Submitted:
03 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Through comprehensive QSAR analyses and molecular docking studies on COX-1 and COX-2, we successfully identified the pharmacophoric structure and essential moieties that account for the recognition of non-selective COX NSAIDs, such as aro-matic-carboxylate moiety anchors and guides the binding with the guanidinium group of arginine residue, in contrast, the accessory moieties modulate the affinity and blocking of the oxidation of substrate via free radicals. Therefore, the carboxylate group and the aromatic ring are not only important for docking and recognition by COXs, but the cap-todative effect between the carboxylate and the electron-donating group at position 2 is relevant for the stabilization of free radicals, mainly in salicylates and fenamates. Finally, the medical significance of these drugs is substantial and diverse, affecting various acute and chronic inflammatory and pain conditions, with their activity primarily depending on COX-1 and COX-2 inhibition. This research established the physicochemical, molec-ular, and intermolecular interaction basis of non-selective NSAIDs that determine their recognition by these enzymes, thereby providing a molecular foundation for the design of COX inhibitor drugs.
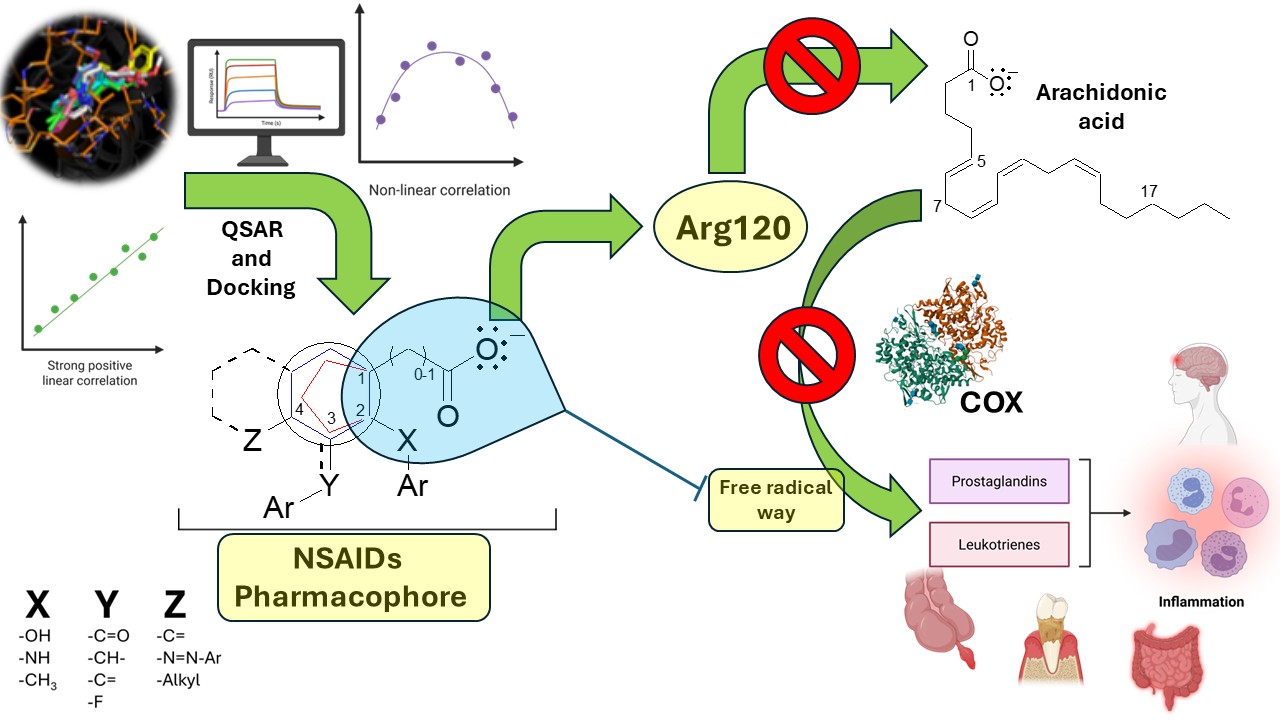
Keywords:
NSAIDs
; QSAR
; captodative effect
; free radicals
; molecular docking
; pharmacophore
1. Introduction
The inflammatory process goes through distinct phases, and the activation of phospholipases is essential for generating mediators for the free radical pathway, such as prostaglandins, lipoxins, thromboxanes, and leukotrienes, including platelet-activating factor (PAF). [1,2]
In contemporary medical practice, Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) represent a fundamental pillar in the treatment of various painful and inflammatory conditions. Since their discovery and subsequent commercialization in the mid-20th century, these drugs have revolutionized the management of chronic and acute pain, as well as inflammatory diseases such as rheumatoid arthritis and osteoarthritis. However, its widespread use is not exempt from critical scrutiny due to its potential adverse effects, particularly at the gastrointestinal and cardiovascular levels, which have motivated a constant search for safer, more effective formulations. [3,4]
The chemical structure of NSAIDs plays a crucial role in their pharmacological activity and selectivity. These compounds usually consist of a central nucleus, frequently an aromatic ring, to which a carboxylic acid or an enol is attached. This basic structure facilitates binding to specific active sites on COX, allowing interference with enzymatic activity, and provides it with physicochemical and molecular properties shared by COX. Over the decades, researchers have explored various modifications to this structure to improve therapeutic efficacy and reduce side effects. [5,6]
Structure-Activity Relationship (SAR) studies have been instrumental in the present contribution, which describes the development and optimization of NSAIDs. These studies aim to identify how subtle modifications in molecular structure can influence the potency, actions, and pharmacokinetics of these drugs. [7,8,9,10,11] For example, it has been shown that the addition of substituent groups at specific positions on the aromatic ring can improve the slight tendency toward COX-2 without significantly affecting COX-1. Therefore, in this work, was identified by molecular docking and SAR/QSAR analyses the necessary structural moieties for the recognition and inhibitory activity on both COXs, were for the recognition being the aryl-carboxylate with captodative properties (primarily shown in salycilates and fenamates) the guide and accessory moieties, which led to establishment the pharmacophore structure for the COX non-selective NSAIDs.
Therefore, it is important to highlight that the present study identified through SAR/QSAR analysis and molecular docking the structural portions necessary for recognition and inhibitory activity on both COX, highlighting a carboxylate on an aryl group with captodative properties (mainly evident in salicylates and fenamates), which led to the establishment of the pharmacophore for non-selective COX NSAIDs.
2. Results and Discussion
2.1. Fenamates and Salicylates
SAR/QSAR analysis is fundamentally valuable for elucidating in greater detail the mechanisms of action of both novel and known molecules. Furthermore, it serves as a critical tool for establishing pharmacophoric groups, which function as foundational elements in drug design, by identifying the physicochemical and molecular rationales supporting such proposals. In this study, we specifically focus on cyclooxygenase inhibitors.
One approach to demonstrate the involvement of specific physicochemical and/or molecular properties in biological activity (COX inhibitory activity) is to construct simple correlations between biological activity and individual properties. These correlations for each COX inhibitor be helpful in identifying the structural requirements essential for biological activity. Consequently, we generated diagrams comparing physicochemical properties across distinct groups of NSAIDs with known experimental activity - specifically examining the relationship between molar refractivity (MR) and the partition coefficient of structurally related salicylate and fenamate derivatives (Figure 1A).
For the salicylate family, structural requirements for COX-1 and COX-2 recognition indicate optimal ranges of logP between 1.0 and 3.5 and molecular size between 33.0 and 102.0. cm3/mol. It is important to emphasize that these drugs are considered non-selective COX inhibitors [12,13], as confirmed in this study, where the average pIC50 values for both enzymes showed no significant difference in vitro and in silico studies (Figure S1). This demonstrates that these physicochemical properties are essential for protein recognition.
For the fenamate family, no significant differences were observed compared to salicylates (Figure S1). The mean inhibitory activities against COX-1 and COX-2 were remarkably similar and did no differ significantly. However, a slight preference for COX-1 affinity wasnoted, suggesting that the molecular requirements for logP and MR properties are more favourable for COX-1 than COX-2. The optimal physicochemical ranges were logP 3.3-4.8 (indicating greater lipophilicity than salicylates) and MR 66.0-75.0 cm³/mol (a narrow mid-range), implying that molecular size has less influence on selectivity than lipophilicity.
The core structures of salicylates and fenamates differ primarily by the isosteric replacement of -OH with -NH₂ [salicylic acid (logP = 1.20, MR = 33.9 cm³/mol) vs. anthranilic acid (logP = 0.79, MR = 36.9 cm³/mol), respectively], conferring similar physicochemical properties. The observed differences in activity and selectivity stem from their respective accessory moieties.
Molecular docking revealed that fenamates form ion-ion interactions between Arg120 and the carboxylate group, except for flufenamic acid (FFA), which interacts with Ala527. Salicylates (sulfasalazine/SSZ and diflunisal/DF) showed ionic interactions with Arg120, while acetylsalicylic acid (ASA) and salicylic acid (SA) formed hydrogen bonds with serine. [14,15,16]
Like the previous analysis, dipole moment (μ) and lipophilicity were evaluated using logP and logμ as key physicochemical descriptors (Figure 1B). The results indicate that active salicylates typically exhibit logP values between 1.0–3.5 and dipole moments (μ) ranging from 0.80–1.4 Debye. In contrast, fenamates display a distinct profile characterized by higher lipophilicity (logP 3.3–4.8) and a narrow dipole moment range (1.0–1.2 Debye).
Notably, the COX-1 inhibitory activity of both drug classes follows a positive parabolic correlation with dipole moment, with optimal compounds (primarily fenamates) clustering in the 1.05–1.20 Debye range – consistent with the logP-logμ diagram. Furthermore, the dipole moment vector is oriented from the accessory ring moiety toward the nitrogen atom of the anthranilic acid group (Figure S2 and S3).
2.2. Figures, Tables, and Schemes
These two drug classes exhibit a negative parabolic correlation with LUMO energy, where specific fenamates (FFA and MCF) demonstrate the highest activity (Figure S4). Conversely, their correlation with TPSA follows a negative linear relationship, with fenamates (MFA, FFA, MCF, NFA) again showing superior activity (Figure S5). Based on these descriptors, we propose the following model (Figure 2):
n = 8, F = 2.88, Q2 = 0.8861
The model indicates that fenamates are more potent inhibitors than salicylates. Fenamates exhibit intermediate dipole moments, low polar surface distribution, and electrophilic character in the carboxylic acid-containing ring. The electron-withdrawing carboxyl group delocalizes a positive charge across this aromatic system.
Most analyzed drugs (DF, MCF, MFA, and NFA – the most active compounds) form ionic interactions between the carboxylate group and Arg120. In contrast, the less active compounds (SSZ, ASA, SA, and FFA) lack this Arg120 interaction. For both fenamates and salicylates, the carboxylate-containing ring interacts with nonpolar amino acids (Leu352, Val523, Gly526, Ala527, etc.), facilitating negative charge transfer to the electrophilic ring, a feature linked to the LUMO orbital.
Therefore, it is interesting that for both enzymes, the substitutions at position 2 with respect to the carboxylate group are essential for the recognition and stabilization of free radicals due to the captodative properties of salicylates and fenamates; the latter being more active since the accessory moieties in this position better stabilize the charge by possessing, and additional aromatic system.
2.2. Acetates
For the acetate-class drugs, correlations were identified between physicochemical properties and binding to both COX-1 and COX-2, consistent with their non-selective inhibition profile observed experimentally and in silico (Figure S6). Regarding COX-1, significant correlations emerged with: partition coefficient (logP), showing a positive parabolic relationship (Figure S7); LUMO energy (ELUMO), showing a negative parabolic relationship (Figure S8); and molecular volume (V), showing a negative linear relationship (Figure S9). These correlations enabled the development of a QSAR model incorporating all three physicochemical properties influencing COX-1 inhibitory activity (Figure 3A):
n = 7, F = 1.774, Q2 = 0.9009
n = 7, F = 224.5, Q2 = 0.9889
According to the model, the partition coefficient significantly influences inhibitory activity. The exponential component indicates an upward-opening parabola with broad curvature, suggesting low system sensitivity. This is consistent with the negative linear coefficient, which positions the parabola in the second quadrant (logP range: 1.6–4.2) – a range compliant with Lipinski's Rule of Five. [17,18] This positive correlation with biological activity reflects interactions with nonpolar amino acids (Leu, Val, and Ala). Notably, diclofenac (DIC), one of the most active compounds in this group, contains a dichlorinated phenyl ring as its accessory moiety, which enhances lipophilicity.
