Submitted:
04 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Inflammatory bowel disease (IBD) is triggered by genetic predisposition and chronic inflammation, with aberrant activation of the innate immune complex NLRP3 inflammasome playing a pivotal role in its pathogenesis. In this study, we investigated the effects of a hot water extract from the brown alga Endarachne binghamiae (EB-WE) on the inhibition of NLRP3 inflammasome activation, with a focus on its antioxidant properties, in various inflammation models. In bone marrow-derived macrophages (BMDMs), NLRP3 inflammasome activation was induced using LPS and ATP, and EB-WE pretreatment (100, 200 µg/mL) significantly reduced the secretion of IL-1β and IL-18. Confocal immunofluorescence analysis further confirmed that EB-WE suppressed the formation of the NLRP3-ASC/caspase-1 complex. Furthermore, the in vivo anti-IBD efficacy of EB-WE was assessed using a DSS-induced mouse model, in which colonic inflammation and NLRP3-mediated responses were prominent. Oral administration of EB-WE (2 or 5 mg/day) markedly ameliorated clinical symptoms, such as weight loss, diarrhea, and rectal bleeding, and significantly reduced the disease activity index (DAI). EB-WE also decreased serum pro-inflammatory cytokine levels and the expression of NLRP3 inflammasome-related molecules in colon tissue at both the gene and protein levels. In both BMDMs and the IBD mouse model, we further analyzed the upstream regulatory pathway involving NOX2 -iNOS. EB-WE efficiently inhibited the activation of the NOX-iNOS axis and NF-κB phosphorylation, thereby alleviating inflammasome activation associated with DSS-induced oxidative stress and neutrophil/macrophage infiltration. Collectively, these results demonstrate that EB-WE effectively suppresses the formation and activation of the NLRP3 inflammasome by modulating the NOX-iNOS axis and the NF-κB pathway, via antioxidant mechanisms. These findings suggest that EB-WE holds promise as a novel marine-derived natural therapeutic agent for the treatment of chronic inflammatory diseases.
Keywords:
IBD
; iNOS-NOX crosstalk
; NLRP3
; p65 RELA
; Brown algae
1. Introduction
Inflammatory bowel disease (IBD) is a severe chronic inflammatory disorder in which the innate immune complex, the NLRP3 inflammasome, plays a central role in disease pathogenesis [1]. Under inflammatory conditions, NLRP3 forms a multiprotein complex with the adaptor ASC and caspase-1, leading to the maturation and secretion of pro-inflammatory cytokines such as IL-1β and IL-18, thereby driving potent immune responses and tissue damage [2,3]. In animal models of colitis, excessive activation of NLRP3 aggravates intestinal inflammation, whereas genetic deletion of NLRP3 markedly attenuates the symptoms and tissue injury induced by DSS [4]. These findings are consistent in patients with Crohn’s disease and ulcerative colitis, further supporting the pivotal role of NLRP3 inflammasome in IBD pathogenesis [5,6]. Therefore, inhibition of the NLRP3 inflammasome pathway has emerged as a promising strategy to prevent the progression of IBD and metabolic diseases.
Traditionally, NLRP3 inflammasome activation has been linked to upstream inflammatory signaling pathways such as TLR4/MyD88/AP1 or apoptotic cascades including BAX/cytochrome c/caspase-3 [7,8]. However, recent studies have revealed that NLRP3 activation can be directly triggered not only by receptor-dependent mechanisms but also fundamentally by increased intracellular oxidative stress [9]. Elevated intracellular ROS activates CaMKII through enhanced calcium influx and calmodulin signaling, independently of TAK1 [10]. This leads to the phosphorylation and subsequent degradation of IκBα, allowing NF-κB (p65/RelA) to translocate into the nucleus, where it promotes the transcriptional priming of NLRP3 and TXNIP. The elevated expression of TXNIP further facilitates NLRP3 activation by linking redox imbalance to inflammasome assembly [11].
This oxidative stress-driven inflammation is orchestrated through the depletion of NADPH, primarily regulated by NADPH oxidases (NOXs) [12,13]. NOX/DUOX (Dual oxidase) Family localized on the plasma and endosomal membranes, utilizes NADPH to generate superoxide (O₂⁻) as a means of host defense [14]. The superoxide is then dismutated to H₂O₂ by SOD [15]. Chronic NOX activation leads to a rapid reduction of intracellular NADPH levels, thereby decreasing the reduced forms of major antioxidant molecules such as CAT, GSH, GRX, TRX, PRDX, and NQO [16]. This redox imbalance facilitates IκB ubiquitination and p65 nuclear translocation, while also enhancing the intracellular activity of NOX4—an isoform distinguished by its ability to directly produce hydrogen peroxide (H₂O₂) rather than superoxide. Unlike other NOX enzymes, NOX4 is constitutively active and typically upregulated in later stages of cellular stress, contributing to sustained oxidative signaling within organelles such as the ER and mitochondria [17]. Nuclear p65 upregulates the expression of COX2, MMP9, various inflammatory cytokines, and inducible nitric oxide synthase (iNOS) [18]. Although H₂O₂ does not directly react with NO to form peroxynitrite (ONOO⁻), its sustained accumulation depletes major antioxidant defenses such as NADPH/GSH, GRX/TRX/PRDX, and catalase. This redox imbalance impairs SOD activity, allowing intracellular superoxide (O₂⁻) to accumulate and react with NO, thereby indirectly accelerating ONOO⁻ formation [19]. ONOO⁻ induces nitration and oxidation of specific tyrosine and cysteine residues in NLRP3, ASC, and caspase-1, directly mediating NLRP3 activation in the later phase [20]. The reduction mechanism for this process is primarily governed by the thioredoxin system; however, NOX-mediated NADPH depletion impairs thioredoxin reductase (TR) activity via TXNIP activation, resulting in increased cellular vulnerability [21].
Recent conceptual frameworks have proposed that NOX–NOS crosstalk represents a fundamental redox signaling axis driving multi-organ inflammatory and metabolic dysregulation [22]. In this context, intestinal inflammation may represent one organ-specific manifestation of this broader redox network. From this perspective, we investigated the IBD-suppressive effects of a hot water extract of the brown alga Endarachne binghamiae (EB-WE), known for its reported antioxidant, anti-inflammatory, and anti-cancer properties. Our previous studies demonstrated that EB-WE potently ameliorates LPS-induced acute pneumonia [23] and significantly improves MASLD induced by a high-fat diet through metabolic and antioxidant mechanisms [24]. Based on UHPLC-QTOF-LC-MS/MS and in silico analyses from our prior research, pyropheophorbide (a chlorophyll a–derived metabolite) and loliolide (a benzofuran compound) were tentatively identified as putative bioactive constituents of EB-WE [24]. Given that both compounds have previously been associated with antioxidant and anti-inflammatory activities, these findings may support the hypothesis that EB-WE exerts broad efficacy in ameliorating both inflammatory and metabolic dysfunctions, likely through a common upstream mechanism involving redox regulation and antioxidant activity [25,26].
Based on this hypothesis, we evaluated the effects of EB-WE in a BMDM (LPS+ATP-induced inflammasome activation) model and a DSS-induced IBD mouse model. The present study aims to elucidate the NLRP3 inflammasome-inhibitory effects and underlying mechanisms of EB-WE, with a particular focus on redox-regulated inflammatory signaling, thereby providing insights into the therapeutic potential of marine algal natural products for inflammatory diseases.
2. Result
2.1. Phytochemical Profile of EB-WE
LC-MS/MS-based metabolite profiling of EB-WE revealed distinct chemical signatures in both positive and negative ionization modes (Figure 1, Section 4.1). In ES+ mode, pyropheophorbide A was identified as the dominant compound, while monogalactosyldiacylglycerol (MGDG) predominated in ES- mode, consistent with typical brown algal lipid profiles. A total of 12 compounds were putatively identified through GNPS molecular networking analysis. Based on the selection criteria described in Section 4.1, pyropheophorbide A and loliolide were prioritized as major putative bioactive constituents for further investigation.
2.2. Inhibition of NLRP3 Inflammasome Formation by EB-WE in BMDMs
To investigate the inhibitory effect of EB-WE on NLRP3 inflammasome activation, bone marrow-derived macrophages (BMDMs) were stimulated with LPS and ATP. Upon activation, BMDMs released large amounts of IL-1β and IL-18. However, pretreatment with EB-WE (100 and 200 µg/mL) significantly and dose-dependently reduced the secretion of IL-1β and IL-18 in response to LPS+ATP stimulation (Figure 2A). Specifically, IL-1β secretion in the EB-WE 200 µg/mL group was reduced to approximately 50% of the inflammasome-activated control, with a similar suppression observed for IL-18 (p < 0.05). In addition, confocal immunofluorescence staining was performed to visualize the formation of NLRP3, ASC, and caspase-1 complexes (Figure 2B; see also Figure S1 for complete images). In the control BMDMs, robust co-localization of NLRP3 and ASC was observed, indicative of inflammasome speck formation, whereas EB-WE treatment markedly decreased the formation of NLRP3-ASC complexes. Similarly, co-localization of NLRP3 and caspase-1 was also significantly reduced in the EB-WE-treated group, providing visual evidence that EB-WE effectively inhibits inflammasome assembly in macrophages under LPS+ATP stimulation. Collectively, these results indicate that EB-WE suppresses both the assembly and activation of the NLRP3 inflammasome in macrophages, thereby inhibiting the maturation and secretion of IL-1β and IL-18.
2.3. Western Blot Analysis of NLRP3-Related Proteins in the BMDM Model
To further validate the ELISA and immunohistochemical findings, Western blotting was performed to assess the effects of EB-WE on the expression and secretion of key inflammatory proteins at the protein level. The results supported the trends observed in section 2.1. In control BMDMs activated with LPS+ATP, the expression of pro-caspase-1 was markedly increased, and substantial amounts of mature IL-1β (p17) and ASC proteins were released into the culture supernatant, reflecting effective pyroptosis via NLRP3 inflammasome activation. In contrast, EB-WE treatment resulted in increased intracellular levels of pro-IL-1β and pro-c0aspase-1, while the amounts of mature IL-1β and ASC released into the supernatant were significantly reduced compared to the control (Figure 3). These findings indicate that EB-WE exerts anti-inflammatory effects by inhibiting the formation of the NLRP3 inflammasome complex.
2.4. EB-WE Attenuates ROS-Mediated NLRP3 Inflammasome Activation in BMDMs
Based on our previous findings demonstrating the antioxidant activity of EB-WE [24], we further investigated the interplay between intracellular ROS generation and NLRP3 inflammasome activation in BMDMs. Live-cell staining with DCF-DA (green) was first performed to detect ROS, followed by fixation and immunofluorescence staining for NLRP3 (red). Nuclei were counterstained with DAPI (blue), and all images were acquired using confocal microscopy. As shown in Figure 4, LPS+ATP treatment marked-ly increased both ROS accumulation and perinuclear clustering of NLRP3 compared to non-treated controls. Notably, EB-WE pretreatment (100 or 200 µg/mL) led to a pronounced re-duction in DCF-DA fluorescence and NLRP3 expression, indicating effective suppression of ROS-mediated inflammasome activation. Merge images clearly demonstrated spatial overlap of ROS and NLRP3 in the LPS+ATP group, which was decresed upon EB-WE treatment. in the LPS+ATP group, which was decreased upon EB-WE treatment. Given the close association between ROS and NLRP3 activation [27], these results imply a potential link between the antioxidant and anti-inflammatory effects of EB-WE.
2.5. Anti-Inflammatory Effects of EB-WE in the IBD Mouse Model
As shown in Figure 5A, DSS administration significantly elevated the serum levels of multiple pro-inflammatory cytokines, including IL-2, IL-1β, IL-18, TNF-α, IFN-γ, and IL-17A, confirming the presence of systemic inflammation. Notably, EB-WE treatment effectively and dose-dependently reduced these cytokines, with the 5 mg/mice group showing the most consistent anti-inflammatory effects. In particular, IL-2, IL-1β, TNF-α, and IFN-γ levels were significantly decreased compared to the DSS group, indicating suppression of both Th1 and general inflammatory responses.
In addtion, to further validate the local anti-inflammatory effects of EB-WE, cytokine levels in colon tissues were assessed (Figure 5B). DSS markedly elevated TNF-α, IL-1β, IL-18, and IL-17A concentrations in the colon, consistent with mucosal inflammation. EB-WE treatment significantly attenuated the expression of TNF-α, IL-1β, and IL-17A, particularly at the 5 mg/mice dose (*p < 0.05 or **p < 0.01 vs. DSS), indicating local immunomodulatory activity within the colonic microenvironment. These findings collectively demonstrate that EB-WE confers both systemic and tissue-level anti-inflammatory protection in DSS-induced colitis.
2.6. Histological and Clinical Changes in the IBD Model
As shown in Figure 6A, DSS administration significantly increased weight loss, stool consistency scores, visible blood in feces, and overall Disease Activity Index (DAI) over time, clearly indicating the progression of colitis. In contrast, EB-WE treatment markedly attenuated these DSS-induced clinical symptoms in a dose-dependent manner. The 5 mg/mice group showed the most pronounced improvement across all parameters, with significantly lower weight loss, improved stool consistency, reduced fecal bleeding, and substantially decreased DAI scores (**p < 0.01 to ***p < 0.001 vs. DSS), suggesting a strong protective effect against DSS-induced colitis.
Histological evaluation further supported these findings (Figure 6B). In the DSS group, severe mucosal damage, destruction of intestinal villi, marked submucosal edema, and massive inflammatory cell infiltration were observed. In the EB-WE 2 mg/mice group, some mucosal damage and inflammatory infiltration remained, but pathological features were notably improved compared to the DSS-only group. Particularly, the EB-WE 5 mg/mice group exhibited near-complete preservation of mucosal and villous structures, with minimal inflammatory signs, closely resembling the normal histological architecture. These results indicate that EB-WE effectively and dose-dependently ameliorates both clinical and histological features of DSS-induced colitis.
