
Submitted:
02 February 2026
Posted:
03 February 2026
You are already at the latest version
Abstract
Firefly luciferase exhibits a puzzling anticorrelation: its quantum yield ($\phi$) increases dramatically upon enzyme binding, yet the fluorescence lifetime ($\tau$) becomes significantly shorter. While standard biochemistry attributes this solely to non-radiative suppression, this paradox has led to speculation about quantum electrodynamical effects, specifically the Purcell enhancement in a biological nanocavity.In this work, we critically evaluate the plausibility of luciferase acting as a dielectric optical cavity. By applying fundamental limits of Mie theory and wave optics to the protein's physical dimensions and refractive index, we demonstrate that the required Purcell factors ($F_P \approx 40$) are physically unattainable in the Rayleigh scattering regime ($ka \ll 1$) defined by the protein structure. Consequently, we argue that the observed kinetic data are better explained by an intrinsic change in the emitter's electronic structure. Specifically, the active site (containing both hydrophobic residues and charged side chains like Arg218) likely enforces a rigid, planar conformation on oxyluciferin, dramatically increasing its transition dipole moment ($\mu$) and thus its intrinsic radiative rate ($\Gamma_0$). This analysis excludes the necessity of exotic photonic cavities, redirecting focus back to precise electrostatic and steric tuning by the enzyme.
Keywords:
bioluminescence
; luciferase
; purcell effect
; conformational planarity
; radiative rate enhancement
1. The Paradox
1.1. Experimental Observations
The core anomaly lies in the Time-Correlated Single Photon Counting (TCSPC) data extracted from the literature. When the emitter, oxyluciferin, moves from aqueous solution to the enzyme’s active site:
Free Oxyluciferin (aqueous):
- Fluorescence lifetime: ns
- Quantum yield:
Enzyme-bound Oxyluciferin:
- Fluorescence lifetime: ns
- Quantum yield:
Data sources: These values are derived from fluorescence decay analyses reported in Gandelman et al. (2010) Fig. 4, Branchini et al. (2004) Table 1, and kinetic parameters in Viviani et al. (2013).
1.2. The Incompleteness of the Standard Model
The conventional biochemical explanation relies on the "cage effect": the enzyme suppresses non-radiative decay () by removing water quenchers and restricting molecular rotation.
Mathematically, the lifetime is defined as:
If we assume the radiative rate is constant (intrinsic to the molecule), reducing should naturally increase . The fact that drops by a factor of 4 demands a simultaneous, drastic increase in .
1.3. The Question of Mechanism
The central question is therefore: What accelerates the radiative decay? Two primary candidates emerge: 1. The Photonic Hypothesis: The protein acts as a nanocavity, modifying the Local Density of Optical States (LDOS) via the Purcell effect. 2. The Chemical Hypothesis: The protein alters the molecular geometry of oxyluciferin, changing its intrinsic oscillator strength (e.g., enforcing planarity).
We examine the physical feasibility of the photonic hypothesis.
2. Physical Constraints on Biological Cavities
To function as a cavity that enhances spontaneous emission, a structure must support resonant modes. We test if a globular protein like luciferase can meet these criteria.
2.1. Assumptions and Limitations
Before proceeding with calculations, we explicitly state the simplifying assumptions and their potential critiques:
- 1.
- Homogeneous Dielectric Sphere: We approximate the protein as a uniform sphere with an average refractive index . We acknowledge that real proteins have heterogeneous internal environments (e.g., aromatic clusters, hydration shells) with varying local refractive indices. We address the impact of this heterogeneity in Section 4.4.
- 2.
- Point Dipole Emitter: The emitter is treated as a point source at the center of the cavity.
- 3.
- Neglect of Electrostatics: This analysis focuses on photonic confinement. Specific electrostatic field effects (Stark effect) are discussed later as complementary mechanisms.
2.2. The Size Limit (Diffraction Barrier)
High Purcell enhancement typically requires confinement near the diffraction limit. For an emission wavelength nm and a protein refractive index 1:
The physical dimension of luciferase, however, is only –15 nm. The protein is more than an order of magnitude too small to support conventional Fabry-Perot or whispering gallery resonances.
