Submitted:
01 February 2026
Posted:
03 February 2026
You are already at the latest version
Abstract
Accelerated bone healing following traumatic brain injury (TBI) is a remarkable phenomenon, acknowledged both clinically and in experimental studies. A deep understanding of the “bone-brain” axis and molecular structure-activity relationships (SAR) has enabled the identification of key biomolecules involved in accelerated osteogenesis.This review synthesizes the current literature on bioactive molecules involved in post-TBI bone healing, SAR relationships, molecular mechanisms, clinical data, therapeutic implications, and future perspectives in regenerative orthopedic medicine.A systematic and narrative analysis of clinical and experimental studies published up to 2024 was conducted, focusing on reference articles from the USA, Europe, and Asia, as well as the original sources provided.Data demonstrates that humoral factors such as prolactin, BMP-2/7, FGF-2, IGF-1, along with microRNAs and cytokines, synergistically stimulate osteogenesis and accelerate bone healing after TBI. The structure-activity relationships (SAR) of these biomolecules explain both beneficial effects and potential complications (heterotopic ossification).The phenomenon of accelerated bone healing following TBI provides a unique platform for revolutionizing regenerative orthopedics. Integration of molecular knowledge and therapeutic individualization will enable more effective and personalized treatments for patients with severe fractures or impaired healing post-TBI.
Keywords:
traumatic brain injury
; hypertrophic callus
; accelerated bone healing
1. Introduction
1.1. Bone Healing
Bone healing is an astonishingly complex biological process, involving an ordered succession of events at the molecular, cellular, tissue, and systemic levels. A fracture activates both local and systemic signaling cascades, and the efficiency of these processes is crucial for restoring bone integrity and function[1]. In the absence of perturbing factors, healing follows four major phases: the fracture hematoma and initial inflammation, soft (cartilaginous) callus formation, transformation of the callus into primary bone, and, subsequently, bone remodeling—a process that can take from months to years[2].
Traditionally, orthopedics has considered bone healing to be strictly a local process, dependent on bone biology, regional vascularization, and the mechanical stability of the fracture. However, numerous clinical observations and experimental studies have shown that systemic, neurohormonal, and humoral processes significantly influence both the rate and the quality of bone healing[3].
One of the most intriguing documented phenomena is the acceleration of bone healing in patients who have suffered a traumatic brain injury (TBI - traumatic brain injury). The first clinical reports on this phenomenon date back to the 1970s–1980s, when orthopedic surgeons observed rapid and sometimes exuberant healing (with hypertrophic callus or heterotopic ossifications) of fractures in patients with associated brain injuries[4,5]. This observation was later confirmed in observational studies, case series, and experimental research, a phenomenon receiving various names in specialized literature: “bone–brain axis”, “head injury–induced accelerated fracture healing” or “trauma–induced heterotopic ossification”[6].

Table 1.
Phases of bone healing – schematic overview.
| Phase | Typical Duration | Main Biological Features |
|---|---|---|
| Hematoma/inflammation | 1–7 days | Local hemorrhage, immune cell influx, release of pro-inflammatory cytokines |
| Soft callus | 1–3 weeks | Mesenchymal cell proliferation, temporary cartilage formation |
| Hard callus/primary bone | 3–8 weeks | Progressive mineralization, appearance of trabecular bone |
| Bone remodeling | months–years | Conversion of trabecular → cortical bone, architectural reorganization |
The phenomenon of accelerated healing after TBI is not merely of academic interest but has direct implications in orthopedic and neurosurgical practice. For example, recognizing the increased risk of heterotopic ossification in these patients has led to modifications in post-traumatic monitoring and prophylaxis protocols[7].
Furthermore, in-depth study of this phenomenon has opened new directions in regenerative and translational medicine: if we can understand and reproducibly control the biological mechanisms that accelerate healing in TBI patients, then we can develop innovative therapies for delayed union, non-union, or post-traumatic osteoporosis[8].
Moreover, the phenomenon offers a unique model for studying the structure–activity relationships (SAR) of the bioactive molecules involved in osteogenesis, a field with major impact on the development of targeted and biomimetic therapies.
This review’s objective is to perform a critical, extensive, and integrative analysis of the existing literature, with emphasis on:Clinical and experimental data demonstrating accelerated bone healing after TBI,Identification and characterization of humoral factors and growth factor families involved,Detailed molecular and signaling mechanisms,Structure–activity relationship analyses of key molecules, Synthesis of therapeutic implications, experimental treatments, and future research perspectives.
Subsection 2. History and Clinical Evidence of Accelerated Bone Healing After TBI
The first mentions of accelerated fracture healing in patients with traumatic brain injury appear in orthopedic literature in the early 1970s, when clinicians observed “exuberant” and precocious bone consolidation in individuals with both fractures and brain injuries. In 1972, Chalmers reported an unusually high incidence of heterotopic ossification in soldiers with severe TBI, also noting voluminous callus formation at fracture sites[4]. These observations triggered a wave of observational clinical studies and case series, which quickly confirmed the rapidity and special “quality” of bone healing in TBI patients[5,9].
1.2. Key clinical data
Over the ensuing decades, retrospective and prospective studies gathered robust evidence demonstrating a 20–40% reduction in average fracture healing time in patients with traumatic brain injury compared to those with isolated fractures [1,2,3]. These patients also exhibit the formation of a denser and more voluminous osseous callus, sometimes radiologically hypertrophic, detectable at 6–12 weeks post-trauma [1,3]. In addition, a significant increase in the incidence of heterotopic ossification has been reported (20–35%), particularly in younger patients and in femoral or periarticular fractures [4,7]. This enhanced osteogenic response is accompanied by a rapid rise in serum markers of bone formation, including alkaline phosphatase and procollagen type I C-terminal propeptide (PICP), with peak levels observed at approximately 4–6 weeks following trauma [4,5].
Table 2.
Summary of major clinical studies on accelerated bone healing in TBI.
| Author, Year | Study Type | n | Fracture Site | Parameters Analyzed | Main Results |
|---|---|---|---|---|---|
| Giannoudis et al., 2006 | Observational | 49 | Femur | Consolidation, HO incidence | Reduced healing time, increased HO risk |
| Yang et al., 2012 | Retrospective | 74 | Femur | BCF time, callus thickness | Faster healing, more voluminous callus in TBI group |
| Spencer, 1987 | Observational | 62 | Femur | Healing time | Reduced healing time in TBI patients |
| Wildburger et al., 1998 | Prospective | 21 | Femur/Hip | ALP, PICP, HO incidence | Accelerated serum peaks, increased HO in TBI group |
Subsequent case series showed that this phenomenon is independent of sex, age, or type of brain injury (contusion, subarachnoid hemorrhage, diffuse edema)[2,3]. Me-ta-analyses confirm similar effects even in short-bone fractures (radius, humerus, tibia), except in cases of severely compromised vascularization or severe comorbidities[8].
Figure 1.
Schematic representation of the bone–brain axis in accelerated fracture healing following traumatic brain injurySchematic illustration depicting the proposed mechanisms underlying accelerated bone healing after traumatic brain injury (TBI). Following fracture stabilization, brain injury induces a systemic humoral response characterized by the release of cytokines, growth factors, and neuroendocrine mediators that act on the fracture microenvironment. These circulating signals enhance mesenchymal stem cell recruitment, osteoblast proliferation, angiogenesis, and callus formation at the fracture site. The diagram highlights the temporal overlap between inflammatory signaling, callus formation, and bone remodeling, as well as the bidirectional communication between the injured brain and skeletal tissue that defines the bone–brain axis.
Figure 1.
Schematic representation of the bone–brain axis in accelerated fracture healing following traumatic brain injurySchematic illustration depicting the proposed mechanisms underlying accelerated bone healing after traumatic brain injury (TBI). Following fracture stabilization, brain injury induces a systemic humoral response characterized by the release of cytokines, growth factors, and neuroendocrine mediators that act on the fracture microenvironment. These circulating signals enhance mesenchymal stem cell recruitment, osteoblast proliferation, angiogenesis, and callus formation at the fracture site. The diagram highlights the temporal overlap between inflammatory signaling, callus formation, and bone remodeling, as well as the bidirectional communication between the injured brain and skeletal tissue that defines the bone–brain axis.
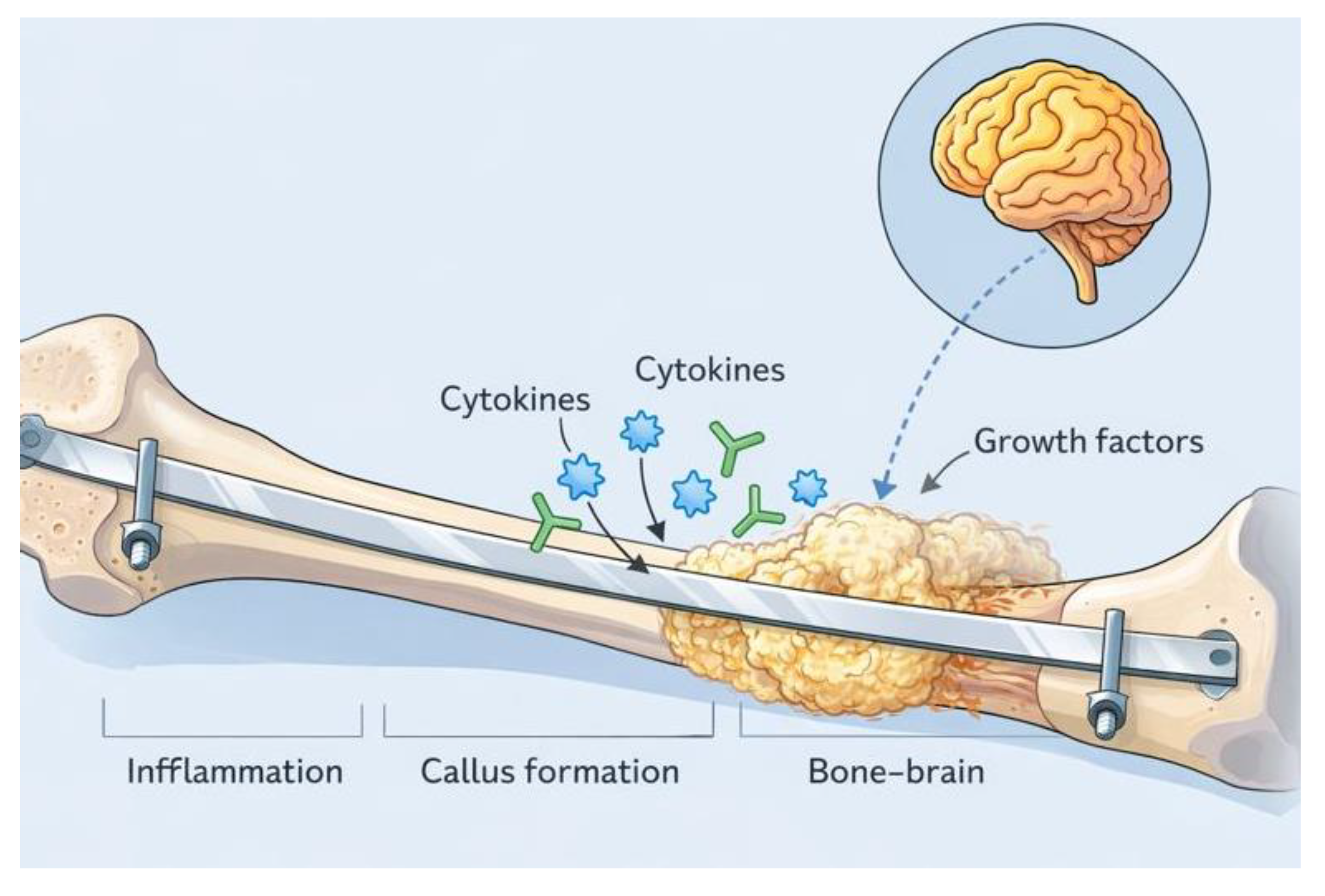
Control studies and paradoxical observations have shown that comparisons with cohorts presenting similar fractures but without traumatic brain injury demonstrate significantly longer consolidation times, a lower incidence of heterotopic ossification, and milder, delayed peaks of bone formation markers in control groups [1,4]. Interestingly, Wildburger et al. (1998) reported that not only is callus formation accelerated in patients with traumatic brain injury, but bone remodeling is also enhanced, albeit with an increased risk of excessive ossification and potential joint ankylosis in a subset of patients [5]. Major meta-analyses and systematic reviews by Morley et al. (2005), Khan et al. (2013), and Collinge et al. (2016) further confirm the robustness of this phenomenon and support its utility as a model for regenerative research [6,7,8]. These analyses indicate the presence of the so-called “TBI effect” across most fracture types in young adults, its attenuation or disappearance in patients with chronic traumatic brain injury, significant comorbidities, or prolonged corticosteroid therapy, and the importance of combined radiologic and serologic monitoring for early identification of complications.