The LUMO orbital energy also shows a parabolic relationship, but with a downward-opening curve. The most active compounds (DIC, KT, and TOL) feature electron-withdrawing groups on their phenyl rings that delocalize partial positive charge, positioning the LUMO orbital at these sites. This facilitates molecular recognition with electron-donating residues (Leu352 and Trp387) (Figure 4A).
The size descriptor analysis reveals that smaller molecular volumes increase drug-receptor recognition. This trend is observed for DIC, KT, and TOL, which are significantly more compact than indoleacetic acid derivatives.
With the acetates, a positive parabolic relationship was observed between the partition coefficient (logP) and COX-1 inhibitory activity, indicating that lipophilicity is a critical determinant of both target recognition and inhibitory potency. The analysis demonstrates two optimal lipophilicity ranges (logP 1.5-2.0 and 3.5-4.0) that maximize inhibitory activity for key acetate-class drugs (ketorolac, tolmetin, diclofenac, and indomethacin), with intermediate values showing reduced efficacy.
In the same vein, correlations were also found with other physicochemical properties for the acetate family against COX-2. In this case, they were with the partition coefficient (logP), the molar refractivity (MR), and the ovality (Ov). A positive parabolic relationship was observed with the partition coefficient (Figure S10), while negative linear correlations were found with molar refractivity and ovality (Figures S11 and S12). From these correlations, a QSAR model was obtained that involves three descriptors that influence the enzyme (COX-2) inhibitory activity (Figure 3B).
The QSAR model reveals that the partition coefficient (logP) significantly influences inhibitory activity, with a tighter parabolic relationship that for COX-1. This suggests enhanced enzyme recognition, where optimal lipophilicity positively correlates with biological activity through interactions with nonpolar residues (Leu, Val, and Ala). Ketorolac (KT), the most potent compound in this series, demonstrates this principle through its phenyl accessory moiety, which forms closer interactions with Leu352 (5.05 Å) and Trp387 (5.10 Å) in COX-2 versus COX-1 (5.43 Å and 5.21 Å, respectively).
Using molar refractivity as a size criterion, it indicates that less bulky drugs are favoured for recognition by the enzyme; once again, KT, TOL, and DIC are the smallest compared with the indoleacetic acid derivatives.
Finally, the molecular shape also influences biological activity, as the compounds should ideally present an ovoid shape, as in KT, TOL, and DIC. However, when ovality is significantly higher, activity decreases due to reduced affinity, as observed for ETD, SUL, OX, and IND (Figure S13).
2.3. Propionates
For the propionate group, correlations were found with specific physicochemical properties, both for drugs binding to COX-1 and to COX-2, which are not selective for either isoform, as shown both experimentally and in molecular docking studies (Figure S14). For COX-1, the identified correlations were with the acidity constant (pKa) and molecular volume (V); the pKa correlation was negative and parabolic (Figure S15), whereas the molecular volume correlation positive and linear (Figure S16). From these correlations, a QSAR model was constructed using two physicochemical properties that influence the enzyme (COX-1) inhibitory activity (Figure 5A).
n = 5, F = 1.23, Q2 = 0.7802
n = 5, F = 1.028, Q2 = 0.7323
According to the QSAR model, pKa has a positive influence on biological activity; in general terms, more acidic pKa values are favoured. Nevertheless, the drugs with the highest inhibitory activity against COX-1 correspond to KP, FEN, and FP (Figure S15). Docking analysis shows that the carboxylate group of KP, FEN, and FP establishes a unique ion–ion interaction with Arg120; whereas in the cases of IBU and NAP, which are the least active, the negative charge of the carboxylate is distributed between Arg120 through an ion–ion interaction and Tyr355 through an ion–dipole interaction (Figure 4B).
The QSAR analysis demonstrates that molecular volume significantly impacts the inhibitory activity of propionate-class drugs against COX-1, with larger molecules exhibiting enhanced potency (Figure S16). This observation is supported by molecular docking studies, which reveal that high-activity compounds (KP, FEN, and FP) possess two aromatic rings that establish extensive hydrophobic interactions with nonpolar residues (Val349, Leu352, Tyr385, Ile523, and Ala527). In contrast, lower-activity compounds such as IBU and NAP exhibit reduced binding efficiency due to structural limitations: IBU contains only a single aromatic ring, while NAP features a fused ring system with restricted conformational flexibility, both resulting in fewer optimal contacts with key hydrophobic residues (Figure 4B).
For COX-2 inhibition, distinct structure-activity relationships emerged. A positive parabolic correlation was observed between biological activity and acidity constant (pKa), indicating an optimal pKa range for maximal inhibition (Figure S17). Additionally, a linear correlation with HOMO energy (EHOMO) was identified, suggesting that electronic properties further modulate inhibitory potency (Figure S18). These relationships were integrated into a robust QSAR model that accounts for the combined influence of these descriptors on COX-2 inhibition (Figure 5B).
This COX-2 QSAR model reveals a compelling relationship between inhibitory activity and two interconnected physicochemical properties: pKa and HOMO energy (EHOMO). The observed trends are mechanistically significant: (1) lower pKa values reflect greater carboxylate ionization at physiological pH, enhancing ionic interactions, and (2) more negative EHOMO values (Figure S18) indicate stronger nucleophilic character, as the HOMO is localized on the carboxylate group. Consequently, compounds with both low pKa and negative EHOMO (FEN, KP, FP) exhibit superior activity compared to less active analogues (IBU, NAP).
Structurally, all drugs interact with Arg120 via ion-ion interactions and with Tyr355 via ion-dipole contacts with the carboxylate. The ionic interaction is governed by carboxylate acidity, while the concurrent nucleophile (HOMO)-electrophile (LUMO of protonated Arg120) interaction further stabilizes binding (Figure 4B). Activity differences within the series arise partly from electronegative substitutions at the 3- and 4-positions of the propionate-bearing ring (Table 1), which fine-tune electronic distribution and thus modulate carboxylate reactivity.
2.4. Pharmacophore Structure
The integrated QSAR and molecular docking studies highlight the critical role of the ionic interaction between Arg120 and the carboxylate group of NSAIDs for initial guidance and recognition by both COX-1 and COX-2, particularly in high-activity compounds. Furthermore, the aryl group containing the carboxylic acid interacts with non-polar amino acids (Table 2), forming a complementary hydrophobic pocket that stabilizes the drug-enzyme complex. These dual ionic interactions at Arg120 and hydrophobic interactions with nonpolar residues are relevant for binding affinity and inhibitory potency. Similarly, the arachidonic acid is oriented from the carboxylic acid to interact with Arg120 and Tyr355 aminoacids. In contrast, the Val349, Ala527, Ser353, Ile523 (in case COX-1) and Val523 (in case COX-2), Ile523 (in case COX-1) interact between 1 and 5 position of arachidonic acid (Figures S19 and S20), which are the same aminoacids that interact with accessory moieties of the NSAIDs, where the most critical aminoacid is Tyr385 that initiates the oxidation via free radicals (Table 2). [2]
The moiety responsible for the anchoring and orientation of NSAIDs on both COXs consists of an aryl group (aromatic generally) which is binding to a carboxylic acid with 0, 1, or 2 carbon atoms of distance of separation (Figure 6). The accessory moieties are characteristic for each drug group, the substitutions in position 2 correspond to the salicylates and fenamates that show relevant captodative properties; while the substitutions in the position 3 is characteristic of the acetates and some propionates; also the substitutions in the position 4 are similar between the acetates, propionates and salicylates (sulfasalazine) drugs (Figure 7), such drugs show decrease in the biological activity on both enzymes.
The core pharmacophore of NSAIDs consists of a carboxylic acid group, connected either directly or via a single carbon bridge, to a 5- or 6-membered aryl ring (Figure 7). This structural arrangement is essential for biological activity, where the carboxylate forms a key ionic interaction with Arg120. At the same time the aromatic ring provides structural rigidity and electron density required for molecular interactions. The accessory moieties of these molecules interact with various amino acids in the COX active site. In position 2, the accessory moieties show plane geometry, while the X-groups are bioisosters between them; such interactions are more specific, particularly aromatic-aromatic contacts with Phe381, Phe518, Tyr385, and Trp387 residues. In positions 3 and 4, the Y and Z substituents establish hydrophobic interactions with nonpolar residues such as Val349, Leu352, and Ala527, which contribute to stabilizing the enzyme-drug complex. This pattern is especially relevant for indoleacetic acid derivatives, where the indole system engages in π-stacking interactions with residues like Tyr385 (Table 2). Therefore, the description in this manuscript show the molecular moieties in the anchoring and orientation with COX-1 and COX-2 enzymes, thus the proposed pharmacophore is applicative for explain the ligand-receptor recognition and the non-selective NSAIDs rational design; also is have established the molecular bases for the search the moieties and stereochemistry necessaries for confer the selectivity on these enzymes, particularly on the COX-2, whose key moiety should be related to the aminoacids that configure the guide molecular portion.
3. Materials and Methods
3.1. Bibliographic Search
By means of an exhaustive bibliographic search of articles published from 1990 to 2023, the median inhibitory concentration (IC50) of COX-1 and COX-2 enzymes was found for each of the Non-steroidal anti-inflammatory drugs (NSAIDs) [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118] most widely prescribed for the pain, inflammatory and antipyretic diseases, which are the first cause of consulting at the first level of care in the world. The databases consulted were IUPHAR Database, Taylor & Francis, Scopus, PubMed, Google Scholar, SciELO, NCBI, Elsevier, UpToDate, Springer Nature, and Wolters Kluwer. The NSAIDs groups most frequently administered for pain and anti-inflammatory diseases are Salicylates (salicylic acid, acetylsalicylic acid, diflunisal, sulfasalazine), Fenamates (mefenamic acid, flufenamic acid, meclofenamic acid, niflumic acid), Acetates (diclofenac, ketorolac, indomethacin, oxamethacin, sulindac, etodolac, tolmentin), Propionates (ibuprofen, ketoprofen, feniprofen, flurbiprofen, naproxene). [119]
3.2. QSAR Analysis
All the IC50 values obtained from the literature were averaged to express the activity of NSAIDs over COX-1 and COX-2 enzymes. These IC50 values were converted to molar concentration (M) to evaluate the structure-activity relationship of each group. The pIC50 values of each drug were correlated with its respective partition coefficient (logP, representing liposolubility) and molar refractivity (MR, indicating polarity). These two physicochemical properties were selected with the ACD/ChemSketch and CS ChemDraw Pro v.6 software. [120] The experimental pKa values (an electronic criterion) were taken from the PubChem platform. [121]
Briefly, each NSAID was built with the Gaussian 16 and GaussView 6 program [122,123], and the conformational analysis search was carried out with molecular mechanics (with the Merck molecular force field, or MMFF) after establishing the most relevant torsions for the molecule. Once the conformer set was defined, the most stable conformer was optimized at the DFT level with the B3LYP 6-31G(d,p) basis set. The molecular volumes, HOMO and LUMO energies, and dipole moment were taken from the output file, and then the dipole moment vector and the HOMO and LUMO orbitals were visualized. [124]
The partition coefficient is an important physicochemical property that corresponds to the ratio of the concentrations of a substance in two immiscible phases in equilibrium in a mixture. Usually, the solvents used are water and n-octanol. This property often serves as a criterion to predict the ease of simple diffusion of the analyte across membranes, particularly the blood-brain barrier [125]. The partition coefficient is estimated according to the following equation:
where P is the partition coefficient, [A]oct is the analyte concentration in the n-octanol phase, and [A]wis the analyte concentration in the aqueous phase. The biological activity is relevant when the logP values are greater than 1.0, indicating that the analytes can probably crossmembranes and interact with their molecular targets.
MR, a measure of the total polarization of one mole of a substance, is related to molecular volume because small molecules are more likely to cross membranes and produce biological activity. This parameter is calculated with the Lorentz-Lorenz equation [126]:
where MR is the molar refractivity, MW is the molecular weight, is the density, and n is the refractive index of the molecule in question.
The values of the Topological Polar Superficial Area (TPSA) and molecular ovality were determined using the Molinspiration Platform [127] and Spartan ’14 software respectively, which are topological descriptors related like a measure of the polarity-area and the deviation from sphericity of a molecule. [128]
The apparent partition coefficient is called the distribution coefficient (log D). It is related to pKa values and is determined by the pH of the system. In the present study, a physiological pH (7.4) was assumed, and log D was calculated from the following equation [129]:
where D is the distribution coefficient, Pn is the partition coefficient for the ionizable form of the analytes in the organic phase, pH is the logarithmic form of the inverse of the H+ concentration, and pKa is the logarithmic form of the inverse of the acidity constant. The log D is the most accurate method for evaluating drug diffusion across membranes, accounting for the medium pH and the ionizable and non-ionizable forms.
