Submitted:
03 February 2026
Posted:
04 February 2026
You are already at the latest version
Abstract
We perform Density Functional Theory calculations to determine adsorption energies of small gas molecules ($\mathrm{H}_2$, $\mathrm{N}_2$, NO, and CO) on defective, vacancy-laden, black phosphorene. Different configurations of single and double vacancies in the monolayer structure are considered, together with several possible adsorption sites onto them. The van der Waals interaction is considered for the exchange-correlation functional. This research aims to provide fundamental insights into how atomic vacancies can be engineered to tune phosphorene's surface reactivity, offering a broader understanding of its multifaceted applications.
Keywords:
black phosphorene
; single and double vacancies
; small gas molecule adsorption
; density functional theory
1. Introduction
The isolation of graphene in 2004 established a new paradigm in materials science, facilitating the broad investigation of atomically thin materials [1,2,3,4]. Within the search for alternative 2D systems, black phosphorus has attracted significant attention, particularly in its monolayer allotrope, phosphorene [5]. In contrast to semi-metallic graphene, phosphorene (also called black phosphorene) exhibits a direct and tunable band gap, a property that makes it suitable for optoelectronic applications, such as field-effect transistors (FETs) with substantial on/off ratios [6,7,8]. Furthermore, its puckered honeycomb lattice results in strong anisotropy in relation to its electronic, optical, and thermal properties, a characteristic that has motivated significant research [9,10,11,12,13]. Recent studies have examined the utility of functional phosphorene in diverse areas, including supercapacitors and nanotherapeutic platforms [14,15,16].
Despite its remarkable potential, the practical implementation of phosphorene is constrained by its inherent instability at ambient conditions. The presence of lone-pair of electrons on the phosphorus atoms heightens chemical reactivity, precipitating degradation upon exposure to oxygen and moisture [17]. However, this intrinsic reactivity invites a paradigm shift towards "defect engineering." Far from being regarded as merely structural aberrations, atomic vacancies are increasingly recognized as mechanisms to precisely modulate material properties at the atomic scale [18,19]. Theoretical frameworks suggest that native vacancies fundamentally reshape the electronic landscape, introducing localized in-gap states and inducing magnetism in the otherwise non-magnetic lattice [20,21,22]. Notably, single and double vacancies stabilize in anionic (-1) charge states, altering local geometries; while these defects may lower the energy barrier for oxidative degradation via dissociation [23], they concurrently offer significant tunability. For example, vacancy-induced magnetic moments can be manipulated by doping with elements such as N, Fe, or Co [24], while the emergence of quasi-flat states near the Fermi level significantly influences optical responses [25].
This ability to tailor the surface reactivity is particularly pertinent to gas-sensing applications. Pristine phosphorene is generally limited by weak physisorption interactions with common analytes, resulting in suboptimal sensitivity [26]. Defect engineering addresses this limitation by introducing active sites—such as vacancies or heteroatom substitutions—that enhance the adsorption strength. While mono- and di-vacancy defects significantly augment gas adsorption, it is noted that the resulting electronic perturbations must be carefully managed to ensure sufficient sensitivity [27]. As it turns out, when optimized, defective phosphorene not only improves adsorption kinetics but also preserves the material’s stability and reusability, solidifying its promise for sensing platforms [28].
First-principles calculations have long predicted that phosphorene could surpass other 2D materials in sensing performance 00[29,30]. The underlying hypothesis posits that structural defects function as reactive centers, facilitating a transition from physisorption to chemisorption characterized by substantial charge transfer. This mechanism is well-documented in analogous systems; for example, vacancies in graphene markedly enhance sensitivity to [31], while specific defect configurations in Tin Monosulfide (SnS) enable tunable selectivity [32]. Similarly, single vacancies in blue phosphorene have been shown to drastically strengthen interactions with pollutant molecules [33].
Despite these advances, a systematic understanding of how specific vacancy configurations in phosphorene govern interactions with small, environmentally relevant molecules remains elusive. Critical questions persist regarding the comparative efficacy of single vacancies (SV) versus double vacancies (DV), and how distinct defect geometries influence the adsorption mechanisms for simple diatomic molecules. Addressing this lack of fundamental comparative insight is essential for overcoming current bottlenecks and designing phosphorene-based sensors with tailored selectivity.
This is precisely the void that the present study aims to fill. Using the SIESTA package, which is based on Density Functional Theory [34], we rigorously investigate the adsorption of key molecules such as , , , and [35]. Our focus is on three surface types: phosphorene with a single vacancy (SV) and two distinct double vacancy (DV) configurations, consistent with the comparison of the results against the pristine phosphorene case. To ensure the precision of the adsorption calculations, the van der Waals interaction is explicitly included in the exchange-correlation functional [36]. The ultimate goal is to provide a detailed understanding of how structural defects govern adsorption, providing a critical piece of the puzzle to unleash the multifaceted potential of phosphorene in chemical sensing.
2. Methodology
Our study employs first-principles calculations within the framework of Density Functional Theory (DFT), using the SIESTA code [34] for all simulations, while the Atomic Simulation Environment (ASE) library [37] was used for workflow management and structural setup. To accurately describe exchange-correlation interactions, particularly the dispersive forces relevant to adsorption, we included the KBM van der Waals functional [36]. The core electrons of the phosphorus atoms were described using norm-conserving pseudopotentials optimized for the KBM functional. Valence states were expanded using a linear combination of numerical atomic orbitals with a double-zeta polarized (DZP) basis set. The spatial precision was defined by a basis-set confinement energy shift of 0.1 eV and a real-space grid cutoff of 250 Ry. Structural relaxations were performed using the conjugate gradient method until the maximum force component on any atom was less than . For the Brillouin zone integration, a Monkhorst-Pack grid was employed for the unit cell, while a sampling (-point) was used for supercell partition. Additionally, spin-polarized calculations were enabled to evaluate the total magnetization in cases where adsorption induces unpaired magnetic moments in the molecule-phosphorene complex.