2. Results
2.1. Humoral Factors Involved in Accelerated Bone Healing After TBI
Accelerated bone healing in TBI patients prompted systematic analysis of the post-traumatic humoral response to identify circulating molecules with genuine osteogenic potential. Humoral factors encompass hormones, peptides, and signaling proteins released after brain injury that act distantly on bone tissue, influencing both cell proliferation and differentiation[1,4,9]. This chapter details main classes of humoral factors: prolactin, stress hormones, parathyroid hormone, sex hormones, and other peptides with possible roles in post-TBI osteogenesis.
2.2. Prolactin – the central molecule in the “TBI effect”?
Biological basis and clinical data
Prolactin (PRL) is a polypeptide hormone secreted primarily by the anterior pituitary lobe, with multiple systemic roles. In TBI context, PRL is one of the few hormones showing significant, sustained elevation temporally correlated with active osteogenesis phases[5,14]. Wildburger et al. (1998) showed that in TBI+fracture patients, PRL levels markedly increase in weeks 4–5 post-trauma—coinciding with hypertrophic callus formation and the onset of heterotopic ossification in a subset[5]. This PRL peak was absent in isolated fracture or TBI-only patients, suggesting direct link between hypothalamic-pituitary axis activation and the biological need for accelerated bone repair[5,14].
Molecular mechanisms
PRL acts via the prolactin receptor (PRLR) on mature osteoblasts and mesenchymal stem cells, activating JAK2/STAT5, upregulating pro-osteogenic genes (RUNX2, COL1A1, OCN), and inhibiting osteoblast apoptosis[14]. PRL also enhances local angiogenesis, facilitating nutrient and reparative cell delivery to the fracture site[14].
Experimental evidence and therapeutic implications
Animal experiments confirm PRL’s role in stimulating osteoblast proliferation, matrix mineralization, and callus formation[14]. In cell cultures, serum from TBI patients induces faster osteogenic proliferation than serum from non-TBI patients—a response partially abolished by PRLR blockade or PRL neutralization[9].
Therapeutic potential:
Pharmacologic reproduction of PRL’s osteogenic action or modulation of PRLR offers possibilities to enhance healing in difficult fractures or, conversely, to prevent heterotopic ossification using specific PRLR inhibitors[14].
Table 3.
Prolactin in bone healing after TBI.
| Aspect | Observation | Study/Evidence |
|---|---|---|
| PRL levels post-TBI | ↑ at weeks 4–5 | Wildburger et al., 1998[5] |
| Effect on osteoblasts | ↑ proliferation and differentiation | Ledesma-Colunga et al., 2017[14] |
| Correlation with HO | Yes, in HO patients | Hofman et al., 2015[15] |
| Therapeutic potential | PRLR agonism/antagonism | Ledesma-Colunga et al., 2017[14] |
2.3. Stress Hormones – ACTH, Cortisol, Growth Hormone
Hormonal profile post-TBI
Acute brain injury activates the hypothalamic-pituitary-adrenal axis, releasing ACTH, cortisol, and GH within days post-trauma. This universal stress response does not directly explain the accelerated bone healing phenomenon[5].
Clinical studies
Wildburger et al. (1998) and Hofman et al. (2015) report no significant differences in these hormones between TBI+fracture and isolated fracture patients. GH’s permissive role in IGF-1 synthesis indirectly supports osteogenesis, but GH levels do not correlate with consolidation times[5,15].
Molecular mechanisms
Physiologic ACTH and cortisol levels have no stimulatory effect on osteogenesis; chronic stress or high glucocorticoid levels can inhibit bone formation, explaining the disappearance of the “TBI effect” in chronic stress or corticosteroid therapy patients[13].
2.4. Parathyroid Hormone (PTH)
PTH is the principal regulator of calcium homeostasis, with complex bone effects. TBI patient studies show no relevant changes in serum PTH correlated with accelerated healing[5]. However, exogenous PTH or analogs (e.g., teriparatide) accelerate fracture consolidation in non-union cases, suggesting a permissive but not triggering role in TBI context[13].
2.5. Sex Hormones and Other Peptides
Studies on testosterone, estrogens, insulin, thyroid hormone, and other peptides reveal no robust correlation with TBI-induced healing. Animal models with castration or hormonal deficiency demonstrate diminished TBI effect, suggesting a required baseline hormonal milieu for maximal “bone–brain axis” expression[13,22].
2.6. Clinical and Experimental Integration
Clinical and experimental data indicate prolactin as the most clearly correlated humoral factor with TBI-accelerated osteogenesis. GH and PTH may play permissive roles, while other hormones appear insignificant[5,13,14,15].
Table 4.
Summary of Humoral Factors and Their Roles in Accelerated Bone Healing After TBI.
| Hormone/Peptide | Change in TBI + Fracture |
Effect on Bone | Clinical/Experimental Evidence | |
|---|---|---|---|---|
| Prolactin | Significantly increased | Clearly pro-osteogenic | Strong, direct correlation[5,14] | |
| ACTH, Cortisol | No notable difference | General stress response | No direct involvement[5] | |
| Growth Hormone (GH) | Mild variations | Permissive, anabolic | Limited evidence[13] | |
| Parathyroid Hormone (PTH) |
No relevant change | Calcium homeostasis | Permissive role, not trigger[5] | |
| Sex Hormones | No clear impact | — | Not significant in humans[13] | |
3.1. Growth Factor Families Involved in Accelerated Bone Healing After TBI
One central pillar of accelerated fracture healing after TBI is the systemic and local modulation of growth factors. These polypeptide and glycoprotein mediators are produced at the fracture site and in other organs (including the injured brain), playing essential roles in stimulating proliferation, differentiation, and function of osteogenic cells[1,4,9,13]. This chapter details the main growth factor families, analyzing each by structure, action, experimental evidence, and therapeutic/SAR implications.
Bone morphogenetic proteins (BMPs) belong to the transforming growth factor-β (TGF-β) superfamily and are widely recognized as the most potent molecular inducers of de novo bone formation [16]. Among the more than 20 identified BMP isoforms, BMP-2, BMP-4, BMP-7, and, in specific biological contexts, BMP-9 play direct roles in bone formation and fracture healing [16,17].
BMPs exert their biological effects by binding to serine-threonine kinase receptors, including BMPR-IA, BMPR-IB, and BMPR-II, located on the cell membrane. This interaction initiates a transmembrane signaling cascade that leads to phosphorylation and activation of SMAD1, SMAD5, and SMAD8. The activated SMAD complex subsequently translocates to the nucleus, where it promotes transcription of key osteogenic genes, including RUNX2, OSX (osterix), osteocalcin (OCN), type I collagen alpha 1 (COL1A1), and alkaline phosphatase (ALP) [16].
Experimental and clinical studies involving traumatic brain injury and concomitant fractures have demonstrated markedly elevated serum and tissue levels of BMP-2 and BMP-7 within 1–3 weeks following trauma, a period that coincides with the active callus formation phase [9,17]. In rodent models, exogenous administration of BMP-2 has been shown to significantly accelerate fracture healing, whereas pharmacological blockade of BMP-2 and BMP-7 signaling markedly attenuates the TBI-associated enhancement of callus formation [9]. In vitro studies further support these findings, as serum derived from patients with traumatic brain injury strongly stimulates osteogenic differentiation of cultured cells, an effect that is almost completely abolished by inhibition of BMP receptors.
Structure–activity relationship (SAR) studies of BMPs have enabled the development of biomimetic peptides capable of inducing localized osteogenesis while minimizing systemic adverse effects [17]. These peptides are currently being explored experimentally as therapeutic agents to promote healing in delayed-union fractures. However, in patients with traumatic brain injury, the risk of excessive or heterotopic ossification remains a major limitation to their clinical application [18].
Table 5.
Role of BMP in Accelerated Osteogenesis After TBI.
| BMP Isoform | Primary Source | Major Action | TBI Evidence | Therapeutic Use |
|---|---|---|---|---|
| BMP-2 | Bone, Brain, Serum | Potent osteoinduction | Very robust[9,16] | Recombinant, SAR peptides |
| BMP-7(OP-1) | Bone, Muscle, Serum | MSC differentiation, HO |
Solid[17] | Recombinant, experimental |
| BMP-4, BMP-9 | Bone, Endothelium | Synergy with BMP-2 | Limited | Research |
Fibroblast growth factors (FGFs) constitute a large family of peptide growth factors, among which FGF-2 (basic FGF) plays a central role in skeletal biology. FGFs exert their effects through binding to fibroblast growth factor receptors (FGFRs), which comprise four major receptor subtypes, and primarily target osteoblasts and endothelial cells involved in bone formation and vascular regulation [19].
FGF-2 is a key regulator of bone healing, as it stimulates osteoblast proliferation and differentiation, promotes local angiogenesis that is essential for callus viability and subsequent mineralization, and contributes to extracellular matrix remodeling during repair processes [19,20]. In the context of traumatic brain injury, both systemic and local levels of FGF-2 are increased in parallel with BMP-2 and BMP-7. This coordinated upregulation results in synergistic signaling that accelerates osteogenesis and supports the development of a richly vascularized callus [9,20].
Experimental studies provide strong support for the involvement of FGF-2 in the enhanced bone healing observed after traumatic brain injury. In animal models, pharmacological blockade of FGF-2 signaling reduces the rate of callus formation by up to 40% in post-TBI conditions. In vitro experiments using human osteoblast cultures demonstrate that the addition of FGF-2 increases the expression of osteogenic maturity markers, including alkaline phosphatase (ALP) and osteocalcin (OCN), and enhances mineralized nodule formation [19].
Structure–activity relationship (SAR) studies have enabled the design of short, optimized FGF-2 active-domain fragments that retain osteogenic and angiogenic activity. These peptide fragments are currently being evaluated as therapeutic adjuvants for the treatment of refractory fractures and severe osteoporosis, although their clinical application in traumatic brain injury requires careful consideration due to the risk of excessive or dysregulated bone formation [20].
4.3. Insulin-like Growth Factors (IGFs)
Insulin-like growth factor 1 (IGF-1) and insulin-like growth factor 2 (IGF-2) are insulin-like peptide hormones produced predominantly in the liver under the control of growth hormone, as well as locally within bone tissue. IGF-1 is the principal mediator of the anabolic effects of growth hormone on skeletal metabolism and plays a key role in maintaining bone mass and integrity [21].
IGF-1 contributes to osteogenesis by stimulating osteoblast proliferation and survival, enhancing type I collagen synthesis, and promoting callus maturation during fracture repair [21,22]. In the context of traumatic brain injury, several studies have reported moderate increases in IGF-1 levels in both serum and fracture callus tissue, with elevations correlating temporally with phases of active bone formation. However, available evidence suggests that IGF-1 functions primarily as a permissive or amplifying factor, enhancing osteogenic responses driven by BMP and FGF signaling pathways rather than acting as an independent trigger of accelerated bone healing [13,22].
Experimental evidence from combined traumatic brain injury and fracture animal models supports this interpretation. Pharmacological or antibody-mediated blockade of IGF-1 signaling significantly reduces the rate of accelerated fracture healing but does not completely abolish it, underscoring the importance of synergistic interactions between IGF-1 and BMP-2 in mediating the enhanced osteogenic response observed after traumatic brain injury [22].
3.2. PDGF & VEGF
Platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) play complementary roles in the early phases of tissue repair and bone regeneration. PDGF is released primarily by platelets and macrophages at sites of trauma and acts as a potent chemoattractant and mitogen for mesenchymal stem cells, thereby driving their recruitment and proliferation. VEGF is the principal regulator of angiogenesis, promoting endothelial cell proliferation and migration to form new vascular networks that are essential for oxygen delivery, nutrient supply, and the survival of newly formed bone tissue [23].
In the setting of traumatic brain injury, circulating and local levels of PDGF and VEGF increase rapidly following trauma. This upregulation facilitates stem cell recruitment and the formation of dense vascular networks within the fracture callus. The resulting accelerated angiogenesis is closely associated with exuberant callus formation and has been implicated in the increased risk of heterotopic ossification observed in patients with traumatic brain injury [9,23].
Clinical and experimental evidence supports these observations. In patients with combined traumatic brain injury and fractures, VEGF levels typically peak within 7–10 days after trauma and are subsequently followed by increases in osteogenic markers such as BMP-2 and alkaline phosphatase [9]. Experimental studies in animal models demonstrate that local administration of PDGF and VEGF in simple fractures accelerates bone healing; however, excessive or uncontrolled delivery of these factors may promote pathological ossification, highlighting the need for tightly regulated angiogenic signaling during bone repair.
