined with DNA sequencing provided further insight into the exonuclease function. While Pol ε was highly accurate, base substitution and frameshift error rates increased significantly upon the selective inhibition of its exonuclease activity [36]. Since base substitution and frameshift errors are the mutations most responsible for the formation of oncogenes and inactivation of tumor suppressors, a hypothesis was formulated that inhibition of the proofreading exonuclease would be carcinogenic.
3.1.5. Inhibition of Proofreading Activity Is Carcinogenic
Evidence that selective inhibition of proofreading exonuclease activity is carcinogenic was first obtained in mouse experiments. Mice carrying a point mutation (D400A) in the proofreading domain of the pol δ gene (POLD), which selectively disrupted exonuclease proofreading activity while preserving polymerase activity, were used as an experimental model. Homozygous D400A mice began developing cancer as early as 2 months of age, with 94% of these mice developing cancer by 18 months. In contrast, only 3–4% of heterozygous or wild-type mice developed cancer. Among the 66 tumors identified in 49 homozygous mice, 40 were epithelial in origin [37].
Similarly, mice with a mutation in POLE, resulting in a Pol ε exonuclease-deficient enzyme with preserved polymerase activity, were developed. Homozygous exonuclease-deficient mice progressively died of cancer between 9 and 24 months of age, with a median survival of 16 months, whereas heterozygotes were indistinguishable from wild-type mice in terms of median survival (median survival = 25 months; P > 0.05). The most common tumors observed were intestinal adenomas and adenocarcinomas, with some animals presenting multiple gastrointestinal tumors [38].
Selective inhibition of proofreading exonuclease activity is carcinogenic in humans. Natural knockout mutations in the exonuclease domains of POLD and especially POLE are strongly linked to the development of cancer. In 2012, the first large-scale cancer genome sequencing study identified tumorigenesis-related POLE mutations. Using whole-exome sequencing (WES) and whole-genome sequencing (WGS) of 276 colorectal cancer (CRC) tumors, researchers revealed a subset (16%) of tumors that were hypermutated (mutation burden ≥10 mut/Mb) and presented microsatellite instability (MSI). However, another smaller subset (3%) of tumors were classified as ultramutated, with a mutation burden exceeding 100 mut/Mb, and POLE mutations in these tumors were clustered in the exonuclease domain [39]. Similarly, in 2013, analysis of 373 endometrial carcinomas (ECs) from The Cancer Genome Atlas (TCGA) revealed POLE exonuclease domain mutations in 7% of tumors, with mutation burdens exceeding 100 mut/Mb [40]. These ultramutated tumors presented high rates of base substitutions made by the DNA polymerases, demonstrating and underscoring the important role of the exonuclease in preventing carcinogenesis.
Further studies, including a 2013 study by Church et al. in which the exonuclease domains of POLE (residues 268–471) and POLD (residues 304–517) were screened in 173 EC cases, identified 14 nonsynonymous variants. Thirteen of these nonsynonymous variants were found in POLE, and one was found in POLD. These variants were linked to the incidence of cancer, with POLE mutations predicted to strongly impair proofreading, leading to hypermutated tumors [41]. WGS by Palles et al. revealed germline heterozygous mutations in the exonuclease domains of POLE and POLD in several multiple-adenoma and CRC patients but not in controls. The POLE p. Leu424Val and POLD p.Ser478Asn variants were identified as susceptibility markers with high penetrance. The POLD variant was also associated with an increased risk of EC. These mutations were mapped to equivalent sites in the proofreading exonuclease domains of POLE and POLD and were predicted to impair the correction of mis-paired bases during DNA replication [42].
A comprehensive review by Rayner et al. in 2016 further established the link between germline POLE and POLD exonuclease mutations and cancer. Germline mutations in these exonuclease domains were present in 0.5–2% of patients with intestinal polyposis and familial CRC, with POLE L424V being the most common deleterious germline variant. This variant not only was associated with CRC but also predisposed patients to EC and increased the risk of other cancers, including breast, stomach, and ovarian carcinomas, as well as brain tumors and duodenal adenomas and carcinomas. Somatic POLE exonuclease domain mutations were reported in 1–2% of CRC tumors and 7–12% of EC tumors, as well as ultra-mutated tumors of the brain, pancreas, ovary, breast, stomach, and uterus [43].
A 2019 study queried 10,967 cases in the cBioPortal database and reported 92 POLE exonuclease domain mutations in hypermutated tumors [44]. These cases occurred in several cancer types, including EC (9.7%); CRC (2.2%); and stomach, adrenocortical, and pancreatic cancers (each with an incidence of 1–2% or lower). Prostate, renal, bladder, and head and neck cancers; cervical squamous cell carcinoma; and melanoma also harbored POLE mutations, albeit at lower frequencies. The prevalence of POLD exonuclease domain mutations followed a similar trend [45]. The cancers were primarily epithelial in origin, like the cancer profile associated with obesity with endometrial (EC) and CRC being especially prominent. Many similar reports followed, and exonuclease domain mutations are established as a pathological and diagnostic cause of cancer.
Both somatic and germline mutations in the proofreading exonuclease domains of POLD and POLE demonstrate errors made by the DNA polymerase as a source of oncogenic mutations and genomic instability. They underscore the role of the proofreading exonuclease activity in the prevention of cancers. The hypothesis that inhibition of proofreading exonuclease activity promotes DNA replication-associated carcinogenesis has been confirmed.
3.2. Energy Metabolism in Obesity and Associated Disorders
3.2.1. The Hallmarks of Metabolic Syndrome
The Metabolic Syndrome (MS) was initially recognized as a cluster of interrelated hallmarks which include: insulin resistance, obesity, dyslipidemia, and hypertension, contributing to an increased risk of T2DM and cardiovascular disease [46]. Ruderman and Prentki (2004) observed that MS typically occurs in obese patients and is strongly associated with reduced or impaired activation of AMP activated protein kinase (AMPK), a lack of physical activity, sedentary behavior, overnutrition, a lack of exercise, all of which contribute to the development of related metabolic disorders. They further noted that multiple interventions known to activate AMPK, -- including exercise, leptin, adiponectin, metformin, 5-aminoimidazole-4-carboxamide riboside, (AICAR) and thiazolidinediones -- have been shown to improve insulin resistance and associated metabolic abnormalities in both animal models and humans. On this basis they proposed impaired AMPK activity as both a central pathogenic factor and promising therapeutic target in these disorders [47].