2.7. EB-WE Suppresses Inflammatory and Apoptotic Signaling while Preserving Epithelial Barrier Integrity in DSS-Induced Colonic Tissue
To further elucidate the protective mechanisms of EB-WE in vivo, Western blot analysis was performed on colon tissues from DSS-induced IBD mice. As shown in Figure 7, the DSS group exhibited significant upregulation of inflammatory signaling proteins, including iNOS, phosphorylated p38 MAPK, p-IKB, and p-p65, along with activation of the NLRP3 inflammasome pathway, as evidenced by increased levels of ASC and cleaved caspase-1. Pro-apoptotic markers (BAX, cleaved caspase-3) were also elevated, while anti-apoptotic BCL2 and tight junction proteins (Occludin, ZO-1) were markedly reduced.
Conversely, EB-WE treatment (2 or 5 mg/mice) dose-dependently suppressed these inflammation- and apoptosis-related proteins, and restored the expression of tight junction proteins. These results collectively indicate that EB-WE effectively inhibits both inflammatory signaling and NLRP3 inflammasome activation, while preserving epithelial barrier integrity in DSS-induced colonic tissue.
2.8. Modulation of Redox Regulatory Proteins by EB-WE in Colon Tissue of the IBD Model
To elucidate the antioxidant mechanisms underlying the anti-inflammatory effects of EB-WE, we performed Western blot analyses targeting key redox-regulating proteins in colon tissue (Figure 8). DSS administration markedly upregulated the expression of NOX2, NOX4, and TXNIP, reflecting increased oxidative stress in the inflamed colon. In contrast, EB-WE treatment dose-dependently reduced the levels of these pro-oxidant proteins. Notably, EB-WE also restored the expression of NQO1, NRF2, and HO-1 antioxidant defense proteins that were suppressed by DSS challenge. These results suggest that EB-WE attenuates colonic oxidative stress not only by inhibiting ROS-generating enzymes (NOX2, NOX4, TXNIP) but also by activating the cellular antioxidant response (NQO1, NRF2, HO-1). Collectively, these data demonstrate that the redox-modulating effects of EB-WE contribute substantially to its anti-inflammatory actions in DSS-induced IBD.
In addition, to understand the impact of EB-WE on cellular redox balance, we analyzed the expression levels of NAD+/NADH and NADP+/NADPH ratios in colon tissues. The DSS-induced rise in the NAD⁺/NADH ratio likely reflects a shift toward enhanced glycolytic and mitochondrial oxidative flux under inflammatory stress [28,29], whereas the concomitant drop in NADP⁺/NADPH indicates exhaustion of the principal reductive reserve needed for glutathione and thioredoxin-dependent detoxification [28,30]. By partially normalizing both cofactor ratios, EB-WE not only tempers excessive pro-oxidant metabolism but also replenishes the reducing power essential for phase II antioxidant enzymes.
In parallel, the downregulation of TXNIP relieves thioredoxin inhibition, synergizing with the upregulation of NQO1, NRF2 and HO-1 to bolster cellular defenses (Figure 8A, B). The unchanged NADK levels, despite restored NADP⁺/NADPH, suggest that EB-WE may activate alternative NADPH-regenerating pathways such as glucose-6-phosphate dehydrogenase or malic enzyme instead of directly modulating NADK expression.
Finally, the rise in NAD⁺ availability may further trigger sirtuin-mediated deacetylation events, promoting mitochondrial biogenesis and anti-inflammatory gene expression. Altogether, these coordinated changes in cofactor balance, enzyme expression, and redox-sensitive signaling highlight EB-WE as a multifaceted modulator of metabolic and antioxidant networks in inflamed colon tissue.
3. Discussion
3.1. ROS and NLRP3 Inflammasome: Priming and Oligomerization
Recent studies have reported that ROS can activate NF-κB (p65) independently of TAK1 and, by disrupting the thioredoxin reductase pathway, promote the formation of the NLRP3 inflammasome [31]. Upon DSS challenge, colonic epithelial cells and immune cells (including macrophages) activate NOX, which produces large amounts of superoxide (O₂⁻) and other ROS to defend against pathogens, but this process rapidly depletes intracellular NADPH [32]. As NADPH is the main source of reducing power for the thioredoxin and glutathione antioxidant systems, its depletion severely impairs antioxidant defense, resulting in the loss of redox homeostasis [33]. Accumulated ROS consequently influence intracellular signaling and can activate p65 RelA independently of the canonical TAK1 pathway [34]. Excessive ROS has been shown to accelerate IκBα degradation, promoting NF-κB (p65/p50 complex) nuclear translocation and upregulation of pro-inflammatory cytokines [35]. DSS promotes IBD by activating NOX in colonic epithelial cells and depleting the thioredoxin system [36]. The oxidative environment, together with macrophage and neutrophil infiltration, cooperatively supports NLRP3 inflammasome activation and perpetuates chronic IBD pathology [37]. Indeed, excessive activation of the NF-κB pathway by oxidative stress has been observed in colitis models and is recognized as a major factor aggravating chronic inflammation.
Furthermore, ROS not only enhance the expression of NLRP3 by promoting p65-mediated transcriptional priming, but also support the oligomerization of ASC and activation of caspase-1, thereby facilitating full inflammasome assembly [38,39] (Figure 9). This dual role of ROS highlights its involvement in both the priming and activation phases of NLRP3 inflammasome signaling.
3.2. Implications for Inflammation-Focused Mechanistic Analysis
In this study, we systematically demonstrated that the hot water extract of the brown alga Endarachne binghamiae (EB-WE) effectively inhibits NLRP3-mediated inflammatory responses in various experimental models. Briefly, EB-WE suppressed LPS+ATP-induced activation of the NLRP3 inflammasome in macrophages, leading to reduced maturation and secretion of IL-1β and IL-18. In the DSS-induced IBD model, EB-WE ameliorated both inflammatory cytokine production and tissue injury, accompanied by a reduction in the expression of NLRP3 inflammasome-related molecules. These consistent findings, validated at the protein level, suggest that the anti-inflammatory efficacy of EB-WE is primarily mediated through the suppression of the NLRP3 inflammasome pathway. In other words, EB-WE demonstrates the potential to attenuate the progression of chronic inflammatory diseases by inhibiting the excessive activation of the NLRP3 inflammasome, a key contributor to disease pathogenesis (Figure 10).
Inflammatory stimuli such as DSS and LPS/ATP activate NOX2 and NOX4, which promote oxidative stress through H₂O₂ production and enhance NF-κB activation. NOX2 also upregulates iNOS expression, leading to excessive peroxynitrite (ONOO⁻) formation via iNOS–H₂O₂ interaction. TXNIP acts as a redox-sensitive mediator that links NOX4-derived ROS to NLRP3 inflammasome activation. This cascade culminates in caspase-1 cleavage and IL-1β maturation, amplifying mucosal inflammation. EB-WE treatment inhibits this vicious cycle by suppressing NOX2/NOX4 expression, reducing TXNIP and iNOS levels, and blocking the oxidative/nitrosative stress axis, thereby attenuating NLRP3 inflammasome activation and downstream inflammation.
3.3. NOX–iNOS Crosstalk and IBD: Inflammation Driven by Nitrosative Stress
Once NF-κB translocates to the nucleus, transcription of various inflammatory genes is upregulated, with a marked increase in iNOS expression [40]. The high levels of NO generated by iNOS rapidly react with NOX1/2-derived O₂⁻ to form peroxynitrite (ONOO⁻) [41]. In addition, chronic activation of NOX in macrophages and colonic epithelium has been observed in DSS-induced inflammatory microenvironments [42]. The activation of NF-κB, including p65 RELA, further contributes to increased NOX4 expression [43], and NOX4 continuously produces H₂O₂, which together with iNOS-derived NO, accelerates ONOO⁻ formation [44]. ONOO⁻, a potent reactive nitrogen species (RNS), not only damages cellular components but also nitrates tyrosine residues of proteins, thereby altering their function [45]. In both DSS-induced colitis models and colonic tissues from IBD patients, iNOS overexpression and increased protein nitration (e.g., nitrotyrosine) have been observed concurrently, indicating that excessive ONOO⁻ generation is a key driver of ongoing oxidative injury [46,47].
3.4. Oxidative Damage and NLRP3: The Intimate Crosstalk Between Inflammation and Oxidative Stress
This nitrosative stress serves as a crucial trigger for the activation of the NLRP3 inflammasome. Additionally, it has been reported that, as cytosolic thioredoxin becomes oxidized, dissociated TXNIP binds directly to NLRP3, thus functioning as a redox-sensitive mechanism for inflammasome activation [48]. In other words, collapse of the thioredoxin system due to NADPH depletion can facilitate the interaction between TXNIP and NLRP3, providing a strong signal for inflammasome activation [49]. Moreover, when peroxynitrite (ONOO⁻) induces nitration of inflammasome components such as NLRP3, ASC, and caspase-1, the resulting modifications favor their oligomerization and activation, thereby promoting the assembly of the NLRP3 inflammasome complex. There is evidence that decomposing or inhibiting the generation of peroxynitrite reduces both priming and activation of the NLRP3 inflammasome, further supporting the direct role of ONOO⁻ in inflammasome activation [50,51].
Overactivation of the NLRP3 inflammasome leads to the processing and massive secretion of mature IL-1β and IL-18 from their precursors, amplifying inflammatory responses in the intestinal mucosa [52]. This, in turn, results in epithelial cell injury, increased intestinal permeability, and reactivation of immune cells, creating a vicious cycle that exacerbates IBD pathology and culminates in pyroptosis [53]. Notably, in chronic DSS-induced colitis models, simultaneous overexpression of NOX, DUOX, and iNOS has been shown to result in the accumulation of excessive ONOO⁻ in local tissues and aggravation of inflammation [54]. This suggests that massive ONOO⁻ generation—stemming from the explosive reaction between NOX-derived ROS and iNOS-derived NO—plays a central role in NLRP3-driven IBD exacerbation [55].
3.5. Translational Implications Supported by Human Transcriptomic Data
To validate the relevance of our findings to human pathology, we independently analyzed publicly available transcriptomic data from colonic tissues of IBD patients (GSE16879) [56,57] (Figure S2, Table S1). This dataset revealed a consistent and coordinated upregulation of 13 genes involved in redox and inflammatory signaling—including DUOX1, DUOX2, iNOS (NOS2), CASP1, and IL1B—as well as accessory or amplifying factors such as DUOXA1/2, TANK, NKAPL, and XDH. Notably, LOXL2, a fibrosis-related oxidase, was also significantly elevated. These findings closely mirror the molecular targets modulated by EB-WE in both murine and cellular models of inflammation. Importantly, PRDX6, a cytosolic antioxidant enzyme, was markedly downregulated, suggesting a compromised ROS-buffering capacity in the IBD mucosa. CAMK2N1, an endogenous inhibitor of calcium/calmodulin-dependent kinase II (CaMKII), was also significantly reduced (p = 0.00067). Given that DUOX1, DUOX2, and NOX5 are the only NOX isoforms directly activated by Ca²⁺ via EF-hand domains, this suppression of CAMK2N1 may point to enhanced calcium–redox crosstalk and a loss of intrinsic regulation of CaMKII-driven inflammation.
Statistical analysis using the Mann–Whitney U test confirmed that all 13 analyzed genes exhibited significant differences (*p < 0.05, **p < 0.01, ***p < 0.001) between control and IBD tissues. These consistent transcriptomic alterations reinforce the translational relevance of our mechanistic model [58], particularly the notion that oxidative and nitrosative stress synergistically activate the NLRP3 inflammasome through the NOX–iNOS–NLRP3 axis. Collectively, our findings support the therapeutic potential of targeting this axis to mitigate chronic mucosal inflammation in human IBD.
4. Materials & Methods
4.1. Preparation and Phytochemical Analysis of EB Hot Water Extract
The EB-WE (hot water extract of Endarachne binghamiae) used in this study was prepared according to the methods described in our previous report [24]. For phytochemical characterization, compounds putatively identified by UHPLC-QTOF-LC-MS/MS were subjected to a secondary selection based on MS/MS fragmentation pattern matching, previous reports of the compounds in marine algae, and documented antioxidant/anti-inflammatory activities. Subsequently, a third selection was performed using in silico analysis via MetFrag [59], GNPS [60], and SIRIUS [61] platforms, applying a cosine similarity threshold of ≥0.85. Through this workflow, 12 tentative candidate bioactive compounds were identified, with pyropheophorbide and loliolide ultimately selected as the major putative active constituents (Table 1).
4.2. BMDM Inflammation Induction and EB-WE Pretreatment—Cell Isolation and Culture
Bone marrow-derived macrophages (BMDMs) were differentiated from three-month-old male C57BL/6 mice. Bone marrow was flushed from the femurs and tibias and cultured in RPMI medium supplemented with 10% fetal bovine serum and L929 cell-conditioned medium (as a source of macrophage colony-stimulating factor) at 37°C in a 5% CO₂ incubator for 7 days to induce differentiation of immature monocytes into macrophages. On day 7, BMDMs were harvested and seeded at 1 × 10⁶ cells/well in 12-well plates, allowed to adhere for 24 hours, and then used for subsequent experiments.
4.3. NLRP3 Inflammasome Activation
To activate the NLRP3 inflammasome in BMDMs, a dual stimulation protocol with LPS and ATP was employed. Cells were primed with LPS (E. coli O111:B4, Sigma) at 100 ng/mL for 5 hours to induce expression of Nlrp3 and Il1b (priming), followed by ATP (Sigma) at 5 mM for 30 minutes to trigger NLRP3 inflammasome assembly and caspase-1 activation. For the EB-WE treatment groups, cells were pretreated with EB-WE at final concentrations of 100 or 200 µg/mL one hour before LPS priming. The EB-WE extract was prepared by suspending freeze-dried E. binghamiae powder in distilled water, filtering through a 0.2 μm membrane, and then using the filtrate for experiments.