To model the adsorption surfaces, we constructed a rectangular phosphorene supercell. Based on this geometry, four distinct systems were generated to assess the impact of structural defects, as schematically shown in Figure 1. These systems include the pristine non-defective monolayer, which serves as a baseline, and three defective configurations: the Single Vacancy (SV), formed by removing a single phosphorus atom; the Divacancy Type 1 (DV1), created by eliminating two neighboring phosphorus atoms located in different (top and bottom) sub-layers; and the Divacancy Type 2 (DV2), generated by removing two adjacent atoms from the same puckered sub-layer. After creating these vacancies, the geometries were fully relaxed to allow local atomic reconstruction around the defect sites.
To quantify the stability of the interaction between molecules and the surface, the adsorption energy () is calculated using the following expression.
where corresponds to the total energy of the layer-adsorbed gas complex, is the energy of the isolated substrate (pristine or defective), and is the energy of the isolated gas phase molecule. To identify the most favorable binding configurations, we explored several potential adsorption sites as illustrated in Figure 2. For the pristine monolayer, high-symmetry sites were considered: the top site (directly above a P atom), the bridge site (midway between two bonded P atoms), and the hollow site (at the center of a phosphorene ring). For the defective surfaces, additional adsorption sites near the low-coordination atoms were thoroughly investigated to account for the unique geometries introduced by the vacancies.
3. Result and Discussion
We first examine the electronic structure of both pristine and defective phosphorene monolayers, with the calculated band structures illustrated in Figure 3. The pristine monolayer (NV) exhibits a direct band gap of ≈ 0.9 eV at the point, a result consistent with previous first-principles studies [21]. The introduction of a single vacancy (SV) breaks the lattice symmetry, leading to the emergence of a highly localized, nearly dispersionless, defect state within the forbidden region. This state is pinned at the Fermi level (0 eV) and lies approximately 0.2 eV above the valence-band maximum. Such states are expected to act as chemically active sites, facilitating molecular adsorption. A similar behavior is observed in the DV-1 configuration, mainly affecting the conduction-band interval; however, a band gap comparable to that of the pristine monolayer is preserved. In contrast, the DV-2 defect retains the semiconducting character but results in a significantly narrowed direct band gap of ≈ 0.3 eV. These findings align well with previous theoretical reports on defective phosphorene [20]. In all defective cases, a significant increase in the number of valence or conduction band states within the same energy interval is observed. These distinct electronic signatures demonstrate that specific defect topologies and coordination environments modulate the material’s electronic landscape, a critical factor that governs subsequent interactions with gas molecules.
We started by studying the adsorption of , , CO, and NO on the pristine phosphorene monolayer to establish a baseline for comparison. For the polar molecules CO and NO, we considered two distinct orientations: with the oxygen atom pointing towards the surface (v1) and with the carbon/nitrogen atom pointing towards the surface (v2). The calculated adsorption energies () and molecular bond lengths are summarized in Table 1.
In general, all configurations exhibit characteristic traits of weak physisorption. The strongest attractive interaction is observed for NO at the top site, yielding a of only meV. For NO, molecular orientation plays a noticeable role; specifically, the configuration with the oxygen atom closer to the surface (NO-v1) is energetically more favorable. In stark contrast, the interaction with the CO molecule is found to be repulsive (positive ) or negligible for both v1 and v2 orientations, indicating that pristine phosphorene has a negligible affinity for carbon monoxide regardless of its orientation. Regarding the molecule, the adsorption energies display typical small values reported for other 2D nanomaterials, such as defective graphene doped with B/N, which exhibits adsorption energies ranging from to eV depending on molecular orientation [13]. This weak-interaction nature is further corroborated by analysis of intramolecular bond lengths. For all adsorbed molecules, these bond lengths remain largely unperturbed, showing minimal variation across different adsorption sites and orientations. Collectively, these findings confirm that the interactions are governed by van der Waals forces with negligible charge transfer, establishing the pristine phosphorene monolayer as an ineffective substrate for the sensing of these small gas molecules.
To enhance the adsorption properties of phosphorene, we systematically evaluated the interaction of gas molecules within the vicinity of the three defective structures illustrated in Figure 1, considering all adsorption positions detailed in Figure 2. Figure 4 summarizes the maximal absolute adsorption energy () calculated for each molecule in different structural configurations.
For homonuclear diatomic molecules, and , the adsorption energetics are largely insensitive to surface defects. As shown in Figure 4, the maximal adsorption energy for remains comparable between the pristine and defective monolayers, peaking slightly at eV in the Single Vacancy (SV) system (Hollow-1 site). A similar trend is observed for , which exhibits a consistent adsorption energy hovering around eV regardless of the type of defect. These low energy values indicate a physisorption mechanism dominated by weak van der Waals interactions, suggesting that neither pristine nor defective phosphorene significantly retains these gases.
In contrast, the adsorption behavior of the heteronuclear molecules, CO and NO, is strongly influenced by the surface topology. Structural defects significantly enhance the interaction strength for these species. For CO, the maximal adsorption energy increases from eV on the pristine surface to eV in the presence of a Single Vacancy (SV, Top-2 site). The effect is most pronounced for NO, where the introduction of defects triggers a dramatic transition from weak physisorption to strong chemisorption. Although NO is weakly adsorbed on the pristine surface ( eV), the binding energy increases to eV for the SV system (Top-1 site) and reaches a maximum of eV for the Divacancy Type 2 (DV-2) system (Hollow-1 site). A comprehensive analysis of the adsorption energies, including values for all considered positions, is provided in the Supplementary Material.
The analysis suggests that while most gas molecules (, , and CO) undergo physisorption with relatively low binding energies, the NO molecule is a notable exception. The substantial energies observed for NO on defective surfaces (exceeding eV) imply the formation of strong chemical bonds. This increased adsorption probability is likely correlated with significant structural distortions in the adsorbate, such as bond elongation, particularly at the highly active sites identified in the SV and DV-2 systems. Consequently, based on these calculations, defective phosphorene monolayers —especially those containing Single Vacancies or Type-2 Divacancies— appear to be highly suitable candidates for the capture or detection of gaseous nitric oxide.