3.2.2. Obesity and Associated Disorders Are Characterized by Impaired Energy Metabolism
Obesity and insulin resistance have been extensively linked to impaired energy metabolism including mitochondrial dysfunction. For example, obese and insulin-resistant individuals exhibit reduced mitochondrial oxidative capacity and defective lipid metabolism, compared to healthy, lean controls [8]. Microarray studies have further elucidated the nature of this mitochondrial dysfunction, demonstrating that genes involved in oxidative metabolism-related genes are downregulated in insulin-resistant subjects compared to healthy controls [48,49]. In addition, the expression of genes involved in fatty acid oxidation is significantly reduced in insulin-resistant individuals. Oxidative metabolism is further compromised by decreased expression of multiple genes involved in glycolysis and tricarboxylic acid cycle. Components of the mitochondrial respiratory chain are also downregulated, including subunits of ATP synthase [49]. Consistent with these transcriptional changes, noninvasive assessment of mitochondrial ATP synthesis rates in young, lean, and sedentary insulin-resistant individuals reveals a 30% reduction compared with insulin-sensitive controls [50].
3.2.3. Impaired Energy Metabolism in [48,49] Obesity: The Central Role of AMPK
Ruderman and colleagues later expanded and the defining hallmarks of the metabolic syndrome as “Insulin resistance and hyperinsulinemia, central adiposity, dyslipidemia, and predisposition to type 2 diabetes, atherosclerotic cardiovascular disease, hypertension, and certain cancers.” [51]. In this updated framework particular emphasis was again placed on the hypothesis that impaired AMPK activity represents a key pathogenic mechanism, as well as an important target in prevention and therapy. Consistent with this expanded disease spectrum, Esposito et al. (2014) in an analysis of 38,940 cancer cases determined that the Metabolic Syndrome (MS) is associated with an increased risk of several primarily epithelial derived malignancies: liver cancer (RR 1.43, P<0.0001), colorectal cancer (1.25, P<0.001), and bladder cancer (1.10, P=0.013) in men. In women MS was associated with endometrial (1.61, P=0.001), pancreatic (1.58, P<0.0001), postmenopausal breast (1.56, P=0.017), rectal (1.52, P=0.005), and colorectal cancer (1.34, P=0.006) [52]. As impaired AMPK activity is proposed as a key pathogenic mechanism in these epithelial primarily cancers, it is necessary to consider the role of AMPK in the production of energy to understand the relation to mutational carcinogenesis.
3.2.4. AMP Regulates Energy Production and Consumption
Adenosine triphosphate (ATP) serves as the primary energy currency in cells, driving a wide array of metabolic processes including DNA replication. Daniel Atkinson proposed that adenine nucleotides regulate branch points between anabolism and catabolism based on observations that energy-producing metabolic enzymes that directly monitor the cellular energy status, such as muscle phosphorylase, fructose bisphosphatase and phosphofructokinase which were allosterically regulated by adenine nucleotides, with AMP and ATP acting in a reciprocal fashion. This concept was formulated as the Energy Charge Hypothesis [53], wherein the energy charge was defined as:
Hans Krebs pointed out that the cellular AMP concentrations fluctuate to a much greater extent than those of ATP or ADP. Normally, cells have a much higher concentration of ATP (5–10 mM) than that of AMP (<0.1 mM). For example: If ATP is consumed such that its concentration drops by 10%, from 5 mM to 4.5 mM, the AMP concentration that was previously 0.1 mM would increase to approximately 0.6 mM, a 600% increase. Although ATP is the primary metabolite that transports high-energy phosphates during metabolic processes via transitioning to ADP or AMP, Krebs concluded that “the absolute concentration of AMP is a much more sensitive controlling agent than the absolute concentration of ATP” [54]. This concept was further established by the discovery of AMP-activated Protein Kinase (AMPK). Acetyl-CoA carboxylase kinase was found to be stimulated by AMP, and it was suggested that this mechanism might inhibit fatty acid synthesis in response to falling energy charge [55], and HMG-Co A reductase kinase also found to be stimulated by AMP [56]. In 1987, Carling et al. reported that a single protein kinase could account for both observations [57]. When it became evident that the kinase had multiple physiological substrates, it was named “AMP-activated protein kinase” (AMPK), after its allosteric activator, AMP [58]. AMPK is activated in an ultrasensitive manner by cellular stresses that deplete ATP and consequently elevate AMP concentrations. AMP binding to AMPK was found to have three regulating effects: (i) a conformational change that activates and makes AMPK a good substrate for phosphorylation at Tyr 172 of the catalytic subunit by another kinase, namely, LKB1, (ii) phosphorylation of this tyrosine residue by LKB1 potentiates further AMP-induced activation of AMPK (10-fold), and (iii) inhibition of Tyr 172 dephosphorylation. These three mechanisms act synergistically and make the system sensitive to small increases in AMP [59]. AMPK activation switches on catabolic ATP synthesis pathways while suppressing many ATP-consuming, anabolic processes. As a result, ATP concentration is increased, while AMP concentration is reduced. The half maximal effective concentration (EC50) for AMP activating AMPK under physiological conditions was determined to be 196 uM [60] implying that this concentration of AMP represents an energy deficient state.
The discovery of AMPK reinforced the Energy Charge Hypothesis, with cellular AMP concentration coordinating energy production and consumption. “Any system that monitors cellular energy status should respond to AMP concentrations” [61]. A relatively high AMP occurs when the energy charge is low and should be conserved and ATP production increased. Conversely, cellular AMP is low when the energy charge, ATP + ½ ADP, content, is high.
In mammals, AMPK is a heterotrimeric complex with an α catalytic subunit as well as β and γ regulatory subunits. Each subunit is encoded by two or three genes (α1, α2, β1, β2, γ1, γ2, γ3), and at least 12 heterotrimeric combinations are possible, providing a diversity of regulatory functions [62]. These combinations confer different properties to AMPK, including subcellular localization and signaling functions. Inhibitory regulation of AMPK by nutrients, lipid overload, high glucose concentrations, glycogen, various amino acids, hormones, cytokines, and especially inflammatory signals have been shown to be relevant to disorders of impaired energy metabolism [63].
In summary, AMPK is the major regulator of cellular energy production and consumption at multiple and diverse levels in the control of whole-body homeostasis and the promotion of cell survival under conditions of low cellular energy with the number of reported targets for AMPK currently more than one hundred including regulation of (a wide array of) other physiological events including) cellular growth and proliferation, mitochondrial function and biogenesis, and factors that have been linked to insulin resistance, including inflammation oxidative stress and autophagy [64].