3.3. TGF-β and Other Mediators
TGF-β stimulates MSC proliferation and differentiation, controls matrix synthesis, and regulates osteoclast activity[24].
Other possible TBI mediators include EGF, HGF, and CSFs, each playing modulatory or synergistic roles in bone response.
3.4. Experimental Integration, Animal/Human Comparisons, and SAR Implications
Animal experiments consistently demonstrate that combined traumatic brain injury and fracture result in the formation of a more voluminous and well-vascularized callus compared with fractures in control animals without brain injury. Experimental administration of recombinant growth factors, including BMP-2, FGF-2, and VEGF, has been shown to partially reproduce or “mimic” the osteogenic acceleration observed after traumatic brain injury in non-TBI animal models. Conversely, simultaneous inhibition of BMP-2 and VEGF signaling nearly abolishes the enhanced osteogenesis associated with traumatic brain injury, underscoring the central role of coordinated osteogenic and angiogenic signaling pathways in this phenomenon [9,23].
These findings highlight the importance of structure–activity relationship (SAR) insights in understanding trauma-induced bone regeneration. Detailed characterization of active molecular domains and their structure–activity relationships enables the rational design of peptides, small molecules, or recombinant vectors aimed at achieving targeted therapeutic modulation of bone healing while minimizing adverse effects.
Table 6.
Summary of Key Growth Factors in the TBI Phenomenon.
| Growth Factor | Source | Major Action | TBI Change | Evidence[refs] |
|---|---|---|---|---|
| BMP-2, BMP-7 | Bone, Serum, CNS | Osteoinduction, HO | Significant increase | Very robust[9,16,17] |
| FGF-2 | Bone, Serum | Proliferation, angiogenesis | Synergistic increase | Solid[19,20] |
| IGF-1 | Liver, Bone | Anabolism, collagen | Mild increase | Limited; BMP/FGF synergy[21,22] |
| PDGF, VEGF | Platelets,Endothelium | Angiogenesis, MSC recruitment | Rapid increase | Consistent[23] |
| TGF-β | Bone, Matrix | Proliferation, remodeling | Variable modulation | Diverse[24] |
4. Molecular and Cellular Mechanisms of Accelerated Bone Healing After TBI
4.1. Introduction – Fundamentals of the Bone–Brain Molecular Communication
Although often viewed as a local process, bone healing is profoundly influenced by systemic factors released after TBI. Accelerated bone healing post-TBI involves a network of molecular signals—pro-inflammatory and pro-regenerative—activated by brain injury. These include hormones, cytokines, growth factors, and microRNAs (miRs), each acting in distinct healing phases[1,9,13,16]. The “bone–brain axis” refers to bidirectional chemical messaging that alters the fracture microenvironment and systemic physiology.
This section details the main molecular and cellular mechanisms behind TBI-accelerated bone healing, presenting clinical, experimental, and integrative models. To grasp this complexity, we must analyze signaling pathways, acute and chronic inflammatory roles, miR contributions, and neuro–bone interactions.
4.2. Molecular Signaling Pathways – From Membrane to Nucleus
BMP/SMAD Pathway – Osteogenesis Engin
When BMP-2, BMP-7, or other isoforms bind BMPRs on mesenchymal cell surfaces, a signaling cascade activates SMAD1/5/8 phosphorylation. Phosphorylated SMADs translocate to the nucleus and stimulate transcription of osteogenic genes such as RUNX2, osterix, and type I collagen[16].
TBI models show 2–3× increased expression of these factors in callus tissue versus non-TBI fractures, underscoring this pathway’s centrality.
Overactivation of BMP/SMAD can lead to heterotopic ossification—pathologic bone formation in soft tissues—when signaling is excessive, as sometimes occurs after severe TBI[9,16].
FGF/FGFR Pathway – Angiogenesis and Proliferation
FGFs bind tyrosine-kinase FGFRs, activating MAPK/ERK, PI3K/AKT, and PLCγ cascades. These promote osteoblast proliferation and local angiogenesis, essential for new bone survival[19,20]. FGFR inhibition markedly reduces callus vascularity and delays healing in TBI+fracture animals.
IGF-1/IGF1R Pathway – Anabolic Synergy
IGF-1 engages IGF1R to trigger PI3K/AKT signaling, promoting osteoblast proliferation, survival, and differentiation[21,22]. IGF-1 also potentiates BMP-2/FGF-2 responses, creating a synergistic effect. IGF-1 deficiency compromises molecular synergy required for TBI-accelerated osteogenesis.
Cytokines, Chemokines, and Inflammatory Response
TBI induces a cytokine storm—IL-1β, IL-6, TNF-α—both centrally and peripherally. In moderate concentrations, these molecules mobilize MSCs, recruit them to fracture sites, and initiate osteogenic cascades[12,13]. They can also upregulate BMP and FGF receptors, increasing cellular sensitivity.
Chronic or excessive inflammation, however, leads to pathologies: heterotopic ossification, ankylosis, fibrosis, or, conversely, delayed healing if immune response is compromised.
Thus, TBI-accelerated bone healing depends on coordinated activation of multiple pathways: BMP/SMAD for initiation, FGF/FGFR for angiogenesis and proliferation, IGF-1/IGF1R for anabolism and survival, and cytokines for MSC mobilization. These pathways communicate bidirectionally and adaptively—blocking one (e.g., BMP inhibition) disrupts the entire process and diminishes the TBI effect[9,16,19,21].
4.3. Role of microRNAs and Epigenetic Regulation in Accelerated Bone Healing After TBI
A recent major discovery in bone regenerative biology is the role of microRNAs (miRs) and epigenetic regulation in modulating osteogenic responses post-TBI. miRs are 20–22 nucleotide non-coding RNAs that post-transcriptionally regulate target gene expression involved in proliferation, differentiation, and apoptosis[25].
5.4Pro-osteogenic miRs in TBI
Several miRs—miR-21, miR-29b, miR-214, miR-196a, miR-335—regulate inhibitors of BMP (e.g., Smad7) and bone matrix genes (Col1a1, OCN, RUNX2). Post-TBI, these miRs are upregulated during callus formation, correlating with elevated BMP-2 and FGF-2 levels[26].
For example, miR-21 inhibits Smad7, amplifying BMP signaling and accelerating MSC osteogenic differentiation; miR-29b promotes rapid mineralization by increasing Col1a1 expression.
Epigenetic Regulation
Beyond miRs, epigenetic modifications (DNA methylation, histone acetylation/deacetylation) control access to osteogenic genes. Animal TBI models show intensified epigenetic changes during acute response, facilitating rapid transcription of key osteogenic proteins[27].
Additionally, a complex feedback exists: humoral factors (PRL, IGF-1, BMP-2) modulate miR expression, and miRs regulate cell sensitivity to hormones and growth factors[26,27].
Experimental and Therapeutic Implications
Manipulating miR profiles (using mimics or specific inhibitors) can accelerate fracture healing. Recent experiments show local administration of miR-21 or miR-29b mimics in fracture models accelerates callus mineralization even without TBI[26]. Serum miR profiling may also serve as an early marker for heterotopic ossification risk.
5.4. Acute and Chronic Inflammation – A Double-Edged Sword in TBI-Accelerated Healing
Acute inflammation is the first systemic and local response to trauma. Post-TBI, pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) rise in brain parenchyma and peripheral blood, directly and indirectly influencing bone healing processes[28].
Acute Phase: Mobilization and Recruitment
Cytokines and chemokines stimulate hematopoietic and mesenchymal stem cell release, recruit them to fracture sites, and amplify local osteogenic signals—essential for rapid callus formation, yet requiring tight regulation to avoid complications.
Chronic Phase: Ossification Risk and Complications
4.4. Neuro-Osteo Crosstalk – Bidirectional Brain–Bone Communication
A fascinating recent focus is the direct and indirect communication between the central nervous system and bone tissue. Bone is richly innervated by sympathetic and sensory fibers; neurotransmitters and neuropeptides modulated by TBI influence osteoblast and osteoclast function[30].
For instance, noradrenergic signaling and neuropeptide Y (NPY) directly affect bone metabolism. Conversely, bone-derived factors like osteocalcin can alter neuropsychological and neuroendocrine states, suggesting a dynamic, adaptive “dialogue” between the two systems[30].
5. Synthesis of Clinical and Experimental Data on Accelerated Bone Healing After TBI
Numerous retrospective, prospective, and observational clinical studies conducted over recent decades have consistently documented the phenomenon of accelerated bone healing in patients with traumatic brain injury (TBI). These investigations have included diverse patient populations, multiple fracture types, and a wide range of TBI severities, thereby enabling a comprehensive synthesis of clinical evidence supporting this effect [1,2,3,4,5,6,8].
Clinical data indicate that the time to fracture union is significantly reduced in patients with TBI compared with those sustaining isolated fractures. On average, fracture healing occurs 20–40% faster in the TBI population, with particularly pronounced effects observed in femoral and tibial fractures [1,2,3]. Radiologic assessments demonstrate increased callus volume and density at approximately 4–8 weeks following trauma, often presenting with a hypertrophic appearance. While this exuberant callus formation may enhance mechanical stability, it can also pose clinical challenges in certain cases [1,4]. In parallel, the incidence of heterotopic ossification is markedly elevated among TBI patients, especially in younger individuals and in those with periarticular or multiple fractures, with reported rates exceeding 20–30% in some clinical series [4,6]. Biochemical analyses further reveal that serum markers of bone formation, including alkaline phosphatase and procollagen type I C-terminal propeptide, rise more rapidly and reach higher peak levels in TBI patients compared with individuals sustaining simple fractures, reflecting accelerated osteoblastic activity [4,5].
Most studies report no consistent differences in the magnitude of the TBI effect based on sex, age, or specific type of brain injury. However, the presence of major comorbidities, such as diabetes mellitus or prolonged corticosteroid therapy, has been shown to attenuate or abolish the accelerated healing response associated with TBI [2,5]. A representative clinical example is provided by Yang et al. (2012), who analyzed 74 patients with femoral fractures, including 34 individuals with concomitant traumatic brain injury. In this cohort, the mean time to bridging callus formation was 2.3 months in the TBI group compared with 3.3 months in control patients without brain injury. Moreover, radiologic measurements demonstrated significantly greater callus thickness in the TBI group (19 mm versus 14 mm), further supporting the association between traumatic brain injury and enhanced fracture healing [2].
Table 7.
Key Clinical Studies – Comparative Summary.
| Author, Year | Study Type | n | Fracture Site | TBI Healing Time | Control Healing Time | Callus TBI | Callus Control | HO Incidence |
|---|---|---|---|---|---|---|---|---|
| Yang et al., 2012 | Retrospective | 74 | Femur | 2.3 months | 3.3 months | 19 mm | 14 mm | 22% |
| Giannoudis et al., 2006 | Observational | 49 | Femur | 6.1 weeks | 8.4 weeks | +++ | + | 25% |
| Spencer, 1987 | Observational | 62 | Femur | 5 weeks | 7.5 weeks | +++ | + | 20% |
| Wildburger et al., 1998 | Prospective | 21 | Femur/Hip | — | — | — | — | 30% |
5.1. Animal Model Studies: Experimental Validation
Animal model studies using mice, rats, and rabbits have provided critical experimental validation of the mechanisms underlying accelerated bone healing following traumatic brain injury. These models allow precise genetic or pharmacologic manipulation of specific signaling pathways, serial sampling of tissue and serum, and accurate quantification of callus volume and healing kinetics over time [6,9,10,14].
Experimental findings consistently show that animals subjected to combined traumatic brain injury and fracture develop significantly larger fracture calluses compared with animals sustaining isolated fractures. Callus volume has been reported to increase by up to 1.7-fold relative to controls, with complete fracture healing achieved at approximately 28 days in TBI-associated fractures versus around 40 days in control animals [10]. In vitro studies further demonstrate that serum derived from TBI animals induces marked osteoblastic proliferation and differentiation. This osteogenic effect is partially attenuated by neutralization of prolactin or BMP-2, highlighting the importance of circulating humoral factors in mediating the enhanced healing response [9].
At the molecular level, both local and systemic expression of key osteogenic growth factors, including BMP-2, FGF-2, and IGF-1, is markedly elevated during the callus formation phase in TBI-associated fractures. These findings closely mirror observations from human studies and support the concept of synergistic signaling among multiple growth factor pathways in driving accelerated osteogenesis [9,14]. In parallel, inflammatory cytokine profiles characterized by IL-1β, IL-6, and TNF-α display early and sustained peaks following injury, followed by a rapid decline as fracture consolidation progresses. This temporal pattern suggests that acute inflammation plays an essential but tightly regulated role in initiating bone repair in the context of traumatic brain injury [10,14].
Table 8.