The polarity of a molecule helps explain several physicochemical properties, such as chromatographic retention on a polar stationary phase [130]. Among the many descriptors proposed to quantify the effects of polarity, the most obvious and most used parameter is the dipole moment of the molecule. This parameter relates to the number of donors and acceptors in the hydrogen bond. Biological activity is higher for molecules with fewer donors and acceptors.
HOMO (the highest occupied molecular orbital) and LUMO (the lowest unoccupied molecular orbital) are quantum chemical descriptors that play a key role in the control of chemical reactions and the determination of electronic band gaps in solids. In the frontier molecular orbital (FMO) theory of chemical reactivity, the formation of a transition state is due to an interaction between the border orbitals (HOMO and LUMO) of the reactive species. [131,132]
While the HOMO energy is directly linked to the ionization potential and characterizes the susceptibility of the molecule to an attack by electrophiles, the LUMO energy is directly connected to electronic affinity. It characterizes the susceptibility of the molecule to an attack by nucleophiles. According to FMO, the electronic density of the frontier orbitals provides essential information for a detailed characterization of donor-acceptor interactions [123], based on the idea that most chemical reactions take place at the position and in the orientation in which the overlap of HOMO and LUMO of the respective reagents reaches a maximum [131]. Whereas the atomic HOMO density is critical for charge transfer in a donor molecule, the atomic coefficients of LUMO are essential for an acceptor molecule [132]. HOMO-LUMO interactions are relevant for understanding ligand-receptor recognition in biological systems.
Correlations were established between the biological activity of the drugs on COX-1 and COX-2 and their physicochemical and molecular properties mentioned above. Correlations with r values > 0.75 were considered significant.
3.3. Molecular Docking
Preparation of the protein (receptor): Searching and obtaining the three-dimensional structure of the protein in the Protein Data Bank platform, the following crystals were selected: for COX-1 (PDB 3N8Z) and COX-2 (PDB 3PGH). In Autodock Tools 1.5.6, water molecules and other substances were removed from the protein structure with which it was crystallized, leaving only the binding site of the endogenous ligand.
Ligand preparation: The structures of the compounds were drawn in their ionized forms, and the ligand was sent to GaussView 6.0. 16 and Gaussian 16, where the optimization was carried out at a DFT level.
Validation: The RMSD (Root Mean Square Distance) of both docked ligands is within the reliable range of 2 Å, with a value for both of 0.00 Å, verifying that flurbiprofen can interact with the 3N8Z and 3PGH crystal structures in a similarly manner to the pre-existing co-crystallized flurbiprofen (Figures S21 and S22).
Grid generation and docking algorithm: It was decided that targeted molecular docking would be carried out, so a three-dimensional grid was defined with the following dimensions: 66 x 66 x 66, 0.375 Å spacing, center Grid Box: X center: 8.833, y center: -5.250, Z center: 24. 833, for COX-1, while for COX-2: They were used with the following dimensions: 66 x 66 x 66, 0.375 Å spacing, center Grid Box: X center: -19.500, y center: -9.333, Z center: -21.528, which formed the grid around the receptor binding site, where ligand complementarity was assessed. These grids guided the search for the ligand within the search space. The Lamarckian algorithm was applied to explore the ligand’s possible conformations at the binding site.
3.5. Statistical Analysis
Linear regression analysis was performed using the least squares method and one-way ANOVA. The values of the ordinate, slope, and linear correlation coefficient were evaluated using Student's t-test. [133]
Second-order polynomial regression was performed using the least-squares method and one-way ANOVA. The values of the ordinate, slope, and correlation coefficient were evaluated using Student's t-test. Calculations were performed from the equations derived from the second-order polynomial regression model for both positive and negative parabolic curves. [134,135,136]
Data were analyzed using least squares regression and one-way ANOVA. The slopes were adjusted to the origin, and the correlation coefficients were evaluated using Student's t-test, with p < 0.05 considered significant in all cases. The QSAR models were examined by multiple regression analysis using the determinant method and one-way ANOVA. Statistical analyses were performed using Sigma Plot 15.0 [137] and the R framework. The external predictive ability of the QSAR models was assessed by the squared predictive correlation coefficient (Q2). [138]
4. Conclusions
Through multiple QSAR and molecular docking analyses of COX-1 and COX-2, the pharmacophoric features that explain the recognition of non-selective COX inhibitors (NSAIDs) were identified. The pharmacophoric structure show two regions: the guide moiety (carboxylate) that directs the binding mode of the NSAIDs and arachidonic acid to the Arg120 through electrostatic interaction; and the accessory moieties essential to blocking substrate oxidation via free radicals for both enzymes that catalyze the same reaction, the salicylate and fenamate families show a relevant captodative effect by electronic interaction between the carboxylate and an electron-donating group in position 2 on the aromatic ring. The general physicochemical and molecular properties that determine such ligand–enzyme recognition correspond to topological descriptors (dipole moment and topological polar surface area), geometric descriptors (volume, molar refractivity, and ovality), thermodynamic descriptors (partition coefficient), and electronic descriptors (acidity constant, HOMO and LUMO energies). Therefore, the elucidation and identification of the pharmacophore of non-selective COX inhibitors opens the door to a new approach for designing drugs with inhibitory activity on these enzymes and lays the foundation for a more detailed understanding of the physicochemical and molecular properties, as well as the intermolecular interactions, that may be involved in isoform selectivity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
E.S.C.R.: Writing-original draft, resources, methodology, investigation. J.R.M.D.: Writing review & editing, formal analysis, docking methodology. J.G.T.F.: Resources, formal analysis, conceptualization. J.A.G.S.: Writing review & editing, investigation, formal analysis, data curation, conceptualization.
Funding
Please add: This research was funded by the Consejo Nacional de Ciencia y Tecnología (CONACyT, grant no. 257364) and the SIP Project of the Instituto Politécnico Nacional (grants # 20151409, 20240792, 20250190, 20254103, 20250201, 20252305).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors on request.
Acknowledgments
E.S.C.R. greatly appreciates the additional support given by the Secretaría de Ciencia, Humanidades, Tecnología e Innovación through the National Scholarship (SECIHTI, grant CVU no. 1313062) and the program of the Beca de Estímulo Institucional de Formación de Investigadores (BEIFI) (grant # 83). J.A.G.S. and J.G.T.F. are members of the EDI fellowship programs of the IPN.
Conflicts of Interest
The authors declare that there was no conflict of interest involved in the study or the writing of the manuscript.
Abbreviations
The following abbreviations are used in this manuscript:
| ANOVA | Analysis of variance |
| COX | Cyclooxygenase |
| D | Distribution coefficient |
| DFT | Density functional theory |
| E | Energy |
| HOMO | High-occupied molecular orbital |
| Ka | Acidity constant |
| LUMO | Low-unoccupied molecular orbital |
| m | Dipolar moment |
| MMFF | Merck molecular force field |
| MR | Molar refractivity |
| MW | Molecular weight |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| P | Partition coefficient |
| PAF | Platelet-activating factor |
| Q2 | Predictability coefficient |
| QSAR | Quantitative structure-activity relationship |
| SAR | Structure-activity relationship |
| TPSA | Topological polar surface area |
| V | Volume |
References
- Brunton, L.L.; Knollman, B.C. Goodman & Gilman: Las bases farmacológicas de la terapéutica, pp 774-775, 829-83, 14ª ed; McGraw-Hill Education: México, 2022. [Google Scholar]
- Hajeyah, A.A.; Griffiths, W.J.; Wang, Y.; Finch, A.J.; O’Donnell, V.B. The Biosynthesis of Enzymatically Oxidized Lipids. Front. Endocrinol. 2020, 11, 591819. [Google Scholar] [CrossRef]
- Wallace, J.L.; Del Soldat, P. The therapeutic potential of No-NSAIDs. Fundam. Clin. Pharmacol. 2003, 17(1), 11–20. [Google Scholar] [CrossRef]
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500 years of aspirin history from its roots – A concise summary. Vasc. Pharmacol. 2019, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Asirvatham, S.; Dhokchawle, B.V.; Tauro, S.J. Quantitative structure activity relationships studies of non-steroidal anti-inflammatory drugs: A review. Arab. J. Chem. 2019, 12(8), 3948–3962. [Google Scholar] [CrossRef]
- Hadjipavlou-Litina, D. Quantitative structure - activity relationship (QSAR) studies on non steroidal anti-inflammatory drugs (NSAIDs). Curr. Med. Chem. 2000, 7(4), 375–388. [Google Scholar] [CrossRef]
- Delgado, A.; Minguillón, C.; Joglar, J. Introducción a la Química terapéutica, 2ª ed; Diaz de Santos, España.
- James, D. The multisystem adverse effects of NSAID therapy. J. Am. Ostepath. Assoc. 1999, 99(11), 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin.Gastroenterol. 2010, 24(2), 121–132. [Google Scholar] [CrossRef]
- Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 2013, 15 Suppl 3(S3), S2. [Google Scholar] [CrossRef] [PubMed]
- Tomić, M.; Micov, A.; Pecikoza, U.; Stepanović-Petrović, R. Clinical uses of nonsteroidal anti-inflammatory drugs (NSAIDs) and potential benefits of NSAIDs modified-release preparations. In Microsized and Nanosized Carriers for Nonsteroidal Anti-Inflammatory Drugs; Bojan, Čalija, Ed.; Elsevier, Serbia, 2017; pp. 1–29. [Google Scholar]
- Alvarado, J.C.B.; Víquez, M.M. Fisiopatología y seguridad del uso de AINEs selectivos y no selectivos: balance de riesgos. Rev. Méd. Univ. Costa Rica. 2011, 5(1), 39–57. [Google Scholar] [CrossRef]
- Marcén, B.; Sostres, C.; Lanas, A. AINE y riesgo digestivo. Aten. Primaria. 2016, 48(2), 73–76. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Boeglin, W.E.; Boutaud, O.; Malkowski, M.G.; Schneider, C. Residual cyclooxygenase activity of aspirin-acetylated COX-2 forms 15R-prostaglandins that inhibit platelet aggregation. The FASEB Journal. 2018, 33(1), 1033–1041. [Google Scholar] [CrossRef]
- Patrono, C. Low-dose aspirin for the prevention of atherosclerotic cardiovascular disease. Eur. Heart J. 2024, 45(27), 2362–2376. [Google Scholar] [CrossRef]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochem. 2016, 55(8), 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23(1-3), 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45(12), 2615–2623. [Google Scholar] [CrossRef]
- Robles-Navarro, A.; Cárdenas, C.; Fuentealba, P. Electronegativity under Confinement. Molecules 2021, 26(22), 6924. [Google Scholar] [CrossRef]
- Matsunaga, N.; Rogers, D.W.; Zavitsas, A.A. Pauling’s Electronegativity Equation and a New Corollary Accurately Predict Bond Dissociation Enthalpies and Enhance Current Understanding of the Nature of the Chemical Bond. J. Org. Chem. 2003, 68(8), 3158–3172. [Google Scholar] [CrossRef]
- Calvello, R.; Panaro, M.A.; Carbone, M.L.; Cianciulli, A.; Perrone, M.G.; Vitale, P.; Malerba, P.; Scilimati, A. Novel selective COX-1 inhibitors suppress neuroinflammatory mediators in LPS-stimulated N13 microglial cells. Pharmacol. Res. The Official Journal of the Italian Pharmacological Society 2012, 65(1), 137–148. [Google Scholar] [CrossRef]
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104(5), 413–421. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Abdellatif, K.R.A.; Dong, Y.; Das, D.; Yu, G.; Velázquez, C.A.; Suresh, M.R.; Knaus, E.E. Synthesis and biological evaluation of salicylic acid and N-acetyl-2-carboxybenzenesulfonamide regioisomers possessing a N-difluoromethyl-1,2-dihydropyrid-2-one pharmacophore: Dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2009, 19(24), 6855–6861. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. PNAS 1993, 90(24), 11693–11697. [Google Scholar] [CrossRef] [PubMed]
- Medchemexpress.com. Available online: https://www.medchemexpress.com/aspirin.html?locale=es-ES (accessed on 15 January 2026).
- (s/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/70260 (accessed on 15 January 2026).
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. PNAS 1999, 96(13), 7563–7568. [Google Scholar] [CrossRef]
- Perrone, M.G.; Centonze, A.; Miciaccia, M.; Ferorelli, S.; Scilimati, A. Cyclooxygenase inhibition safety and efficacy in inflammation-based psychiatric disorders. Molecules 2020, 25(22), 5388. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, M.; Percival, M.D. Effect of inhibitor time-dependency on selectivity towards cyclooxygenase isoforms. Biochem. J. 1995, 306 Pt 1, 247–251. [Google Scholar] [CrossRef] [PubMed]
- (S/f). Medkoo.com. Available online: https://www.medkoo.com/products/45720 (accessed on 17 January 2026).