3.2.5. Impaired AMPK Activity Is Linked with Carcinogenesis
Impaired AMPK activity is a key pathogenic mechanism in certain cancers. The Peutz–Jeghers syndrome (PJS), links impaired AMPK regulated energy metabolism with (epithelial cell) carcinogenesis. The RR of epithelial-derived carcinomas, similar to that of obesity, is 15.2 times greater in patients with familial PJS than the general population [65]. In 1998, Hemminski et al. [66], and Jenne et al. [67], reported that PJS is caused by inactivating mutations in the serine‒threonine kinase LKB1, indicating that LKB1 is a tumor suppressor. In 2003, Hawley et al. [68], Woods et al. [69], and Shaw et al. [70], reported that LKB1 is the upstream kinase required for AMPK activation, thus linking impaired AMPK regulation with carcinogenesis. Hardie noted that “the most significant impact of this discovery was the connection between LKB1, a tumor suppressor involved in cancer, and AMPK, a protein kinase previously recognized for its role in regulating metabolism, particularly in disorders like diabetes” [71]. Thus, LKB1 deficiency impairs AMPK and establishes a mechanistic connection between impaired energy metabolism and epithelial cell carcinogenesis.
3.2.6. AMPK Activation Prevents These Cancers
AMPK activity in the adipose tissue of obese, insulin- resistant individuals before undergoing bariatric surgery was found to be reduced by thirty to fifty percent [72,73]. Weight-loss by procedures such as bariatric surgery reverse insulin resistance and the metabolic disorder associated with MS. Notably, bariatric surgery was found to lower the prevalence of solid (obesity associated) cancers by 70% within five years [74].
Similarly, Hardie hypothesized that “if evidence suggests that the LKB1-AMPK pathway is indeed a tumor-suppressing pathway, then AMPK-activating drugs might be expected to provide protection against the development of cancer” [71]. A 2005 study reported that patients with T2DM receiving metformin, a known AMPK activator, had a significantly reduced incidence of cancer was referenced [75]. This finding was subsequently validated in other T2DM patient cohorts, as a meta-analysis indicated an overall cancer RR of 0.61 (95% confidence interval [CI] [0.54–0.70]) in metformin users compared with sulfonylurea users [76].
Hardie also proposed that AMPK activation can explain some of the beneficial effects of salsalate and aspirin. These drugs break down into salicylate, which activates AMPK by binding at an allosteric site, inhibiting the dephosphorylation of AMPK at Thr172 [77]. The cancer-preventive effect of aspirin was first reported after long-term follow-up in large trials designed to evaluate its cardiovascular benefits. Data from 51 trials, including more than 77,000 participants, demonstrated that individuals had a reduced risk of cancer after five years of aspirin administration (hazard ratio [HR]: 0.81, 95% CI: 0.72–0.93) [78].
Obesity and EC are strongly and significantly associated, suggesting that this relationship is suitable for exploring the potential application of AMPK activators for cancer prevention. Several large studies have demonstrated the effectiveness of aspirin in preventing EC, particularly in obese patients [79]. For example, the Australian National EC Study, comprising 1,398 cases, revealed a nearly 50% risk reduction (OR = 0.54, 95% CI: 0.38–0.78) in women reporting the use of ≥2 aspirin tablets per week. A meta-analysis of eight previous studies combined with these data suggested that aspirin use was associated with a lower cancer risk, with a pooled risk estimate for obese women (BMI > 30 kg/m²) of 0.72 (95% CI: 0.58–0.90). However, no significant association between aspirin use and cancer risk was observed among nonobese women (BMI < 30 kg/m²) [80].
Another meta-analysis of 13 observational studies, including 11,323 cases, demonstrated that regular aspirin use was associated with a decreased risk of EC. When patients with the highest frequency of use were compared with nonusers, a risk reduction of 37% (OR = 0.63, 95% CI: 0.45–0.88) was observed. Analysis with respect to BMI revealed an inverse association of EC risk reduction among women with a BMI > 30 kg/m² (case‒control: 44% risk reduction, OR = 0.56, 95% CI: 0.33–0.95; cohort: 20% risk reduction, OR = 0.80, 95% CI: 0.60–1.07) [81]. Further analysis revealed a greater risk reduction with increasing aspirin dose and frequency, with long-term use conferring protection from EC mainly in women with obesity [82].
Finally, subgroup analysis of data from over 7,000 women with EC from the Epidemiology of Endometrial Cancer Consortium revealed that the use of aspirin at least once weekly was associated with a risk reduction (OR = 0.86, 95% CI: 0.76–0.98) in overweight and obese women, whereas no effect was observed in women of normal weight [83].
Aspirin has also been shown to reduce the risk of CRC, particularly in obese patients. In the Colorectal Adenoma/Carcinoma Prevention Program (CAPP), individuals with Lynch syndrome were randomly assigned to 600 mg/day aspirin or a placebo. Long-term analysis demonstrated a marked reduction in the incidence of CRC (incidence rate ratio: 0.37) in participants taking aspirin for two or more years compared with those receiving a placebo [72]. Subgroup analysis suggested that the preventive benefits of aspirin in Lynch syndrome may be most pronounced in individuals with obesity [84].
A recent meta-analysis confirmed the association of regular aspirin use with a significantly reduced risk of cancers, including CRC (RR=0.73, 95% CI=0.69–0.78, 45 studies), squamous cell esophageal cancer (RR=0.67, 95% CI=0.57–0.79, 13 studies), adenocarcinoma of the esophagus and gastric cardia (RR=0.61, 95% CI=0.49–0.77, 10 studies), stomach cancer (RR=0.64, 95% CI=0.51–0.82, 14 studies), hepato-biliary tract cancer (RR=0.62, 95% CI=0.44– 0.86, five studies), and pancreatic cancer (RR=0.78, 95% CI=0.68–0.89, 15 studies [85]. A dose- and time-dependent linear response was observed for CRC, with a significant 10% reduction with 75 mg/day aspirin increasing to 50% with 500 mg/day aspirin. Patients exhibited a time-dependent RR of 4%, 19%, and 29% at 1, 5, and 10 years, respectively [85]. Obesity was not included in the analysis. However, in a complementary cohort study of 107,655 men and women receiving aspirin and followed up for more than 3 decades, aspirin reduced the absolute risk of CRC primarily in individuals with obesity supporting the thesis that AMPK activation in obesity reduces the risk of the associated carcinogenesis [7].