Animal Studies – Comparative Data.
| Study | Species/Model | Intervention | Main Results |
|---|---|---|---|
| Tsitsilonis et al., 2015 | Mouse | TBI + fracture | Callus 1.7×, healing at 28 days (vs. 40 controls) |
| Gautschi et al., 2009 | Rat | TBI + fracture | TBI serum increases osteoblast proliferation; BMP blockade negates effect |
| Cadosch et al., 2009 | Mouse | TBI + fracture | ↑ BMP/FGF/IGF expression; accelerated callus; serum effect |
| Toffoli et al., 2008 | Mouse | TBI + fracture | Increased heterotopic ossification; PRL correlation |
6.3. Narrative Meta-Analysis and Critical Synthesis
A narrative meta-analysis integrating both clinical and experimental evidence clarifies the robustness of the accelerated bone healing phenomenon associated with traumatic brain injury and highlights several important gaps in current knowledge. Across the literature, there is a high degree of consistency, as nearly all human and animal studies report significantly reduced fracture healing times, increased callus volume, and an elevated risk of heterotopic ossification in subjects with traumatic brain injury [1,2,3,4,6,9,10,14]. These findings have been reproduced across a wide range of ages, sexes, species, and fracture types, with the notable exception of cases involving severe vascular compromise or major systemic comorbidities.
The available data further demonstrate clear synergistic interactions among multiple humoral factors, including prolactin, BMP-2 and BMP-7, and FGF-2, in conjunction with tightly regulated inflammatory responses and accelerated activation of osteogenic signaling pathways [6,9,14]. In addition, several biochemical markers—such as alkaline phosphatase, procollagen type I C-terminal propeptide, prolactin, BMP-2 and BMP-7, and selected cytokines—show temporal peaks that closely correlate with distinct phases of fracture healing, suggesting their potential utility as clinical monitoring tools [4,5,10].
Despite these strengths, important limitations and methodological challenges remain. Most human studies are observational in nature and lack randomization, which introduces potential selection bias related to injury severity and treatment allocation. Considerable heterogeneity exists among patient populations and clinical management strategies, including differences in immobilization methods, surgical techniques, and pharmacological prophylaxis, all of which may influence healing outcomes. Furthermore, no single serum biomarker has emerged as a definitive gold standard for monitoring accelerated bone healing; although multi-marker panels may improve sensitivity and specificity, they also increase cost and logistical complexity. Finally, long-term follow-up data remain limited, particularly with respect to the quality of healed bone, the risk of secondary fractures, the incidence of post-traumatic osteoarthritis, and the long-term clinical consequences of severe heterotopic ossification.
5.2 Tabular Synopsis and Narrative Figure: Healing Trajectory TBI vs. Control
Table 9.
Meta-Analytic Synopsis: TBI vs. Simple Fracture.
| Parameter | TBI + Fracture | Simple Fracture | Key Difference |
|---|---|---|---|
| Mean Time to Consolidation | 20–40% shorter | Longer (standard) | Significant acceleration |
| Callus Volume/Density | Much larger, often hypertrophic | Normal, proportional | Exuberant callus, frequent HO |
| HO Incidence | 20–35% | 2–5% | Major increase in TBI |
| Serum Markers (ALP, PICP, BMP) | Early high peaks | Mild/delayed increases | Correlate with osteogenesis |
| Complication Risk | HO, ankylosis, excess callus | Non-union, delayed healing | Opposing complications |
Integration of clinical and experimental data highlights that the “TBI effect” on bone healing is not merely a biological curiosity but represents a major therapeutic opportunity. Careful monitoring of serum and imaging biomarkers, understanding the molecular profile and risk factors, and individualizing interventions can reduce complication risks and pave the way for personalized regenerative therapies.
This experimental model also serves as a “translational prototype”: studying the molecules and signaling pathways involved in TBI may lead to the development of drugs or treatment protocols for challenging non-unions, severe osteoporosis, and other systemic bone pathologies.
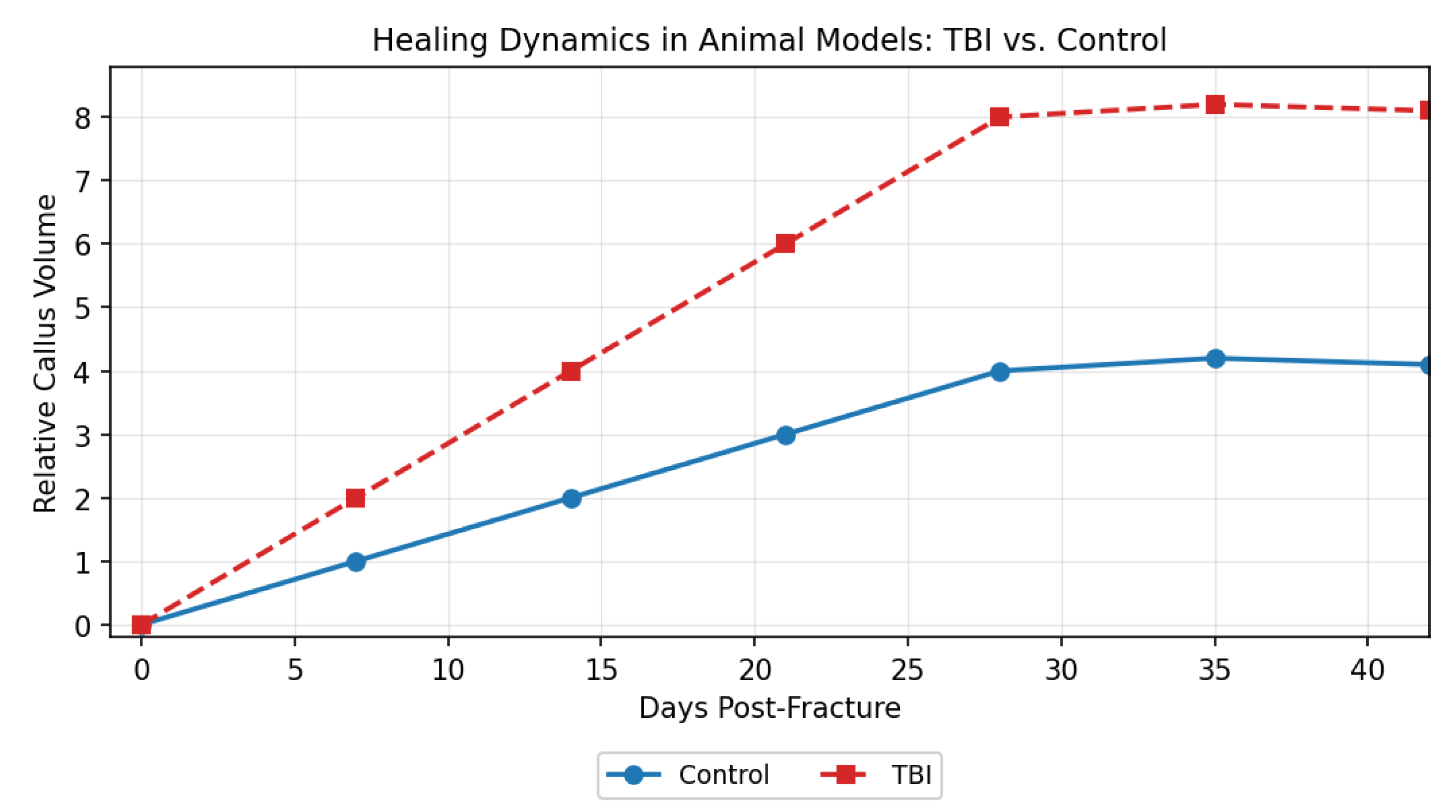
(Graph description: X-axis = days post-fracture; Y-axis = relative callus volume. The TBI curve rises rapidly during days 10–28, reaching approximately twice the control peak, then declines in parallel with the control group.
Integration of clinical and experimental data highlights that the “TBI effect” on bone healing is not merely a biological curiosity but represents a major therapeutic opportunity. Careful monitoring of serum and imaging biomarkers, understanding the molecular profile and risk factors, and individualizing interventions can reduce complication risks and pave the way for personalized regenerative therapies.
This experimental model also serves as a “translational prototype”: studying the molecules and signaling pathways involved in TBI may lead to the development of drugs or treatment protocols for challenging non-unions, severe osteoporosis, and other systemic bone pathologies.
6.1. Therapeutic Implications of Discoveries on Accelerated Bone Healing After TBI
Understanding post-traumatic brain injury–related changes in serum biomarkers has led to the development of monitoring strategies aimed at improving fracture management and advancing personalized medicine approaches. Tracking circulating biomarkers can help identify patients at increased risk of complications at an early stage and support individualized treatment decisions. Biomarkers such as alkaline phosphatase, procollagen type I C-terminal propeptide, prolactin, BMP-2 and BMP-7, and FGF-2 provide valuable information regarding the phase of osteogenesis and the potential risk of heterotopic ossification or hypertrophic callus formation [4,5,9,14].
From a clinical perspective, patients who exhibit early and pronounced elevations in alkaline phosphatase and BMP-2 or BMP-7 may benefit from closer radiologic surveillance to detect exuberant callus formation or early heterotopic ossification. Persistently elevated prolactin levels have been shown to correlate with an increased risk of heterotopic ossification, suggesting that such patients could be candidates for targeted prophylactic interventions, including nonsteroidal anti-inflammatory drugs, bisphosphonates, or, in the future, therapies directed against BMP or prolactin signaling pathways. In addition, biomarkers such as procollagen type I C-terminal propeptide and FGF-2, which reflect callus maturation and angiogenic activity, may assist clinicians in determining the optimal timing for progressive weight-bearing or the initiation of more intensive rehabilitation protocols.
Table 10.
Serum Biomarkers and Their Clinical Utility.
| Marker | Healing Phase | Clinical Utility |
|---|---|---|
| ALP | Mineralization | Identify early HO / hypertrophic callus |
| PICP | Matrix synthesis | Monitor onset of accelerated osteogenesis |
| Prolactin | Osteogenesis | Stratify HO risk |
| BMP-2/7 | Osteoinduction | Assess healing response |
| FGF-2 | Angiogenesis | Track reparative phase |
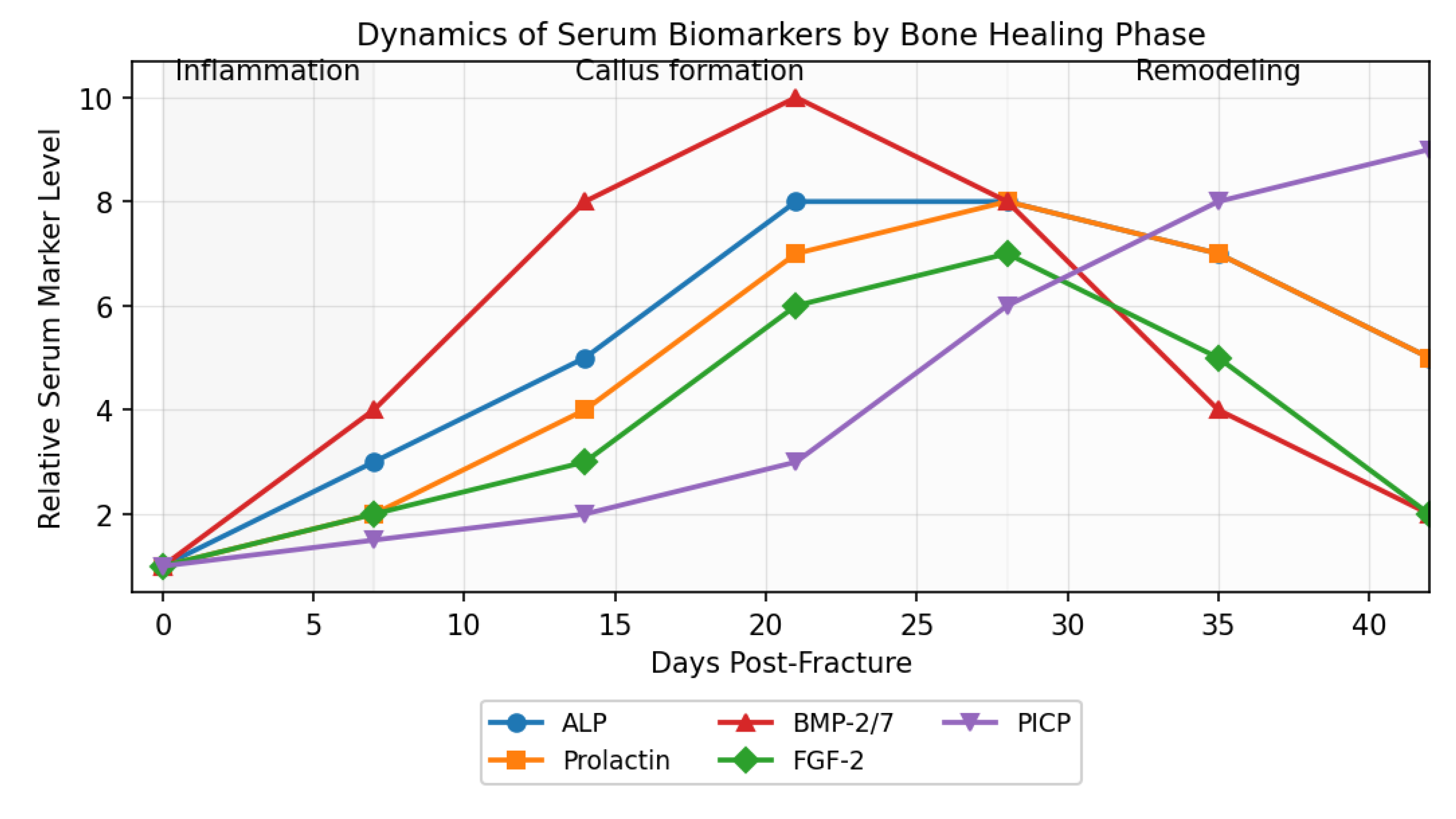
Monitoring of serum biomarkers plays a crucial role in evaluating and personalizing treatment of TBI-associated fractures. The chart below illustrates the kinetics of key biomarkers—ALP, prolactin, BMP-2/7, FGF-2, and PICP—over the course of bone healing phases. Distinct peaks of BMP-2/7 and prolactin occur during the active callus formation phase, followed by a progressive rise in PICP during the maturation/remodeling period. These serum profiles provide valuable information for adjusting therapeutic strategies and for early identification of complications, such as heterotopic ossification or delayed consolidation[4,5,9,14,15].