- (s/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/21447/flufenamic-acid. (accessed on 17 January 2026).
- Drago, S.; Imboden, R.; Schlatter, P.; Buylaert, M.; Krähenbühl, S.; Drewe, J. Pharmacokinetics of transdermal etofenamate and diclofenac in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2017, 121(5), 423–429. [Google Scholar] [CrossRef]
- Madhava, G.; Ramana, K.V.; Sudhana, S.M.; Rao, D.S.; Kumar, K.H.; Lokanatha, V.; Rani, A.U.; Raju, C.N. Aryl/heteroaryl substituted celecoxib derivatives as COX-2 inhibitors: Synthesis, anti-inflammatory activity, and molecular docking studies. Med. Chem. (Shariqah (United Arab Emirates)) 2017, 13(5), 484–497. [Google Scholar] [CrossRef]
- Narsinghani, T.; Chaturvedi, S.C. QSAR analysis of meclofenamic acid analogues as selective COX-2 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16(2), 461–468. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Rowlinson, S.W.; Crews, B.C.; Marnett, L.J. Amide derivatives of meclofenamic acid as selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12(4), 521–524. [Google Scholar] [CrossRef]
- Du, L.; Du, S.; Li, J.; Wang, H. Design, synthesis and biological evaluation of novel 2- (indole arylamide) benzoic acid analogs as dual COX-2 / 5-LOX inhibitors. Research Square 2022, preprint. [Google Scholar] [CrossRef]
- (S/f-b). Medchemexpress.com. Available online: https://www.medchemexpress.com/mefenamic-acid.html (accessed on 17 January 2026).
- (s/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/70550/meclofenamate-(sodium-salt) (accessed on 17 January 2026).
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104(5), 413–421. [Google Scholar] [CrossRef] [PubMed]
- Huntjens, D.R.H.; Danhof, M.; Della; Pasqua, O.E. Pharmacokinetic–pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology 2005, 44(7), 846–859. [Google Scholar] [CrossRef] [PubMed]
- (s/f). Scbt.com. Available online: https://www.scbt.com/es/p/mefenamic-acid-61-68-7 (accessed on 18 January 2026).
- (s/f). Glpbio.com. Available online: https://www.glpbio.com/sp/mefenamic-acid.html (accessed on 18 January 2026).
- Lees, P.; Landoni, M.F.; Giraudel, J.; Toutain, P.L. Pharmacodynamics and pharmacokinetics of nonsteroidal anti-inflammatory drugs in species of veterinary interest. J. Vet. Pharmacol. Ther. 2004, 27(6), 479–490. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Cho, D.-Y.; Choi, S.-R.; Lee, J.-Y.; Choi, D.-K.; Kim, E.; Park, J.-Y. Synthesis and biological evaluation of salicylic acid analogues of celecoxib as a new class of selective cyclooxygenase-1 inhibitor. Biol. Pharm. Bull. 2021, 44(9), 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; Oancea, C.; Heinssmann, M.; Khan, D.; Held, H.; Kahnt, A.S.; Capelo, R.; la Buscató, E.; Proschak, E.; Puccetti, E.; Steinhilber, D.; Fleming, I.; Maier, T.J.; Ruthardt, M. 5-Lipoxygenase is a candidate target for therapeutic management of stem cell–like cells in acute myeloid leukemia. Cancer Res. 2014, 74(18), 5244–5255. [Google Scholar] [CrossRef]
- Riendeau, D.; Percival, M.D.; Brideau, C.; Charleson, S.; Dubé, D.; Ethier, D.; Falgueyret, J.P.; Friesen, R.W.; Gordon, R.; Greig, G.; Guay, J.; Mancini, J.; Ouellet, M.; Wong, E.; Xu, L.; Boyce, S.; Visco, D.; Girard, Y.; Prasit, P.; Chan, C.C. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J. Pharmacol Exp. Ther. 2001, 296(2), 558–566. [Google Scholar] [CrossRef] [PubMed]
- Munir, A.; Khushal, A.; Saeed, K.; Sadiq, A.; Ullah, R.; Ali, G.; Ashraf, Z.; Ullah; Mughal; Saeed, E.; Jan, M.; Rashid, U.; Hussain, I.; Mumtaz, A. Synthesis, in-vitro, in-vivo anti-inflammatory activities and molecular docking studies of acyl and salicylic acid hydrazide derivatives. Bioorg. Chem. 2020, 104(104168), 104168. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Defining the COX inhibitor selectivity of NSAIDs: implications for understanding toxicity. Expert Rev. Clin. Pharmacol. 2010, 3(6), 769–776. [Google Scholar] [CrossRef]
- Dannhardt, G.; Ulbrich, H. In-vitro test system for the evaluation of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) inhibitors based on a single HPLC run with UV detection using bovine aortic coronary endothelial cells (BAECs). Inflamm. Res. 2001, 50(5), 262–269. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.H.; Hawcroft, G.; Hull, M.A. Effect of nonsteroidal anti-inflammatory drugs on β-catenin protein levels and catenin-related transcription in human colorectal cancer cells. Br. J. Cancer. 2004, 91(1), 153–163. [Google Scholar] [CrossRef]
- Kim, S.J.; Reddy, R. Critical appraisal of ophthalmic ketorolac in treatment of pain and inflammation following cataract surgery. Clin. Ophthalmol. 2011, 5, 751–758. [Google Scholar] [CrossRef]
- Pradilla, O.E. Ciclooxigenasa 3: La nueva iso-enzima en la familia. MedUNAB 2004, 7(21), 181–4. [Google Scholar]
- (S/f). Caymanchem.com. Available online: https://cdn.caymanchem.com/cdn/insert/19187.pdf (accessed on 18 January 2026).
- Mohanapriya, A.; Achuthan, D. Comparative QSAR analysis of cyclo-oxygenase 2 inhibiting drugs. Bioinformation 2012, 8(8), 353–358. [Google Scholar] [CrossRef]
- Hardin, H. Etoricoxib: A new COX-2 inhibitor. Farma Note 2004, 20(3), 1–6. [Google Scholar]
- (S/f-c). Medchemexpress.com. Available online: https://www.medchemexpress.com/etodolac.html (accessed on 18 January 2026).
- (S/f). Axonmedchem.com. Available online: https://www.axonmedchem.com/product/3451 (accessed on 18 January 2026).
- (S/f-d). Abcam.com. Available online: https://www.abcam.com/en-mx/products/biochemicals/etodolac-cox-2-inhibitor-nsaid-ab141086. (accessed on 22 February 2024).
- Kato, M.; Nishida, S.; Kitasato, H.; Sakata, N.; Kawai, S. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs: investigation using human peripheral monocytes. J. Pharm. Pharmacol. 2001, 53(12), 1679–1685. [Google Scholar] [CrossRef]
- Roberts, J.S.; Ma, C.; Robertson, S.Y.T.; Kang, S.; Han, C.S.; Deng, S.X.; Zheng, J.J. R-etodolac is a more potent Wnt signaling inhibitor than enantiomer, S-etodolac. Biochem. Biophys. Rep. 2022, 30(101231), 101231. [Google Scholar] [CrossRef]
- (S/f-c). Medchemexpress.com. Available online: https://www.medchemexpress.com/etodolac.html (accessed on 18 January 2026).
- Etodolac, *!!! REPLACE !!!*. (s/f). Axonmedchem.com. Available online: https://www.axonmedchem.com/product/3451 (accessed on 18 January 2026).
- S/f-d). Abcam.com. Available online: https://www.abcam.com/en-mx/products/biochemicals/etodolac-cox-2-inhibitor-nsaid-ab141086 (accessed on 22 February 2024).
- (s/f-b). Glpbio.com. Available online: https://www.glpbio.com/sp/etodolac.html (accessed on 18 January 2026).
- Cheng, Z.; Nolan, A.M.; McKellar, Q.A. Measurement of cyclooxygenase inhibition in vivo: a study of two non-steroidal anti-inflammatory drugs in sheep. Inflammation 1998, 22(4), 353–366. [Google Scholar] [CrossRef] [PubMed]
- Beretta, C.; Garavaglia, G.; Cavalli, M. COX-1 and COX-2 inhibition in horse blood by phenylbutazone, flunixin, carprofen and meloxicam: an in vitro analysis. Pharmacol. Res. 2005, 52(4), 302–306. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.; Doble, M. COX-2 enzyme and its inhibitors. Curr. Bioact. Compd. 2006, 2(2), 161–178. [Google Scholar] [CrossRef]
- Riendeau, D.; Percival, M.D.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Falgueyret, J.P.; Ford-Hutchinson, A.W.; Gordon, R.; Greig, G.; Gresser, M.; Guay, J.; Kargman, S.; Léger, S.; Mancini, J.A.; O’Neill, G.; Ouellet, M.; Chan, C.-C. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br. J. Pharmacol. 1997, 121(1), 105–117. [Google Scholar] [CrossRef]
- flurbiprofen (CAS 5104-49-4). (s/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/70250 (accessed on 18 January 2026).
- (S)-Flurbiprofen. (s/f). Apexbt.com. Available online: https://www.apexbt.com/s-flurbiprofen.html (accessed on 18 January 2026).
- (S/f-c). Medchemexpress.com. Available online: https://www.medchemexpress.com/flubiprofen.html (accessed on 18 January 2026).
- (S/f-f). Tocris.com. Available online: https://www.tocris.com/products/flurbiprofen_1769 (accessed on 18 January 2026).
- (S/f-c). Medchemexpress.com. Available online: https://www.medchemexpress.com/ibuprofen.html (accessed on 18 January 2026).
- (S/f-g). Selleckchem.com. Available online: https://www.selleckchem.com/products/Ibuprofen(Advil).html (accessed on 18 January 2026).
- Dvorakova, M.; Langhansova, L.; Temml, V.; Pavicic, A.; Vanek, T.; Landa, P. Synthesis, inhibitory activity, and in silico modeling of selective COX-1 inhibitors with a quinazoline core. ACS Med. Chem. Lett. 2021, 12(4), 610–616. [Google Scholar] [CrossRef] [PubMed]
- (S/f-h). Selleckchem.com. Available online: https://www.selleckchem.com/products/Indomethacin(Indocid).html (accessed on 18 January 2026).
- (S/f-c). Medchemexpress.com. Available online: https://www.medchemexpress.com/Indomethacin.html (accessed on 18 January 2026).
- (S/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/16407/(s)-ketoprofen. (accessed on 20 January 2026).
- (S/f). Glpbio.com. Available online: https://www.glpbio.com/sp/s-ketoprofen.html (accessed on 20 January 2026).
- (S/f-i). Medchemexpress.com. Available online: https://www.medchemexpress.com/S-__addition__-Ketoprofen.html?locale=es-ES (accessed on 20 January 2026).
- Rao, R.; Kumar, R.; Sarwal, A.; Sinha, V.R. Ocular Inflammation and NSAIDs: An Overview with Selective and Non-Selective COX Inhibitors. p. Corpus ID: 40708527. Available online: https://thepharmstudent.com/issue_2015/8.Rao_et_al.pdf?i=1 (accessed on 20 January 2026).
- Gouda, A.M.; Ali, H.I.; Almalki, W.H.; Azim, M.A.; Abourehab, M.A.S.; Abdelazeem, A.H. Design, synthesis, and biological evaluation of some novel pyrrolizine derivatives as COX inhibitors with anti-inflammatory/analgesic activities and low ulcerogenic liability. Molecules 2016, 21(2), 201. [Google Scholar] [CrossRef]
- Waterbury, L.D.; Silliman, D.; Jolas, T. Comparison of cyclooxygenase inhibitory activity and ocular anti-inflammatory effects of ketorolac tromethamine and bromfenac sodium. Curr Med Res Opin. 2006, 22(6), 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- (S/f-j). Selleckchem.com. Available online: https://www.selleckchem.com/products/Ketorolac-Tromethamine(Toradol).html (accessed on 20 January 2026).
- (S/f-k). Medchemexpress.com. Available online: https://www.medchemexpress.com/ketorolac-d5.html?locale=es-ES (accessed on 20 January 2026).
- (S/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/11348/(s)-ketorolac (accessed on 20 January 2026).