A consideration of the prominence of epithelial cancer in obesity and associated disorders is warranted. Epithelial tissues are characterized by continuous and highly ordered cell turnover and proliferation, requiring tightly synchronized cycles of DNA replication, mitosis, and differentiation to maintain tissue integrity and barrier functions. Large populations of cells enter the cell cycle in a coordinated fashion in response to physiological cues. As a result, epithelial cells operate near the upper limits of replicative capacity under normal conditions, leaving little tolerance for perturbations in cell-cycle control, nucleotide availability, or energy homeostasis. Their failure to participate could have immediate life-threatening consequences. Consequently, epithelial replication fidelity is intrinsically linked to cellular energy status and to the metabolic pathways that sustain nucleotide pool balance and ATP/AMP homeostasis.
Endometrial epithelial cells are prototypical in this regard. Across the menstrual cycle, the endometrium undergoes repeated rounds of hormonally driven proliferation, differentiation, shedding, and regeneration. During the proliferative phase, endometrial epithelial cells engage in rapid and synchronized DNA synthesis, making them exquisitely dependent on intact energy metabolism and robust nucleotide biosynthesis. This recurrent, large-scale replicative activity renders the endometrium particularly vulnerable to fluctuations in energy charge, and mechanisms that coordinate energy availability with DNA replication fidelity.
4. Discussion
The hypothesis that selective inhibition of proofreading exonuclease activity leads to carcinogenesis by failing to correct DNA polymerase base substitutions and frameshift errors has been confirmed. The hypotheses that impaired AMPK regulated energy production in obesity contributes to carcinogenesis and conversely that AMPK activators can reduce the risk of obesity-associated cancers also have been validated. These concepts are interconnected through increased AMP which under conditions of low cellular energy inhibits the proofreading exonuclease to conserve energy and promote cell survival. Increased AMP also attempts to increases energy production and decrease consumption; however, AMPK is impaired. Therefore, we postulate that AMP regulated proofreading activity underlies obesity associated carcinogenesis, and we propose the following mechanism and synthesis.
In obesity, impaired AMPK activity leads to decreased ATP levels, causing a relatively much greater increase in AMP levels, as described by Krebs. Increased AMP inhibits DNA polymerase proofreading exonuclease activity, reducing the correction of misincorporated bases, some of which give rise to oncogenic mutations or disrupt tumor suppressor genes. Conversely, if AMPK is reactivated, increasing ATP production and decreasing AMP levels, allows proofreading activity to resume, thereby reducing the accumulation of carcinogenic mutations, and lowering cancer risk over time. The AMP concentration selectively inhibits DNA replication proofreading activity in accordance with available energy levels which are reduced in the circumstance of impaired AMPK in obesity and associated disorders.
The selective inhibition of exonuclease-mediated proofreading by AMP differs from inhibition caused by exonuclease domain mutations. AMP-mediated inhibition is conditional rather than permanent, fluctuating across different cells and timeframes, depending on metabolic conditions and energy availability. In contrast, loss of exonuclease activity due to domain mutations is permanent and pathological. Despite differing mechanisms, both AMP-driven inhibition and exonuclease gene mutations contribute to mutational carcinogenesis in a similar manner by allowing base substitutions and frameshift errors made by the DNA polymerase to remain uncorrected and accumulate within the genome.
Regulating the mutation rate in relation to available energy introduces a third function to the DNA polymerase catalytic core, alongside DNA synthesis and exonuclease proofreading. Thus, selective inhibition of proofreading activity by AMP establishes both a metabolic and physical link between energy metabolism and mutational carcinogenesis. This metabolic linkage should be a primary target for intervention. Specifically, increasing cellular ATP concentrations to lower AMP levels and restore proofreading exonuclease activity by activating impaired AMPK represents a viable strategy to enhance DNA replication fidelity and mitigate obesity-associated carcinogenesis. The referenced clinical studies support the carcinogenic preventive effectiveness of this approach.
This analysis reveals two key insights:
- The mutation rate is metabolically regulated via energy-sensitive proofreading modulation.
- This regulation is integrated within the single 122 kDa core of the DNA polymerase complex, linking impaired energy metabolism directly to genomic instability.
Together they indicate that the prevention of mutational oncogenesis requires an integration of the disciplines of energy metabolism and DNA replication enzymology both in the research laboratory and the clinic. These findings explain the epidemiological connection between obesity and cancer and offer a mechanistic framework for understanding how impaired energy metabolism may promote carcinogenesis. The implications for prevention and therapeutic intervention are considerable, particularly, but not exclusively, for obesity-associated cancers.
Within the Hallmarks of Cancer paradigm, this model positions impaired energy metabolism not merely as an adaptive feature of cancer cells, but as a causal upstream driver of mutational oncogenesis. It explains how metabolic dysfunction characteristic of obesity can precede and promote tumor initiation by enabling genome instability, thereby accelerating cancer evolution.
Within the Hallmarks of Cancer paradigm, this synthesis includes two “enabling characteristics”:
- “Genomic Instability and mutation”. Impaired energy metabolism, increasing AMP-mediated inhibition of DNA polymerase proofreading accelerates stochastic mutation accumulation and genomic instability.
- “Dysregulated cellular energetics”, obesity-associated AMPK impairment represents a primary metabolic lesion that proceeds and facilitates genomic instability.
And Core Hallmarks are also influenced indirectly.
- “Sustaining proliferative signaling”: an increased mutational burden raises the probability of activating driver mutations.
- “Resisting Cell Death”: Energy stress-induced prioritization of survival over fidelity favors short-term viability at the cost of long-term genomic integrity.
- “Tumor evolution and heterogeneity”: A higher mutation rate increases clonal diversity and accelerates evolutionary selection.
This synthesis is also consistent with the second law of thermodynamics: Increasing informational order, in this case, the accurate copying of genetic information, necessarily requires the dissipation of free energy. In practical terms, each incremental reduction in mutation frequency requires a disproportionate increase in energy expenditure, producing a nonlinear approximately logarithmic relationship between replication fidelity and energetic cost, as emphasized by Loeb. Thus, the maintenance of genomic stability is contingent upon adequate energy availability, whereas energy limitation favors a shift towards lower fidelity DNA replication that conserves ATP at the expense of increased mutational burden. Consequently, DNA replication fidelity emerges as a metabolically regulated parameter rather than a fixed biochemical constant. Under conditions of impaired energy metabolism such as those observed in obesity and insulin resistance, energetic constraints may selectively relax proofreading and repair mechanisms, thereby increasing mutation rates and promoting genomic instability. This energetic perspective provides a unifying physical and biochemical framework linking cellular metabolism, thermodynamic constraints, and carcinogenic mutation accumulation. At last, there is a metabolically reasonable explanation for the selective inhibition of the proofreading exonuclease by a prevalent regulatory metabolite.