To gain a deeper understanding of the electronic consequences of adsorption, we calculated the Total Density of States (TDOS) for the previously identified most stable configurations. Figure 5 presents the DOS for the molecule-phosphorene complexes. The shadowed gray regions at the bottom of each panel represent the reference DOS of the isolated phosphorene substrates (pristine or defective) prior to adsorption.
Upon gas adsorption, the electronic structure undergoes discernible modifications. For homonuclear molecules ( and ), the interaction remains largely non-invasive regarding the metallicity of the system. As shown in the upper panels of Figure 5, the presence of induces a rigid shift of the energy states; however, the band gap is preserved across all defect types (NV, SV, DV-1, and DV-2), indicating that the system retains its semiconductor character with the Fermi level () located within the gap. A comparable behavior is observed for , where the DOS profiles show that the semiconducting nature of the phosphorene monolayer is largely unaffected by the physical adsorption of nitrogen.
In heteronuclear molecules, the electronic response is more complex. For CO adsorption, the system generally maintains a semiconductor band gap, similar to the pristine case. However, significant changes occur with NO adsorption. The NO molecule introduces states within the band gap, and, notably, induces a symmetry breaking in the electronic states. As evidenced in the bottom-right panel of Figure 5, the DOS for NO becomes spin-polarized (asymmetry between solid and dashed lines), particularly for defective substrates. This interaction leads to the emergence of impurity states near the Fermi level, effectively doping the material.
This spin polarization is quantified in Table 2. Although adsorption of , , and CO results in non-magnetic ground states () across all structural defects, the NO molecule induces a significant magnetic moment. This confirms that the distinct split observed in the NO DOS plots corresponds to a net magnetization arising from the unpaired electrons of the nitric oxide molecule interacting with the localized defect states of the phosphorene lattice.
The optical properties of the phosphorene-molecule complexes were analyzed by calculating the imaginary part of the dielectric function , which is directly related to the optical absorption spectra. This analysis provides a spectroscopic signature that complements the discussion of electronic structure provided above. Figure 6 displays the absorption spectra for both X (armchair) and Y (zigzag) light polarizations, summing over spin components to capture the total optical response.
Consistent with the intrinsic anisotropy of black phosphorene, all systems exhibit a marked polarization-dependent behavior. The absorption profiles along the X-direction generally show broad absorption bands starting in the visible range, while the Y-direction is characterized by sharper features and distinct low-energy transitions.
For the physisorbed molecules (, ) and CO, the optical spectra largely mirror the behavior of the host phosphorene lattice. As observed in Figure 6, the absorption edges for these species are located above eV. This is in excellent agreement with the DOS analysis, where these systems were identified as semiconductors with preserved band gaps. The lack of optical absorption in the infrared region ( eV) confirms that the adsorption of these gases does not introduce new intragap states capable of mediating low-energy excitations. Consequently, the optical transparency of phosphorene in the low-energy regime remains intact upon exposure to , , and CO.
A strikingly different behavior is observed for the NO molecule. Correlating with the DOS results, where NO adsorption induced spin-polarized impurity states within the band gap and near the Fermi level, the optical spectra exhibit distinct new absorption peaks in the low-energy range ( eV). These features are particularly prominent in the Y-polarization for the defective systems (SV, DV-1, and DV-2), appearing as sharp peaks or "pre-edges" below the main absorption band. These transitions correspond to electronic excitations involving the localized mid-gap states generated by the strong chemical interaction between nitric oxide and the defective phosphorene surface. The emergence of these unique optical signatures in the infrared region suggests that optical spectroscopy could be an effective method to selectively detect NO, distinguishing it from other common gases.
4. Conclusions
We have performed DFT calculations to investigate the features of small gas-molecule adsorption on black phosphorene. In particular, we have assumed that the adsorbent surfaces can be defect-laden, with single and double atomic vacancies in the two-dimensional lattice of otherwise pristine monolayers. Given the structure’s geometry, several adsorption sites can be identified, which are more numerous when double vacancies are present. In all cases, the adsorption energy was evaluated for each of the gases considered: , , CO, and NO. It has been found that adsorption energies tend to be small (only a few tens of meV) for three of the species included in the study. The only one that exhibits rather significant values of the adsorption energy is NO, and this happens only in the case of a defective adsorbent surface with single phosphorus atom vacancies. Here, the obtained values suggest that this particular defective substrate is suitable for practical nitric oxide capture. In addition, magnetic response due to spin polarization is predicted for NO adsorption on double-vacancy-laden black phosphorene monolayers.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
The contributions of the authors are as follows: A.S.G.-N.: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Data curation, Writing—Review and Editing, Visualization. J.D.C.: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Data curation, Writing—Review and Editing, Visualization. M.E.M-R.: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Data curation, Writing—Review and Editing, Visualization. A.L.M.: Conceptualization, Formal Analysis, Writing—Review and Editing, Visualization. C.A.D.: Conceptualization, Formal Analysis, Writing— Review and Editing, Visualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
ASGN and CAD thank the University of Antioquia for support through the Young Researchers Program fellowship. JDC and MEMR are grateful to Mexican SECIHTI for support through Grant CBF-2025-I-1058.