4.4. ELISA Analysis
Culture supernatants from BMDMs were collected to quantify IL-1β and IL-18 levels using ELISA. After centrifugation to remove cellular debris, cytokine concentrations were measured using Mouse IL-1β/IL-18 DuoSet ELISA kits (R&D Systems) following the manufacturer’s protocol. Absorbance was read at 450 nm with a microplate reader, and concentrations were calculated based on standard curves.
For intracellular redox cofactor analysis, NAD⁺/NADH and NADP⁺/NADPH levels were quantified using WST-8-based colorimetric assay kits (NAD⁺/NADH: Elabscience, Cat# E-BC-K804-M; NADP⁺/NADPH: Elabscience, Cat# E-BC-K803-M). BMDMs were lysed in extraction buffer provided by the manufacturer, and supernatants were used for enzymatic cycling reactions followed by WST-8 colorimetric detection. Absorbance was measured at 450 nm using a microplate reader. The resulting values were normalized to the mean ratio of the Normal group (set as 1.0) to allow comparison across groups.
4.5. Immunofluorescence (Confocal Microscopy)
For visualization of NLRP3 inflammasome complex formation, BMDMs were seeded at 2 × 10⁵ cells/well in 8-well chamber slides and subjected to the same treatments (LPS+ATP ± EB-WE pretreatment). After stimulation, cells were fixed with 4% paraformaldehyde for 15 minutes, washed with PBS, and permeabilized with 0.1% Triton X-100. Non-specific binding was blocked with 1% BSA for 30 minutes. Cells were incubated overnight at 4 °C with primary antibodies against NLRP3 (Abcam) and either ASC or caspase-1 p20 (Cell signaling). The next day, Alexa Fluor-conjugated secondary antibodies were applied for 1 hour at room temperature. Nuclei were counterstained with DAPI. Slides were mounted and examined by confocal laser scanning microscopy. Inflammasome speck formation (co-localization of NLRP3 with ASC or caspase-1) was assessed in at least five random fields per group.
In parallel, a separate set of BMDMs was used to visualize the spatial overlap between ROS and NLRP3. After treatment, cells were incubated with 10 µM DCF-DA in serum-free medium for 30 minutes at 37 °C to detect intracellular ROS. Cells were then washed, fixed with 4% paraformaldehyde, permeabilized, and subjected to immunostaining for NLRP3 (Abcam) overnight at 4 °C. Alexa Fluor 594-conjugated secondary antibodies were applied for 1 hour at room temperature, and nuclei were counterstained with DAPI. Confocal images were acquired using a Zeiss LSM, and co-localization of green (ROS) and red (NLRP3) signals was analyzed in five random fields per group.
4.6. Western Blot Analysis
To analyze the expression of inflammatory, redox, and barrier-related proteins in BMDMs and colon tissue, Western blotting was performed. For in vitro analysis, BMDMs were stimulated with LPS (1 µg/mL, 5 h) and ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL). After treatment, whole-cell lysates were prepared using RIPA buffer containing protease and phosphatase inhibitors, and culture supernatants were concentrated using TCA precipitation. For in vivo analysis, colon tissue was homogenized in RIPA buffer after DSS treatment and EB-WE administration. Protein concentrations were quantified using a BCA assay, and 20μg of protein per lane was loaded onto SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked with 5% skim milk or 5% BSA (for phospho-proteins) for 1 hour at room temperature and incubated overnight at 4 °C with the following primary antibodies:
Inflammatory signaling: pro-IL-1β, cleaved IL-1β (p17), pro–caspase-1, cleaved caspase-1 (p10), ASC, iNOS, p-p65 (Ser536), total p65, p-IKBα, IKBα, p-p38 MAPK, total p38 MAPK
Redox markers: NOX2, NOX4, TXNIP, NQO1, HO-1, NRF2, NADK
Apoptosis markers: BAX, BCL2, pro-caspase-3, cleaved caspase-3
Barrier-related proteins: Occludin, ZO-1
After washing, membranes were incubated with HRP-conjugated secondary antibodies for 1 hour at room temperature. Protein bands were detected using enhanced chemiluminescence (ECL) reagents and visualized using a chemiluminescent imaging system. Densitometric analysis was performed using ImageJ software. Band intensities were normalized to β-actin and expressed as fold change relative to the control group. All Western blot experiments were performed in triplicate (n = 3), and results are presented as mean ± standard deviation (SD).
4.7. In Vivo Induction of DSS-Induced Colitis and EB-WE Administration
Eight-week-old male C57BL/6 mice (20–22 g) were used in this study. Animals were randomly assigned to four groups (n = 10 per group): normal control, DSS control, EB-WE 2 mg group, and EB-WE 5 mg group. To induce colitis, dextran sulfate sodium (DSS, molecular weight ~36–50 kDa; MP Biomedicals) was dissolved in drinking water at a concentration of 2.5% (w/v) and administered ad libitum for 5 days. The DSS solution was then replaced with regular drinking water for an additional 2-day recovery period, resulting in a total of 7 days of acute colitis induction. In the EB-WE treatment groups, EB-WE was administered by oral gavage once daily, starting one day prior to DSS exposure and continuing throughout the DSS treatment period. EB-WE was suspended in 0.2 mL PBS at doses of 2 mg/day or 5 mg/day. Mice in the normal control and DSS control groups received the same volume of PBS only.
4.8. Clinical Monitoring and Disease Activity Index (DAI)
Body weight was measured daily, and stool consistency as well as the presence of fecal blood were monitored throughout the experimental period to assess colitis progression. The disease activity index (DAI) was calculated by summing scores for weight loss, stool consistency, and bleeding. Weight loss was scored as follows: 1–5% loss = 1 point; 5–10% = 2 points; 10–20% = 3 points; >20% = 4 points. Stool consistency was scored as: normal firm stool = 0; slightly soft = 1; semi-formed (incipient diarrhea) = 2; complete diarrhea = 3. Bleeding was scored as: no visible blood = 0; mild blood = 1; overt blood = 2; rectal bleeding = 3. The DAI for each mouse was calculated daily, and group means were determined.
4.9. Sample Collection and Analysis
On day 7 of DSS administration, all mice were euthanized, and blood and colon tissues were collected. Whole blood was centrifuged at 3,000 rpm for 15 minutes at 4°C to obtain serum. Serum cytokine concentrations were measured using commercial ELISA kits (R&D Systems) for TNF-α, IFN-γ, IL-2, and IL-4, and all samples were analyzed in duplicate to determine mean values. Colon segments (~1 cm in length) were fixed in 10% neutral-buffered formalin for histological analysis, while the remaining colon tissue was homogenized and processed for Western blot as described in section 4.5. Fixed colon tissues were embedded in paraffin, sectioned at 7 μm thickness, and stained with hematoxylin and eosin (H&E). Stained slides were examined under a microscope to assess the degree of mucosal injury and inflammatory cell infiltration, and representative pathological findings were documented by photomicrography.
4.10. Ethical Approval
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Konyang University (Approval No. 24-19-A-08). All procedures were conducted in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition), and efforts were made to minimize animal suffering throughout the study.
4.11. Statistical Analysis
All data are expressed as mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism and SAS 9.4 software. Student’s t-test was applied for comparisons between two groups, and one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used for multiple group comparisons. For time-dependent variables such as body weight and disease activity index (DAI), repeated measures two-way ANOVA was performed to evaluate treatment effects. A p-value < 0.05 was considered statistically significant. In all figures and tables, statistical significance is indicated using the following symbols: Asterisks (*, **, ***) indicate comparisons versus the LPS/ATP-treated group (for in vitro data) or the DSS-treated group (for in vivo data). The levels of significance are denoted as follows: * p < 0.05; ** p < 0.01; *** p < 0.001. Daggers (†, ††, †††) indicate comparisons versus the normal untreated control group.The levels of significance are denoted as follows: † p < 0.05; †† p < 0.01; ††† p < 0.001.
4.12. Abbreviations
The following abbreviations are used in this study (Table 2).
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, S.-S.L.; methodology, S.-S.L.; software, S.-S.L. and S.-Y.K.; validation, S.-H.L. and S.-Y.K.; formal analysis, S.-S.L. and S.-H.L.; investigation, S.-S.L. and S.-Y.K.; resources, B.-H.L.; data curation, S.-S.L., S.-H.L. and Y.-C.Y.; writing—original draft preparation, S.-S.L.; writing—review and editing, S.-H.L. and Y.-C.Y.; visualization, S.-S.L.; supervision, Y.-C.Y.; project administration, Y.-C.Y.; funding acquisition, Y.-C.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Bio and Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. 2022M3A9E4095340), and partly by R&D Program for Forest Science Technology (Project No. RS-2024-00404789) provided by Korea Forest Service (Korea Forestry Promotion Institute).
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
References
- Ren, W.; Sun, Y.; Zhao, L.; Shi, X. NLRP3 inflammasome and its role in autoimmune diseases: A promising therapeutic target. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2024, 175, 116679. [Google Scholar] [CrossRef]
- Zhang, W. J.; Li, K. Y.; Lan, Y.; Zeng, H. Y.; Chen, S. Q.; Wang, H. NLRP3 Inflammasome: A key contributor to the inflammation formation. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 2023, 174, 113683. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cellular & molecular immunology 2021, 18(9), 2114–2127. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H. A.; Fitzgerald, K. A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59(9), 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, H.; Guo, Y.; Zhang, W.; Jiang, Q.; Zhang, S.; Han, L.; Chen, S.; Xue, R. Activation of Platelet NLRP3 Inflammasome in Crohn's Disease. Frontiers in pharmacology 2021, 12, 705325. [Google Scholar] [CrossRef]
- Ali, F. E. M.; Ibrahim, I. M.; Ghogar, O. M.; Abd-Alhameed, E. K.; Althagafy, H. S.; Hassanein, E. H. M. Therapeutic interventions target the NLRP3 inflammasome in ulcerative colitis: Comprehensive study. World journal of gastroenterology 2023, 29(6), 1026–1053. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K. I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer's Disease. Frontiers in immunology 2020, 11, 724. [Google Scholar] [CrossRef]
- Vince, J. E.; De Nardo, D.; Gao, W.; Vince, A. J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L. M.; Bouillet, P.; Rizzacasa, M. A.; Man, S. M.; Silke, J.; Masters, S. L.; Lessene, G.; Huang, D. C. S.; Gray, D. H. D.; Kile, B. T.; Shao, F.; Lawlor, K. E. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell reports 2018, 25(9), 2339–2353.e4. [Google Scholar] [CrossRef]
- Singh, D. D. NLRP3 inflammasome: structure, mechanism, drug-induced organ toxicity, therapeutic strategies, and future perspectives. In RSC medicinal chemistry; Advance online publication, 2025; p. 10.1039/d5md00167f. [Google Scholar] [CrossRef]
- Wang, Q.; Hernández-Ochoa, E. O.; Viswanathan, M. C.; Blum, I. D.; Do, D. C.; Granger, J. M.; Murphy, K. R.; Wei, A. C.; Aja, S.; Liu, N.; Antonescu, C. M.; Florea, L. D.; Talbot, C. C., Jr.; Mohr, D.; Wagner, K. R.; Regot, S.; Lovering, R. M.; Gao, P.; Bianchet, M. A.; Wu, M. N.; Anderson, M. E. CaMKII oxidation is a critical performance/disease trade-off acquired at the dawn of vertebrate evolution. Nature communications 2021, 12(1), 3175. [Google Scholar] [CrossRef]
- Yosri, H.; El-Kashef, D. H.; El-Sherbiny, M.; Said, E.; Salem, H. A. Calycosin modulates NLRP3 and TXNIP-mediated pyroptotic signaling and attenuates diabetic nephropathy progression in diabetic rats; An insight. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2022, 155, 113758. [Google Scholar] [CrossRef]
- Sylvester, A. L.; Zhang, D. X.; Ran, S.; Zinkevich, N. S. Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies. Biomolecules 2022, 12(6), 823. [Google Scholar] [CrossRef]
- Taylor, J. P.; Tse, H. M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox biology 2021, 48, 102159. [Google Scholar] [CrossRef] [PubMed]