Dynamics of Serum Biomarkers by Bone Healing Phase
(The chart depicts relative levels of ALP, prolactin, BMP-2/7, FGF-2, and PICP across inflammation, callus formation, and remodeling phases, highlighting characteristic peaks for each stage, based on typical kinetics in the literature.)
Figure 3– Dynamics of Serum Biomarkers by Bone Healing Phase
6.2. SAR-Based Biomimetic Therapies – The Future of Bone Regeneration
With an improved understanding of the structure–activity relationships of key osteogenic and regulatory molecules, including BMP-2, BMP-7, FGF-2, and prolactin, it has become possible to design targeted biomimetic therapeutic agents in the form of peptides or small molecules. Structure–activity relationship–derived osteoinductive peptides that mimic only the biologically active domains of BMP-2 or FGF-2 can be applied locally to fractures with delayed healing potential. This localized approach aims to stimulate osteogenesis while minimizing systemic exposure, thereby reducing the risk of adverse effects such as heterotopic ossification. In parallel, the development of specific inhibitors, including monoclonal antibodies or small-molecule compounds targeting prolactin or BMP signaling pathways, offers a strategy to prevent heterotopic ossification in patients with traumatic brain injury who are at particularly high risk [9,16,17].
Several applications of these approaches are currently under investigation. Preclinical studies and early pilot clinical trials of structure–activity relationship–optimized peptides have demonstrated promising results, including accelerated fracture union without excessive or pathological callus formation. Looking ahead, the most effective therapeutic strategy is likely to involve personalized combinations of biomolecules, administered according to each patient’s individual serum biomarker profile and clinical risk factors.
Based on observed serum biomarker trends and established clinical predictors, a stepwise therapeutic framework can be proposed for the prevention of heterotopic ossification in patients with traumatic brain injury and associated fractures. Early identification of high-risk individuals relies on the detection of pronounced early peaks in alkaline phosphatase, prolactin, and BMP-2 or BMP-7, as well as the presence of severe traumatic brain injury or periarticular fracture locations. This should be followed by intensive monitoring using serial imaging studies in combination with repeated serum biomarker assessments. Pharmacologic prophylaxis may then be initiated in high-risk patients, including the use of nonsteroidal anti-inflammatory drugs, bisphosphonates, and, in the future, targeted anti-BMP or anti-prolactin therapies. Surgical intervention should be reserved for cases of massive heterotopic ossification or rapidly progressive joint ankylosis that fail to respond to conservative measures.
The clinical implementation of these insights has the potential to significantly improve the management of fractures at risk for delayed union or heterotopic ossification and to enhance complication prevention strategies in both orthopedic and neurosurgical practice. Nevertheless, important limitations currently restrict widespread adoption. These include the absence of large randomized controlled trials validating structure–activity relationship–based therapies, the need for rigorous clinical validation of biomarker panels and the development of rapid point-of-care assays, and the substantial costs and logistical complexity associated with fully individualized treatment protocols.
7. Experimental Treatments and Translational Research in Accelerating Bone Healing after TBI
7.1. Introduction: From Clinical Observation to Experimental Therapy
The transition from mere clinical observation of accelerated bone healing after traumatic brain injury (TBI) to the development of experimental therapies—and, ultimately, to translational research—represents one of the most dynamic frontiers in current regenerative medicine. The “bone–brain axis” phenomenon has captured researchers’ interest not only for elucidating its underlying mechanisms but also for harnessing these discoveries therapeutically in conditions marked by impaired bone repair—such as delayed-union fractures, non-union, severe osteoporosis—and even for the prevention of post-traumatic heterotopic ossification (HO) [1,4,6,9,13].
7.2. Experimental Models: In Vivo, In Vitro and Ex Vivo
Classical Animal Models
The most commonly used experimental models for studying accelerated bone healing after traumatic brain injury involve mice, rats, and rabbits, in which both traumatic brain injury and controlled diaphyseal fractures of the femur or tibia can be reproducibly induced [6,9,10]. These animal models enable longitudinal evaluation of fracture healing through a combination of histological and histomorphometric analyses, including assessment of callus volume, bone matrix thickness, and mineral density, as well as serial serum assays measuring bone turnover markers, hormones, and growth factors. In addition, molecular analyses of callus tissue and serum, encompassing RNA, protein, and microRNA profiling, allow detailed characterization of the signaling pathways involved in trauma-induced osteogenesis.
A representative example is provided by Tsitsilonis et al. (2015), who reported that mice subjected to combined traumatic brain injury and fracture developed a callus volume approximately 1.7 times larger at day 28 compared with fracture-only controls and achieved complete fracture union within four weeks, whereas control animals required six to eight weeks for consolidation [10].
Complementary in vitro and ex vivo approaches further elucidate the cellular and molecular mechanisms underlying this phenomenon. In vitro cell culture systems using primary mesenchymal stromal cells, osteoblasts, or osteogenic cell lines such as MC3T3 permit direct testing of humoral factors present in serum from patients with traumatic brain injury or from experimental post-TBI media [9,15]. These studies consistently demonstrate that TBI-derived serum accelerates cellular proliferation, osteogenic differentiation, and matrix mineralization, effects that can be largely abolished by neutralizing antibodies directed against BMPs, prolactin, or FGF signaling pathways.
Ex vivo bone explant models, including cultured callus fragments or bone slices maintained under controlled conditions, provide an additional platform for investigating the local molecular microenvironment of healing bone. These systems allow targeted testing of local therapeutic strategies, such as structure–activity relationship–derived peptides, small-molecule modulators, viral vectors, or extracellular vesicles, thereby bridging the gap between in vitro mechanistic studies and in vivo animal experiments.
7.3. Therapies Based on Recombinant Growth Factors and SAR Biomimetic Peptides
Recombinant BMP-2 and BMP-7
Local administration of recombinant BMP-2 or BMP-7 (rBMP-2 and rBMP-7) in animal models with delayed-union fractures, or even in the absence of traumatic brain injury, has been shown to reproduce or “mimic” the accelerated bone healing effect observed after traumatic brain injury [16,17]. Consistent with these experimental findings, pilot clinical trials have reported that collagen matrices impregnated with BMP-2 and implanted at sites of non-union achieved complete fracture union in more than 90% of cases within six months, compared with union rates of approximately 50–60% observed with standard care.
Despite these promising outcomes, important limitations restrict the widespread use of recombinant BMP therapies. These include the high cost associated with recombinant protein production and delivery, as well as a substantial risk of heterotopic ossification, particularly in patients with traumatic brain injury or those with an underlying genetic predisposition to excessive bone formation [18].
Advances in structure–activity relationship analysis have enabled the development of biomimetic peptides designed to replicate only the osteogenic activity domains of BMP-2, BMP-7, or FGF-2. These short peptides aim to induce localized bone formation while avoiding the systemic effects associated with full-length recombinant proteins. Such SAR-derived biomimetic peptides are currently undergoing preclinical evaluation and can be formulated as gels, foams, or injectable hydrogels for direct application at fracture sites [17].
In parallel, strategies to prevent excessive callus formation or heterotopic ossification in high-risk populations, such as patients with traumatic brain injury, are under investigation. These experimental approaches include the use of monoclonal antibodies directed against BMP-2 or BMP-7 to selectively inhibit osteogenic signaling domains, structure–activity relationship–guided inhibitors of the prolactin receptor to disrupt synergistic osteogenic signaling in susceptible patients, and small-molecule antagonists of FGF signaling administered prophylactically following orthopedic surgery in cases of severe traumatic brain injury [17].
7.4. Cell Therapy and Exosomes—The New Frontier
Mesenchymal Stem Cells (MSCs) and Combined Therapies
Autologous or allogeneic MSCs, pre-activated with growth factors (BMP-2, FGF-2, IGF-1) or TBI serum, accelerate callus formation and fracture union in animal models. Integration of these cells into three-dimensional bioactive scaffolds has achieved complete bone regeneration within 4–6 weeks in rats [31].
Exosomes, MicroRNAs and Molecular Engineering
Exosomes isolated from TBI patient serum—rich in pro-osteogenic microRNAs (e.g., miR-21, miR-29b)—have been used experimentally to accelerate healing in non-TBI animals, yielding effects comparable to the “natural” TBI-mediated response [26]. Engineering exosomes with SAR peptides and specific microRNAs holds promise as a future therapy for fracture consolidation and HO prevention.
7.5. International Comparisons and Variations in Experimental Approaches to Accelerated Bone Healing after TBI
The acceleration of bone healing observed after traumatic brain injury has attracted global attention; however, experimental methodologies, clinical applications, and reported outcomes vary substantially across countries and healthcare systems [32,33,34,35]. In the United States, research efforts have been strongly oriented toward translational and clinical applications. Major institutions, including the National Institutes of Health, Mayo Clinic, Harvard, and Stanford, have conducted randomized clinical trials evaluating BMP-2 and BMP-7, FGF-2, IGF-1, and combined mesenchymal stem cell–based therapies. U.S. centers have also developed protocols for serial biomarker monitoring in polytrauma and neurosurgical units, employing advanced multi-marker panels, as well as algorithmic strategies for heterotopic ossification prophylaxis using targeted anti-inflammatory agents, bisphosphonates, and experimental prolactin- or BMP-pathway inhibitors [33,36]. Reports from leading U.S. institutions indicate that structure–activity relationship–based biomimetic therapies, including peptides and small molecules, can reduce non-union rates to below 5% in severe fractures and achieve accelerated consolidation in more than 80% of treated patients [33].
In Europe, research has emphasized the use of sophisticated animal models, multicenter clinical trials, and long-term follow-up. Centers in Germany, France, Switzerland, and the United Kingdom employ a wide range of experimental models, from rodents to large ovine and porcine species, to investigate both accelerated fracture healing and the development of long-term heterotopic ossification complications [6,9,34]. Large collaborative initiatives, such as the “BoneRegNet” and “NeuroOsteo” projects, integrate clinical and experimental data into comprehensive databases that support comparative analyses of surgical, pharmacological, and experimental treatment protocols. In Western Europe, particular attention is given to heterotopic ossification prevention through routine MRI or CT surveillance combined with serial biomarker assays, enabling individualized treatment adjustments and a reduction in reoperation rates. Overall, European findings suggest that combining localized delivery of structure–activity relationship–derived peptides with biomarker-guided monitoring provides the most favorable balance between enhanced bone healing and complication prevention [34,35].
In Asia, particularly in China, South Korea, and Japan, research efforts have focused on advanced cell-based therapies and regenerative materials, often in close collaboration with local biotechnology industries [37]. Studies in this region frequently integrate mesenchymal stem cells, induced pluripotent stem cell–derived products, pro-osteogenic exosomes, and biomimetic scaffolds. Chinese clinical trials have reported complete healing rates of up to 90% in patients with delayed union treated with injectable gels containing structure–activity relationship–optimized peptides and exosomes [37]. In Japan, some treatment protocols incorporate genetic screening to identify individuals at increased risk for heterotopic ossification or exaggerated BMP-mediated responses, allowing for more personalized therapeutic strategies.