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Tang, S.; Cao, Z. Adsorption and dissociation of ammonia on graphene oxides: A first-principles study. J. Phys. Chem. C 2012, 116, 8778–8791. [Google Scholar] [CrossRef]
- Mattson, E.C.; Pande, K.; Unger, M.; Cui, S.; Lu, G.; Gajdardziska-Josifovska, M.; Weinert, M.; Chen, J.; Hirschmugl, C.J. Exploring adsorption and reactivity of NH3 on reduced graphene oxide. J. Phys. Chem. C 2013, 117, 10698–10707. [Google Scholar] [CrossRef]
- Zhu, S.; Sun, H.; Liu, X.; Zhuang, J.; Zhao, L. Room-temperature NH3 sensing of graphene oxide film and its enhanced response on the laser-textured silicon. Sci. Rep. 2017, 7, 14773. [Google Scholar] [CrossRef] [PubMed]
- Batmunkh, M.; Bat-Erdene, M.; Shapter, J.G. Phosphorene and phosphorene-based materials–prospects for future applications. Adv. Mater. 2016, 28, 8586–8617. [Google Scholar] [CrossRef]
- Li, L.; Yu, Y.; Ye, G.J.; Ge, Q.; Ou, X.; Wu, H.; Feng, D.; Chen, X.H.; Zhang, Y. Black phosphorus field-effect transistors. Nat. Nanotechnol. 2014, 9, 372–377. [Google Scholar] [CrossRef]
- Mohammadi, M.; Tavangar, Z. Adsorption of aromatic molecules on a black phosphorene surface: A first-principles study. New J. Chem. 2023, 47, 1842–1851. [Google Scholar] [CrossRef]
- Talukdar, D.; Mohanta, D.; Ahmed, G.A. Nitrogen doped compound defect in black phosphorene for enhanced gas sensing. Surf. Interfaces 2024, 51, 104699. [Google Scholar] [CrossRef]
- Tariq, M.; Khattak, J.I.; Iqbal, M.; Ullah, R.; Zeb, A.; Khan, M.; Khan, A.; Mahmood, T.; Ahmad, I. DFT study of the therapeutic potential of phosphorene as a drug delivery system for chlorambucil to treat cancer. RSC Adv. 2019, 9, 24325–24332. [Google Scholar] [CrossRef]
- Pica, M.; D’Amato, R. Chemistry of Phosphorene: Synthesis, Functionalization and Biomedical Applications in an Update Review. Inorganics 2020, 8, 29. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, M.; Kumar, M. Recent advances in phosphorene-based gas sensors. Sens. Actuators A Phys. 2019, 295, 523–537. [Google Scholar]
- de Sousa, F.E.B. Electronic, Transport and Optical Properties in Multilayer Phosphorene. Ph.D. Thesis, Federal University of Ceará, Brazil, 2023. [Google Scholar]
- Akilan, R.; Vinnarasi, S.; Mohanapriya, S.; Shankar, R. Adsorption of H2 molecules on B/N-doped defected graphene sheets—A DFT study. Struct. Chem. 2020, 31, 2413–2434. [Google Scholar] [CrossRef]
- Halder, P.; Mondal, I.; Kundu, M.; Ghosh, A.; Paul, B.K.; Biswas, S.; Sau, S.; Chattopadhyay, B.; Mondal, D.; Das, S. Optimizing phosphorene nanosheets for high performance all-solid asymmetric supercapacitors: A theoretical and experimental insight. J. Energy Storage 2024, 94, 112451. [Google Scholar] [CrossRef]
- Cui, X.; Tang, X.; Niu, Y.; Tong, L.; Zhao, H.; Yang, Y.; Jin, G.; Li, M.; Han, X. Functional phosphorene: Burgeoning generation, two-dimensional nanotherapeutic platform for oncotherapy. Coord. Chem. Rev. 2024, 507, 215744. [Google Scholar] [CrossRef]
- Zhu, Y.; Xie, Z.; Li, J.; Liu, Y.; Li, C.; Liang, W.; Huang, W.; Kang, J.; Cheng, F.; Kang, L.; Al-Hartomy, O.A.; Al-Ghamdi, A.; Wageh, S.; Xu, J.; Li, D.; Zhang, H. From phosphorus to phosphorene: Applications in disease theranostics. Coord. Chem. Rev. 2021, 446, 214110. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, G.; Wang, W.; Wang, Y.; Liu, F.; Wu, M. Black phosphorus: Synthesis, stability, and applications. iScience 2021, 24, 103116. [Google Scholar] [CrossRef] [PubMed]
- Kaewmaraya, T.; Singh, D.; Kumar, A.; Park, S.; Lee, H. Drastic Improvement in Gas-Sensing Characteristics of Phosphorene Nanosheets under Vacancy Defects and Elemental Functionalization. J. Phys. Chem. C 2018, 122, 19864–19874. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhou, M.; Zeng, M.; Fu, L. Atomic-scale structural modification of 2D materials. Adv. Sci. 2019, 6, 1801501. [Google Scholar] [CrossRef]
- Hu, W.; Yang, J. Defects in phosphorene. J. Phys. Chem. C 2015, 119, 20474–20480. [Google Scholar] [CrossRef]
- Pantha, N.; Chauhan, B.; Sharma, P.; Adhikari, N.P. Tuning structural and electronic properties of phosphorene with vacancies. J. Nepal Phys. Soc. 2020, 6, 7–15. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, S.; Gao, J.; Zhang, G.; Zhang, Y.W. Evolution of intrinsic vacancies and prolonged lifetimes of vacancy clusters in black phosphorene. Nanoscale 2019, 11, 20987–20995. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Xu, W.; Zou, R.; Yang, J.; Fan, J.; Wu, X.; Wang, R. Interplay of charged states and oxygen dissociation induced by vacancies in phosphorene. J. Phys. Chem. C 2019, 123, 27080–27087. [Google Scholar] [CrossRef]