- Krzyżak, E.; Szkatuła, D.; Wiatrak, B.; Gębarowski, T.; Marciniak, A. Synthesis, Cyclooxygenases Inhibition Activities, and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives. Molecules 2020, 25(12), 2934. [Google Scholar] [CrossRef]
- Chan, C.C.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Ford-Hutchinson, A.W.; Forrest, M.J.; Gauthier, J.Y.; Gordon, R.; Gresser, M.; Guay, J.; Kargman, S.; Kennedy, B.; Leblanc, Y.; Leger, S.; Mancini, J.; O’Neill, G.P.; Oullet, M.; Patrick, D.; Percival, M.D.; Perrier, H.; Pasit, P.; Rodger, I.; et al. Rofecoxib [Vioxx, MK-0966; 4-(4-Methylsulfonylphenyl)-3- phenyl-2-(5H)-furanone]: A Potent and Orally Active Cyclooxygenase-2 Inhibitor. J Pharmacol Exp Ther. 1999, 290(2), 551–560. [Google Scholar] [CrossRef] [PubMed]
- Escolar, M.; Sádaba, B.; Honorato, J. Meloxicam. Revista De Medicina De La Universidad De Navarra 2017, 41(2), 51–55. [Google Scholar] [CrossRef]
- Hinz, B.; Cheremina, O.; Besz, D.; Zlotnick, S.; Brune, K. Impact of naproxen sodium at over-the-counter doses on cyclooxygenase isoforms in human volunteers. Int. J. Clin. Pharmacol. Ther. 2008, 46(4), 180–186. [Google Scholar] [CrossRef]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2010, 285(45), 34950–9. [Google Scholar] [CrossRef]
- (S/f-p). Selleckchem.com. Available online: https://www.selleckchem.com/datasheet/Naproxen-Sodium(Aleve)-S162601-DataSheet.html (accessed on 20 January 2026).
- (S/f-q). Medchemexpress.com. Available online: https://www.medchemexpress.com/Naproxen.html?locale=es-ES (accessed on 20 January 2026).
- (S/f). Scbt.com. Available online: https://www.scbt.com/es/p/naproxen-22204-53-1 (accessed on 20 January 2026).
- Franzone, J.S.; Natale, T.; Cirillo, R. Effect of a new anti-inflammatory drug (oxametacine) on the prostaglandin biosynthesis. Farmaco Sci. 1980, 35(6), 498–503. [Google Scholar] [PubMed]
- Vergin, H.; Ferber, H.; Brunner, F.; Kukovetz, W.R. Pharmakokinetik und Biotransformation von Oxametacin bei gesunden Probanden [Pharmacokinetics and biotransformation of oxametacine in healthy volunteers (author's transl)]. In Arzneimittelforschung; German, 1981; Volume 31, 3, pp. 513–518. [Google Scholar] [PubMed]
- Mitchell, J. A.; Akarasereenont, P.; Thiemermann, C.; Flower, R. J.; Vane, J. R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. PNAS 1993, 90(24), 11693–11697. [Google Scholar] [CrossRef]
- Botting, R.M.; Harvey, T.W.; Vane, J.R. Inhibitors of cyclooxygenases: mechanisms, selectivity and uses. J. Physiol. Pharmacol. 2006, 57 Suppl 5, 113–24. [Google Scholar] [PubMed]
- Liedtke, A.J.; Crews, B.C.; Daniel, C.M.; Blobaum, A.L.; Kingsley, P.J.; Ghebreselasie, K.; Marnett, L.J. Cyclooxygenase-1-Selective Inhibitors Based on the (E)-2′-Des-methyl-sulindac Sulfide Scaffold. J. Med. Chem. 2012, 55(5), 2287–2300. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Mathew, B.; Chen, X.; Maxuitenko, Y.Y.; Li, N.; Lowe, W.M.; Whitt, J.D.; Zhang, W.; Gary, B.D.; Keeton, A.B.; Grizzle, W.E.; Grubbs, C.J.; Reynolds, R.C.; Piazza, G.A. Novel non-cyclooxygenase inhibitory derivative of sulindac inhibits breast cancer cell growth in vitro and reduces mammary tumorigenesis in rats. Cancers 2023, 15(3), 646. [Google Scholar] [CrossRef]
- Vitale, P.; Panella, A.; Scilimati, A.; Perrone, M.G. COX-1 inhibitors: Beyond structure toward therapy. Med. Res. Rev. 2016, 36(4), 641–671. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, F.; Warner, T.D. Ex vivo assay to determine the cyclooxygenase selectivity of non-steroidal anti-inflammatory drugs. Br. J. Pharmacol. 1999, 126(8), 1824–1830. [Google Scholar] [CrossRef]
- (S/f-m). Medchemexpress.com. Available online: https://www.medchemexpress.com/tolmetin.html?locale=es-ES (accessed on 20 January 2026).
- (S/f). Targetmol.com. Available online: https://www.targetmol.com/compound/Tolmetin (accessed on 20 January 2026).
- (S/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/18195 (accessed on 20 January 2026).
- (S/f). Cambridge Bioscience Limited. Available online: https://www.bioscience.co.uk/product~98035 (accessed on 22 January 2026).
- (S/f). Caymanchem.com. Available online: https://www.caymanchem.com/product/70650/niflumic-acid. (accessed on 22 January 2026).
- S/f. Laboratory Chemicals-FUJIFILM Wako Chemicals U.S.A. Corporation. Available online: https://labchem-wako.fujifilm.com/us/product/detail/W01W0114-0734.html (accessed on 22 January 2026).
- Kim, B.M.; Maeng, K.; Lee, K.-H.; Hong, S.H. Combined treatment with the Cox-2 inhibitor niflumic acid and PPARγ ligand ciglitazone induces ER stress/caspase-8-mediated apoptosis in human lung cancer cells. Cancer Lett. 2011, 300(2), 134–144. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11(2), 81s–110s. [Google Scholar] [CrossRef] [PubMed]
- Journal of Chemical Software, Vol.5, No.3 (1999). (s. f.). Available online: https://www.sccj.net/CSSJ/jcs/v5n4/a2/text.html (accessed on 22 January 2026).
- Vishwakarma, R.; Negi, D.S. The development of COX-1 and COX-2 inhibitors: A review. Int. J. Pharm. Sci. Res. 2020, 11(8), 3544–3555. [Google Scholar]
- (S/f). Medchemexpress.com. Available online: https://www.medchemexpress.com/niflumic-acid.html?locale=es-ES (accessed on 22 January 2026).
- Kucherenko, Y.V.; Lang, F. Niflumic acid affects store-operated Ca2+-permeable (SOC) and Ca2+-dependent K+ and Cl− ion channels and induces apoptosis in K562 cells. J. Membr. Biol. 2014, 247(7), 627–638. [Google Scholar] [CrossRef]
- El-Dash, Y.; Khalil, N.A.; Ahmed, E.M.; Hassan, M.S.A. Synthesis and biological evaluation of new nicotinate derivatives as potential anti-inflammatory agents targeting COX-2 enzyme. Bioorg. Chem. 2021, 107, 104610. [Google Scholar] [CrossRef]
- Gomaa, M.; Gad, W.; Hussein, D.; Pottoo, F.H.; Tawfeeq, N.; Alturki, M.; Alfahad, D.; Alanazi, R.; Salama, I.; Aziz, M.; Zahra, A.; Hanafy, A. Sulfadiazine exerts potential anticancer effect in HepG2 and MCF7 cells by inhibiting TNFα, IL1b, COX-1, COX-2, 5-LOX gene expression: Evidence from in vitro and computational studies. Pharmaceuticals 2024, 17(2), 189. [Google Scholar] [CrossRef]
- Bertolini, A.; Ottani, A.; Sandrini, M. Selective COX-2 inhibitors and dual acting anti-inflammatory drugs: Critical remarks. Curr. Med. Chem. 2002, 9(10), 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Parmar, D.K.; Das, D. Recent applications of azo dyes: A paradigm shift from medicinal chemistry to biomedical sciences. Mini-Rev. Med. Chem. 2021, 21(9), 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Blanca-López, N.; Soriano, V.; Martin, E.G.; Canto, G.; Blanca, M. NSAID-induced reactions: classification, prevalence, impact, and management strategies. J. Asthma Allergy. 2019, 12, 217–233. [Google Scholar] [CrossRef]
- ChemDraw Prime. Versión 17.1; PerkinElmer, 2018. [Google Scholar]
- PubChem. (s. f.). PubChem. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/.
- Dennington, R; Keith, T.A.; Millam, J.M. GaussView; version 6.1. In Semichem Inc.; Shawnee Mission, KS, 2016. [Google Scholar]
- Gaussian, Inc. Wallingford CT, 2016.
- BIOVIA Discovery Studio 2024, v2024.
- Agatonovic-Kustrin, S.; Chan, C.K.Y.; Gegechkori, V.; Morton, D.W. Models for skin and brain penetration of major components from essential oils used in aromatherapy for dementia patients. J. Biol. Struct. Dyn. 2019, 38(8), 2402–2411. [Google Scholar] [CrossRef]
- Kragh, H. The Lorenz-Lorentz Formula: Origin and Early History. Substantia 2018, 2(2), 7–18. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: https://www.molinspiration.com/.
- Spartan ’14, Version 1.2.0; Wavefunction, Inc.: Irvine, CA, USA, 2014.
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71(6), 525–616. [Google Scholar] [CrossRef]
- Plachká, K.; Pilařová, V.; Gazárková, T.; Švec, F.; Garrigues, J.; Nováková, L. Advancing Fundamental Understanding of Retention Interactions in Supercritical Fluid Chromatography Using Artificial Neural Networks: Polar Stationary Phases with –OH Moieties. Anal. Chem. 2024, 96(31), 12748–12759. [Google Scholar] [CrossRef]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. Synop. 2021, 45, 147–158. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21(6), 748. [Google Scholar] [CrossRef] [PubMed]
- Talavera-Piña, J.O.; Antonio-Ocampo, A.; Castellanos-Olivares, A.; Wacher-Rodarte, N.H. Regresión lineal simple. Rev. Med. IMSS 1995, 33(3), 347–51. [Google Scholar]
- Madroñero, D.M.; Mondragón, E.I.; Vergel-Ortega, M. Análisis estadístico para validar parámetros de modelos matemáticos por medio método de mínimos cuadrados. Revista Boletín Redipe 2021, 10(5), 343–359. [Google Scholar] [CrossRef]
- Morán-Díaz, J.R.; Jiménez-Vázquez, H.A.; Gómez-Pliego, R.; Arellano-Mendoza, M.G.; Quintana-Zavala, D.; Guevara-Salazar, J.A. Correlation study of antibacterial activity and spectrum of penicillins through a structure-activity relationship analysis. Med. Chem. Res. 2019, 28, 1529–1546. [Google Scholar] [CrossRef]
- Morán-Díaz, J.R.; Neveros-Juárez, F.; Arellano-Mendoza, M.G.; Quintana-Zavala, D.; Lara-Salazar, O.; Trujillo-Ferrara, J.G.; Guevara-Salazar, J.A. QSAR analysis of five generations of cephalosporins to establish the structural basis of activity against methicillin-resistant and methicillin-sensitive Staphylococcus aureus. Mol. Divers. 2024, 28(5), 3027–3043. [Google Scholar] [CrossRef] [PubMed]
- SigmaPlot, Version 15.0; Systat Software, Inc.: San Jose, CA, USA, 2020.
- Schüürmann, G.; Ebert, R.; Chen, J.; Wang, B.; Kühne, R. External Validation and Prediction Employing the Predictive Squared Correlation Coefficient — Test Set Activity Mean vs Training Set Activity Mean. J. Chem. Inf. Model. 2008, 48(11), 2140–2145. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) The logP-logMR diagram for salicylates and fenamates families was constructed considering the average inhibitory activity of each family against COX-1 and COX-2 enzymes. (B) logP-logμ diagram of salicylates and fenamates families showing their average inhibitory activity against COX-1 and COX-2 enzymes. Value above: experimental pIC50 of COX-1; Value down: experimental pIC50 of COX-2. ASA, acetylsalicylic acid; SA, salicylic acid; SSZ, sulfasalazine; DF, diflunisal; NFA, niflumic acid; FFA, flufenamic acid; MFA, mefenamic acid; and MCF, meclofenamic acid.
Figure 1.
(A) The logP-logMR diagram for salicylates and fenamates families was constructed considering the average inhibitory activity of each family against COX-1 and COX-2 enzymes. (B) logP-logμ diagram of salicylates and fenamates families showing their average inhibitory activity against COX-1 and COX-2 enzymes. Value above: experimental pIC50 of COX-1; Value down: experimental pIC50 of COX-2. ASA, acetylsalicylic acid; SA, salicylic acid; SSZ, sulfasalazine; DF, diflunisal; NFA, niflumic acid; FFA, flufenamic acid; MFA, mefenamic acid; and MCF, meclofenamic acid.