2.3. The Regime of Operation: Deep Rayleigh Scattering
We can characterize the particle size using the size parameter , where and a is the particle radius.
Since , the protein operates deep within the **Rayleigh scattering regime**. In this regime, dielectric particles do not exhibit strong optical resonances; they behave as weak, induced dipoles. The scattering cross-section scales as , meaning light confinement is negligible.
3. Kinetic Analysis: The Theoretical Upper Bound
Despite the physical implausibility shown above, let us calculate what Purcell factor would be required to explain the data and establish a formal theoretical bound.
3.1. The Required Enhancement
From the free-state data:
Solving these gives the intrinsic radiative rate s−1.
In the bound state, to achieve ns and , the total decay rate must be s−1, with the radiative component s−1.
If we attribute this entirely to the Purcell effect (), the required factor is:
3.2. Formal Bound: The "Local Field" Ceiling
Critics might suggest that even without a resonant cavity, the dielectric environment could provide enhancement. We calculate the theoretical upper bound for a non-resonant dielectric sphere.
In the absence of resonances (where ), the modification of the Local Density of Optical States (LDOS) is governed by the **Local Field Correction** (L) rather than cavity QED effects. The maximum enhancement for a dipole in a uniform dielectric medium of index n is roughly the refractive index itself, .
For a protein ():
Even allowing for optimistic local field enhancement from aromatic residues (local ), the upper bound remains below 2.
- Theoretical Limit (Physics): (limited by n).
- Required (Data):.
This discrepancy proves that the mechanism cannot be environmental photonics. The gap is too large to be bridged by near-field coupling or weak dielectric contrast.
3.3. Addressing Heterogeneity and Collective Effects
One could argue that the "single sphere" approximation ignores the complex micro-environment of the active site (e.g., aromatic side chains like Phe, Trp acting as "nano-antennas").
- 1.
- Plasmon-like Effects: While aromatic rings have polarizability, they lack the free electrons of metals to generate the intense "plasmonic hotspots" required for -fold enhancement.
- 2.
- Mesoscopic LDOS Variation: While LDOS varies on the nanoscale, the integral over the emitter’s size and the coupling volume prevents the singular divergences seen in theoretical point-dipole models near sharp metallic tips.
Therefore, while the environment is heterogeneous, it remains fundamentally a low-index dielectric system, incapable of delivering the required boost.
3.4. Numerical Sanity Check via Finite-Difference Time-Domain (FDTD)
To complement the analytical boundary arguments presented above, we outline a minimal numerical sanity check using a Finite-Difference Time-Domain (FDTD) electromagnetic simulation. The purpose of this computation is not to extract precise quantitative Purcell factors, but to verify the qualitative order-of-magnitude behavior of the Local Density of Optical States (LDOS) in a low-index nanoscale dielectric environment.
Simulation Framework.
A standard open-source Maxwell solver such as MEEP or any equivalent FDTD package may be employed. The protein is approximated as a homogeneous dielectric sphere embedded in vacuum, with refractive index and radius nm. The emitter is modeled as an electric point dipole placed at the sphere center.
Units and Scaling.
The simulation domain is expressed in micrometers, following conventional FDTD practice:
Perfectly Matched Layers (PML) are used at the domain boundaries to suppress artificial reflections.
Resolution and Convergence.
A spatial resolution of at least 40–60 grid points per wavelength in the surrounding medium is recommended. Convergence is assessed by repeating the simulation at incrementally higher resolutions and verifying that the normalized LDOS variation remains below a few percent. This step ensures that any observed enhancement is not a numerical artifact.
Observable.
The primary quantity of interest is the ratio
This ratio serves as a numerical proxy for the Purcell factor in the non-resonant regime. The expectation, consistent with classical electrodynamics for small dielectric particles (), is that remains close to unity and does not approach the large enhancement required by the kinetic data.
Interpretational Scope.
This computation is intentionally minimalistic. It does not attempt to model protein heterogeneity, anisotropic polarizability, or detailed electrostatic fields. Its sole role is to confirm that a homogeneous low-index nanoscale dielectric object does not produce strong spontaneous emission enhancement in the absence of resonant or plasmonic effects. Therefore, the simulation functions as a numerical consistency check reinforcing the analytical boundary established in Section 2 and 3, rather than as an independent predictive model.