- Srivastava, P.; Hembram, K.P.S.S.; Mizuseki, H.; Lee, K.-R.; Han, S.S.; Kim, S. Tuning the electronic and magnetic properties of phosphorene by vacancies and adatoms. J. Phys. Chem. C 2015, 119, 6530–6538. [Google Scholar] [CrossRef]
- de Sousa, F.E.B.; Araújo, F.R.V.; Farias, G.A.; de Sousa, J.S.; da Costa, D.R. Effects on the electronic properties of multilayer phosphorene due to periodic arrays of vacancies: Band unfolding formalism. Physica E 2023, 152, 115750. [Google Scholar] [CrossRef]
- Li, J.-x.; Zhang, Q.; Ma, D.-w.; Liu, C.-k.; Li, F. Adsorption properties of CO2, NO2 and SO2 on pristine/defective/non-metallic element doping phosphorene by DFT method. SSRN Electron. J. 2024. [Google Scholar]
- Singsen, S.; Watwiangkham, A.; Ngamwongwan, L.; Fongkaew, I.; Jungthawan, S.; Suthirakun, S. Defect Engineering of Green Phosphorene Nanosheets for Detecting Volatile Organic Compounds: A Computational Approach. ACS Appl. Nano Mater. 2023, 6, 1496–1506. [Google Scholar] [CrossRef]
- Ghadiri, M.; Ghashghaee, M.; Ghambarian, M. Defective phosphorene for highly efficient formaldehyde detection: Periodic density functional calculations. Phys. Lett. A 2020, 384, 126792. [Google Scholar] [CrossRef]
- Kou, L.; Frauenheim, T.; Chen, C. Phosphorene as a superior gas sensor: Selective adsorption and distinct I-V response. J. Phys. Chem. Lett. 2014, 5, 2675–2681. [Google Scholar] [CrossRef] [PubMed]
- Donarelli, M.; Ottaviano, L. 2D materials for gas sensing applications: A review on graphene oxide, MoS2, WS2 and phosphorene. Sensors 2018, 18, 3638. [Google Scholar] [CrossRef]
- Khudair, S.A.M.; Mohaimeed, A.A. Gas Sensor Investigations through Adsorption of Toxic Gas Molecules on Single and Double Vacancy Graphene. NeuroQuantology 2020, 18, 87–95. [Google Scholar] [CrossRef]
- Qin, Y.; Cao, M.; Lei, M.; Feng, W. First-principles study on the selective gas adsorption of defective SnS. Vacuum 2021, 183, 109792. [Google Scholar] [CrossRef]
- Corona-García, C.A.; Lucas, H.R.; Ramírez-Pimentel, J.G.; Cruz-Martínez, H.; Cocoletzi, G.H. Adsorption of small pollutant molecules on monolayer aluminum-doped and single-vacancy blue phosphorene. J. Mol. Model. 2021, 27, 141. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Zuluaga-Hernandez, E.A.; Flórez, E.; Dorkis, L.; Mora-Ramos, M.E.; Correa, J.D. Small molecule gas adsorption onto blue phosphorene oxide layers. Appl. Surf. Sci. 2020, 530, 147039. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 2009, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the four phosphorene supercells studied. From left to right: the pristine (non-defective) Mono Layer; the Single Vacancy (SV) system, created by removing one P atom; the Divacancy Type 1 (DV-1), formed by removing two adjacent atoms from different sub-layers; and the Divacancy Type 2 (DV-2), formed by removing two adjacent atoms from the same sub-layer.
Figure 1.
Schematic representation of the four phosphorene supercells studied. From left to right: the pristine (non-defective) Mono Layer; the Single Vacancy (SV) system, created by removing one P atom; the Divacancy Type 1 (DV-1), formed by removing two adjacent atoms from different sub-layers; and the Divacancy Type 2 (DV-2), formed by removing two adjacent atoms from the same sub-layer.
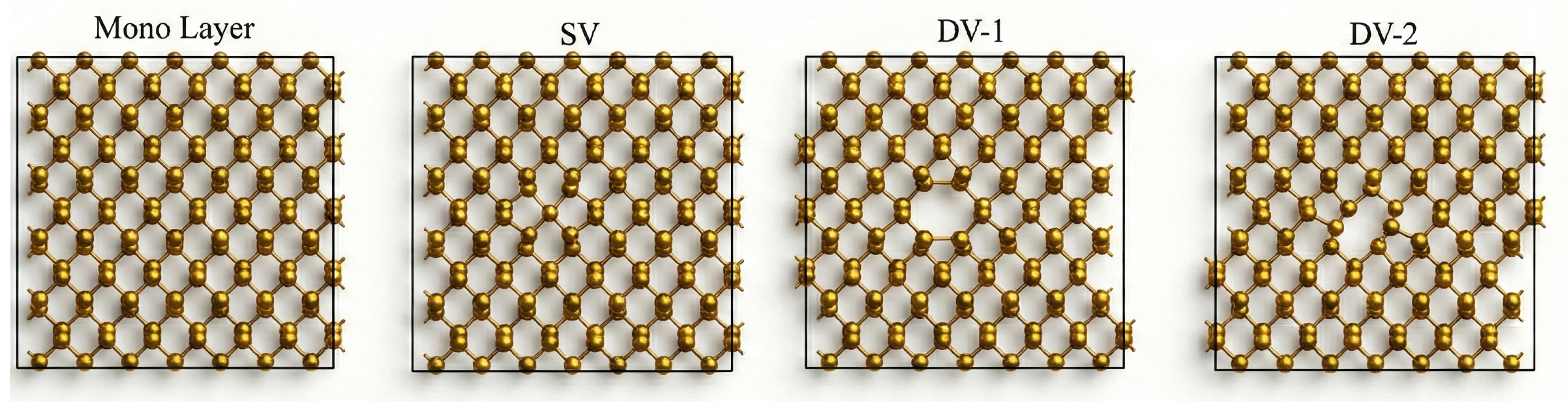
Figure 2.
Top-down view of the adsorption sites investigated for small gas molecules on each phosphorene system. For the pristine Mono Layer, standard top, bridge, and hollow sites were considered. For the defective systems SV, DV-1, and DV-2), additional unique sites were explored in the immediate vicinity of the vacancy to account for the locally altered chemical environment.
Figure 2.
Top-down view of the adsorption sites investigated for small gas molecules on each phosphorene system. For the pristine Mono Layer, standard top, bridge, and hollow sites were considered. For the defective systems SV, DV-1, and DV-2), additional unique sites were explored in the immediate vicinity of the vacancy to account for the locally altered chemical environment.