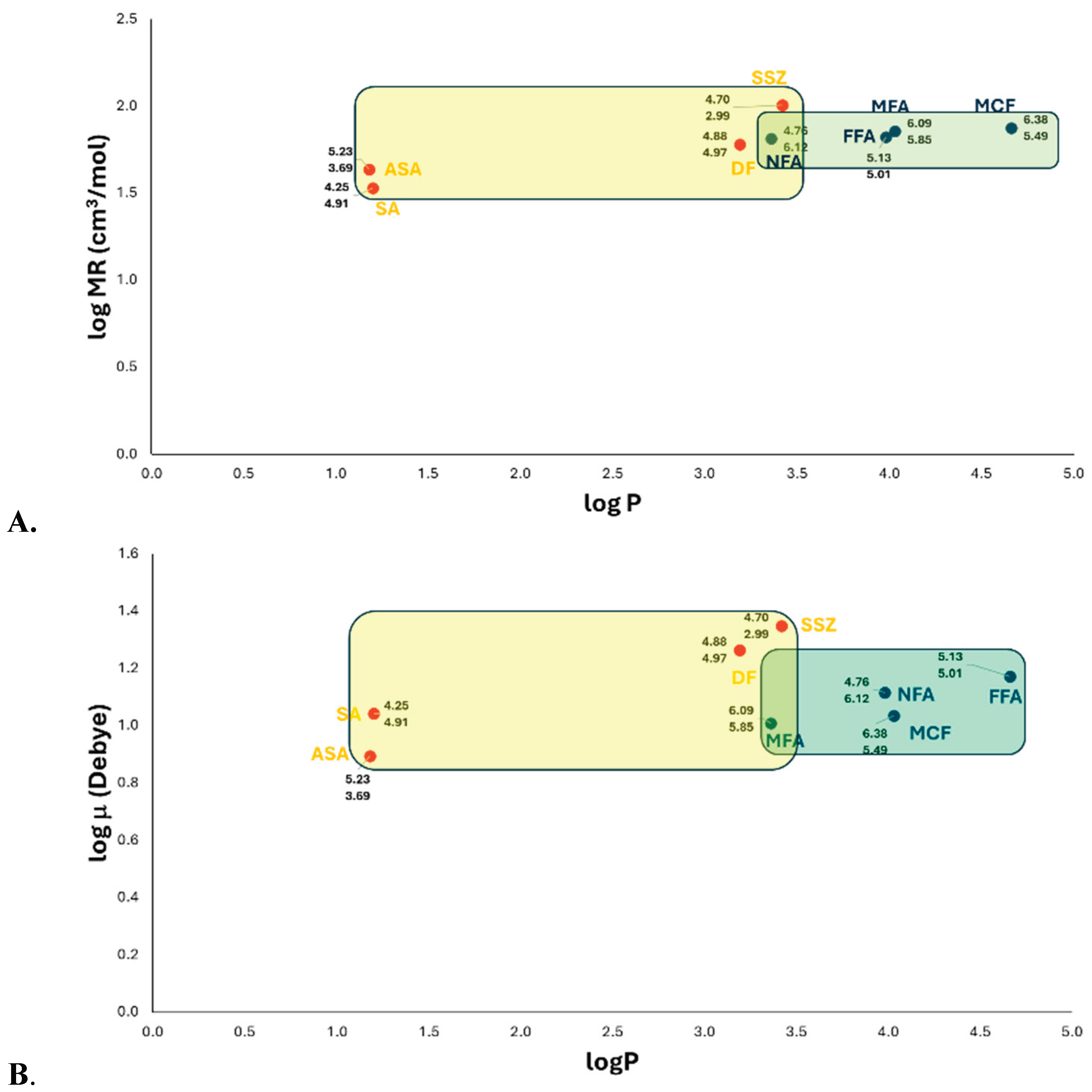
Figure 2.
Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the fenamates and salicylates NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 8) for the QSAR model: pIC50 = alogμ2-blogμ-cELUMO2+dELUMO-elogTPSA+f: a = -6.832 ± 18.864 (p = 0.2776), b = 14.065 ± 42.0015 (p = 0.2776), c = 492.539 ± 373.575 (p = 0.2776), d = -116.515 ± 74.833 (p = 0.2776), e = -10.152 ± 4.006 (p = 0.2776), f = 20.599 ± 28.161 (p = 0.2776) and, r = 0..9413 (p = 0.2776). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. ASA, acetylsalicylic acid; DF, diflunisal; FFA, flufenamic acid; MFA, mefenamic acid; MCF, meclofenamic acid; NFA, niflumic acid; SA, salicylic acid; SSZ, sulfasalazine.
Figure 2.
Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the fenamates and salicylates NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 8) for the QSAR model: pIC50 = alogμ2-blogμ-cELUMO2+dELUMO-elogTPSA+f: a = -6.832 ± 18.864 (p = 0.2776), b = 14.065 ± 42.0015 (p = 0.2776), c = 492.539 ± 373.575 (p = 0.2776), d = -116.515 ± 74.833 (p = 0.2776), e = -10.152 ± 4.006 (p = 0.2776), f = 20.599 ± 28.161 (p = 0.2776) and, r = 0..9413 (p = 0.2776). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. ASA, acetylsalicylic acid; DF, diflunisal; FFA, flufenamic acid; MFA, mefenamic acid; MCF, meclofenamic acid; NFA, niflumic acid; SA, salicylic acid; SSZ, sulfasalazine.
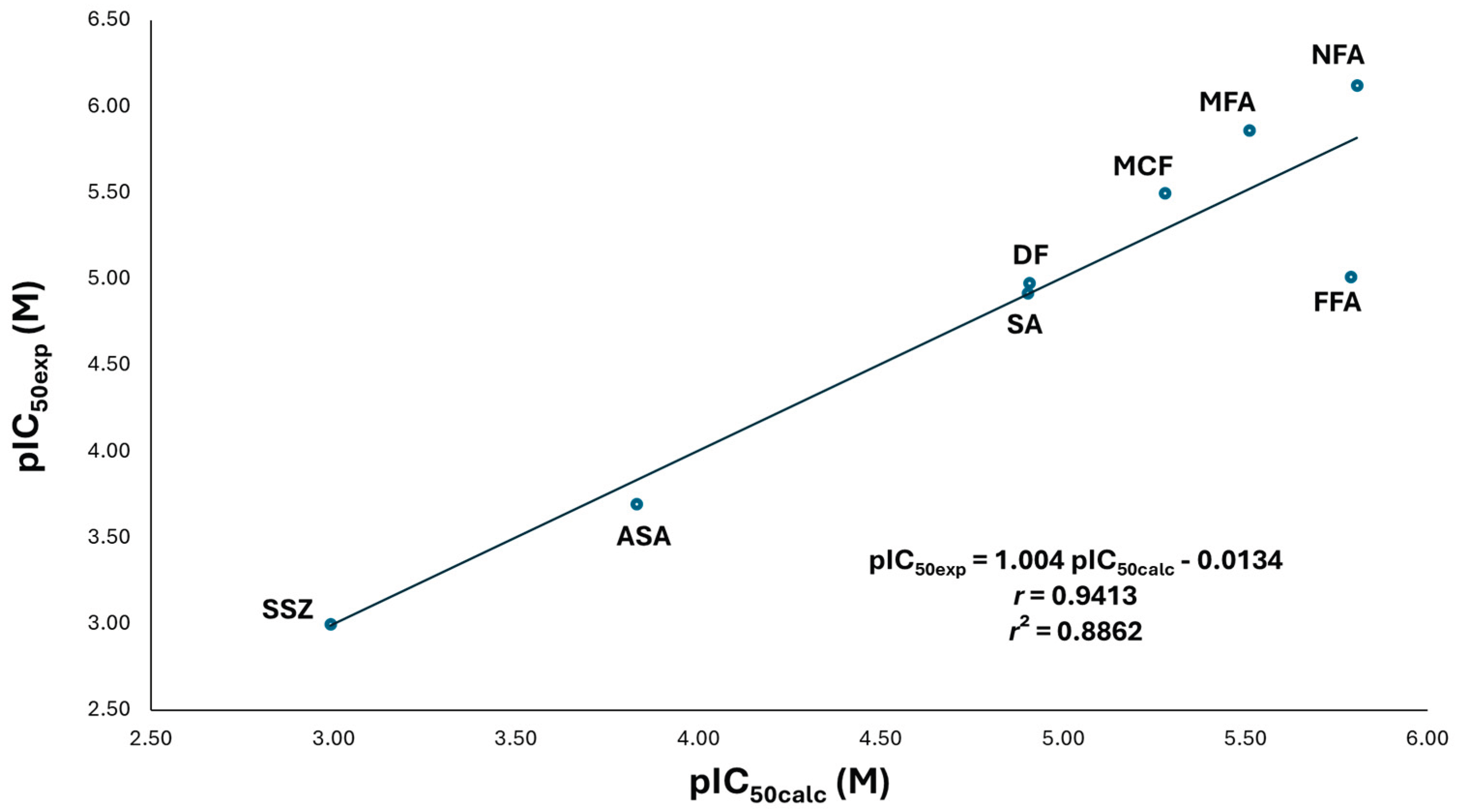
Figure 3.
(A) Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the acetate NSAIDs on COX-1. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 7) for the QSAR model: pIC50 = alogP2-blogP-cELUMO2+dELUMO-elogV+f: a = 0.2714 ± 0.61725 (p = 0.5134), b = -1.6557 ± 3.5867 (p = 0.5134), c = -1678.293 ± 981.2554 (p = 0.5134), d = 192.665 ± 132.41415 (p = 0.5134), e = -9.2588 ± 6.1734 (p = 0.5134), f = 25.5944 ± 14.029 (p = 0.5134) and, r = 0.9492 (p = 0.5134). (B) Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the acetate NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 7) for the QSAR model: pIC50 = alogP2-blogP-clogMR+dlogOv+e: a = 0.54913 ± 0.0299 (p = 0.0044), b = -3.00186 ± 0.1767 (p = 0.0044), c = -190.3265 ± 0.5554 (p = 0.0044), d = 14.0818 ± 0.5454 (p = 0.0044), e = 24.82122 ± 0.38403 (p = 0.0044) and, r = 0.9971 (p = 0.0044). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. DIC, diclofenac; ETD, etodolac; IND, indomethacin; KT, ketorolac; OX, oxamethacin; SUL, sulindac; TOL, tolmentine.
Figure 3.
(A) Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the acetate NSAIDs on COX-1. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 7) for the QSAR model: pIC50 = alogP2-blogP-cELUMO2+dELUMO-elogV+f: a = 0.2714 ± 0.61725 (p = 0.5134), b = -1.6557 ± 3.5867 (p = 0.5134), c = -1678.293 ± 981.2554 (p = 0.5134), d = 192.665 ± 132.41415 (p = 0.5134), e = -9.2588 ± 6.1734 (p = 0.5134), f = 25.5944 ± 14.029 (p = 0.5134) and, r = 0.9492 (p = 0.5134). (B) Quantitative Structure-activity relationship (QSAR) for the model between experimental inhibitory activity (pIC50exp) and the calculated inhibitory activity value (pIC50calc) of the acetate NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 7) for the QSAR model: pIC50 = alogP2-blogP-clogMR+dlogOv+e: a = 0.54913 ± 0.0299 (p = 0.0044), b = -3.00186 ± 0.1767 (p = 0.0044), c = -190.3265 ± 0.5554 (p = 0.0044), d = 14.0818 ± 0.5454 (p = 0.0044), e = 24.82122 ± 0.38403 (p = 0.0044) and, r = 0.9971 (p = 0.0044). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. DIC, diclofenac; ETD, etodolac; IND, indomethacin; KT, ketorolac; OX, oxamethacin; SUL, sulindac; TOL, tolmentine.