Limitations and corollary questions are evident. Research on energy metabolism has focused on specific tissues, including skeletal muscle, adipose tissue, and liver, with findings frequently extrapolated to carcinogenic epithelial tissues due to the separation of the two disciplines. A central goal of our work is to ensure that these limitations and mechanistic uncertainties are recognized and addressed in future research through integrated, unified studies of energy metabolism with cancer prevention.
The cause of impaired AMPK activity in obesity represents an important corollary question. proinflammatory cytokines associated with obesity, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and others, have been implicated, this remains an area of active investigation and evolving science.
The scientific and pharmacologic discovery of AMPK activation is an area of substantial interest, with numerous natural products and several synthetic compounds under consideration for further development. There is intense interest in the pharmacologic activation of AMPK as a therapeutic strategy for multiple disease states, including obesity, diabetes, and cancer [86].
5. Conclusions
This synthesis encompasses a number of practical implications. Impaired AMPK in obesity provides a mechanistically coherent link between disordered energy metabolism and carcinogenesis. This framework is intuitive, biologically grounded, and supports existing recommendations for weight control, diet quality, and physical activity, potentially improving long-term adherence. Importantly, the predicted reduction in cancer risk operates on an extended time horizon: if carcinogenesis is driven by stochastic DNA replication errors whose frequency is modulated by cellular energy status, then metabolic correction reduces the rate of mutation accumulation rather than reversing existing damage. Consequently, measurable oncologic benefit may not be evident for many years.
In contrast, improvements in cardiometabolic health, including insulin sensitivity, hepatic steatosis, glycemic control, and systemic inflammation often/can occur within weeks to months and should be emphasized as primary motivators. Cancer reduction should be framed as a long -term consequence of sustained metabolic normalization.
Pharmacologic agents such as GLP-receptor agonists likely will reduce obesity cancer risk indirectly through sustained weight loss. Since their primary actions are centrally mediated, tissue-specific consequences preclude simple mechanistic predictions.
Finally, continuous AMPK overstimulation may have adverse tissue-specific consequences, underscoring that prevention aims to restore physiologic, context-dependent AMPK regulation rather than induce chronic activation. The history of low-dose aspirin in primary prevention provides a useful, and relevant analogy: modest long-term benefit can be offset by cumulative adverse effects, underscoring the need for balance and selectivity in preventive strategies.
We hope this synthesis of cellular energy metabolism and its relationship to obesity-associated carcinogenesis, particularly in terms of preventive strategies, will encourage its further development and its application to alleviate the projected rise in cancer cases.
“However, in real life it is survival, not fidelity, that is the ultimate virtue” (Radman) [87].
Funding
From 1973 through 1995, the Veterans Administration provided research support to Dr. John Byrnes in the form of Career Development Support including a Clinical Investigatorship, and subsequently through the Merit Review Award System.
Data Availability Statement
All data is available in the attached reference list.
Acknowledgments
Mary Anne Milone, APRN; PhD for exemptional administrative and editorial support.
Conflicts of Interest
John J. Byrnes, the author declares no conflicts of interest.
References
- Calle, E. E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M. J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med From NLM Medline. 2003, 348(17), 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D. A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet From NLM Medline. 2014, 384(9945), 755–765. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; Working, G.; International Agency for Research on Cancer Handbook. Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med From NLM Medline. 2016, 375(8), 794–798. [Google Scholar] [CrossRef]
- Win, A. K.; Dowty, J. G.; Antill, Y. C.; English, D. R.; Baron, J. A.; Young, J. P.; Giles, G. G.; Southey, M. C.; Winship, I.; Lipton, L.; et al. Body mass index in early adulthood and endometrial cancer risk for mismatch repair gene mutation carriers. Obstet Gynecol From NLM Medline. 2011, 117(4), 899–905. [Google Scholar] [CrossRef]
- Win, A. K.; Dowty, J. G.; English, D. R.; Campbell, P. T.; Young, J. P.; Winship, I.; Macrae, F. A.; Lipton, L.; Parry, S.; Young, G. P.; et al. Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer From NLM Medline. 2011, 105(1), 162–169. [Google Scholar] [CrossRef]
- Li, X.; Jansen, L.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Risk of Colorectal Cancer Associated With Lifetime Excess Weight. JAMA Oncol From NLM Medline. 2022, 8(5), 730–737. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R. L.; Rosenberg, P. S.; Jemal, A. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health From NLM Medline. 2019, 4(3), e137–e147. [Google Scholar] [CrossRef]