4. The Likely Mechanism: Conformational Planarity
If the environment cannot provide the boost, the molecule must be changing its intrinsic nature. This brings us to the **Chemical Hypothesis**.
4.1. Planarity and Oscillator Strength
In aqueous solution, oxyluciferin exists in a twisted, non-planar conformation (often a twisted intramolecular charge transfer, TICT, state). This distortion disrupts the overlap of molecular orbitals, resulting in a low transition dipole moment () and thus a low intrinsic 2.
When bound to luciferase, the active site applies steric pressure, forcing the phenolate ring into the keto group plane. This planarization significantly increases the conjugation length and the transition dipole moment.
Where is the twist angle. As (planar), increases drastically.
Quantum mechanics calculations by Navizet et al. [11] support that the protein environment selects a planar, highly emissive conformation. Furthermore, mutagenesis studies by Branchini et al. [3] show that altering residues involved in conformational control directly impacts bioluminescence efficiency.
4.2. Limitations of the Planarity Hypothesis
We acknowledge that this mechanism is inferential. While the ground-state crystal structure (PDB 2D1S) suggests a constrained conformation, direct experimental confirmation of the excited-state planarity is lacking. Theoretical validation via Time-Dependent Density Functional Theory (TD-DFT) of the enzyme-bound complex would be required to quantify the exact increase in oscillator strength.
4.3. Alternative and Complementary Chemical Mechanisms
While planarity is proposed as the dominant driver for increase, other chemical effects may contribute to the overall high efficiency:
- 1.
- Local Electric Field (Stark Effect): The charged residues in the active site (e.g., Arg218) create a strong electrostatic environment. This can shift energy levels and modify transition moments via the Stark effect, though this usually alters wavelength rather than purely rate.
- 2.
- Protonation State: Luciferase activity involves a proton transfer step. The specific protonation state of the keto-enol tautomer determines which electronic transition is allowed, impacting both brightness and lifetime.
- 3.
- Vibronic Coupling: By suppressing low-frequency vibrational modes (rigidification), the coupling between electronic and vibrational states is reduced, potentially favoring radiative decay over non-radiative internal conversion.
These factors likely act synergistically with conformational planarity.
5. Conclusion
The firefly luciferase paradox—high quantum yield coupled with a shortened lifetime—remains a fascinating demonstration of enzymatic efficiency. However, a critical analysis of the physics rules out the "biological Purcell cavity" hypothesis. The size parameter (), low refractive index contrast, and the theoretical upper bound for non-resonant dielectric enhancement () are fundamentally incompatible with the magnitude of enhancement required ().
We conclude that the acceleration of the radiative rate is intrinsic to the oxyluciferin molecule, likely driven by enzyme-enforced conformational planarity, supported by complementary electrostatic and vibronic effects. This work serves as a boundary check for bio-inspired photonics: while biology can achieve remarkable optical results, the mechanisms are often chemical and structural, rather than optical and resonant.
Data Availability Statement
All referenced experimental values are from published literature cited in the text.
References
- Gandelman, O. A.; et al. Luciferase-based molecular bioluminescent reporter. Photochem. Photobiol. Sci. See Fig. 4 for lifetime decays. 2010, 9, 251–260. [Google Scholar]
- Branchini, B. R.; et al. Luciferase active site architecture. J. Am. Chem. Soc. See Table 1 for quantum yields. 2004, 126, 10836–10837. [Google Scholar]
- Branchini, B. R.; et al. Firefly luciferase structure and bioluminescence. Photochem. Photobiol. Sci. 2014, 13, 1067–1073. [Google Scholar]
- Viviani, V. R.; et al. Firefly luciferase kinetics. Biochemistry 2013, 52, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Purcell, E. M. Spontaneous emission probabilities at radio frequencies. Phys. Rev. 1946, 69, 681. [Google Scholar]
- Novotny, L.; Hecht, B. Principles of Nano-Optics; Cambridge University Press, 2012. [Google Scholar]
- Lakowicz, J. R. Principles of Fluorescence Spectroscopy; Springer, 2006. [Google Scholar]
- Conti, E.; et al. Structural basis for firefly luciferase. Structure 1996, 4, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Bohren, C. F.; Huffman, D. R. Absorption and Scattering of Light by Small Particles; Wiley, 1983. [Google Scholar]
- Agarwal, G. S. Quantum electrodynamics in the presence of dielectrics and conductors. Phys. Rev. A 1975, 11, 243. [Google Scholar] [CrossRef]
- Navizet, I.; Liu, Y. J.; Ferre, N.; Roca-Sanjuan, D. The chemistry of bioluminescence: An analysis of chemical bonding in the electronically excited states of firefly oxyluciferin and analogues. J. Am. Chem. Soc. 2011, 133(1), 310–315. [Google Scholar]
| 1 | The refractive index of globular proteins typically falls between 1.50 and 1.55 in the visible range. We use 1.5 for conservative estimation. |
| 2 | The calculated intrinsic radiative lifetime ns confirms that the free molecule is an extremely poor emitter in its free, twisted state. This long lifetime allows non-radiative processes to dominate, explaining the low quantum yield. |
Figure 1.