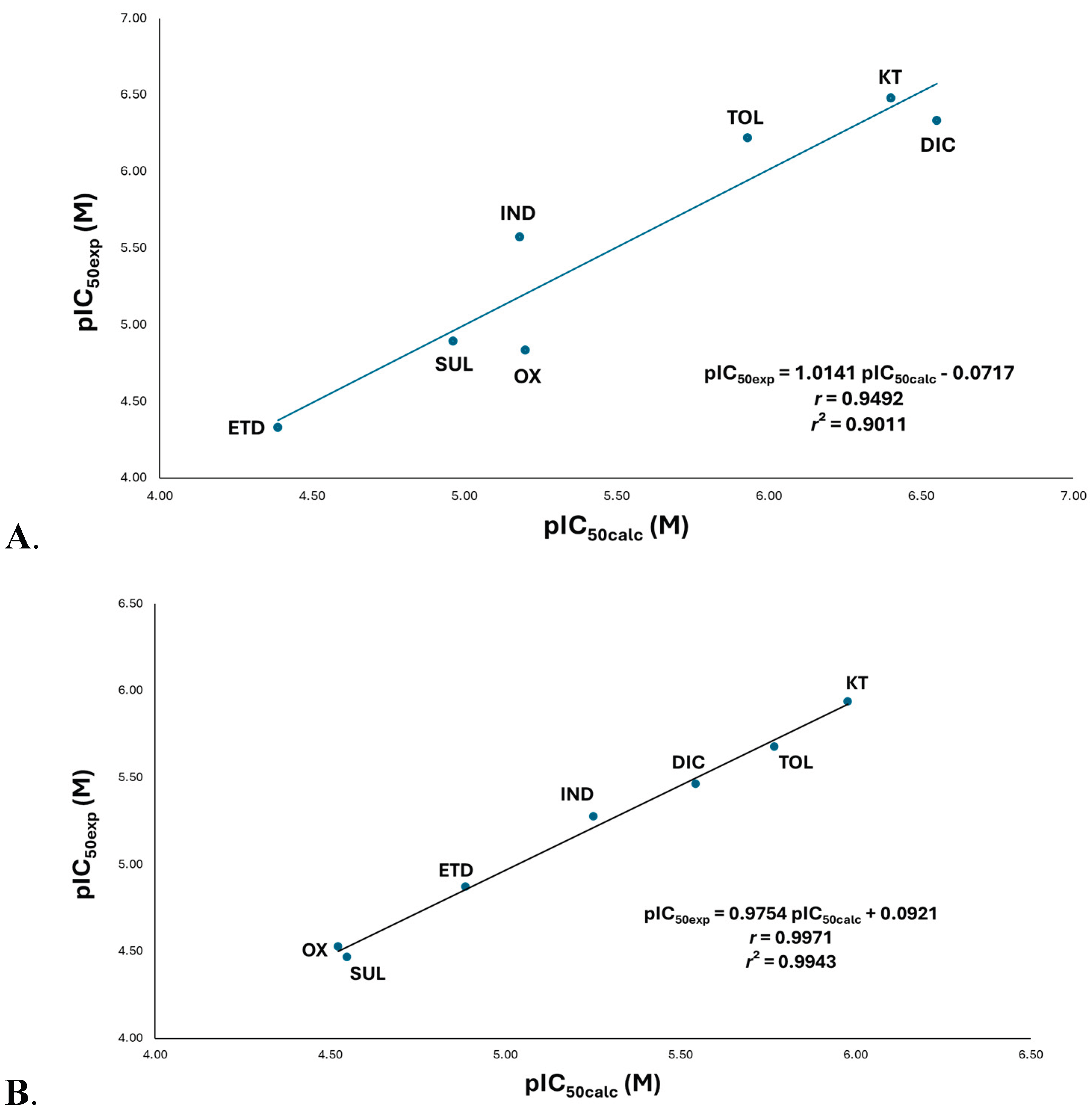
Figure 4.
(A) Binding Mode of DIC, KT, and TOL in the COX-1 Active Site: The analyzed compounds (diclofenac/DIC, ketorolac/KT, and tolmetin/TOL) exhibit conserved interactions in two key regions: Pharmacophoric region: Ionic bonding between the carboxylate group and Arg120 and the accessory moiety: Van der Waals contacts with nonpolar residues (Leu352, Trp387, and other hydrophobic amino acids). (B) Intermolecular interactions of propionates NSAIDs on COX-1, where the carboxylate group interacts ion-ion with Arg120 and Tyr355 with ion-dipole interaction.
Figure 4.
(A) Binding Mode of DIC, KT, and TOL in the COX-1 Active Site: The analyzed compounds (diclofenac/DIC, ketorolac/KT, and tolmetin/TOL) exhibit conserved interactions in two key regions: Pharmacophoric region: Ionic bonding between the carboxylate group and Arg120 and the accessory moiety: Van der Waals contacts with nonpolar residues (Leu352, Trp387, and other hydrophobic amino acids). (B) Intermolecular interactions of propionates NSAIDs on COX-1, where the carboxylate group interacts ion-ion with Arg120 and Tyr355 with ion-dipole interaction.
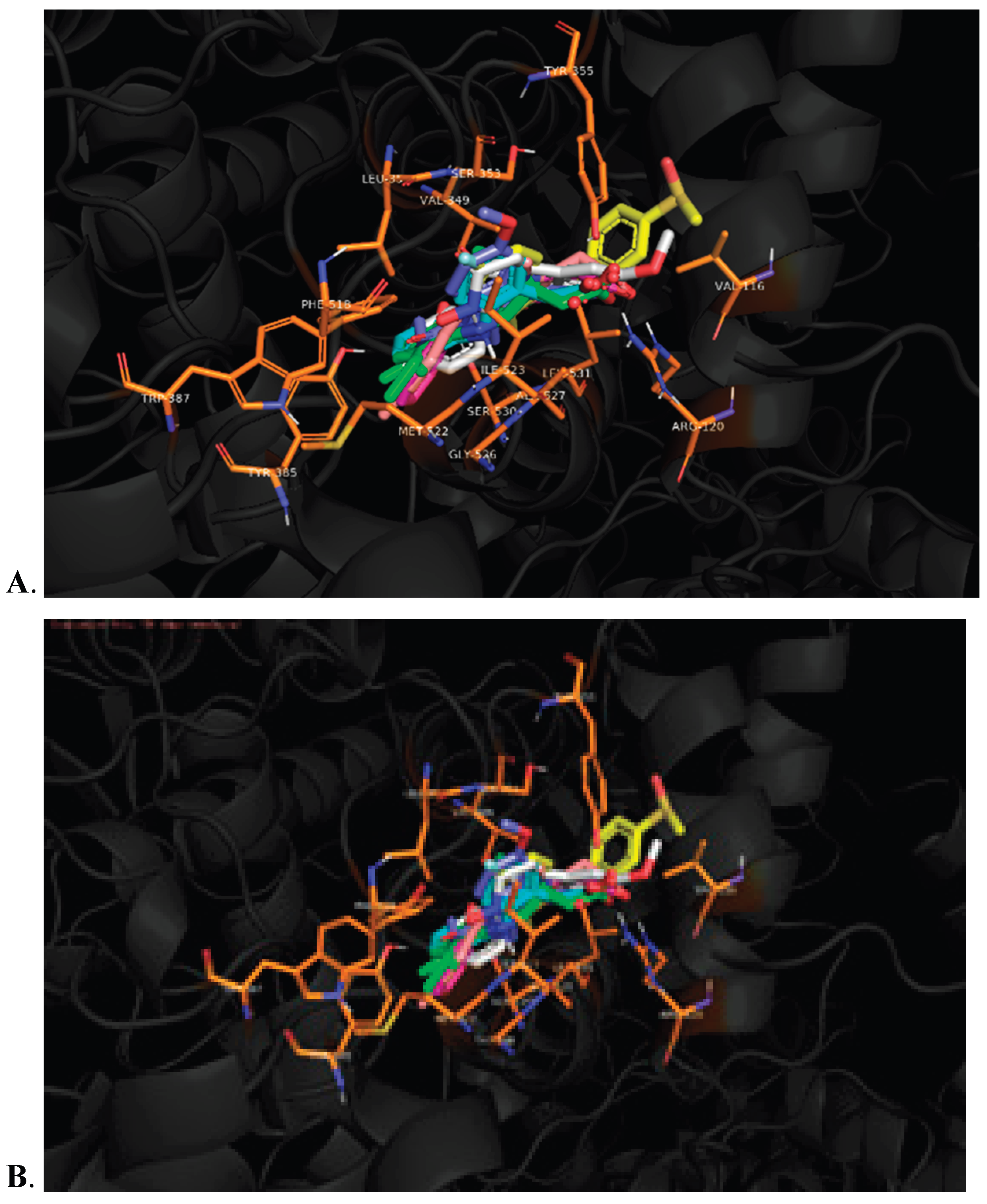
Figure 5.
(A) Quantitative Structure-activity relationship (QSAR) model between experimental inhibitory activity (pIC50exp) and calculated inhibitory activity (pIC50calc) for propionates NSAIDs on COX-1. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 5) for the QSAR model: pIC50 = apKa2-bpKa-clogV+d: a = -44.912 ± 28.1305 (p = 0.5663), b = 426.233 ± 267.5175 (p = 0.5663), c = -7.201 ± 11.907 (p = 0.5663), d = -987.923 ± 608.3335 (p = 0.5663), and, r = 0.8834 (p = 0.5663). (B) Quantitative Structure-activity relationship (QSAR) model between experimental inhibitory activity (pIC50exp) and calculated inhibitory activity (pIC50calc) for propionates NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 5) for the QSAR model: pIC50 = apKa2-bpKa-cEHOMO+d: a = -44.912 ± 28.1305 (p = 0.5663), b = 426.233 ± 267.5175 (p = 0.5663), c = -7.201 ± 11.907 (p = 0.5663), d = -987.923 ± 608.3335 (p = 0.5663), and, r = 0.8834 (p = 0.5663). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. FEN, fenoprofen; FP, flurbiprofen; IBU, ibuprofen; KP, ketoprofen; NAP, naproxene.
Figure 5.
(A) Quantitative Structure-activity relationship (QSAR) model between experimental inhibitory activity (pIC50exp) and calculated inhibitory activity (pIC50calc) for propionates NSAIDs on COX-1. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 5) for the QSAR model: pIC50 = apKa2-bpKa-clogV+d: a = -44.912 ± 28.1305 (p = 0.5663), b = 426.233 ± 267.5175 (p = 0.5663), c = -7.201 ± 11.907 (p = 0.5663), d = -987.923 ± 608.3335 (p = 0.5663), and, r = 0.8834 (p = 0.5663). (B) Quantitative Structure-activity relationship (QSAR) model between experimental inhibitory activity (pIC50exp) and calculated inhibitory activity (pIC50calc) for propionates NSAIDs on COX-2. Multiple linear regression analysis, which analyzed by determinants method and one-way ANOVA; the ordinate values and slopes were examined by the Student’s t test (n = 5) for the QSAR model: pIC50 = apKa2-bpKa-cEHOMO+d: a = -44.912 ± 28.1305 (p = 0.5663), b = 426.233 ± 267.5175 (p = 0.5663), c = -7.201 ± 11.907 (p = 0.5663), d = -987.923 ± 608.3335 (p = 0.5663), and, r = 0.8834 (p = 0.5663). Significance was set at p < 0.05 to achieve a 95.0 % confidence interval. FEN, fenoprofen; FP, flurbiprofen; IBU, ibuprofen; KP, ketoprofen; NAP, naproxene.