- Simoneau, J. A.; Veerkamp, J. H.; Turcotte, L. P.; Kelley, D. E. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. FASEB J From NLM Medline. 1999, 13(14), 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D. M.; Archer, M. C.; Bergenstal, R. M.; Gapstur, S. M.; Habel, L. A.; Pollak, M.; Regensteiner, J. G.; Yee, D. Diabetes and cancer: a consensus report. Diabetes Care From NLM Medline. 2010, 33(7), 1674–1685. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. A. The hallmarks of cancer. Cell From NLM Medline. 2000, 100(1), 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. A. Hallmarks of cancer: the next generation. Cell From NLM Medline. 2011, 144(5), 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov From NLM Medline. 2022, 12(1), 31–46. [Google Scholar] [CrossRef]
- Loeb, L. A.; Springgate, C. F.; Battula, N. Errors in DNA replication as a basis of malignant changes. In Cancer Res; From NLM Medline, 1974; Volume 34, 9, pp. 2311–2321. [Google Scholar]
- Loeb, L. A. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer Res From NLM Medline. 2016, 76(8), 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L. B.; Nik-Zainal, S.; Wedge, D. C.; Aparicio, S. A.; Behjati, S.; Biankin, A. V.; Bignell, G. R.; Bolli, N.; Borg, A.; Borresen-Dale, A. L.; et al. Signatures of mutational processes in human cancer. Nature From NLM Medline. 2013, 500(7463), 415–421. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E. R.; Hamilton, S. R.; Kern, S. E.; Preisinger, A. C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A. M.; Bos, J. L. Genetic alterations during colorectal-tumor development. N Engl J Med From NLM Medline. 1988, 319(9), 525–532. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B.; Parmigiani, G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A From NLM Medline. 2013, 110(6), 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science From NLM Medline. 2015, 347(6217), 78–81. [Google Scholar] [CrossRef]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science From NLM Medline. 2017, 355(6331), 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V. E.; Zhou, S.; Diaz, L. A., Jr.; Kinzler, K. W. Cancer genome landscapes. Science From NLM Medline. 2013, 339(6127), 1546–1558. [Google Scholar] [CrossRef]
- Byrnes, J. J.; Downey, K. M.; Black, V. L.; So, A. G. A new mammalian DNA polymerase with 3' to 5' exonuclease activity: DNA polymerase delta. Biochemistry From NLM Medline. 1976, 15(13), 2817–2823. [Google Scholar] [CrossRef]
- Goscin, L. P.; Byrnes, J. J. DNA polymerase delta: one polypeptide, two activities. Biochemistry From NLM Medline. 1982, 21(10), 2513–2518. [Google Scholar] [CrossRef]
- Lee, M. Y.; Tan, C. K.; So, A. G.; Downey, K. M. Purification of deoxyribonucleic acid polymerase delta from calf thymus: partial characterization of physical properties. Biochemistry From NLM Medline. 1980, 19(10), 2096–2101. [Google Scholar] [CrossRef]
- Bravo, R.; Frank, R.; Blundell, P. A.; Macdonald-Bravo, H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature From NLM Medline. 1987, 326(6112), 515–517. [Google Scholar] [CrossRef]
- Prelich, G.; Tan, C. K.; Kostura, M.; Mathews, M. B.; So, A. G.; Downey, K. M.; Stillman, B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta auxiliary protein. Nature From NLM Medline. 1987, 326(6112), 517–520. [Google Scholar] [CrossRef]
- Syvaoja, J.; Linn, S. Characterization of a large form of DNA polymerase delta from HeLa cells that is insensitive to proliferating cell nuclear antigen. In J Biol Chem; From NLM Medline, 1989; Volume 264, 5, pp. 2489–2497. [Google Scholar]
- Syvaoja, J.; Suomensaari, S.; Nishida, C.; Goldsmith, J. S.; Chui, G. S.; Jain, S.; Linn, S. DNA polymerases alpha, delta, and epsilon: three distinct enzymes from HeLa cells. Proc Natl Acad Sci U S A From NLM Medline. 1990, 87(17), 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P. M.; Bambara, R. A.; Campbell, J. L.; Chang, L. M.; Downey, K. M.; Hubscher, U.; Lee, M. Y.; Linn, S. M.; So, A. G.; Spadari, S. Revised nomenclature for eukaryotic DNA polymerases. Eur J Biochem From NLM Medline. 1990, 191(3), 617–618. [Google Scholar] [CrossRef]
- Lu, C. D.; Byrnes, J. J. PCNA-dependent DNA polymerase delta from rabbit bone marrow. Biochemistry From NLM Medline. 1992, 31(49), 12403–12409. [Google Scholar] [CrossRef]
- Chung, D. W.; Zhang, J. A.; Tan, C. K.; Davie, E. W.; So, A. G.; Downey, K. M. Primary structure of the catalytic subunit of human DNA polymerase delta and chromosomal location of the gene. Proc Natl Acad Sci U S A From NLM Medline. 1991, 88(24), 11197–11201. [Google Scholar] [CrossRef] [PubMed]
- Kesti, T.; Frantti, H.; Syvaoja, J. E. Molecular cloning of the cDNA for the catalytic subunit of human DNA polymerase epsilon. In J Biol Chem; From NLM Medline, 1993; Volume 268, 14, pp. 10238–10245. [Google Scholar]
- Bebenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA replication-a matter of proofreading. Curr Genet From NLM Medline. 2018, 64(5), 985–996. [Google Scholar] [CrossRef]
- Byrnes, J. J.; Downey, K. M.; Que, B. G.; Lee, M. Y.; Black, V. L.; So, A. G. Selective inhibition of the 3' to 5' exonuclease activity associated with DNA polymerases: a mechanism of mutagenesis. Biochemistry From NLM Medline. 1977, 16(17), 3740–3746. [Google Scholar] [CrossRef] [PubMed]
- Que, B. G.; Downey, K. M.; So, A. G. Mechanisms of selective inhibition of 3' to 5' exonuclease activity of Escherichia coli DNA polymerase I by nucleoside 5'-monophosphates. Biochemistry From NLM Medline. 1978, 17(9), 1603–1606. [Google Scholar] [CrossRef]
- Kunkel, T. A.; Sabatino, R. D.; Bambara, R. A. Exonucleolytic proofreading by calf thymus DNA polymerase delta. Proc Natl Acad Sci U S A From NLM Medline. 1987, 84(14), 4865–4869. [Google Scholar] [CrossRef]