Figure 3.
Band structures for pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2) phosphorene monolayers.
Figure 3.
Band structures for pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2) phosphorene monolayers.
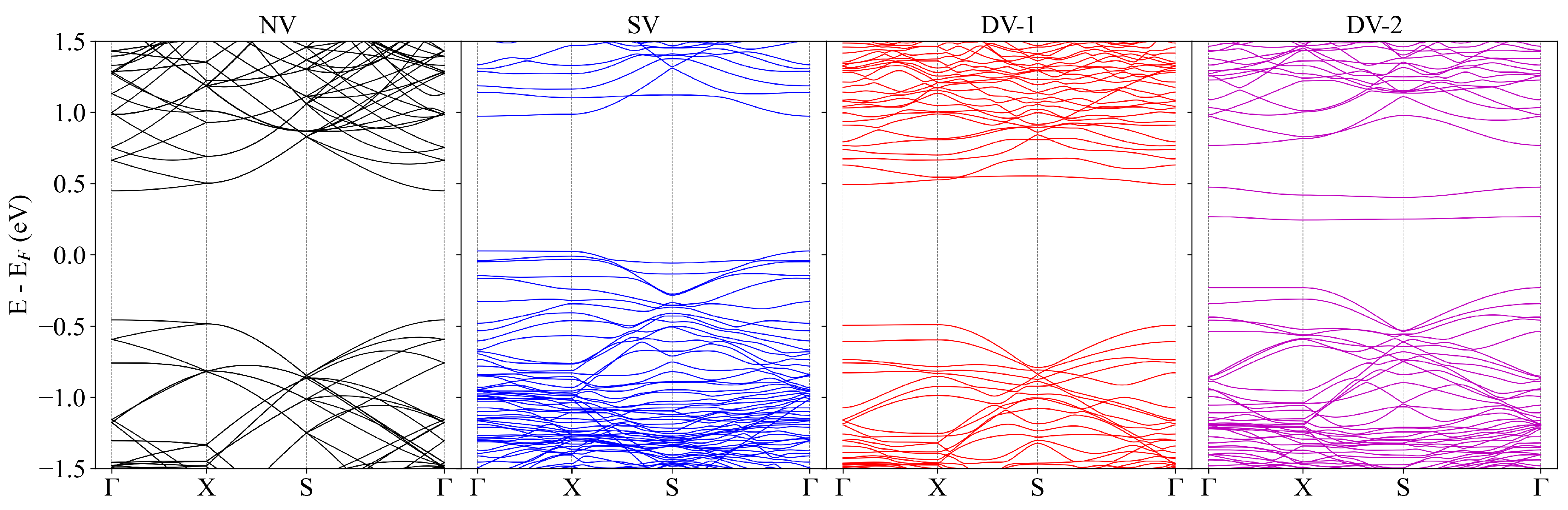
Figure 4.
Absolute adsorption energy ( in eV) for , , CO, and NO molecules on Pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2) black phosphorene monolayers. The height of the bars represents the absolute magnitude, while the labels above indicate the exact calculated adsorption energy values and the corresponding most stable adsorption site.
Figure 4.
Absolute adsorption energy ( in eV) for , , CO, and NO molecules on Pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2) black phosphorene monolayers. The height of the bars represents the absolute magnitude, while the labels above indicate the exact calculated adsorption energy values and the corresponding most stable adsorption site.
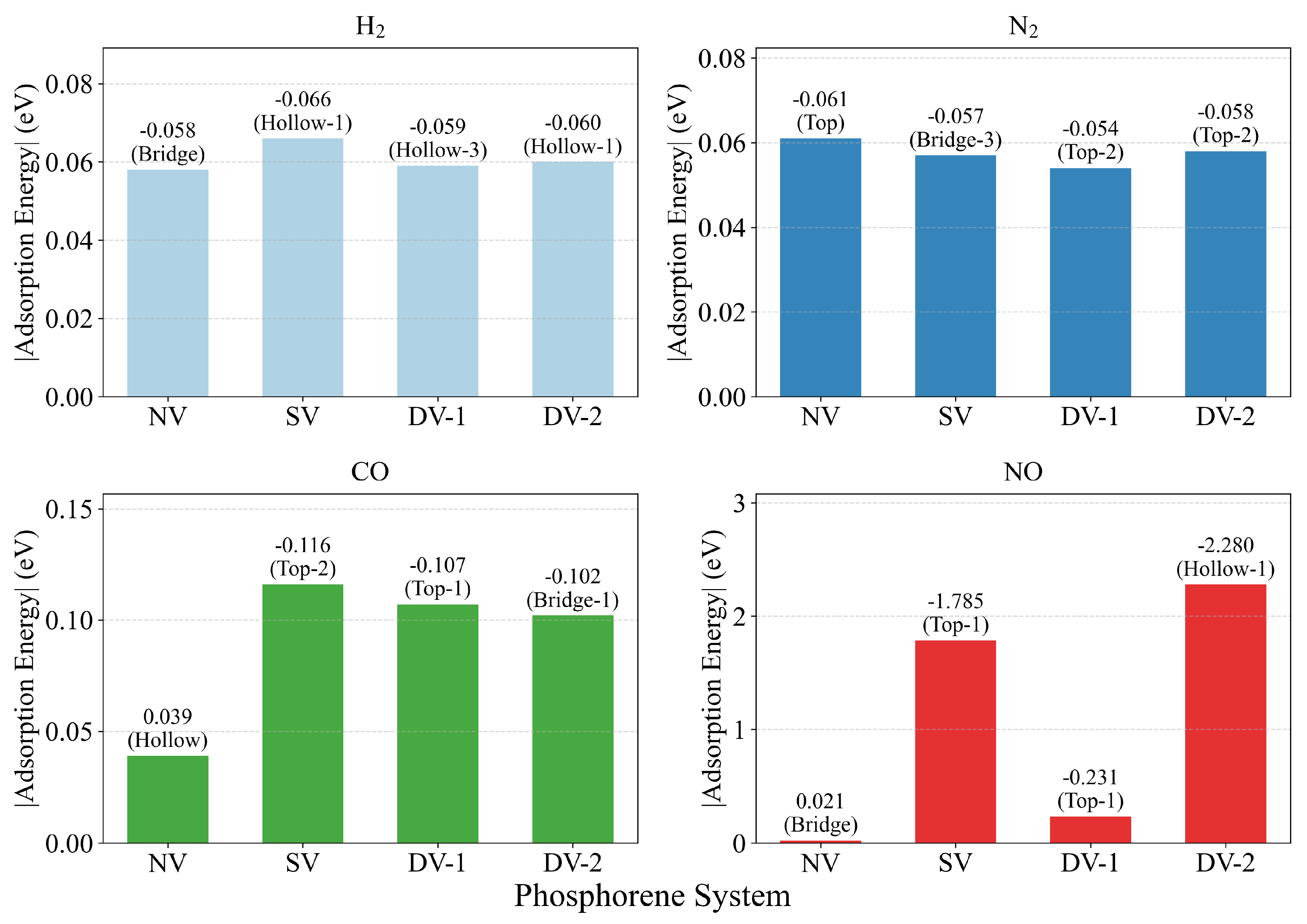
Figure 5.