- Sirokmány, G.; Donkó, Á.; Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends in pharmacological sciences 2016, 37(4), 318–327. [Google Scholar] [CrossRef]
- Sakamoto, T.; Imai, H. Hydrogen peroxide produced by superoxide dismutase SOD-2 activates sperm in Caenorhabditis elegans. The Journal of biological chemistry 2017, 292(36), 14804–14813. [Google Scholar] [CrossRef]
- Muro, P.; Zhang, L.; Li, S.; Zhao, Z.; Jin, T.; Mao, F.; Mao, Z. The emerging role of oxidative stress in inflammatory bowel disease. Frontiers in endocrinology 2024, 15, 1390351. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B. A.; Cosentino-Gomes, D.; Lambeth, J. D. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53(31), 5111–5120. [Google Scholar] [CrossRef]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A. G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2(2), 48–78. [Google Scholar] [CrossRef]
- Robinson, K. M.; Beckman, J. S. Synthesis of peroxynitrite from nitrite and hydrogen peroxide. Methods in enzymology 2005, 396, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C. M. C.; Pérez de la Lastra, J. M.; Andrés Juan, C.; Plou, F. J.; Pérez-Lebeña, E. Impact of Reactive Species on Amino Acids-Biological Relevance in Proteins and Induced Pathologies. International journal of molecular sciences 2022, 23(22), 14049. [Google Scholar] [CrossRef] [PubMed]
- Choi, E. H.; Park, S. J. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Experimental & molecular medicine 2023, 55(7), 1348–1356. [Google Scholar] [CrossRef]
- Lee, S. S.; Yoo, Y. C. NOX-NOS crosstalk in the liver-brain axis: Novel insights for redox regulation and neurodegenerative diseases. Redox biology 2025, 86, 103807. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. H.; Lee, S. S.; Lee, G. Y.; Han, S. Y.; Kim, D. S.; Lee, B. H.; Yoo, Y. C. Endarachne binghamiae Extract Ameliorates Inflammatory Responses in Macrophages Through Regulation of MAPK, NF-kB and PI3K/AKT Pathways, and Prevents Acute Lung Injury in Mice. Life (Basel, Switzerland) 2025, 15(1), 88. [Google Scholar] [CrossRef]
- Lee, S.-S.; Lee, S.-H.; Kim, S.-Y.; Lee, G.-Y.; Han, S.-Y.; Lee, B.-H.; Yoo, Y.-C. Endarachne binghamiae Ameliorates Hepatic Steatosis, Obesity, and Blood Glucose via Modulation of Metabolic Pathways and Oxidative Stress. International Journal of Molecular Sciences 2025, 26(11), 5103. [Google Scholar] [CrossRef]
- Han, E.-J.; Fernando, I. P. S.; Kim, H.-S.; Lee, D.-S.; Kim, A.; Je, J.-G.; Seo, M.-J.; Jee, Y.-H.; Jeon, Y.-J.; Kim, S.-Y.; Ahn, G. (–)-Loliolide Isolated from Sargassum horneri Suppressed Oxidative Stress and Inflammation by Activating Nrf2/HO-1 Signaling in IFN-γ/TNF-α-Stimulated HaCaT Keratinocytes. Antioxidants 2021, 10(6), 856. [Google Scholar] [CrossRef]
- Islam, M. N.; Ishita, I. J.; Jin, S. E.; Choi, R. J.; Lee, C. M.; Kim, Y. S.; Jung, H. A.; Choi, J. S. Anti-inflammatory activity of edible brown alga Saccharina japonica and its constituents pheophorbide a and pheophytin a in LPS-stimulated RAW 264.7 macrophage cells. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 2013, 55, 541–548. [Google Scholar] [CrossRef]
- Zhuang, W.; Wang, C.; Shi, X.; Qiu, S.; Zhang, S.; Xu, B.; Chen, M.; Jiang, W.; Dong, H.; Qiao, Y. MCMV triggers ROS/NLRP3-associated inflammasome activation in the inner ear of mice and cultured spiral ganglion neurons, contributing to sensorineural hearing loss. International journal of molecular medicine 2018, 41(6), 3448–3456. [Google Scholar] [CrossRef]
- Blacker, T. S.; Duchen, M. R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free radical biology & medicine 2016, 100, 53–65. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R. S.; Handy, D. E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxidants & redox signaling 2018, 28(3), 251–272. [Google Scholar] [CrossRef]
- Mercola, J. Reductive stress and mitochondrial dysfunction: The hidden link in chronic disease. Free radical biology & medicine 2025, 233, 118–131. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxidative Medicine and Cellular Longevity 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Yu, T.; Wan, P.; Zhu, X. D.; et al. Inhibition of NADPH oxidase activities ameliorates DSS-induced colitis. Biochemical Pharmacology 2018, 158, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Oberacker, T.; Kraft, L.; Schanz, M.; Latus, J.; Schricker, S. The importance of thioredoxin-1 in health and disease. Antioxidants 2023, 12(5), 1078. [Google Scholar] [CrossRef]
- Morgan, M. J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research 2011, 21(1), 103–115. [Google Scholar] [CrossRef]
- Pan, L. L.; Ren, Z.; Liu, Y.; et al. A novel danshensu derivative ameliorates experimental colitis by modulating NADPH oxidase 4-dependent NLRP3 inflammasome activation. Journal of Cellular and Molecular Medicine 2020, 24(22), 12955–12969. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology 2010, 11(2), 136–140. [Google Scholar] [CrossRef]
- Chen, H.; Guan, B.; Chen, S.; Yang, D.; Shen, J. Peroxynitrite activates NLRP3 inflammasome and contributes to hemorrhagic transformation and poor outcome in ischemic stroke with hyperglycemia. Free Radical Biology and Medicine 2021, 165, 171–183. [Google Scholar] [CrossRef]
- Ziehr, B. K.; MacDonald, J. A. Regulation of NLRPs by reactive oxygen species: A story of crosstalk. Biochimica et biophysica acta. Molecular cell research 2024, 1871(8), 119823. [Google Scholar] [CrossRef]
- Long, Y.; Liu, X.; Tan, X. Z.; Jiang, C. X.; Chen, S. W.; Liang, G. N.; He, X. M.; Wu, J.; Chen, T.; Xu, Y. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118- induced pyroptosis in endothelial cells. Ecotoxicology and environmental safety 2020, 189, 109937. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Y.; Zhang, X.; Liu, X.; Qi, J. Regulation of iNOS expression by NF-κB in human lens epithelial cells treated with high levels of glucose. Investigative ophthalmology & visual science 2013, 54(7), 5070–5077. [Google Scholar] [CrossRef]
- Singer, I. I.; Kawka, D. W.; Scott, S.; et al. Inducible nitric oxide synthase and nitrotyrosine expression in colonic epithelium in inflammatory bowel disease. Gastroenterology 1996, 111(4), 871–885. [Google Scholar] [CrossRef]
- Banskota, S.; Wang, H.; Kwon, Y. H.; Gautam, J.; Haq, S.; Grondin, J.; Steinberg, G. R.; Khan, W. I. Inhibition of NADPH Oxidase (NOX) 2 Mitigates Colitis in Mice with Impaired Macrophage AMPK Function. Biomedicines 2023, 11(5), 1443. [Google Scholar] [CrossRef]
- Li, X.; Cai, W.; Lee, K.; Liu, B.; Deng, Y.; Chen, Y.; Zhang, X.; He, J. C.; Zhong, Y. Puerarin attenuates diabetic kidney injury through the suppression of NOX4 expression in podocytes. Scientific reports 2017, 7(1), 14603. [Google Scholar] [CrossRef]
- Matrullo, G.; Filomeni, G.; Rizza, S. Redox regulation of focal adhesions. Redox biology 2025, 80, 103514. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J. O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185(16), 2853–2878. [Google Scholar] [CrossRef]
- Schreiber, O.; Petersson, J.; Waldén, T.; Ahl, D.; Sandler, S.; Phillipson, M.; Holm, L. iNOS-dependent increase in colonic mucus thickness in DSS-colitic rats. PloS one 2013, 8(8), e71843. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, G.; Moshage, H.; van Dullemen, H. M.; de Jager-Krikken, A.; Tiebosch, A. T.; Kleibeuker, J. H.; Jansen, P. L.; van Goor, H. Expression of nitric oxide synthases and formation of nitrotyrosine and reactive oxygen species in inflammatory bowel disease. The Journal of pathology 1998, 186(4), 416–421. [Google Scholar] [CrossRef]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Frontiers in immunology 2014, 4, 514. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Tao, A.; Xu, X.; Kvietys, P.; Rui, T. Peroxynitrite/PKR Axis Modulates the NLRP3 Inflammasome of Cardiac Fibroblasts. Frontiers in immunology 2020, 11, 558712. [Google Scholar] [CrossRef]
- Linillos-Pradillo, B.; Paredes, S. D.; Ortiz-Cabello, M.; Schlumpf, M.; Lichtensteiger, W.; Vara, E.; Tresguerres, J. A. F.; Rancan, L. Activation of NLRP3 Inflammasome in Liver of Long Evans Lactating Rats and Its Perinatal Effects in the Offspring after Bisphenol F Exposure. International journal of molecular sciences 2023, 24(18), 14129. [Google Scholar] [CrossRef]
- Qiang, R.; Li, Y.; Dai, X.; Lv, W. NLRP3 inflammasome in digestive diseases: From mechanism to therapy. Frontiers in immunology 2022, 13, 978190. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Frontiers in immunology 2019, 10, 276. [Google Scholar] [CrossRef]
- Yu, T.; Wan, P.; Zhu, X. D.; Ren, Y. P.; Wang, C.; Yan, R. W.; Guo, Y.; Bai, A. P. Inhibition of NADPH oxidase activities ameliorates DSS-induced colitis. Biochemical pharmacology 2018, 158, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Herfindal, A. M.; Rocha, S. D. C.; Papoutsis, D.; Bøhn, S. K.; Carlsen, H. The ROS-generating enzyme NADPH oxidase 1 modulates the colonic microbiota but offers minor protection against dextran sulfate sodium-induced low-grade colon inflammation in mice. Free radical biology & medicine 2022, 188, 298–311. [Google Scholar] [CrossRef]
- McCafferty, D. M. Peroxynitrite and inflammatory bowel disease. Gut 2000, 46(3), 436–439. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S. E.; Ledoux, P.; Evangelista, C.; Kim, I. F.; Tomashevsky, M.; Marshall, K. A.; Phillippy, K. H.; Sherman, P. M.; Holko, M.; Yefanov, A.; Lee, H.; Zhang, N.; Robertson, C. L.; Serova, N.; Davis, S.; Soboleva, A. NCBI GEO: archive for functional genomics data sets--update. Nucleic acids research 2013, 41(Database issue), D991–D995. [Google Scholar] [CrossRef]
- Arijs, I.; De Hertogh, G.; Lemaire, K.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; Cleynen, I.; Van Assche, G.; Vermeire, S.; Geboes, K.; Schuit, F.; Rutgeerts, P. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PloS one 2009, 4(11), e7984. [Google Scholar] [CrossRef]
- Nashef, A.; Matthias, M.; Weiss, E.; Loos, B. G.; Jepsen, S.; van der Velde, N.; Uitterlinden, A. G.; Wellmann, J.; Berger, K.; Hoffmann, P.; Laudes, M.; Lieb, W.; Franke, A.; Dommisch, H.; Schäfer, A.; Houri-Haddad, Y.; Iraqi, F. A. Translation of mouse model to human gives insights into periodontitis etiology. Scientific reports 2020, 10(1), 4892. [Google Scholar] [CrossRef]
- Ruttkies, C.; Schymanski, E. L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: incorporating strategies beyond in silico fragmentation. Journal of Cheminformatics 2016, 8(1), 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J. J.; Phelan, V. V.; Sanchez, L. M.; Garg, N.; Peng, Y.; Dorrestein, P. C. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nature Biotechnology 2016, 34(8), 828–837. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A. A.; Melnik, A. V.; Meusel, M.; Böcker, S. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nature Methods 2019, 16(4), 299–302. [Google Scholar] [CrossRef]
Figure 1.
LC-MS/MS-based phytochemical profiling of Endarachne binghamiae hot water extract (EB-WE). (A) Total ion chromatogram (TIC) in positive ionization mode (ES+). (B) TIC in negative ionization mode (ES−). Major peaks are annotated with putatively identified compound names. (C) Representative extracted ion chromatograms (EICs) and MS/MS spectral matching for digiprolactone (loliolide) and pyropheophorbide A. Left panels show EICs with retention times; middle panels display experimental MS/MS spectra; right panels show reference spectra from the GNPS spectral library (CCMSLIB00004680136 and CCMSLIB00010128703, respectively). Blue and red asterisks indicate matched fragment ions between experimental and library spectra.
Figure 1.
LC-MS/MS-based phytochemical profiling of Endarachne binghamiae hot water extract (EB-WE). (A) Total ion chromatogram (TIC) in positive ionization mode (ES+). (B) TIC in negative ionization mode (ES−). Major peaks are annotated with putatively identified compound names. (C) Representative extracted ion chromatograms (EICs) and MS/MS spectral matching for digiprolactone (loliolide) and pyropheophorbide A. Left panels show EICs with retention times; middle panels display experimental MS/MS spectra; right panels show reference spectra from the GNPS spectral library (CCMSLIB00004680136 and CCMSLIB00010128703, respectively). Blue and red asterisks indicate matched fragment ions between experimental and library spectra.
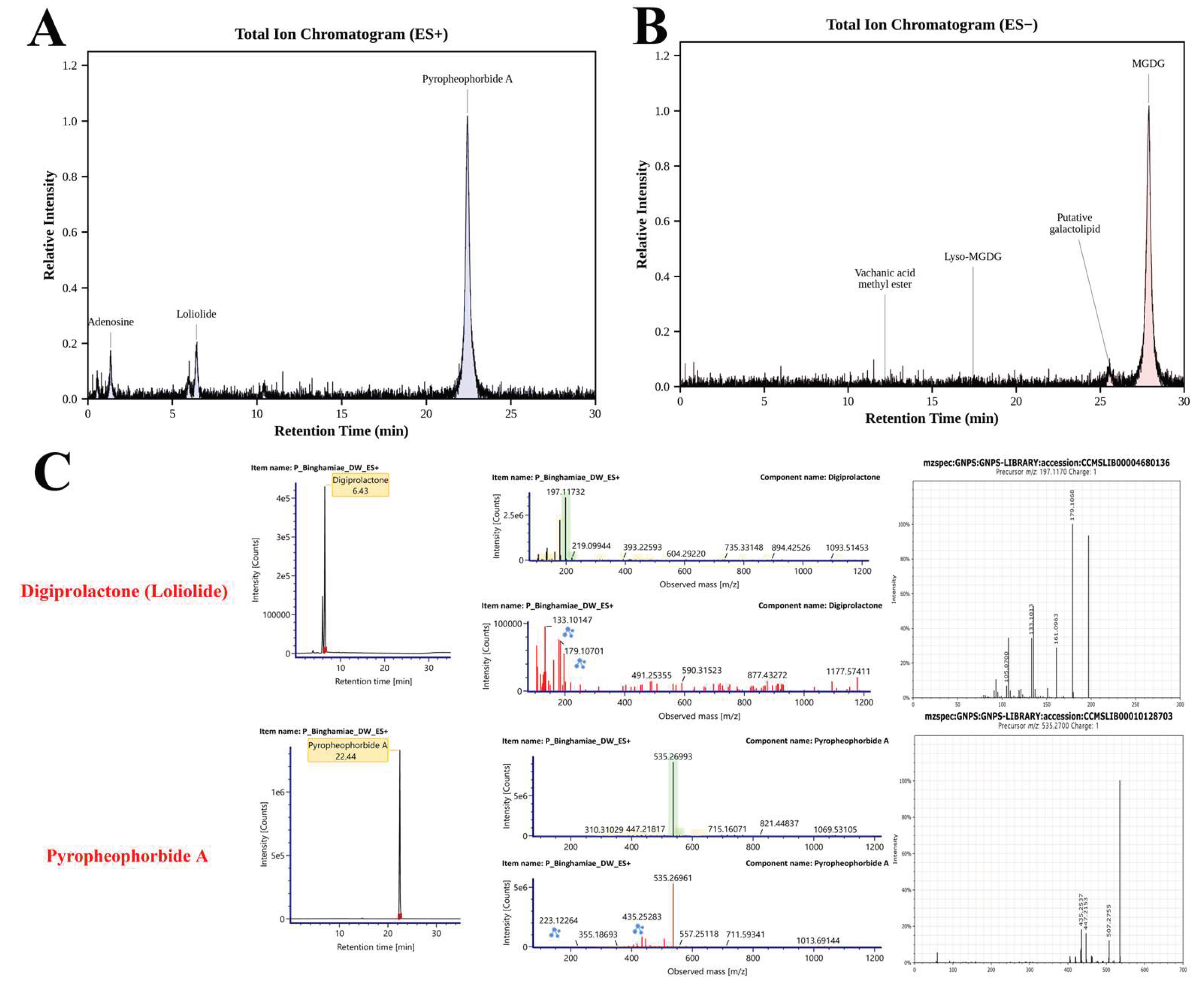
Figure 2.