- Thomas, D. C.; Roberts, J. D.; Sabatino, R. D.; Myers, T. W.; Tan, C. K.; Downey, K. M.; So, A. G.; Bambara, R. A.; Kunkel, T. A. Fidelity of mammalian DNA replication and replicative DNA polymerases. Biochemistry From NLM Medline. 1991, 30(51), 11751–11759. [Google Scholar] [CrossRef]
- Goldsby, R. E.; Lawrence, N. A.; Hays, L. E.; Olmsted, E. A.; Chen, X.; Singh, M.; Preston, B. D. Defective DNA polymerase-delta proofreading causes cancer susceptibility in mice. Nat Med 2001, 7(6), 638–639. [Google Scholar] [CrossRef]
- Albertson, T. M.; Ogawa, M.; Bugni, J. M.; Hays, L. E.; Chen, Y.; Wang, Y.; Treuting, P. M.; Heddle, J. A.; Goldsby, R. E.; Preston, B. D. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A From NLM Medline. 2009, 106(40), 17101–17104. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature From NLM Medline. 2012, 487(7407), 330–337. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A. D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A. G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature From NLM Medline. 2013, 497(7447), 67–73. [Google Scholar] [CrossRef]
- Church, D. N.; Briggs, S. E.; Palles, C.; Domingo, E.; Kearsey, S. J.; Grimes, J. M.; Gorman, M.; Martin, L.; Howarth, K. M.; Hodgson, S. V.; et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet From NLM Medline. 2013, 22(14), 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J. B.; Howarth, K. M.; Domingo, E.; Jones, A. M.; Broderick, P.; Kemp, Z.; Spain, S. L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet From NLM Medline. 2013, 45(2), 136–144. [Google Scholar] [CrossRef]
- Rayner, E.; van Gool, I. C.; Palles, C.; Kearsey, S. E.; Bosse, T.; Tomlinson, I.; Church, D. N. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer From NLM Medline. 2016, 16(2), 71–81. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B. E.; Sumer, S. O.; Aksoy, B. A.; Jacobsen, A.; Byrne, C. J.; Heuer, M. L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012, 2(5), 401–404. [Google Scholar] [CrossRef]
- Park, V. S.; Pursell, Z. F. POLE proofreading defects: Contributions to mutagenesis and cancer. In DNA Repair (Amst); From NLM Medline, 2019; Volume 76, pp. 50–59. [Google Scholar] [CrossRef]
- Reaven, G. M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes From NLM Medline. 1988, 37(12), 1595–1607. [Google Scholar] [CrossRef]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov From NLM Medline. 2004, 3(4), 340–351. [Google Scholar] [CrossRef]
- Mootha, V. K.; Lindgren, C. M.; Eriksson, K. F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet From NLM Medline. 2003, 34(3), 267–273. [Google Scholar] [CrossRef]
- Patti, M. E.; Butte, A. J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A From NLM Medline. 2003, 100(14), 8466–8471. [Google Scholar] [CrossRef]
- Petersen, K. F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G. I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med From NLM Medline. 2004, 350(7), 664–671. [Google Scholar] [CrossRef]
- Ruderman, N. B.; Carling, D.; Prentki, M.; Cacicedo, J. M. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest From NLM Medline. 2013, 123(7), 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care From NLM Medline. 2012, 35(11), 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Hathaway, J. A.; Atkinson, D. E. Adenylate as a Metabolic Regulator. Effect on Yeast Phosphofructokinase Kinetics. J Biol Chem From NLM Medline. 1964, 239, 3619–3622. [Google Scholar] [CrossRef]
- Krebs, H. The Croonian Lecture, 1963. Gluconeogenesis. Proc R Soc Lond B Biol Sci From NLM Medline. 1964, 159, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Yeh, L. A.; Lee, K. H.; Kim, K. H. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. In J Biol Chem; NLM Medline, 1980; Volume 255, 6, pp. 2308–2314. [Google Scholar]
- Ferrer, A.; Caelles, C.; Massot, N.; Hegardt, F. G. Activation of rat liver cytosolic 3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by adenosine 5'-monophosphate. Biochem Biophys Res Commun From NLM Medline. 1985, 132(2), 497–504. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Zammit, V. A.; Hardie, D. G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett From NLM Medline. 1987, 223(2), 217–222. [Google Scholar] [CrossRef]
- Hardie, D. G.; Carling, D. The AMP-activated protein kinase: a multisubstrate regulator of lipid metabolism. Trends Biochem Sci 1989, 14(1), 20–23. [Google Scholar] [CrossRef]
- Hawley, S. A.; Selbert, M. A.; Goldstein, E. G.; Edelman, A. M.; Carling, D.; Hardie, D. G. 5'-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem From NLM Medline. 1995, 270(45), 27186–27191. [Google Scholar] [CrossRef]
- Gowans, G. J.; Hawley, S. A.; Ross, F. A.; Hardie, D. G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. In Cell Metab; From NLM Medline, 2013; Volume 18, 4, pp. 556–566. [Google Scholar] [CrossRef]
- Hardie, D. G.; Hawley, S. A. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays From NLM Medline. 2001, 23(12), 1112–1119. [Google Scholar] [CrossRef]
- Ross, F. A.; MacKintosh, C.; Hardie, D. G. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J From NLM Medline. 2016, 283(16), 2987–3001. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol From NLM Medline. 2010, 45(4), 276–295. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D. G. AMP-activated protein kinase - a journey from 1 to 100 downstream targets. Biochem J From NLM Medline. 2022, 479(22), 2327–2343. [Google Scholar] [CrossRef]