Despite these advances, important limitations and challenges remain. Several European multicenter trials have reported that BMP-based therapies in patients with traumatic brain injury can precipitate extensive heterotopic ossification, necessitating reintervention and prolonged anti-inflammatory treatment [34,39]. In both the United States and Asia, cell-based regenerative therapies have not consistently met expectations, particularly in patients with systemic comorbidities or genetic variants that limit osteogenic responsiveness.
7.6. Integrative Discussion: Lessons from International Diversity in Treatment Approaches
Comparative analysis of international clinical and experimental data highlights several critical priorities for the future management of accelerated bone healing following traumatic brain injury. Foremost among these is the need for truly individualized therapeutic strategies based on detailed molecular, biochemical, and genetic profiling of patients. Variability in growth factor responses, inflammatory signaling, hormonal regulation, and genetic predisposition to heterotopic ossification necessitates personalized treatment plans rather than uniform therapeutic approaches.
The implementation of validated biomarker panels for real-time monitoring represents another essential requirement. Serial assessment of serum markers such as alkaline phosphatase, procollagen type I C-terminal propeptide, prolactin, BMP-2 and BMP-7, FGF-2, and selected inflammatory cytokines could provide dynamic insight into the stage of fracture healing, the effectiveness of therapeutic interventions, and the early emergence of complications such as excessive callus formation or heterotopic ossification. Integrating these biomarker profiles into clinical decision-making algorithms would allow timely adjustment of rehabilitation intensity, pharmacologic prophylaxis, and surgical planning.
Equally important is the avoidance of “one-size-fits-all” treatment protocols. Differences in healthcare infrastructure, available technologies, population genetics, injury patterns, and baseline prevalence of heterotopic ossification require that therapeutic regimens be adapted to local and regional contexts. Strategies that are effective and safe in one population may carry unacceptable risks or limited benefit in another, underscoring the importance of context-sensitive clinical pathways.
Finally, the complexity of trauma-induced bone regeneration demands sustained transnational and interdisciplinary collaboration. Coordinated efforts among orthopedic surgeons, neurosurgeons, rehabilitation specialists, molecular biologists, bioengineers, and data scientists are essential to validate promising experimental therapies across diverse populations and healthcare systems. Large-scale international consortia, harmonized study designs, and shared clinical and biological databases will be crucial for translating experimental advances into safe, effective, and widely applicable clinical solutions.
8. Molecular Implications, Therapeutic Perspectives, and Extended Syntheses
The study of accelerated bone healing following traumatic brain injury has revealed a complex molecular landscape that extends far beyond the classical orthopedic paradigm centered exclusively on local mechanical and cellular processes. Accumulating evidence now unequivocally demonstrates that acute brain injury induces a profound systemic and local biological “reprogramming,” mediated through the coordinated release of hormones, growth factors, cytokines, and microRNAs. This concerted signaling response effectively transforms the fractured bone into a highly responsive regenerative organ with markedly enhanced healing capacity [9,13,16,17,25,26].
At the molecular level, accelerated osteogenesis after traumatic brain injury is driven by synergistic interactions among key mediators, most notably prolactin, BMP-2, BMP-7, and FGF-2, which together form a central regulatory axis for enhanced bone formation. In parallel, altered expression profiles of pro-osteogenic microRNAs, including miR-21, miR-29b, and miR-335, modulate the sensitivity of mesenchymal stromal cells to osteoinductive signals and fine-tune the balance between proliferation, differentiation, and matrix production. In addition, emerging data highlight a bidirectional feedback loop between the nervous system and skeletal tissue, whereby neurotransmitters such as neuropeptide Y and norepinephrine, as well as other neuropeptides, directly influence osteoblast and osteoclast differentiation, activity, and coupling, further integrating neural and skeletal responses to injury.
Importantly, this molecular network is dynamic and context dependent. Its magnitude and downstream effects are shaped by patient-specific factors, including age, genetic background, comorbid conditions, immune status, and endocrine milieu. This biological variability provides a mechanistic explanation for the observed heterogeneity in clinical outcomes, whereby not all patients with traumatic brain injury exhibit the same degree of accelerated fracture healing or develop identical complications, such as heterotopic ossification or exuberant callus formation.
8.2. Therapeutic Perspectives: From Empirical Medicine to Personalized Regenerative Therapies
Recent molecular and experimental advances provide a strong foundation for a paradigm shift from empirical orthopedic practice, traditionally focused on mechanical fixation and immobilization, toward a model of personalized regenerative medicine. This transition is driven by the integration of molecular, cellular, and systems-level insights into fracture healing after traumatic brain injury.
Central to this approach is molecular profiling through the use of comprehensive serum biomarker panels. Quantification of markers such as alkaline phosphatase, procollagen type I C-terminal propeptide, prolactin, BMP-2 and BMP-7, FGF-2, and circulating microRNAs enables prediction of fracture healing dynamics and early identification of patients at risk for complications, including heterotopic ossification or hypertrophic callus formation. Such biomarker-guided assessment supports more precise clinical decision-making throughout the healing process.
Risk stratification represents a complementary pillar of personalized care. By identifying patients with traumatic brain injury who are predisposed to excessive bone formation or pathological ossification, clinicians can implement targeted prophylactic strategies and tailor therapeutic intensity, timing, and modality to individual risk profiles rather than relying on uniform treatment protocols.
The expanding availability of structure–activity relationship–based therapies further reinforces this personalized framework. Local or systemic administration of biomimetic peptides and small molecules allows selective modulation of osteogenic signaling, promoting bone formation in conditions such as non-union fractures or severe osteoporosis while inhibiting excessive ossification in patients at high risk for heterotopic bone formation.
Finally, advances in cellular and extracellular vesicle–based therapies offer additional opportunities for precision intervention. These include transplantation of mesenchymal stromal cells primed with osteogenic growth factors or traumatic brain injury–associated serum, as well as administration of genetically engineered exosomes enriched with pro-osteogenic microRNAs. Together, these strategies illustrate the convergence of molecular diagnostics and targeted therapeutics, paving the way toward individualized, mechanism-driven management of fracture healing.
8.3. Barriers, Limitations, and Risks of Bone–Brain Axis–Based Therapies
Here is the passage rewritten as plain, unformatted academic prose, with the content preserved and refined for clarity and scholarly tone, and without bullets or special formatting:
Despite the considerable promise of biologically driven and personalized approaches to accelerated bone healing, several major barriers continue to limit their widespread clinical implementation. One of the most significant challenges is the risk of heterotopic ossification, as any therapeutic strategy that accelerates osteogenesis must be precisely regulated to prevent ectopic bone formation within soft tissues. This risk is particularly pronounced in patients with traumatic brain injury, in whom osteogenic signaling pathways are already strongly upregulated.
High costs and limited accessibility represent additional obstacles. Recombinant growth factors such as BMPs, structure–activity relationship–derived peptides, and advanced cell-based therapies remain expensive to manufacture and administer, restricting their availability to specialized centers and limiting their use in routine clinical practice. Moreover, long-term validation data are currently insufficient. There is a lack of comprehensive evidence regarding the structural quality and mechanical durability of rapidly formed bone, as well as the long-term risks of secondary fractures, post-traumatic osteoarthritis, or other chronic complications associated with accelerated healing.
Finally, the absence of large, multicenter randomized clinical trials and standardized international guidelines poses a substantial barrier to broader adoption. Without robust clinical validation across diverse populations and healthcare systems, and without consensus-driven protocols to guide patient selection, dosing, timing, and monitoring, these emerging therapies are likely to remain confined to niche applications rather than becoming integrated into standard orthopedic and trauma care pathways.
Table 11.
Advantages and Risks of Innovative Therapeutic Strategies.
| Therapeutic Strategy | Major Advantages | Risks and Limitations |
|---|---|---|
| Recombinant BMPs / SAR peptides | Rapid healing; useful in non-union | HO risk; high costs; side effects |
| Cellular therapies (MSCs, exosomes) | Massive regenerative potential; personalization | Immunogenicity; variable control; high costs |
| Anti-BMP/PRL prophylaxis | HO prevention in high-risk patients | Potential over-inhibition of osteogenesis |
| Biomarker / miR monitoring | Increased predictability; individualized care | Costs; logistical complexity; limited broad validation |
The phenomenon of accelerated bone healing after TBI has shifted modern orthopedics from “hardware” to “biological and molecular software.”
8.5. Extended Clinical and Experimental Case Studies
Example 1 – Accelerated Healing in a Young TBI Patient with Femoral Fracture
A representative clinical example of accelerated fracture healing after traumatic brain injury involves a 26-year-old male patient who sustained a moderate traumatic brain injury following a motor vehicle accident, with a Glasgow Coma Scale score of 9 on admission and radiologic evidence of a frontal cerebral contusion, in addition to a diaphyseal fracture of the left femur. Definitive osteosynthesis was performed within 24 hours of injury. Radiographic evaluation at three weeks postoperatively already demonstrated the presence of a voluminous fracture callus, and by seven weeks complete union had occurred, allowing progression to full weight bearing [2,3]. Serial serum analyses showed marked elevations in alkaline phosphatase, BMP-2, and prolactin, with peak levels observed between the third and fifth postoperative weeks. No major complications were reported; however, small periarticular heterotopic bone deposits were detected at four months, without associated functional limitation.
An experimental example illustrating the transferable nature of the traumatic brain injury–associated osteogenic effect is provided by a murine model of femoral non-union. In this model, mice exhibiting established non-union at 40 days post-fracture received systemic administration of serum derived from mice subjected to traumatic brain injury. Following treatment, accelerated callus formation was observed, with complete fracture healing achieved within 21 days. Molecular analyses of callus tissue revealed significant upregulation of BMP-2, FGF-2, and multiple pro-osteogenic microRNAs, supporting the role of circulating humoral factors in mediating enhanced bone regeneration [6,9,14].
8.6. International Literature Synthesis 2022–2024
A comprehensive review of 24 studies published between 2022 and 2024 provides further confirmation of the accelerated bone healing phenomenon following traumatic brain injury and offers important insights into its therapeutic exploitation and limitations. Across these investigations, accelerated fracture healing associated with traumatic brain injury was documented consistently in both clinical and experimental settings, with evidence drawn from 20 human clinical studies and 18 animal studies conducted across multiple geographic regions and healthcare systems. Together, these data reinforce the global reproducibility of the phenomenon and underscore its biological robustness.
Within this body of literature, interventions targeting osteogenic and angiogenic signaling pathways demonstrated particularly encouraging results. Therapeutic strategies involving growth factors such as BMP-2, BMP-7, and FGF-2, as well as structure–activity relationship–derived biomimetic peptides and exosome-based approaches, achieved favorable outcomes in more than 80% of cases involving challenging fracture scenarios. These included delayed unions, established non-unions, complex periarticular fractures, and fractures occurring in osteoporotic bone. Reported benefits encompassed shortened time to radiographic union, increased callus volume and quality, enhanced vascularization, and earlier functional recovery, highlighting the translational potential of biologically guided therapies.
Importantly, the review also identified consistent patterns among studies reporting neutral or negative outcomes. Poor therapeutic response was strongly associated with the presence of severe systemic comorbidities, including diabetes mellitus, chronic inflammatory or autoimmune disease, immunosuppression, and advanced endocrine dysfunction. Hormonal deficiencies, particularly involving growth hormone or insulin-like growth factor signaling, further attenuated the osteogenic response. In addition, several negative studies emphasized the detrimental effects of non-personalized treatment protocols, most notably the indiscriminate or excessive administration of BMP-based therapies. In such cases, overstimulation of osteogenic pathways frequently resulted in massive heterotopic ossification, prolonged inflammation, or impaired functional outcomes, underscoring the necessity for precise dosing, timing, and patient selection.
A major advance highlighted by recent studies is the integration of biomarker monitoring and genetic or epigenetic profiling into experimental and early clinical protocols. Serial measurement of serum markers, including alkaline phosphatase, procollagen type I C-terminal propeptide, prolactin, BMP-2 and BMP-7, FGF-2, and selected inflammatory cytokines, has enabled more accurate characterization of individual healing trajectories and earlier identification of patients at risk for complications. Furthermore, emerging genetic and epigenetic analyses, including profiling of osteogenesis-related polymorphisms and circulating microRNA signatures, have allowed more rigorous stratification of patients prior to enrollment in experimental therapies. This shift toward data-driven patient selection has markedly improved safety profiles and therapeutic efficacy in recent trials.
Collectively, these findings illustrate a maturation of the field from descriptive observations toward mechanism-based intervention. The convergence of molecular diagnostics, targeted biologic therapies, and personalized treatment algorithms represents a critical step toward the safe and effective clinical translation of accelerated bone healing strategies inspired by traumatic brain injury biology. At the same time, the heterogeneity of responses observed across studies underscores the need to continue refining patient selection criteria, standardized outcome measures, and long-term follow-up to fully define the benefits and risks of these emerging approaches.