Downregulation of pro-inflammatory cytokines and inhibition of NLRP3 inflammasome formation by EB-WE. (A) BMDMs were primed with LPS (1 µg/mL, 4 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL). ELISA analysis showed that LPS+ATP markedly increased IL-1β and IL-18 secretion, while EB-WE pretreatment significantly reduced both cytokines in a dose-dependent manner. (B) Confocal immunofluorescence analysis of NLRP3 inflammasome complex formation. NLRP3 (red) was co-stained with caspase-1 (green) or ASC (green), and nuclei were counterstained with DAPI (blue). LPS+ATP stimulation induced robust co-localization of NLRP3 with both caspase-1 and ASC, consistent with inflammasome assembly and pyroptotic speck formation. EB-WE pretreatment markedly reduced NLRP3 co-localization with caspase-1 and ASC, indicating inhibition of inflammasome assembly. Scale bar = 20 µm. These results suggest that EB-WE suppresses both the activation and assembly of the NLRP3 inflammasome, thereby inhibiting pro-inflammatory cytokine maturation and pyroptosis in macrophages. **p<0.01 vs LPS+ATP Data are expressed as mean ± SD (n = 3).
Figure 2.
Downregulation of pro-inflammatory cytokines and inhibition of NLRP3 inflammasome formation by EB-WE. (A) BMDMs were primed with LPS (1 µg/mL, 4 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL). ELISA analysis showed that LPS+ATP markedly increased IL-1β and IL-18 secretion, while EB-WE pretreatment significantly reduced both cytokines in a dose-dependent manner. (B) Confocal immunofluorescence analysis of NLRP3 inflammasome complex formation. NLRP3 (red) was co-stained with caspase-1 (green) or ASC (green), and nuclei were counterstained with DAPI (blue). LPS+ATP stimulation induced robust co-localization of NLRP3 with both caspase-1 and ASC, consistent with inflammasome assembly and pyroptotic speck formation. EB-WE pretreatment markedly reduced NLRP3 co-localization with caspase-1 and ASC, indicating inhibition of inflammasome assembly. Scale bar = 20 µm. These results suggest that EB-WE suppresses both the activation and assembly of the NLRP3 inflammasome, thereby inhibiting pro-inflammatory cytokine maturation and pyroptosis in macrophages. **p<0.01 vs LPS+ATP Data are expressed as mean ± SD (n = 3).
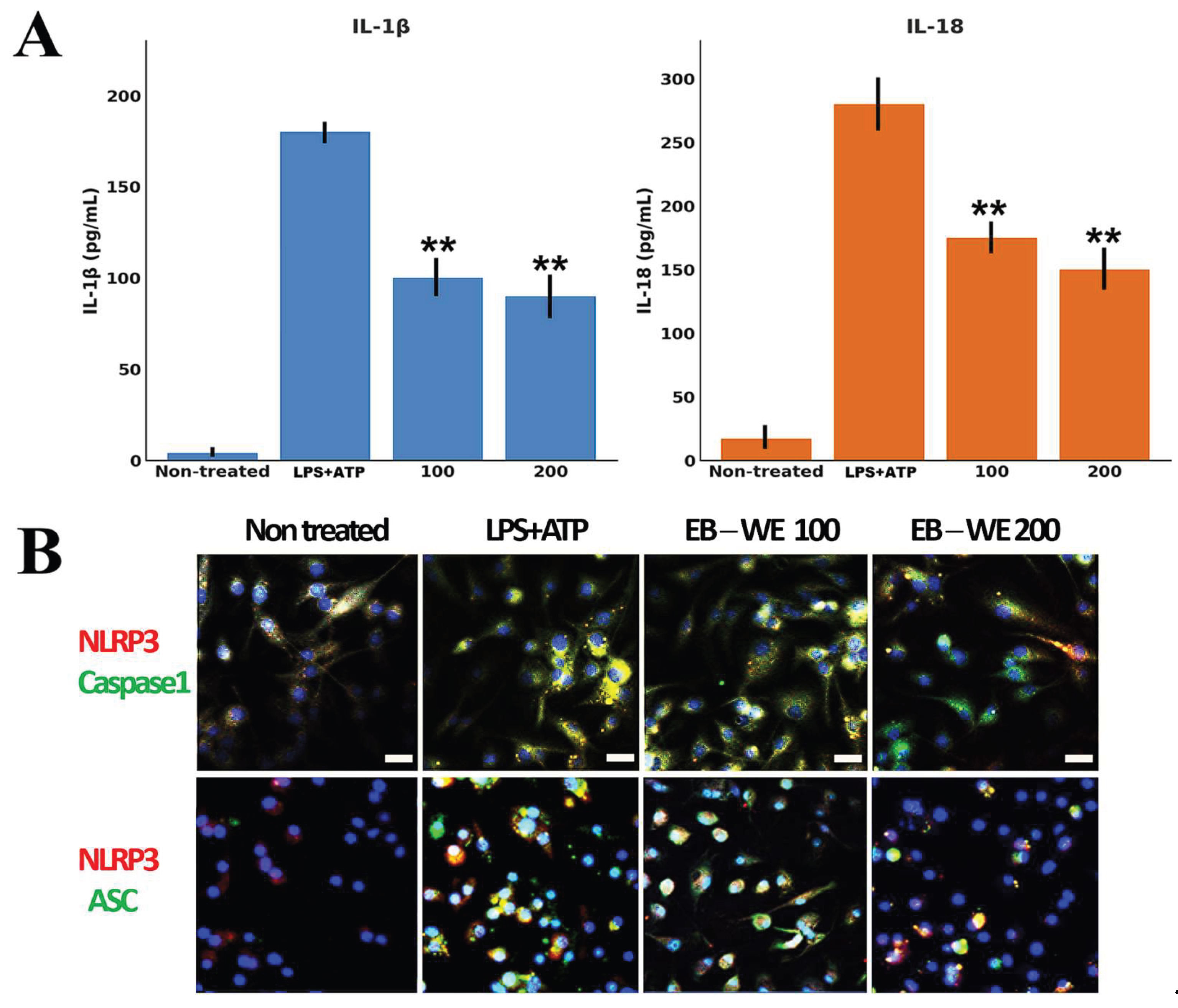
Figure 3.
Western blot analysis in BMDMs. BMDMs were primed with LPS (1 µg/mL, 5 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL). Cell lysates and culture supernatants were analyzed for pro-IL-1β (30 kDa), pro–caspase-1 (45 kDa), mature caspase-1 (p10), mature IL-1β (p17), and ASC (22 kDa) by immunoblotting. β-actin served as the loading control. Densitometric quantification is shown for IL-1β, caspase-1, and ASC (normalized to β-actin; mean ± SD, n=3). LPS+ATP markedly increased pyroptotic protein release, while EB-WE reduced mature IL-1β and ASC levels in the supernatant, indicating inhibition of NLRP3 inflammasome activation. p < 0.05, p < 0.01 vs. LPS+ATP group. Data are expressed as mean ± SD (n = 3).
Figure 3.
Western blot analysis in BMDMs. BMDMs were primed with LPS (1 µg/mL, 5 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL). Cell lysates and culture supernatants were analyzed for pro-IL-1β (30 kDa), pro–caspase-1 (45 kDa), mature caspase-1 (p10), mature IL-1β (p17), and ASC (22 kDa) by immunoblotting. β-actin served as the loading control. Densitometric quantification is shown for IL-1β, caspase-1, and ASC (normalized to β-actin; mean ± SD, n=3). LPS+ATP markedly increased pyroptotic protein release, while EB-WE reduced mature IL-1β and ASC levels in the supernatant, indicating inhibition of NLRP3 inflammasome activation. p < 0.05, p < 0.01 vs. LPS+ATP group. Data are expressed as mean ± SD (n = 3).
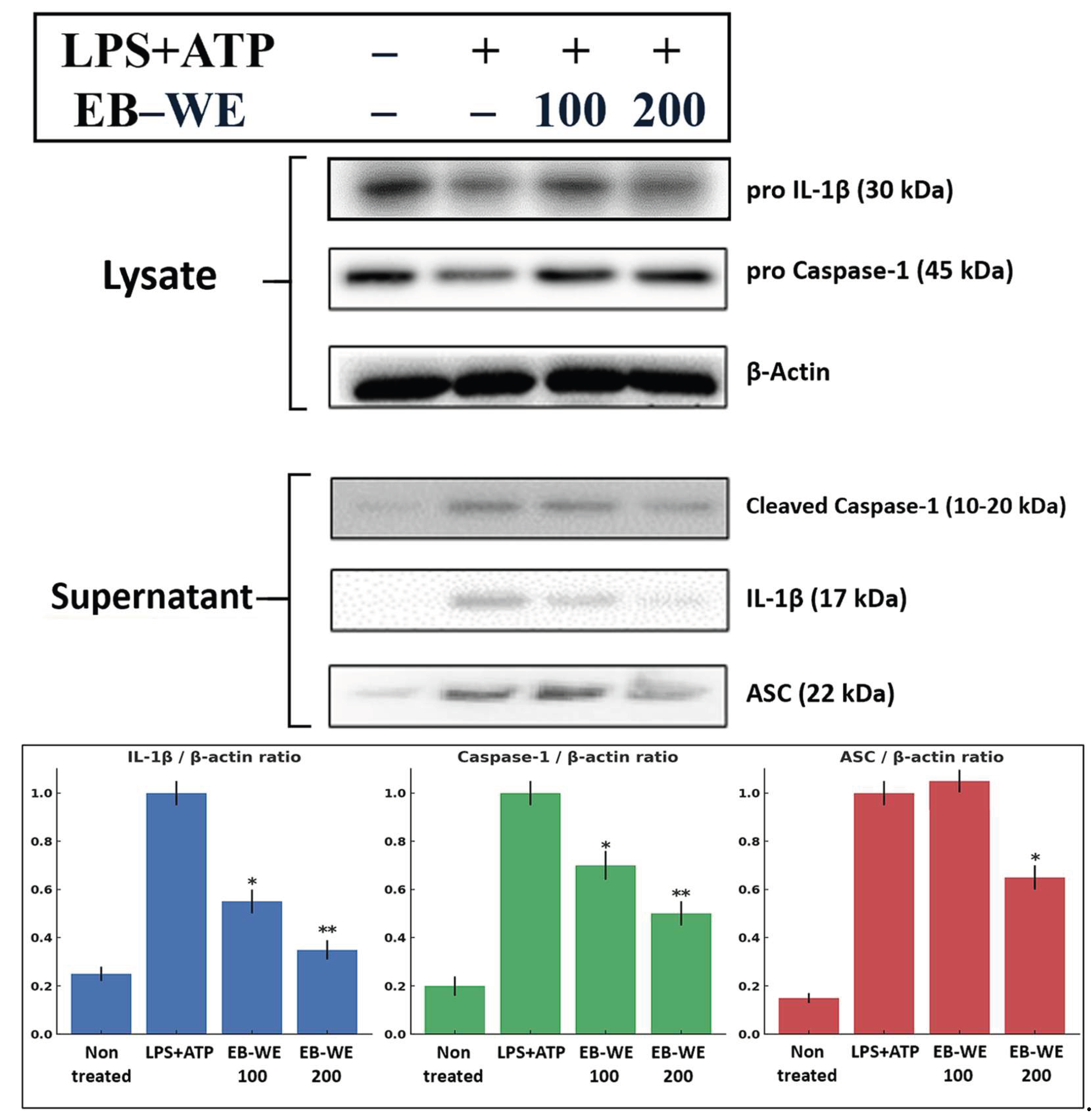
Figure 4.
Confocal immunofluorescence images showing the intracellular localization of ROS (DCF-DA, green), NLRP3 (red), and nuclei (DAPI, blue) in bone marrow-derived macrophages (BMDMs). Cells were primed with LPS (1 µg/mL, 5 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL, 24h). LPS+ATP stimulation induced strong DCF-DA fluorescence and perinuclear NLRP3 clustering, which were both markedly reduced by EB-WE. Merge images reveal co-localization of ROS and NLRP3 in the activated state, potentially indicating that its anti-inflammatory effect is linked to antioxidant activity. Scale bar = 20 µm.