- Giardiello, F. M.; Brensinger, J. D.; Tersmette, A. C.; Goodman, S. N.; Petersen, G. M.; Booker, S. V.; Cruz-Correa, M.; Offerhaus, J. A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology From NLM Medline. 2000, 119(6), 1447–1453. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391(6663), 184–187. [Google Scholar] [CrossRef]
- Jenne, D. E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet From NLM Medline. 1998, 18(1), 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S. A.; Boudeau, J.; Reid, J. L.; Mustard, K. J.; Udd, L.; Makela, T. P.; Alessi, D. R.; Hardie, D. G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol From NLM Medline. 2003, 2(4), 28. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S. R.; Dickerson, K.; Leiper, F. C.; Fryer, L. G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol From NLM Medline. 2003, 13(22), 2004–2008. [Google Scholar] [CrossRef]
- Shaw, R. J.; Kosmatka, M.; Bardeesy, N.; Hurley, R. L.; Witters, L. A.; DePinho, R. A.; Cantley, L. C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A From NLM Medline. 2004, 101(10), 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D. G.; Alessi, D. R. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol From NLM Medline. 2013, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M. S.; O'Brien, E. L.; Bigornia, S.; Mott, M.; Cacicedo, J. M.; Xu, X. J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun From NLM Medline. 2011, 404(1), 382–387. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. J.; Gauthier, M. S.; Hess, D. T.; Apovian, C. M.; Cacicedo, J. M.; Gokce, N.; Farb, M.; Valentine, R. J.; Ruderman, N. B. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res From NLM Medline. 2012, 53(4), 792–801. [Google Scholar] [CrossRef]
- Sjostrom, L.; Gummesson, A.; Sjostrom, C. D.; Narbro, K.; Peltonen, M.; Wedel, H.; Bengtsson, C.; Bouchard, C.; Carlsson, B.; Dahlgren, S.; et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol From NLM Medline. 2009, 10(7), 653–662. [Google Scholar] [CrossRef]
- Evans, J. M.; Donnelly, L. A.; Emslie-Smith, A. M.; Alessi, D. R.; Morris, A. D. Metformin and reduced risk of cancer in diabetic patients. BMJ From NLM Medline. 2005, 330(7503), 1304–1305. [Google Scholar] [CrossRef]
- Soranna, D.; Scotti, L.; Zambon, A.; Bosetti, C.; Grassi, G.; Catapano, A.; La Vecchia, C.; Mancia, G.; Corrao, G. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis. Oncologist From NLM Medline. 2012, 17(6), 813–822. [Google Scholar] [CrossRef]
- Hawley, S. A.; Fullerton, M. D.; Ross, F. A.; Schertzer, J. D.; Chevtzoff, C.; Walker, K. J.; Peggie, M. W.; Zibrova, D.; Green, K. A.; Mustard, K. J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science From NLM Medline. 2012, 336(6083), 918–922. [Google Scholar] [CrossRef]
- Rothwell, P. M.; Fowkes, F. G.; Belch, J. F.; Ogawa, H.; Warlow, C. P.; Meade, T. W. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet From NLM Medline. 2011, 377(9759), 31–41. [Google Scholar] [CrossRef]
- Joharatnam-Hogan, N.; Cafferty, F. H.; Macnair, A.; Ring, A.; Langley, R. E. The role of aspirin in the prevention of ovarian, endometrial and cervical cancers. Womens Health (Lond) From NLM Medline. 2020, 16, 1745506520961710. [Google Scholar] [CrossRef]
- Neill, A. S.; Nagle, C. M.; Protani, M. M.; Obermair, A.; Spurdle, A. B.; Webb, P. M.; Australian National Endometrial Cancer Study; G. Aspirin, nonsteroidal anti-inflammatory drugs, paracetamol and risk of endometrial cancer: a case-control study, systematic review and meta-analysis. Int J Cancer From NLM Medline. 2013, 132(5), 1146–1155. [Google Scholar] [CrossRef]
- Verdoodt, F.; Friis, S.; Dehlendorff, C.; Albieri, V.; Kjaer, S. K. Non-steroidal anti-inflammatory drug use and risk of endometrial cancer: A systematic review and meta-analysis of observational studies. Gynecol Oncol From NLM Medline. 2016, 140(2), 352–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Bai, B.; Xi, Y.; Zhao, Y. Can Aspirin Reduce the Risk of Endometrial Cancer?: A Systematic Review and Meta-analysis of Observational Studies. Int J Gynecol Cancer From NLM Medline. 2016, 26(6), 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Webb, P. M.; Na, R.; Weiderpass, E.; Adami, H. O.; Anderson, K. E.; Bertrand, K. A.; Botteri, E.; Brasky, T. M.; Brinton, L. A.; Chen, C.; et al. Use of aspirin, other nonsteroidal anti-inflammatory drugs and acetaminophen and risk of endometrial cancer: the Epidemiology of Endometrial Cancer Consortium. Ann Oncol From NLM Medline. 2019, 30(2), 310–316. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, M.; Bishop, D. T.; Macrae, F.; Mecklin, J. P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D. G.; Maher, E. R.; Bertario, L.; et al. Obesity, Aspirin, and Risk of Colorectal Cancer in Carriers of Hereditary Colorectal Cancer: A Prospective Investigation in the CAPP2 Study. J Clin Oncol From NLM Medline. 2015, 33(31), 3591–3597. [Google Scholar] [CrossRef]
- Bosetti, C.; Santucci, C.; Gallus, S.; Martinetti, M.; La Vecchia, C. Aspirin and the risk of colorectal and other digestive tract cancers: an updated meta-analysis through 2019. Ann Oncol From NLM Medline. 2020, 31(5), 558–568. [Google Scholar] [CrossRef]
- Steinberg, G. R.; Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov From NLM Medline. 2019, 18(7), 527–551. [Google Scholar] [CrossRef]
- Radman, M. Fidelity and infidelity. Nature From NLM Medline. 2001, 413(6852), 115. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Error Rate vs. Required Energy Difference in DNA Replication.
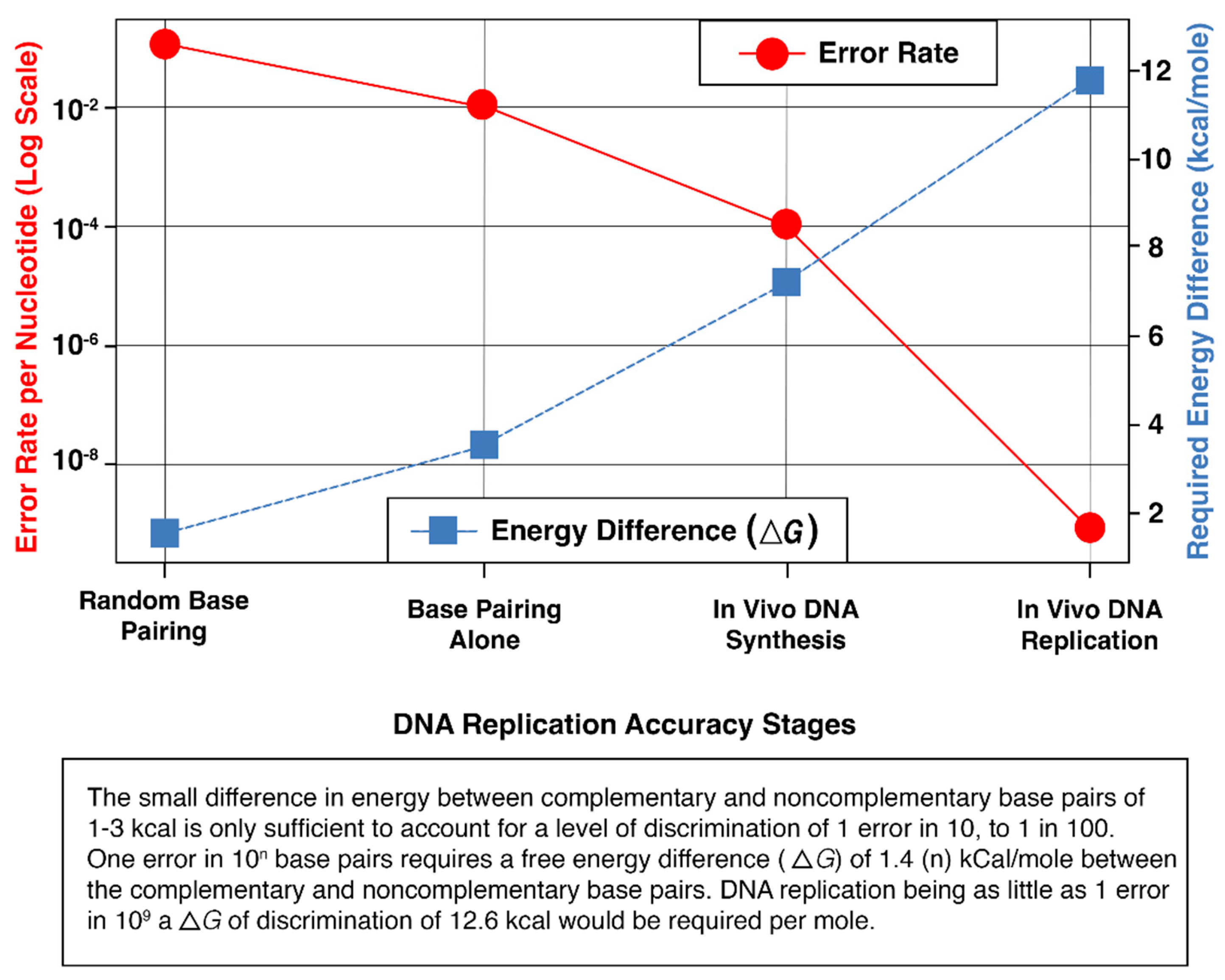
Figure 2.
Stepwise Reduction of DNA Replication Errors.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