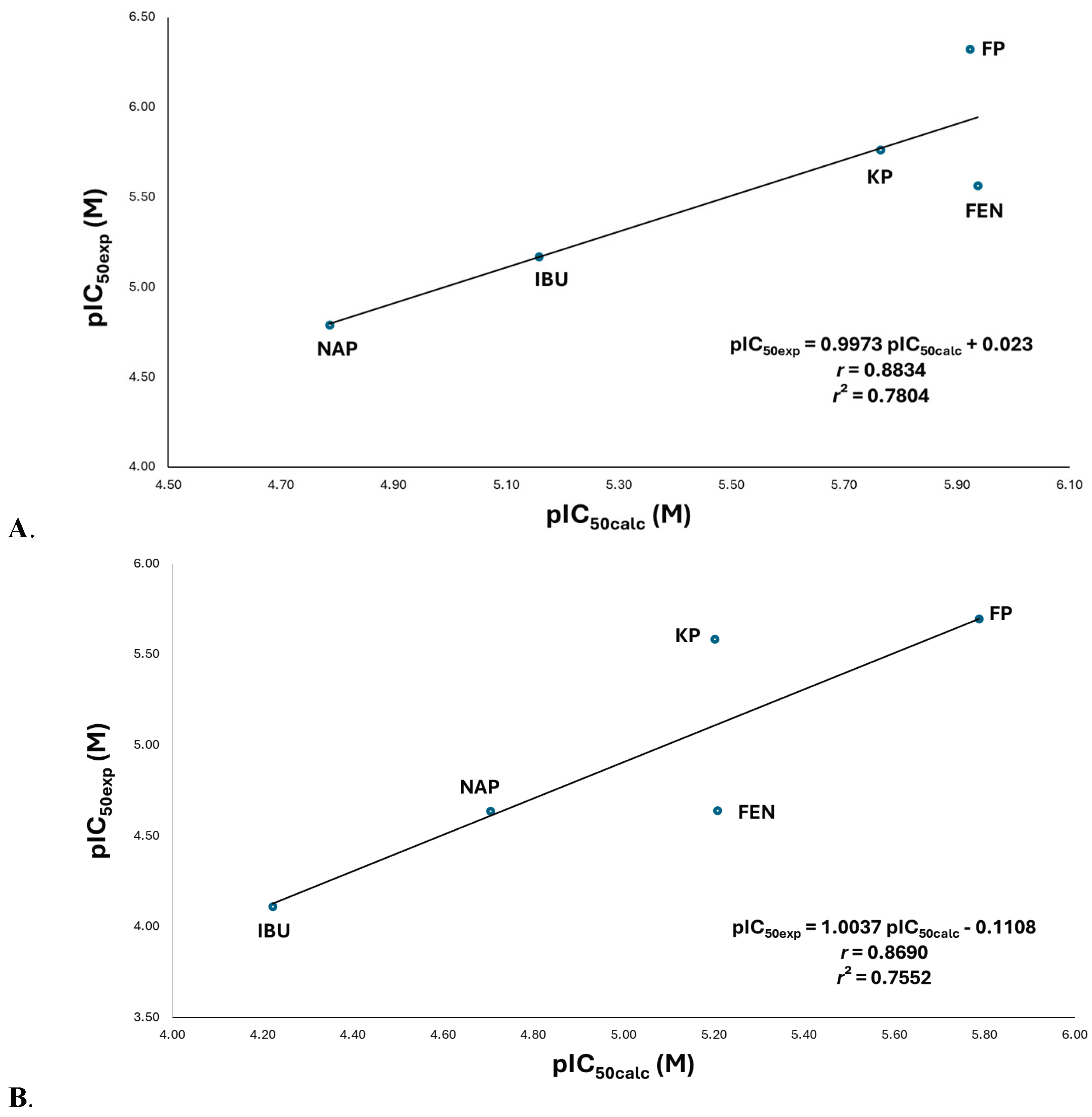
Figure 6.
Molecular moieties identified in the active-site recognition on COX-1 and COX-2. Lilac: Dock and orientation moiety; Yellow: Position 2 moiety; Green: position 3 moiety; Grey: Position 4 moiety.
Figure 6.
Molecular moieties identified in the active-site recognition on COX-1 and COX-2. Lilac: Dock and orientation moiety; Yellow: Position 2 moiety; Green: position 3 moiety; Grey: Position 4 moiety.
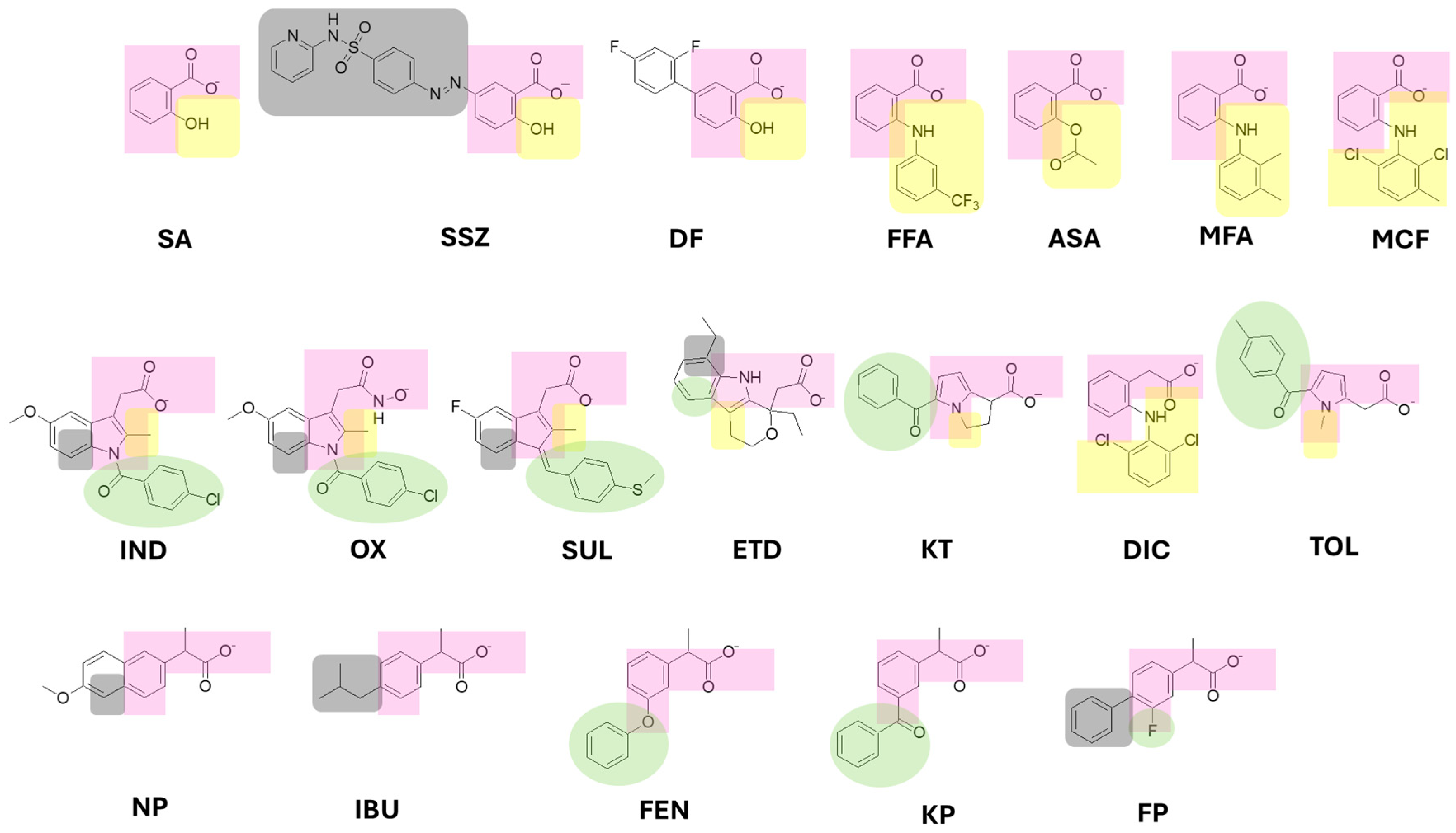
Figure 7.
Pharmacophoric structure and accessory moieties of non-selective COX inhibitors (NSAIDs). In rose: guide moiety; Yellow, green, and grey: accessory moieties.
Figure 7.
Pharmacophoric structure and accessory moieties of non-selective COX inhibitors (NSAIDs). In rose: guide moiety; Yellow, green, and grey: accessory moieties.
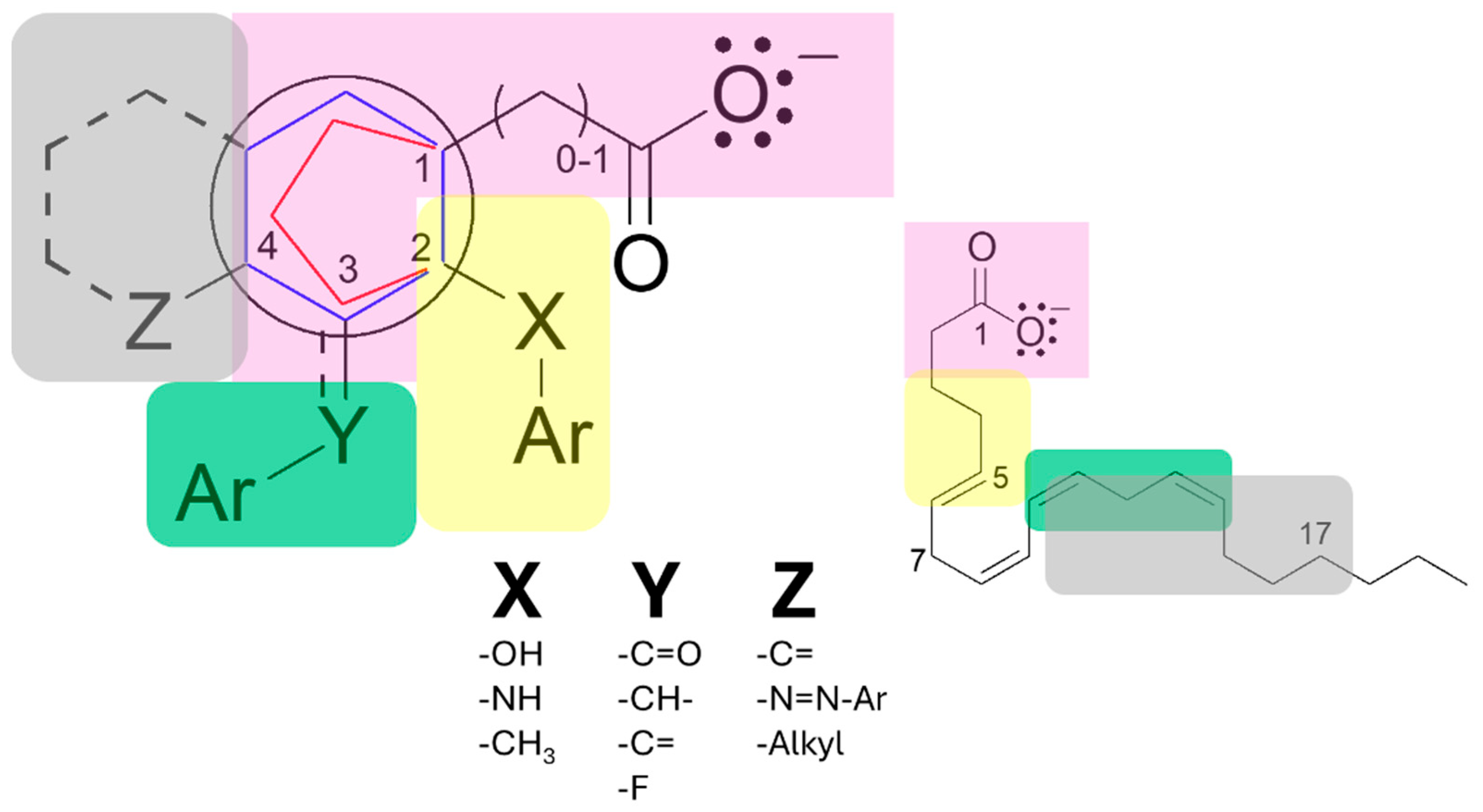

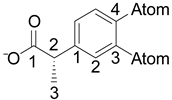
Table 1.
Electronegativity of the atoms at positions 3 and 4 of the phenyl ring in the 2-phenylpropionic acid moiety.
Table 1.
Electronegativity of the atoms at positions 3 and 4 of the phenyl ring in the 2-phenylpropionic acid moiety.
 |
|||
|---|---|---|---|
| Propionate | Position 3, χ | Position 4, χ | χ sum |
| NAP | C, 2.55 | C, 2.55 | 5.10 |
| IBU | H, 2.20 | C, 2.55 | 4.75 |
| KP | C=O, 2.55-3.44 | H, 2.20 | 5.19 |
| FEN | O, 3.44 | H, 2.20 | 5.64 |
| FP | F, 3.98 | C, 2.55 | 6.53 |
Table 2.
Most frequent intermolecular interactions between COX-1 and COX-2 amino acids and the pharmacophoric structure of NSAIDs and arachidonic acid.
Table 2.
Most frequent intermolecular interactions between COX-1 and COX-2 amino acids and the pharmacophoric structure of NSAIDs and arachidonic acid.
| PHARMACOPHORIC STRUCTURE OF NSAIDs ON COX-1 AND COX-2 | ||||
| Guide moiety | Accessory moiety | |||
| NSAIDs | ||||
| Carboxylate | Aryl contains carboxylate | Position 2 | Position 3 | Position 4 |
| Ser119, Arg120, Tyr355, Ser530 | Val349, Ala527, Ile523 (COX-1), Val523 (COX-2) | Phe381, Phe518, Tyr385, Trp387, Ala527 |
Leu352, Tyr385, Ile523 (COX-1), Val523 (COX-2), Ala527 |
Val349, Leu352, Tyr385, Trp387, Met522, Ile523 (COX-1), Val523 (COX-2), Ala527 |
Arachidonic acid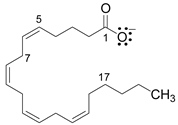
| ||||
| 1: Arg120, Tyr355 | 1-5: Val349, Ala527, Ser353, Ile523 (COX-1) | 7-17: Leu352, Phe381, Tyr385, Met522 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