Figure 4.
Confocal immunofluorescence images showing the intracellular localization of ROS (DCF-DA, green), NLRP3 (red), and nuclei (DAPI, blue) in bone marrow-derived macrophages (BMDMs). Cells were primed with LPS (1 µg/mL, 5 h) and stimulated with ATP (5 mM, 30 min), with or without EB-WE pretreatment (100 or 200 µg/mL, 24h). LPS+ATP stimulation induced strong DCF-DA fluorescence and perinuclear NLRP3 clustering, which were both markedly reduced by EB-WE. Merge images reveal co-localization of ROS and NLRP3 in the activated state, potentially indicating that its anti-inflammatory effect is linked to antioxidant activity. Scale bar = 20 µm.
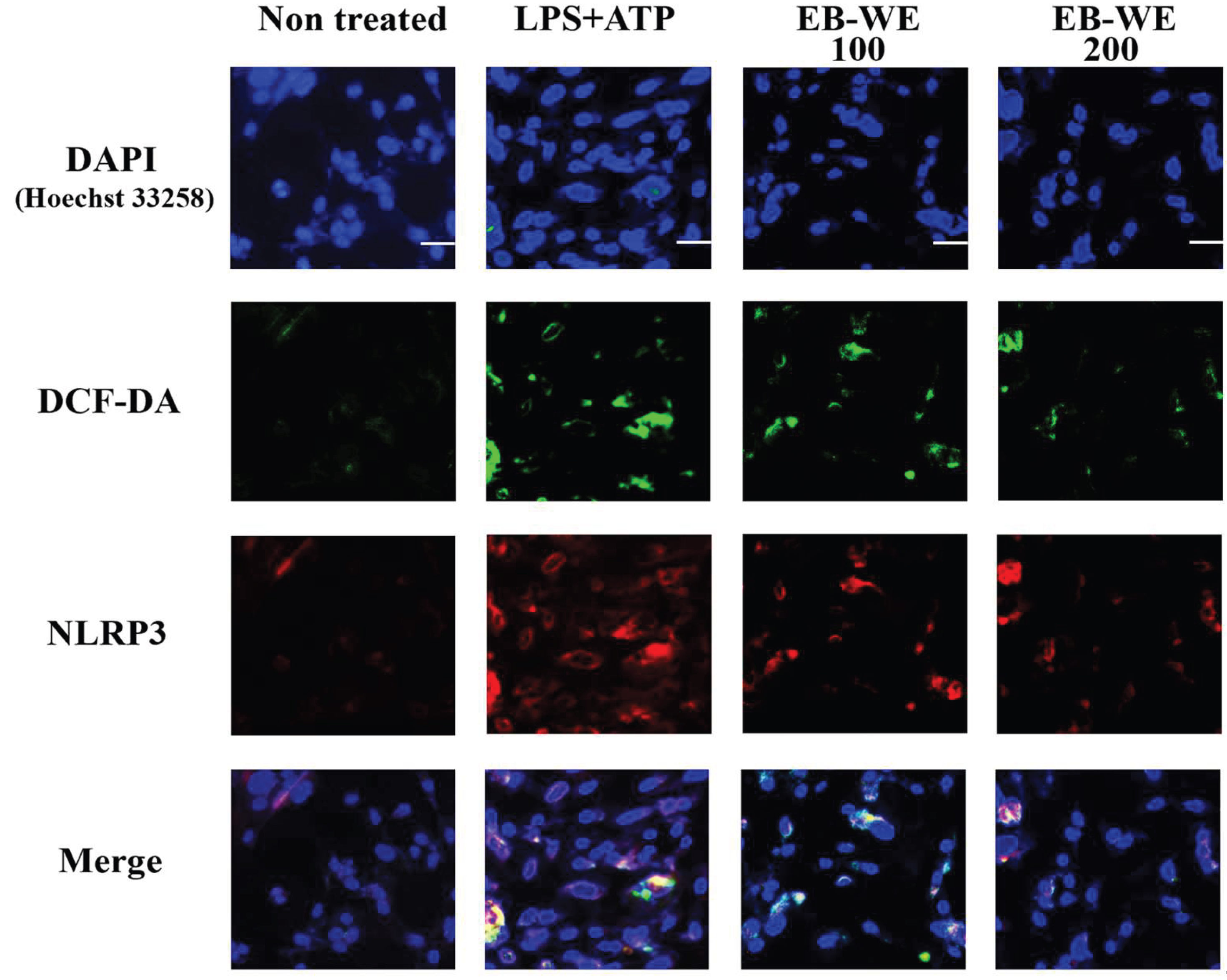
Figure 5.
EB-WE suppresses systemic and local inflammation in DSS-induced colitis. (A) Serum cytokine levels of IL-2, IL-1β, IL-18, TNF-α, IFN-γ, and IL-17A were measured by ELISA. DSS significantly increased all six pro-inflammatory cytokines, while EB-WE administration, especially at 5 mg/mice, markedly reduced IL-2, IL-1β, TNF-α, and IFN-γ levels. (B) Colonic cytokine levels were similarly elevated by DSS, indicating strong mucosal inflammation. EB-WE treatment significantly reduced TNF-α, IL-1β, and IL-17A expression in a dose-dependent manner. †p < 0.05, ††p < 0.01, †††p < 0.001 vs. Normal; *p < 0.05, **p < 0.01, ***p < 0.001 vs. DSS.
Figure 5.
EB-WE suppresses systemic and local inflammation in DSS-induced colitis. (A) Serum cytokine levels of IL-2, IL-1β, IL-18, TNF-α, IFN-γ, and IL-17A were measured by ELISA. DSS significantly increased all six pro-inflammatory cytokines, while EB-WE administration, especially at 5 mg/mice, markedly reduced IL-2, IL-1β, TNF-α, and IFN-γ levels. (B) Colonic cytokine levels were similarly elevated by DSS, indicating strong mucosal inflammation. EB-WE treatment significantly reduced TNF-α, IL-1β, and IL-17A expression in a dose-dependent manner. †p < 0.05, ††p < 0.01, †††p < 0.001 vs. Normal; *p < 0.05, **p < 0.01, ***p < 0.001 vs. DSS.
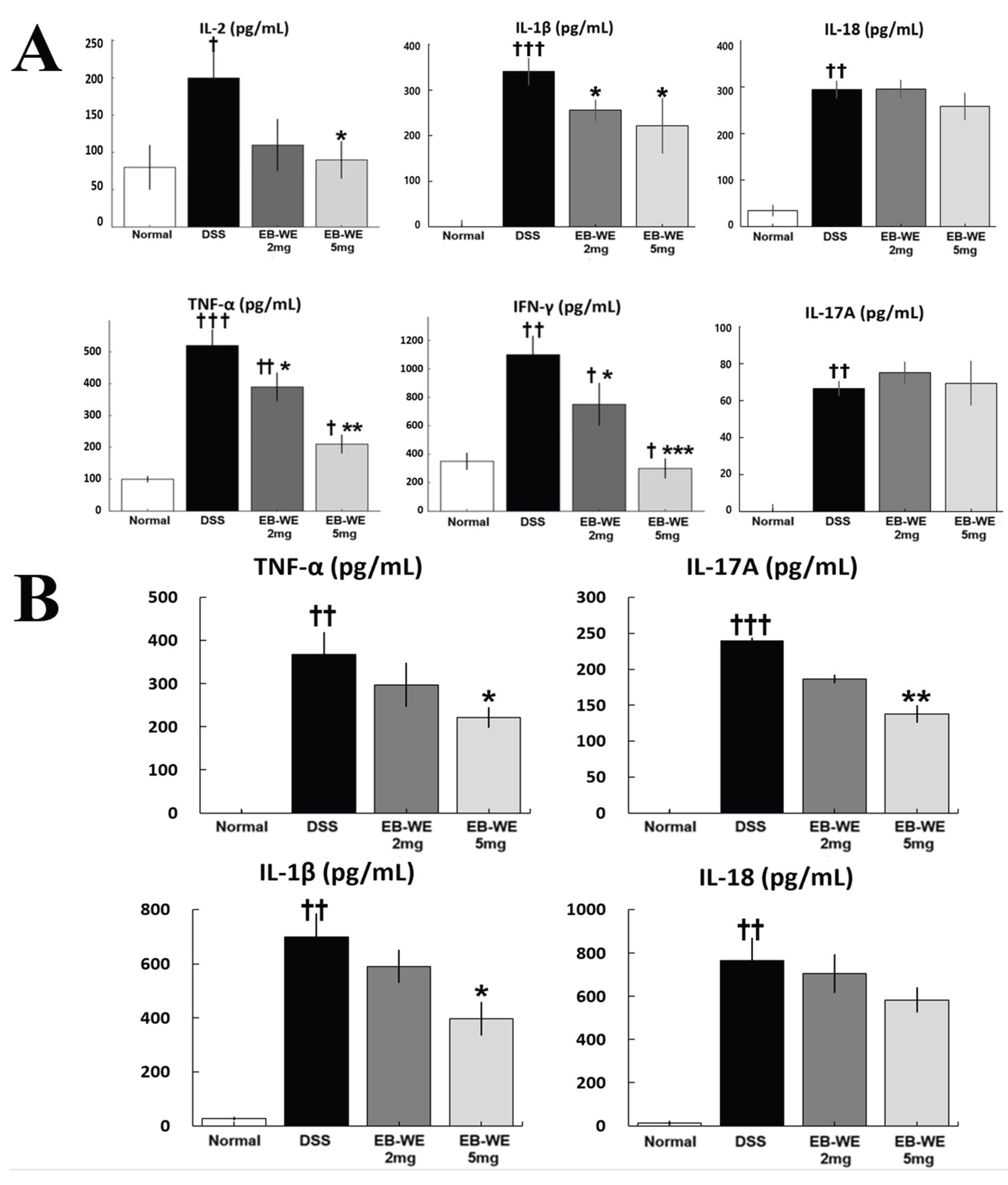
Figure 6.
Anti-IBD effects of EB-WE in colon tissue. (A) EB-WE treatment significantly alleviated DSS-induced clinical symptoms, including body weight loss, increased stool consistency scores, visible blood in feces, elevated Disease Activity Index (DAI) scores, and colon shortening. The 5 mg/mice group exhibited the most pronounced improvements across all parameters, indicating a dose-dependent therapeutic effect of EB-WE. (B) Representative histological images of colon tissue stained with H&E. DSS treatment induced severe epithelial disruption, submucosal edema, and extensive inflammatory cell infiltration (indicated by arrows). EB-WE administration mitigated these pathological features in a dose-dependent manner. The 5 mg/mice group demonstrated nearly intact mucosal and crypt structures, comparable to the normal group. Scale bar = 100 µm. †p < 0.05, ††p < 0.01, †††p < 0.001 vs. Normal; *p < 0.05, **p < 0.01, ***p < 0.001 vs. DSS.
Figure 6.
Anti-IBD effects of EB-WE in colon tissue. (A) EB-WE treatment significantly alleviated DSS-induced clinical symptoms, including body weight loss, increased stool consistency scores, visible blood in feces, elevated Disease Activity Index (DAI) scores, and colon shortening. The 5 mg/mice group exhibited the most pronounced improvements across all parameters, indicating a dose-dependent therapeutic effect of EB-WE. (B) Representative histological images of colon tissue stained with H&E. DSS treatment induced severe epithelial disruption, submucosal edema, and extensive inflammatory cell infiltration (indicated by arrows). EB-WE administration mitigated these pathological features in a dose-dependent manner. The 5 mg/mice group demonstrated nearly intact mucosal and crypt structures, comparable to the normal group. Scale bar = 100 µm. †p < 0.05, ††p < 0.01, †††p < 0.001 vs. Normal; *p < 0.05, **p < 0.01, ***p < 0.001 vs. DSS.
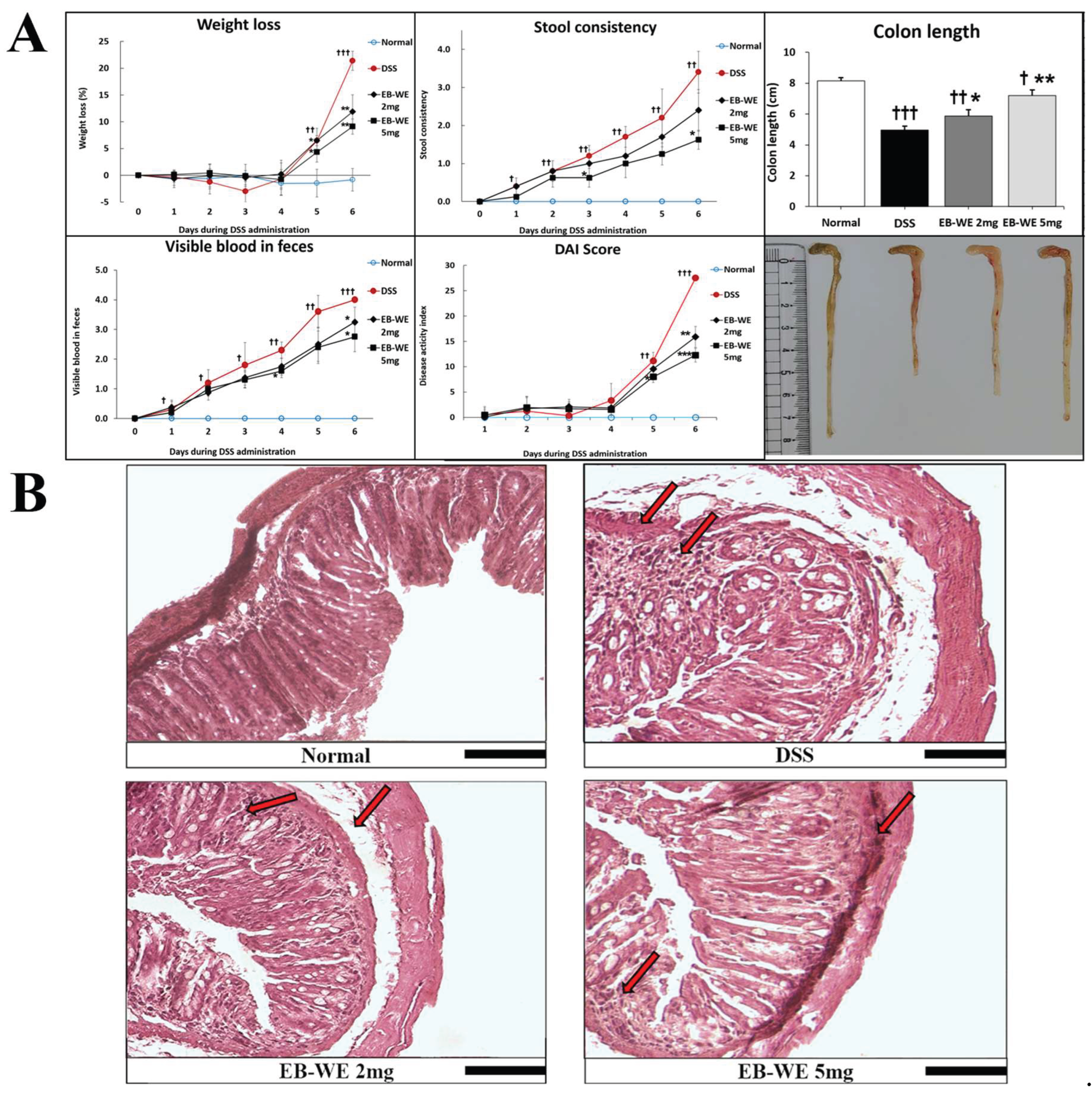
Figure 7.