Calculated density of states (DOS) for all considered black phosphorene monolayer/adsorbed molecule complexes. The solid lines represent the total DOS (or Spin Up for magnetic systems), while the dash-dotted lines represent the Spin Down component, and the dashed lines represent the reference DOS for the clean substrates.
Figure 5.
Calculated density of states (DOS) for all considered black phosphorene monolayer/adsorbed molecule complexes. The solid lines represent the total DOS (or Spin Up for magnetic systems), while the dash-dotted lines represent the Spin Down component, and the dashed lines represent the reference DOS for the clean substrates.
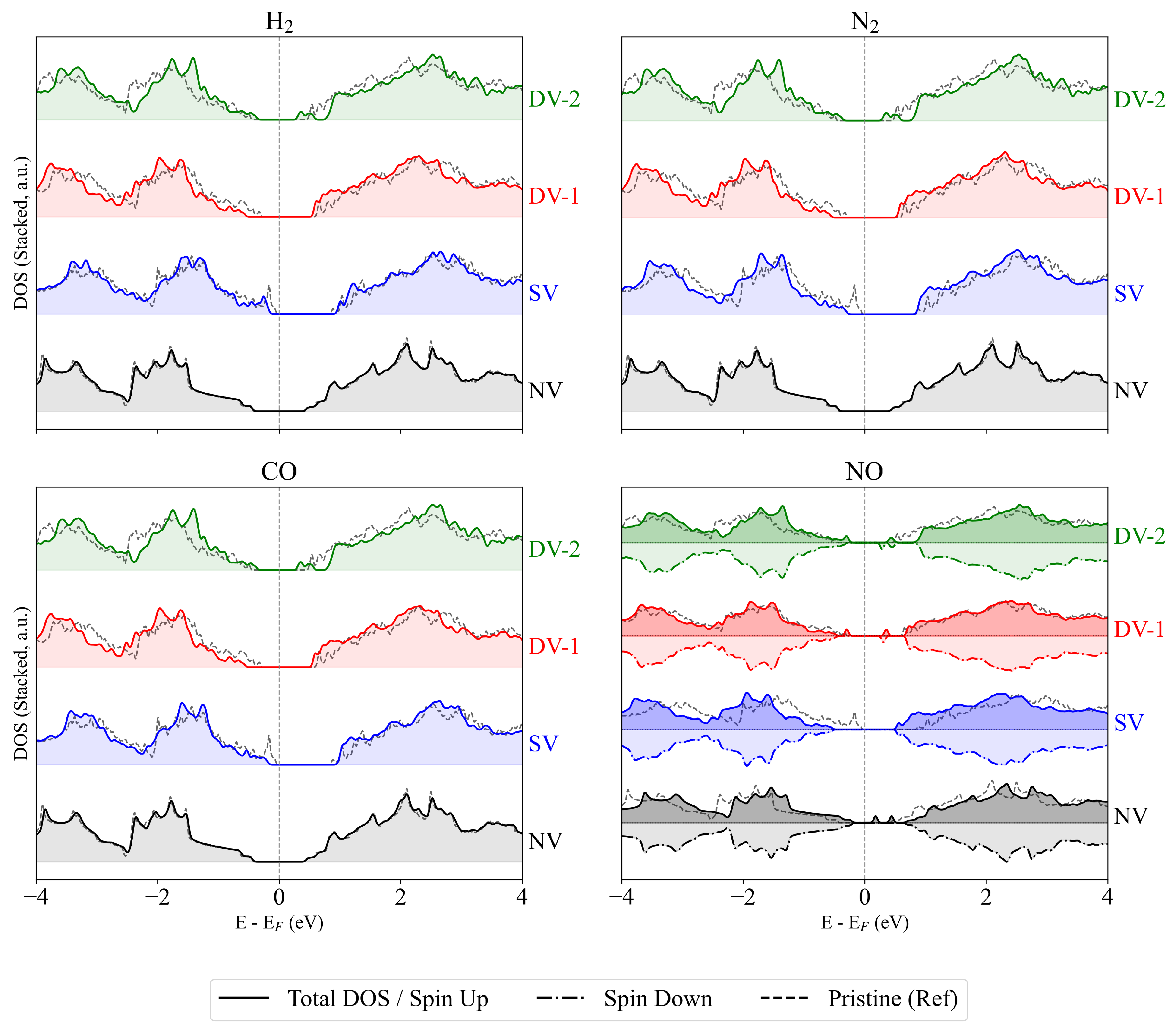
Figure 6.