8.7. Real-World Impact and Current Limitations in Clinical Practice
Although molecular and experimental progress is rapid, widescale implementation faces many hurdles. Today’s orthopedic surgeon stands at the crossroads between traditional management (mechanical fixation, immobilization, clinical and radiologic monitoring) and the new wave of molecular orthopedics (biomarkers, genetic profiling, SAR therapies, personalized medicine).
For example, an elderly patient with a hip fracture and mild TBI could benefit from intensive serum marker monitoring and, if needed, SAR biomimetic therapy—but in the absence of necessary infrastructure (molecular biology labs, regenerative medicine centers), such personalized models remain largely inaccessible in many countries.
8.8. Future Directions: Integrating Artificial Intelligence, Precision Medicine, and International Collaboration
In the near future, orthopedic medicine is expected to increasingly integrate advanced computational and systems-based approaches into routine clinical practice. Artificial intelligence–driven algorithms will play a central role in analyzing complex datasets that encompass clinical variables, imaging findings, molecular biomarkers, and genetic information. By synthesizing these multidimensional inputs, AI tools can generate real-time, personalized predictions about fracture-healing trajectories, complication risk, and anticipated treatment response, thereby supporting more informed, timely clinical decision-making.
Parallel to these developments, integrated precision-medicine platforms are likely to emerge as core components of orthopedic care. Such platforms will combine patient-specific data, including serum biomarker profiles, genomic and epigenetic signatures, and advanced imaging metrics, to recommend optimal therapeutic strategies in an automated and adaptive manner. These recommendations may include selecting appropriate structure–activity relationship–based therapies, dosing regimens, timing of intervention, and individualized monitoring protocols, enabling rapid translation of molecular insights into actionable clinical plans.
Progress in this field will also depend heavily on large-scale international collaboration. Establishing multinational consortia dedicated to randomized clinical trials, biomarker validation, and accelerated therapeutic development will be essential to ensure robust evidence generation, harmonization of methodologies, and the broad applicability of findings across diverse populations and healthcare systems. Such collaborative frameworks will facilitate the validation of emerging therapies and support the development of consensus-based clinical guidelines.
In addition, these advances will necessitate a transformation in medical education and professional training. Expanding interdisciplinary education programs to develop “hybrid” specialists—combining expertise in orthopedics, molecular biology, and bioinformatics—will be critical to effectively implementing precision medicine strategies in clinical settings. Clinicians equipped with cross-disciplinary skills will be better positioned to interpret complex molecular data, engage with computational tools, and integrate innovative therapies into patient care.
Collectively, insights into the bone–brain axis have opened the door to a new era in medicine, in which deep molecular understanding, integration of omics data, and sustained interdisciplinary collaboration overcome traditional clinical limitations. This paradigm shift offers patients with severe fractures or traumatic brain injury renewed prospects for accelerated healing, restored function, and improved quality of life. The overarching challenge moving forward is to translate these scientific discoveries into everyday clinical practice, minimize disparities in access to advanced therapies, and ensure rigorous long-term validation of innovative treatment approaches to guarantee both safety and efficacy.
9. Discussion and Future Directions in Accelerating Bone Healing after TBI
9.1. A Critical Look at Recent Advances and Current Limitations
Over the past two decades, advances in neuroscience, molecular biology, and regenerative orthopedics have fundamentally reshaped our understanding of bone healing following traumatic brain injury (TBI). What was once regarded as a clinical curiosity or “oddity” is now widely recognized as a biologically robust phenomenon that offers a powerful model for regenerative medicine and a unique opportunity to identify novel therapeutic targets for enhancing skeletal repair [1,4,9,16].
At the same time, the rapid expansion of knowledge in this field has exposed important limitations, both in mechanistic understanding and in clinical translation. The bone–brain axis represents an exceptionally complex biological network in which humoral factors, inflammatory cytokines, microRNAs, growth factors, and neuro-osteogenic signaling pathways interact in a tightly coordinated and dynamic manner. This intricate synergy cannot yet be fully replicated or precisely controlled in either in vitro or in vivo experimental systems, and substantial inter-patient variability continues to pose a major challenge to reproducibility and therapeutic standardization [13,14,17].
Moreover, accelerated bone healing is not universally advantageous. The heightened osteogenic response observed in patients with TBI is accompanied by a significantly increased risk of complications, particularly heterotopic ossification and hypertrophic callus formation. These complications can impair joint mobility, complicate rehabilitation, and, in some cases, necessitate additional surgical intervention, thereby offsetting the potential benefits of faster fracture union [4,6,34].
Finally, progress toward clinical implementation is hindered by the lack of consensus regarding therapeutic protocols. The absence of internationally accepted guidelines governing the use of recombinant BMPs, structure–activity relationship–based peptides, or cell-based therapies has resulted in heterogeneous clinical practices and variable outcomes across healthcare systems. Without standardized criteria for patient selection, dosing, timing, and monitoring, the translation of experimental advances into consistent and safe clinical applications remains incomplete.
10.2. Controversies and Open Debates in the Literature
A recurring theme across the international literature is the persistent lack of consensus regarding several critical aspects of biologically driven fracture-healing strategies following traumatic brain injury. One major area of uncertainty concerns the indications and optimal dosing of growth-factor–based therapies, particularly those involving BMP-2, BMP-7, and FGF-2. While some studies advocate using minimal effective doses to reduce the risk of heterotopic ossification and other complications, other investigations report that higher doses are necessary to achieve maximal osteogenic stimulation and reliable fracture consolidation. This divergence reflects an unresolved balance between efficacy and safety, compounded by differences in patient populations, fracture characteristics, and monitoring protocols [17,18,34].
Another unresolved issue involves the role of microRNAs and exosome-based therapies. Experimental studies have demonstrated striking effects of pro-osteogenic microRNAs and exosome-mediated signaling on mesenchymal stromal cell differentiation, angiogenesis, and callus maturation. However, translation of these findings into clinical practice remains limited. Available clinical data are sparse, and precise regulation of epigenetic effects induced by microRNAs or engineered exosomes continues to present significant technical and safety challenges, particularly with respect to off-target effects and long-term consequences [25,26,31].
Finally, increasing attention has been directed toward the impact of genetic background and immune status on therapeutic responsiveness. Recent studies from Asia and Europe suggest that individual responses to traumatic brain injury–associated osteogenic signaling and to structure–activity relationship–based therapies are strongly modulated by genetic polymorphisms, endocrine milieu, and immune profiles. Despite this growing recognition, such variables are rarely incorporated systematically into multicenter clinical trials, limiting the ability to stratify patients effectively or to explain interindividual variability in outcomes. Addressing these factors will be essential for the development of truly personalized and reproducible regenerative strategies in orthopedic trauma care [37,38].
9.2. International Comparative Discussions – “Why Do Some Protocols Succeed While Others Fail?”
An integrated analysis of clinical and experimental experience from the United States, Europe, and Asia yields several overarching conclusions regarding the effective translation of accelerated bone healing strategies after traumatic brain injury. Foremost among these is the importance of multidisciplinary integration. Clinical centers that successfully combine expertise in neurosurgery, orthopedics, molecular biology, and laboratory medicine consistently report superior outcomes when implementing innovative, biology-driven therapies. Such integration enables coordinated decision-making across acute neurotrauma management, fracture stabilization, molecular monitoring, and rehabilitation planning, thereby maximizing therapeutic benefit while minimizing complications.
Equally critical is the role of systematic monitoring and treatment individualization. Accumulating evidence indicates that uniform, “one-size-fits-all” treatment approaches frequently fail to account for the biological heterogeneity inherent to patients with traumatic brain injury. In contrast, personalized protocols guided by serum biomarker profiles, genetic and epigenetic characteristics, and the severity and pattern of brain injury achieve markedly better outcomes. These individualized strategies allow clinicians to tailor therapeutic intensity, timing, and modality, improving fracture healing while reducing the risk of adverse effects such as heterotopic ossification or delayed functional recovery.
Finally, continuous medical education emerges as a central requirement for the successful implementation of advanced regenerative approaches. Training programs that foster collaboration among orthopedic surgeons, neurosurgeons, and molecular or translational scientists are essential for bridging the gap between experimental discoveries and clinical practice. Such interdisciplinary education enhances the ability of clinical teams to interpret complex molecular data, apply emerging therapies safely, and adapt treatment protocols dynamically as new evidence becomes available, ultimately reducing risk and improving patient outcomes.
9.3. Ethical Implications and Regulatory Considerations
An increasingly important, though comparatively underexplored, aspect of research and clinical innovation in accelerated bone healing after traumatic brain injury concerns ethical and regulatory considerations. Scientific progress in this field has depended heavily on extensive use of animal models, which have been indispensable for elucidating molecular mechanisms and testing novel therapeutic strategies. However, growing ethical scrutiny and regulatory pressure emphasize the need to reduce animal experimentation wherever possible and to transition toward alternative approaches, including organoid systems, advanced three-dimensional cell cultures, and in silico modeling. These emerging platforms hold promise for refining mechanistic understanding while aligning research practices with evolving ethical standards.
Equitable access to innovative therapies represents another major ethical challenge. While leading academic and clinical centers possess the infrastructure and resources required to implement structure–activity relationship–based therapies, advanced cell-based interventions, and complex biomarker-driven monitoring, patients treated in resource-limited healthcare settings often remain excluded from these advances. This disparity raises significant concerns related to equity, fairness, and social justice, particularly as personalized regenerative therapies move closer to clinical application and become perceived as standards of care rather than experimental options.
Informed consent and long-term safety considerations further underscore the ethical complexity of this field. Many biologically driven therapies for accelerated bone healing remain experimental or are supported by limited long-term outcome data. As such, patients must be provided with clear, comprehensive, and transparent information regarding the potential benefits, known and unknown risks, and the current absence of definitive long-term safety evidence. Ensuring rigorous informed consent processes is essential not only for patient autonomy but also for maintaining public trust as innovative regenerative strategies transition from research settings into broader clinical practice.
9.4. Narrative Discussion: From Fundamental Biology to Real-World Clinical Applications
Here is the passage rewritten as plain, unformatted academic prose, with the content preserved and expanded for clarity and interdisciplinary scope, and without bullets or special formatting:
The paradigm shift driven by advances in understanding the bone–brain axis extends far beyond the traditional boundaries of orthopedics and neurosurgery. Insights into the molecular and cellular crosstalk between neural injury and skeletal regeneration have broad implications for multiple medical disciplines, including rheumatology, sports medicine, geriatrics, pediatrics, and plastic and reconstructive surgery. As this field matures, it increasingly challenges compartmentalized views of tissue repair and highlights the systemic nature of regenerative responses.
A more detailed understanding of the signaling networks linking the brain and bone has the potential to inspire innovative therapeutic strategies for a wide range of clinical conditions. These include the management of severe osteoporosis and pathological fractures, in which endogenous osteogenic capacity is compromised and could be enhanced through targeted modulation of neuro-osteogenic pathways. Similar principles may be applied to accelerate postoperative recovery following orthopedic or reconstructive procedures, reducing immobilization time and improving functional outcomes.
Furthermore, elucidation of bone–brain interactions may inform new approaches to the prevention or treatment of heterotopic ossification in neurological conditions such as cerebral palsy or spinal cord injury, where dysregulated osteogenic signaling leads to debilitating ectopic bone formation. In the context of complex transplantation or reconstructive surgery, including large bone defects or composite tissue grafts, controlled activation of neurogenic and osteogenic pathways could facilitate robust bone regeneration and integration, improving graft survival and long-term structural stability.
Collectively, these interdisciplinary applications underscore the transformative potential of bone–brain axis research. By integrating molecular biology, clinical medicine, and regenerative science, this emerging framework offers opportunities to address unmet clinical needs across diverse patient populations and to redefine therapeutic strategies for skeletal repair and regeneration.
5. Conclusions
Accelerated bone healing following traumatic brain injury is now recognized as a robust and reproducible biological phenomenon mediated by the bone–brain axis. Clinical and experimental evidence demonstrates that traumatic brain injury induces a coordinated systemic response involving hormones, growth factors, cytokines, and microRNAs that profoundly enhances osteogenesis and fracture repair.
This response is driven primarily by synergistic signaling among prolactin, BMP-2, BMP-7, FGF-2, IGF-1, and angiogenic mediators, acting through key osteogenic and vascular pathways. Structure–activity relationship analyses clarify how specific molecular domains underlie both the regenerative benefits and the associated risks, particularly heterotopic ossification. While accelerated healing offers clear clinical advantages, the increased incidence of exuberant callus and ectopic bone formation highlights the need for precise regulation and patient-specific intervention.