EB-WE suppresses inflammatory signaling, NLRP3 inflammasome activation, and apoptosis in colon tissue of DSS-induced IBD mice. (A) Representative Western blot analysis of inflammatory signaling proteins (iNOS, p-p38 MAPK, p38 MAPK, p-IKB, IKB, p-p65 RELA, p65 RELA) in colon tissue. (B) Representative blots for inflammasome (pro-caspase-1, cleaved caspase-1, ASC), apoptotic (BCL2, BAX, pro-caspase-3, cleaved caspase-3), and barrier-related proteins (Occludin, ZO-1). Actin was used as a loading control. Colon tissues were collected from normal (Sham), DSS-treated, and EB-WE–treated (2 and 5 mg/mice) groups. *p<0.05, **p<0.01 vs DSS. Data are expressed as mean ± SD (n = 3).
Figure 7.
EB-WE suppresses inflammatory signaling, NLRP3 inflammasome activation, and apoptosis in colon tissue of DSS-induced IBD mice. (A) Representative Western blot analysis of inflammatory signaling proteins (iNOS, p-p38 MAPK, p38 MAPK, p-IKB, IKB, p-p65 RELA, p65 RELA) in colon tissue. (B) Representative blots for inflammasome (pro-caspase-1, cleaved caspase-1, ASC), apoptotic (BCL2, BAX, pro-caspase-3, cleaved caspase-3), and barrier-related proteins (Occludin, ZO-1). Actin was used as a loading control. Colon tissues were collected from normal (Sham), DSS-treated, and EB-WE–treated (2 and 5 mg/mice) groups. *p<0.05, **p<0.01 vs DSS. Data are expressed as mean ± SD (n = 3).
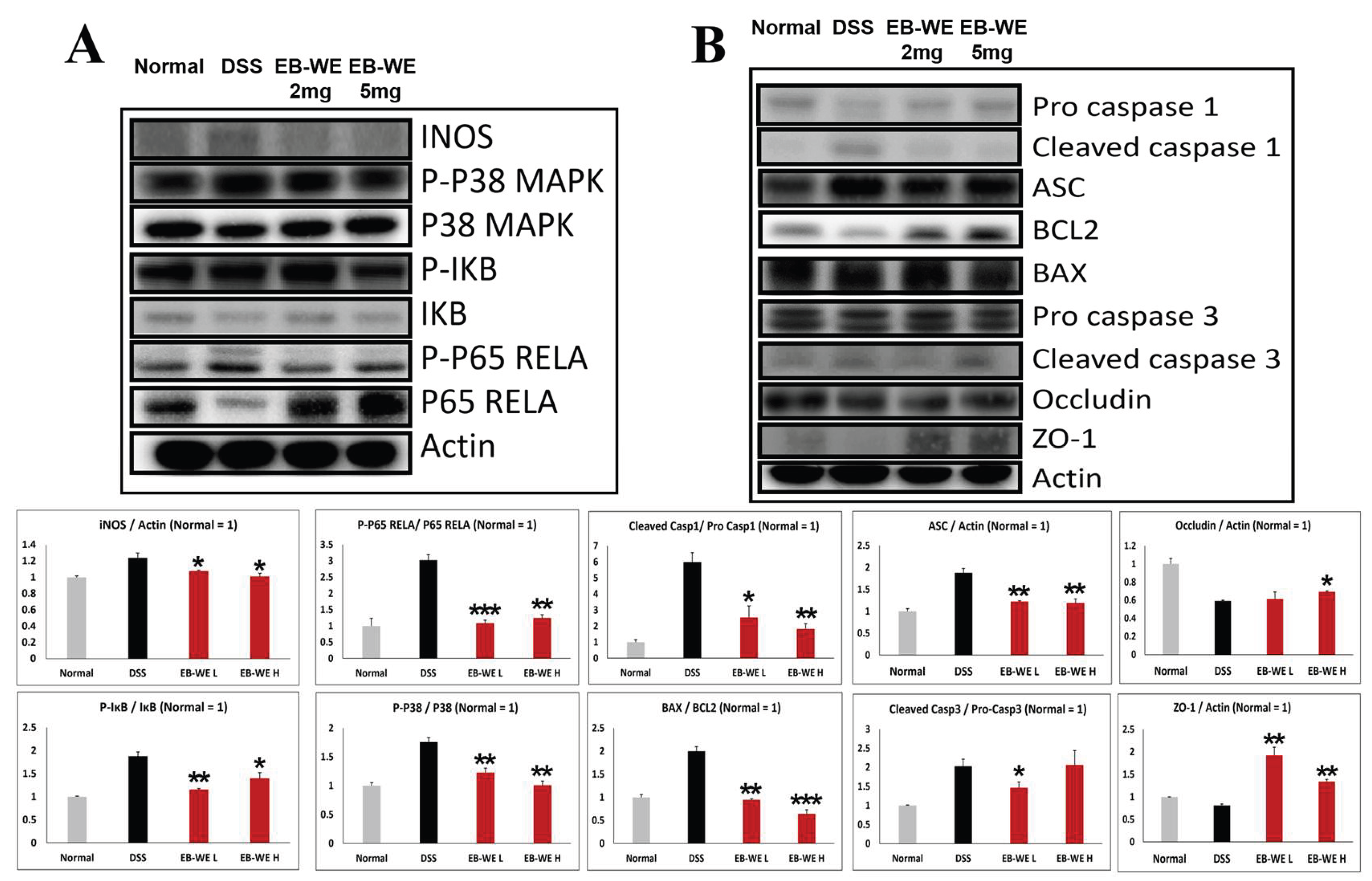
Figure 8.
EB-WE modulates redox-regulating proteins and redox cofactor ratios in colon tissue of DSS-induced IBD mice. (A) Representative Western blot analysis of pro-oxidant (NOX2, NOX4, TXNIP) and antioxidant (NQO1, NRF2, HO-1) proteins with Actin loading control. (B) Quantitative analysis of protein expression normalized to Actin. (C) Ratios of NAD+/NADH and NADP+/NADPH measured in colon tissues from normal, DSS-treated, and EB-WE-treated (2 and 5 mg/mice) groups. †p < 0.05, ††p < 0.01 vs. Normal; *p<0.05, **p<0.01, ***p<0.001 vs DSS. Data are expressed as mean ± SD (n = 3).
Figure 8.
EB-WE modulates redox-regulating proteins and redox cofactor ratios in colon tissue of DSS-induced IBD mice. (A) Representative Western blot analysis of pro-oxidant (NOX2, NOX4, TXNIP) and antioxidant (NQO1, NRF2, HO-1) proteins with Actin loading control. (B) Quantitative analysis of protein expression normalized to Actin. (C) Ratios of NAD+/NADH and NADP+/NADPH measured in colon tissues from normal, DSS-treated, and EB-WE-treated (2 and 5 mg/mice) groups. †p < 0.05, ††p < 0.01 vs. Normal; *p<0.05, **p<0.01, ***p<0.001 vs DSS. Data are expressed as mean ± SD (n = 3).
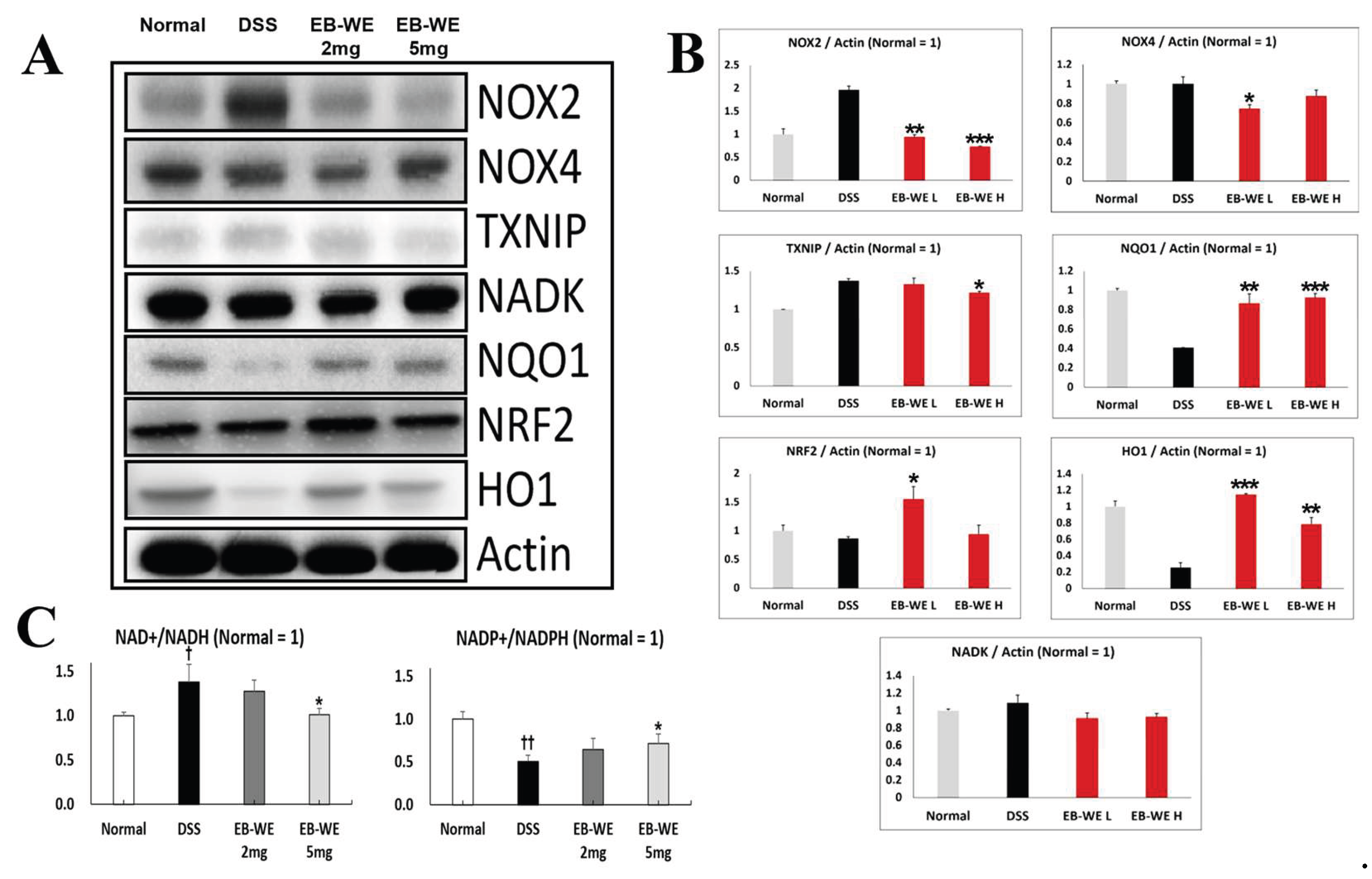
Figure 9.
Dual Contribution of NOX–ROS and iNOS to NLRP3 Inflammasome Priming and Activation. As shown in the figure, both NF-κB (p65) and MAPK signaling pathways mediate the transcriptional priming of NLRP3 and iNOS under NOX1/2-derived oxidative stress. Meanwhile, NOX4 and iNOS contribute to mitochondrial ROS production, facilitating NLRP3 activation (oligomerization) through TXNIP dissociation and ion flux (e.g., Ca²⁺ influx, Na⁺ influx, K⁺ efflux). Upon inflammasome assembly, caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature forms, ultimately triggering pyroptosis.
Figure 9.
Dual Contribution of NOX–ROS and iNOS to NLRP3 Inflammasome Priming and Activation. As shown in the figure, both NF-κB (p65) and MAPK signaling pathways mediate the transcriptional priming of NLRP3 and iNOS under NOX1/2-derived oxidative stress. Meanwhile, NOX4 and iNOS contribute to mitochondrial ROS production, facilitating NLRP3 activation (oligomerization) through TXNIP dissociation and ion flux (e.g., Ca²⁺ influx, Na⁺ influx, K⁺ efflux). Upon inflammasome assembly, caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature forms, ultimately triggering pyroptosis.