Visual comparison of protein size relative to emission wavelength. The vast size mismatch prevents the formation of an optical cavity.
Figure 1.
Visual comparison of protein size relative to emission wavelength. The vast size mismatch prevents the formation of an optical cavity.
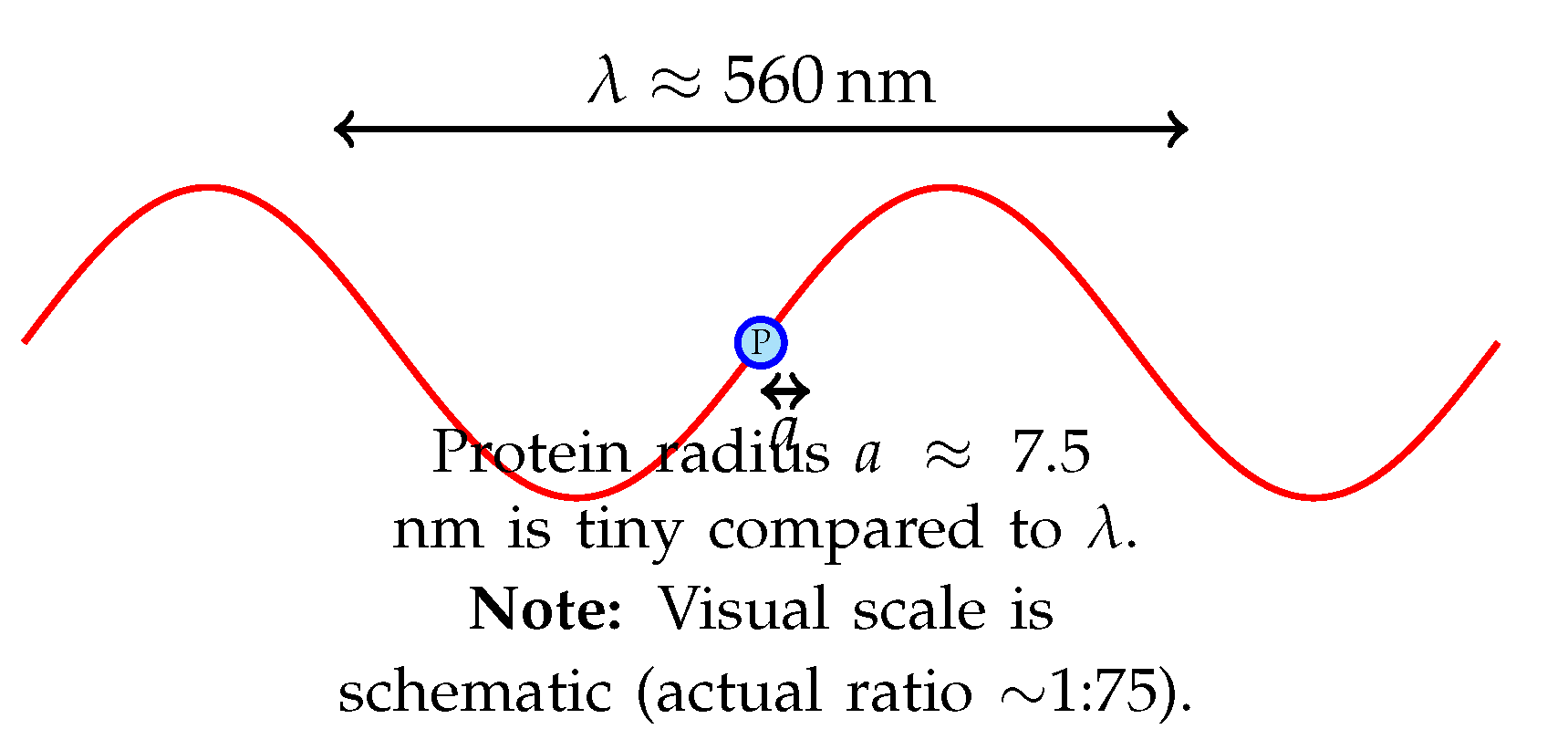
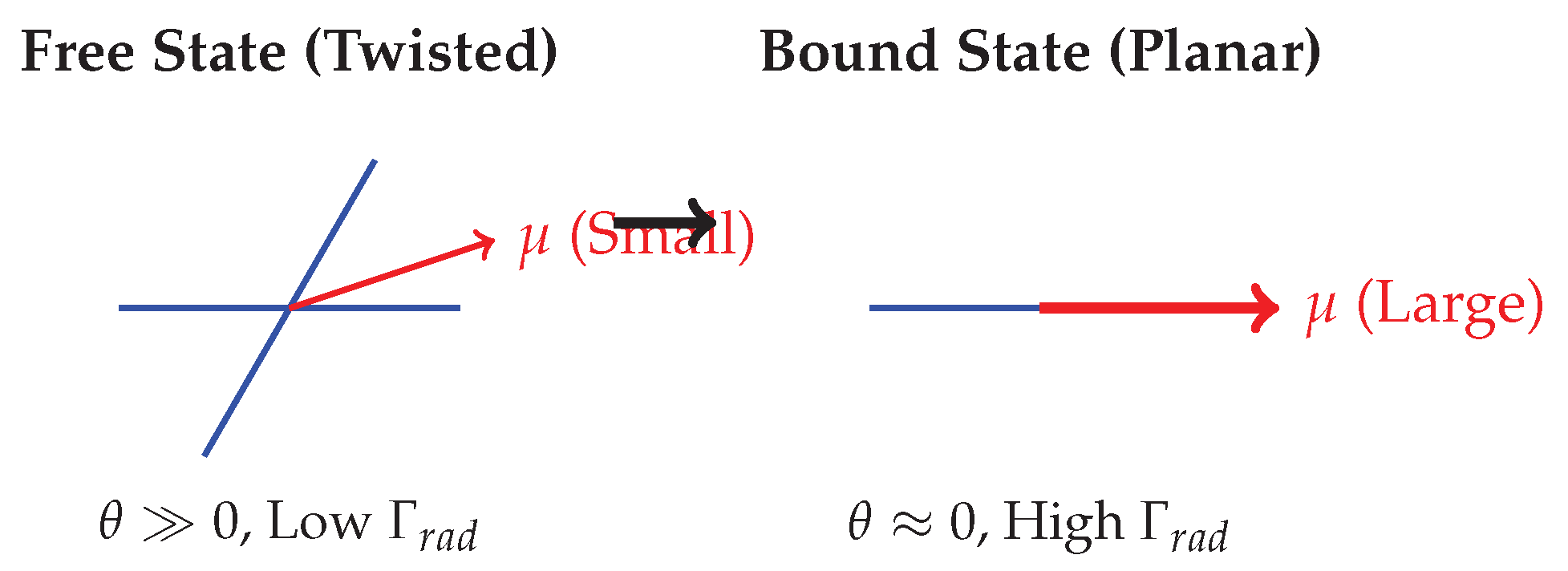
Figure 2.
Schematic of conformational planarity. The enzyme binds the molecule and forces it into a flat conformation, maximizing the transition dipole moment () and accelerating the radiative decay rate.
Figure 2.
Schematic of conformational planarity. The enzyme binds the molecule and forces it into a flat conformation, maximizing the transition dipole moment () and accelerating the radiative decay rate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