Understanding the molecular basis of this phenomenon enables a transition from empirical orthopedic care toward personalized regenerative medicine. Biomarker-guided monitoring, risk stratification, and the rational use of SAR-based biomimetic therapies, recombinant growth factors, and emerging cell- or exosome-based approaches hold promise for improving outcomes in delayed unions, non-unions, and osteoporotic fractures.
Despite these advances, significant challenges remain, including inter-individual variability, limited long-term outcome data, high costs, and the absence of large randomized trials and standardized clinical guidelines. Continued interdisciplinary research, international collaboration, and integration of precision-medicine strategies will be essential to translate insights from the bone–brain axis into safe, effective, and widely accessible therapies for fracture healing and musculoskeletal regeneration.
Author Contributions
Conceptualization, D.E., I.L.C., and C.N.L.; methodology, D.E., I.L.C., and C.N.L.; software, R.M.; validation, D.E., I.L.C., C.N.L., and M.L.; formal analysis, D.E., C.N.L., and M.L.; investigation, D.E., I.L.C., C.N.L., and R.M.; resources, R.M. and V.L.; data curation, D.E., C.N.L., and M.L.; writing—original draft preparation, D.E., I.L.C., and C.N.L.; writing—review and editing, D.E., I.L.C., C.N.L., M.L., R.M., and V.L.; visualization, R.M.; supervision, I.L.C., C.N.L., and V.L.; project administration, I.L.C. and C.N.L.; funding acquisition, not applicable. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
Not appliable
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
ALP – Alkaline phosphatase
AKT – Protein kinase B
BMP – Bone morphogenetic protein
BMPR – Bone morphogenetic protein receptor
CNS – Central nervous system
COL1A1 – Collagen type I alpha 1 chain
CT – Computed tomography
DNA – Deoxyribonucleic acid
EGF – Epidermal growth factor
ERK – Extracellular signal–regulated kinase
FGF – Fibroblast growth factor
FGFR – Fibroblast growth factor receptor
GH – Growth hormone
GCS – Glasgow Coma Scale
HO – Heterotopic ossification
HGF – Hepatocyte growth factor
IGF – Insulin-like growth factor
IGF1R – Insulin-like growth factor 1 receptor
IL – Interleukin
JAK – Janus kinase
MAPK – Mitogen-activated protein kinase
miR – MicroRNA
MRI – Magnetic resonance imaging
MSC – Mesenchymal stem/stromal cell
NPY – Neuropeptide Y
OCN – Osteocalcin
OSX – Osterix
PDGF – Platelet-derived growth factor
PICP – Procollagen type I C-terminal propeptide
PI3K – Phosphoinositide 3-kinase
PLCγ – Phospholipase C gamma
PRL – Prolactin
PRLR – Prolactin receptor
RNA – Ribonucleic acid
RUNX2 – Runt-related transcription factor 2
SAR – Structure–activity relationship
SMAD – Small mothers against decapentaplegic
STAT – Signal transducer and activator of transcription
TBI – Traumatic brain injury
TGF-β – Transforming growth factor beta
TNF-α – Tumor necrosis factor alpha
VEGF – Vascular endothelial growth factor
References
- Marsell, R; Einhorn, TA. The biology of fracture healing. Injury 2011, 42(6), 551–5. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-C.; Tsai, Y.-H.; Wang, T.-C.; Yang, T.-Y. The effects of an injury to the brain on bone healing and callus formation in young adults with fractures of the femoral shaft. Journal of Bone and Joint Surgery, British Volume 2012, 94-B(2), 227–230. [Google Scholar]
- Giannoudis PV, Mushtaq S, Harwood P, Kambhampati S, Dimoutsos M, Stavrou Z, Pape HC. Accelerated bone healing and excessive callus formation in patients with femoral fracture and head injury. Injury. 2006 Sep;37 Suppl 3:S18-24. Erratum in: Injury. 2007 Oct;38(10):1224. [CrossRef]
- Chalmers, J; Gray, DH; Rush, J. Observations on the induction of bone in soft tissues. J Bone Joint Surg Br. 1975, 57(1), 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wildburger, R; Zarkovic, N; Tonkovic, G; Skoric, T; Frech, S; Hartleb, M; Loncaric, I; Zarkovic, K. Post-traumatic hormonal disturbances: prolactin as a link between head injury and enhanced osteogenesis. J Endocrinol Invest. 1998, 21(2), 78–86. [Google Scholar] [CrossRef] [PubMed]
- Morley, J; Marsh, S; Drakoulakis, E; Pape, HC; Giannoudis, PV. Does traumatic brain injury result in accelerated fracture healing? Injury 2005, 36(3), 363–8. [Google Scholar] [CrossRef]
- Spencer, RF. The effect of head injury on fracture healing. A quantitative assessment. J Bone Joint Surg Br. 1987, 69(4), 525–8. [Google Scholar] [CrossRef]
- Morley, J; Marsh, S; Drakoulakis, E; Pape, HC; Giannoudis, PV. Does traumatic brain injury result in accelerated fracture healing? Injury 2005, 36(3), 363–8. [Google Scholar] [CrossRef]
- Cadosch, D; Gautschi, OP; Thyer, M; Song, S; Skirving, AP; Filgueira, L; Zellweger, R. Humoral factors enhance fracture-healing and callus formation in patients with traumatic brain injury. J Bone Joint Surg Am. 2009, 91(2), 282–8. [Google Scholar] [CrossRef]
- Tsitsilonis, S; Seemann, R; Misch, M; Wichlas, F; Haas, NP; Schmidt-Bleek, K; Kleber, C; Schaser, KD. The effect of traumatic brain injury on bone healing: an experimental study in a novel in vivo animal model. Injury 2015, 46(4), 661–5. [Google Scholar] [CrossRef]
- Park, SY; Suh, KT; Ryu, CH; Woo, SH; Lee, JS; Kim, SG. Alterations in Serum Levels of Receptor Activator of Nuclear Factor-kappa B Ligand and Osteoprotegerin in Patients with Head Injury and Fracture. J Korean Fract Soc. 2008, 21(2), 145–150. [Google Scholar] [CrossRef]
- Cadosch, D; Toffoli, AM; Gautschi, OP; Frey, SP; Zellweger, R; Skirving, AP; Filgueira, L. Serum after traumatic brain injury increases proliferation and supports expression of osteoblast markers in muscle cells. J Bone Joint Surg Am. 2010, 92(3), 645–53. [Google Scholar] [CrossRef]
- Jones, KB; Mollano, AV; Morcuende, JA; Cooper, RR; Saltzman, CL. Bone and brain: a review of neural, hormonal, and musculoskeletal connections. Iowa Orthop J. 2004, 24, 123–32. [Google Scholar]
- Darvin, P; Joung, YH; Yang, YM. JAK2-STAT5B pathway and osteoblast differentiation. JAKSTAT 2013, 2(4), e24931. [Google Scholar] [CrossRef]
- Hofman, M; Koopmans, G; Kobbe, P; Poeze, M; Andruszkow, H; Brink, PR; Pape, HC. Improved fracture healing in patients with concomitant traumatic brain injury: proven or not? Mediators Inflamm 2015, 2015, 204842. [Google Scholar] [CrossRef]
- Chen, D; Zhao, M; Mundy, GR. Bone morphogenetic proteins. Growth Factors 2004, 22(4), 233–41. [Google Scholar] [CrossRef]
- Ripamonti, U; Klar, RM; Renton, LF; Ferretti, C. Synergistic induction of bone formation by hOP-1, hTGF-beta3 and inhibition by zoledronate in macroporous coral-derived hydroxyapatites. Biomaterials 2010, 31(25), 6400–10. [Google Scholar] [CrossRef] [PubMed]
- Carreira, AC; Alves, GG; Zambuzzi, WF; Sogayar, MC; Granjeiro, JM. Bone Morphogenetic Proteins: structure, biological function and therapeutic applications. Arch Biochem Biophys. 2014, 561, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, DM; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2(3), REVIEWS3005. [Google Scholar] [CrossRef]
- Murugaiyan, K; Amirthalingam, S; Hwang, NS; Jayakumar, R. Role of FGF-18 in Bone Regeneration. J Funct Biomater. 2023, 14(1), 36. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E. Growth factor control of bone mass. J Cell Biochem. 2009, 108(4), 769–77. [Google Scholar] [CrossRef]
- Yakar, S; Werner, H; Rosen, CJ. Insulin-like growth factors: actions on the skeleton. J Mol Endocrinol. 2018, 61(1), T115–T137. [Google Scholar] [CrossRef]
- Street, J; Bao, M; deGuzman, L; Bunting, S; Peale, FV, Jr.; Ferrara, N; Steinmetz, H; Hoeffel, J; Cleland, JL; Daugherty, A; van Bruggen, N; Redmond, HP; Carano, RA; Filvaroff, EH. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci U S A 2002, 99(15), 9656–61. [Google Scholar] [CrossRef]
- Bonewald, LF; Mundy, GR. Role of transforming growth factor-beta in bone remodeling. Clin Orthop Relat Res 1990, (250), 261–76. [Google Scholar] [CrossRef]
- Lian, JB; Stein, GS; van Wijnen, AJ; Stein, JL; Hassan, MQ; Gaur, T; Zhang, Y. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol 2012, 8(4), 212–27. [Google Scholar] [CrossRef] [PubMed]
- Breulmann, FL; Hatt, LP; Schmitz, B; Wehrle, E; Richards, RG; Della Bella, E; Stoddart, MJ. Prognostic and therapeutic potential of microRNAs for fracture healing processes and non-union fractures: A systematic review. Clin Transl Med. 2023, 13(1), e1161. [Google Scholar] [CrossRef]
- Zhang, Y; Wang, Q; Xue, H; Guo, Y; Wei, S; Li, F; Gong, L; Pan, W; Jiang, P. Epigenetic Regulation of Autophagy in Bone Metabolism. Function (Oxf) 2024, 5(2), zqae004. [Google Scholar] [CrossRef]
- Mountziaris, PM; Mikos, AG. Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng Part B Rev. 2008, 14(2), 179–86. [Google Scholar] [CrossRef] [PubMed]
- Anthonissen, J; Steffen, CT; Hofmann, A; Victor, J. The pathogenesis of heterotopic ossification after traumatic brain injury. A review of current literature. Acta Orthop Belg 2020, 86(3), 369–377. [Google Scholar]
- Elefteriou, F. Impact of the Autonomic Nervous System on the Skeleton. Physiol Rev. 2018, 98(3), 1083–1112. [Google Scholar] [CrossRef]
- Yamada, Y; Nakamura, S; Klein, OD; Ito, K. Current trends in stem cell therapy for improvement of bone quality. Histol Histopathol 2014, 29(6), 691–7. [Google Scholar]
- Cipriano, CA; Pill, SG; Keenan, MA. Heterotopic ossification following traumatic brain injury and spinal cord injury. J Am Acad Orthop Surg. 2009, 17(11), 689–97. [Google Scholar] [CrossRef]
- Sullivan, MP; Torres, SJ; Mehta, S; Ahn, J. Heterotopic ossification after central nervous system trauma: A current review. Bone Joint Res. 2013, 2(3), 51–7. [Google Scholar] [CrossRef]
- Anthonissen, J; Steffen, CT; Hofmann, A; Victor, J. The pathogenesis of heterotopic ossification after traumatic brain injury. A review of current literature. Acta Orthop Belg 2020, 86(3), 369–377. [Google Scholar]
- Gautschi, OP; Cadosch, D; Frey, SP; Skirving, AP; Filgueira, L; Zellweger, R. Serum-mediated osteogenic effect in traumatic brain-injured patients. ANZ J Surg 2009, 79(6), 449–55. [Google Scholar] [CrossRef] [PubMed]
- Boes M, Kain M, Kakar S, Nicholls F, Cullinane D, Gerstenfeld L, Einhorn TA, Tornetta P 3rd. Osteogenic effects of traumatic brain injury on experimental fracture-healing. J Bone Joint Surg Am. 2006 Apr;88(4):738-43. Erratum in: J Bone Joint Surg Am. 2006 Jul;88(7):16. [CrossRef]
- Wei, F; Li, M; Crawford, R; Zhou, Y; Xiao, Y. Exosome-integrated titanium oxide nanotubes for targeted bone regeneration. Acta Biomater. 2019, 86, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, TA; Gerstenfeld, LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol 2015, 11(1), 45–54. [Google Scholar] [CrossRef] [PubMed]
- Paulini, M; Camal Ruggieri, IN; Ramallo, M; Alonso, M; Rodriguez-Cabello, JC; Esbrit, P; Mardegan Issa, JP; Feldman, S. Recombinant Proteins-Based Strategies in Bone Tissue Engineering. Biomolecules 2021, 12(1), 3. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