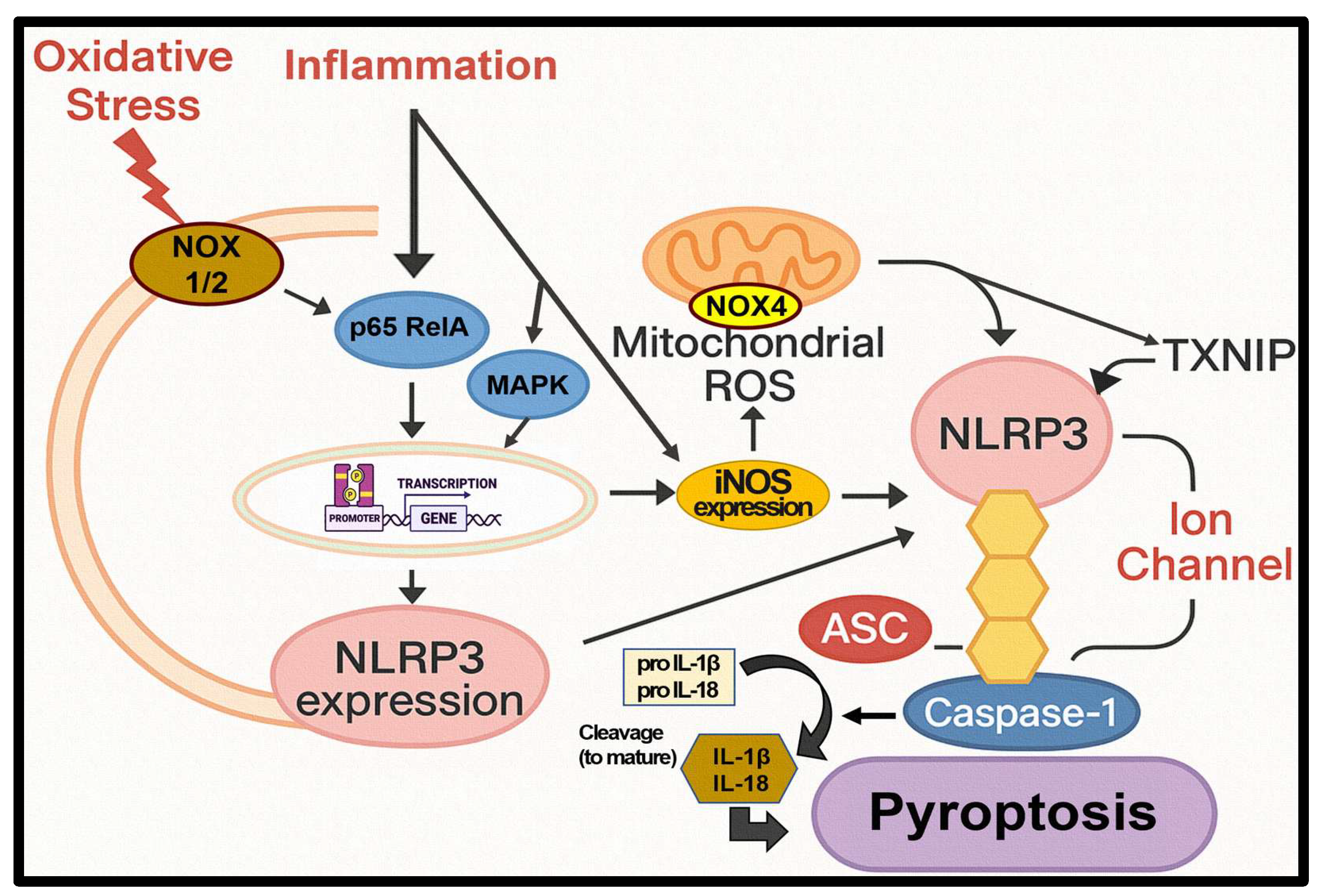
Figure 10.
Proposed schematic model illustrating the anti-inflammatory mechanism of EB-WE in DSS-induced colitis.
Figure 10.
Proposed schematic model illustrating the anti-inflammatory mechanism of EB-WE in DSS-induced colitis.
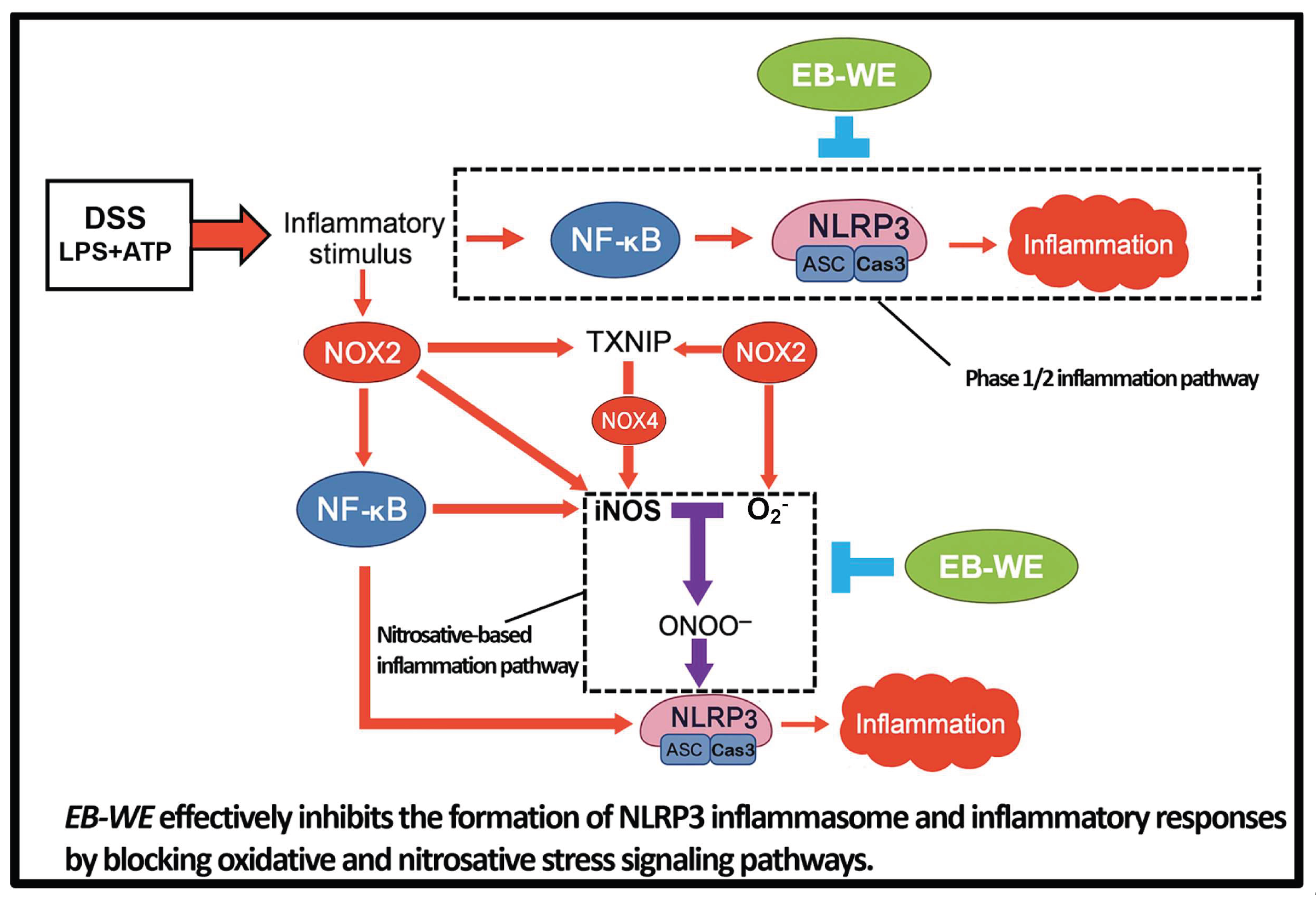

Table 1.
Putative identification of compounds from EB-WE by UHPLC-QTOF-MS/MS coupled with DB and molecular networking analysis.
Table 1.
Putative identification of compounds from EB-WE by UHPLC-QTOF-MS/MS coupled with DB and molecular networking analysis.
| Identified Compounds | Monoisotopic Mass (Da) | Formula | Observed Mass (Da) | Observed m/z | Mass Error (mDa) | Mass Error (ppm) | RT (min) | Detector Counts | Adducts | Score | MS/MS Fragments |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ES+ (Positive Ionization Mode) | |||||||||||
| Pyropheophorbide A | 534.2631 | C₃₃H₃₄N₄O₃ | 534.2627 | 535.2699 | -0.4 | -0.8 | 22.44 | 556,053 | +H, +Na | 0.875 | 223.12, 355.19, 435.25, 447.22 |
| Digiprolactone (Loliolide) | 196.1099 | C₁₁H₁₆O₃ | 196.11 | 197.1173 | 0.1 | 0.5 | 6.43 | 102,239 | +H, +Na | 0.906 | 105.12, 133.1, 179.11 |
| Adenosine | 267.0968 | C₁₀H₁₃N₅O₄ | 267.0968 | 268.1041 | 0.1 | 0.2 | 1.36 | 86,691 | +H, +Na | 0.98 | 136.06 |
| Allitol | 182.079 | C₆H₁₄O₆ | 182.0789 | 205.0681 | -0.1 | -0.6 | 0.57 | 35,257 | +Na | 0.736 | 111.05, 182.09 |
| 6α-Acetoxy-5-epilimonin | 542.2516 | C₃₀H₃₈O₉ | 542.2542 | 565.2434 | 2.6 | 4.6 | 22.8 | 26,387 | +Na | 0.85 | 256.2, 268.2, 503.2 |
| 3-Tert-butyl-4-methoxyphenol | 180.115 | C₁₁H₁₆O₂ | 180.1151 | 181.1224 | 0.1 | 0.3 | 10.43 | 25,850 | +H, +Na | 0.78 | 108, 137.2, 166.8 |
| ES- (Negative Ionization Mode) | |||||||||||
| Monogalactosyldiacylglycerol | 766.4867 | C₄₂H₇₀O₁₂ | 766.4898 | 765.4825 | 3.1 | 4 | 27.89 | 20,566,572 | -H | 0.89 | 225, 317, 537.3 |
| Putative galactolipid (MGDG/DGDG-related) | 764.4711 | C₄₂H₆₈O₁₂ | 764.4747 | 763.4675 | 3.7 | 4.8 | 25.55 | 1,220,709 | -H | 0.86 | 125, 303.2, 561.2 |
| Vachanic acid methyl ester | 266.1882 | C₁₆H₂₆O₃ | 266.1883 | 311.1865 | 0.1 | 0.3 | 12.2 | 161,942 | +HCOO | 0.82 | 155.1, 247.2, 267.2 |
| 18:3 Lyso-MGDG | 598.3142 | C₃₄H₄₆O₉ | 598.3114 | 597.3041 | -2.8 | -4.7 | 17.44 | 163,431 | -H | 0.86 | 153, 281.2, 481.2 |
| 2-Pentadecanone | 226.2297 | C₁₅H₃₀O | 226.2295 | 271.2277 | -0.2 | -0.7 | 19.23 | 103,008 | +HCOO, -H | 0.65 | 221.2, 225.2 |
| Helveticoside | 534.6527 | C₂₉H₄₂O₉ | 579.2838 | 579.2838 | 2.7 | 4.7 | 16.75 | 88,100 | +HCOO | 0.77 | 225, 279.23, 319.23 |
Abbreviations: RT, retention time; Da, Dalton; ppm, parts per million; GNPS, Global Natural Products Social Molecular Networking.
Table 2.
Abbreviations used throughout this manuscript.
| Abbreviation | Full Term |
|---|---|
| AP1 | Activator Protein 1 |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine Triphosphate |
| BAX | Bcl-2-associated X protein |
| BCL2 | B-cell lymphoma 2 |
| BMDM | Bone Marrow-Derived Macrophage |
| CAT | Catalase |
| COX2 | Cyclooxygenase 2 |
| CaMKII | Ca2+/Calmodulin-dependent Protein Kinase II |
| Caspase-1 | Cysteine-aspartic protease 1 |
| DAI | Disease Activity Index |
| DAMPs | Damage-Associated Molecular Patterns |
| DCFDA | 2′,7′-Dichlorodihydrofluorescein diacetate |
| DSS | Dextran Sulfate Sodium |
| EB-WE | Endarachne binghamiae Water Extract |
| ERK | Extracellular signal-Regulated Kinase |
| GRX | Glutaredoxin |
| GSH | Glutathione |
| IBD | Inflammatory Bowel Disease |
| IFN-γ | Interferon gamma |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1 beta |
| IκBα | Inhibitor of kappa B alpha |
| JNK | c-Jun N-terminal Kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinase |
| MMP9 | Matrix Metalloproteinase 9 |
| NADK | NAD + kinase |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate (reduced form) |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| NOX | NADPH Oxidase |
| NQO | NAD(P)H Quinone Dehydrogenase |
| ONOO⁻ | Peroxynitrite |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PRDX | Peroxiredoxin |
| ROS | Reactive Oxygen Species |
| RelA | v-rel avian reticuloendotheliosis viral oncogene homolog A |
| SOD | Superoxide Dismutase |
| SREBP2 | Sterol Regulatory Element-Binding Protein 2 |
| TLR4 | Toll-like Receptor 4 |
| TNF-α | Tumor Necrosis Factor alpha |
| TRX | Thioredoxin |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| TXNIP | Thioredoxin-interacting protein |
| UHPLC-QTOF-LC-MS/MS | Ultra-High Performance Liquid Chromatography-Quadrupole Time-of-Flight Liquid Chromatography-Mass Spectrometry/Mass Spectrometry |
| iNOS | inducible Nitric Oxide Synthase |
| p38 | p38 Mitogen-Activated Protein Kinase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