Calculated optical absorption spectra for , , CO, and NO molecules adsorbed on Pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2). The left and right panels correspond to light polarized along the X-direction (armchair) and Y-direction (zigzag), respectively.
Figure 6.
Calculated optical absorption spectra for , , CO, and NO molecules adsorbed on Pristine (NV), Single Vacancy (SV), Divacancy Type 1 (DV-1), and Divacancy Type 2 (DV-2). The left and right panels correspond to light polarized along the X-direction (armchair) and Y-direction (zigzag), respectively.
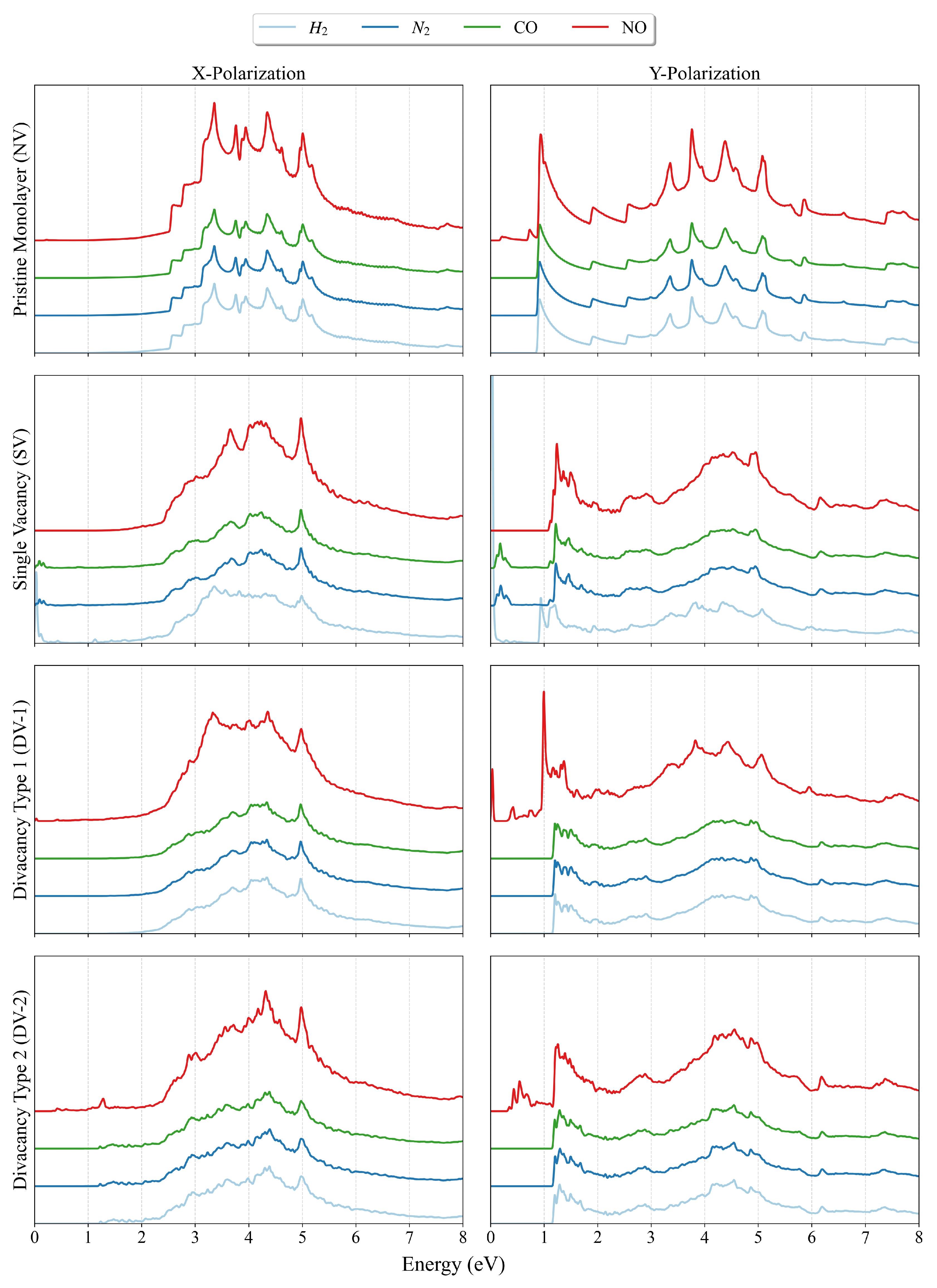

Table 1.
Adsorption energies ( in eV) and bond lengths (d in Å) for various molecules on pristine black phosphorene.
Table 1.
Adsorption energies ( in eV) and bond lengths (d in Å) for various molecules on pristine black phosphorene.
| Molecule | Hollow | Bridge | Top | |||
|---|---|---|---|---|---|---|
| d | d | d | ||||
| -0.057 | 0.778 | -0.058 | 0.778 | -0.048 | 0.778 | |
| -0.031 | 1.118 | -0.042 | 1.119 | -0.060 | 1.119 | |
| CO-v1 | -0.009 | 1.151 | 0.021 | 1.151 | 0.019 | 1.152 |
| NO-v1 | -0.391 | 1.178 | -0.383 | 1.179 | -0.341 | 1.179 |
| CO-v2 | 0.039 | 1.151 | 0.016 | 1.151 | 0.007 | 1.152 |
| NO-v2 | -0.396 | 1.182 | -0.374 | 1.183 | -0.396 | 1.185 |
Table 2.
Total magnetic moment () for molecule adsorption at the most stable sites on defective phosphorene monolayers.
Table 2.
Total magnetic moment () for molecule adsorption at the most stable sites on defective phosphorene monolayers.
| Defect Type | CO | NO | ||
|---|---|---|---|---|
| SV | 0.0 | 0.0 | 0.0 | 1.000 |
| DV-1 | 0.0 | 0.0 | 0.0 | 1.000 |
| DV-2 | 0.0 | 0.0 | 0.0 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



