
Submitted:
01 February 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Infectious laryngotracheitis virus (ILTV) is a highly contagious respiratory pathogen of poultry that causes significant economic losses to the poultry industry worldwide. Live attenuated vaccines have been used for decades to control ILTV outbreaks. Among the ILTV live attenuated vaccines, SA2 and A20 have been frequently used as commercial vaccines. Frequent changes occur in the ILTV genome due to its possibility of genome drift and recombination. The major goal of this study is to monitor genomic variations in recently circulating ILTV field isolates recovered from vaccinated chicken flocks. Tissue samples collected from an infected chickens flock suspected of ILTV were examined by real-time PCR. Samples with high viral load were selected for next-generation sequencing (NGS). Full length genome of three ILTV isolates (B1, B3, and B4) were obtained and submitted to GenBank with accession numbers PX492157, PX496590, and PX522223, respectively. The complete viral genome size of ILTV-B1, ILTV-B3, and ILTV-B4 was 152975, 152978, and 152978 nucleotides, respectively. These three ILTV isolates show 99.9% similarity with the Australian vaccinal strain SA2. The multiple sequence alignment showed that ILTV-B1, ILTV-B3, and ILTV-B4 belong to wild-type genotype VI-IX, which clustered together with Austrian isolates SA2 and A20, and American isolates S2.816 and 6.48.88 isolated in 2017. However, ILTV-B1, ILTV-B3, and ILTV-B4 also showed amino acid substitutions in the gB and gJ glycoproteins when compared with SA2, A20, and the USDA reference strain. Furthermore, most notable mutations were reported in the gB, gJ, ICP4, gG, gD, and gI when compared with American, Australian, and European vaccinal strains. Together, this study indicates the circulation of novel ILTV field isolates with the possible genomic divergence potentially associated with live vaccine-derived drift.
Keywords:
ILTV
; chicken
; vaccine
; respiratory isolate
; RT-qPCR
; NGS
; phylogenetic analysis
; genetic drift
1. Introduction
The infectious laryngotracheitis virus was first reported in Canada in 1925 [1]. ILTV is classified as a member of the family Herpesviridae, subfamily Alphaherpesvirinae, genus Iltovirus, and species Gallid alphaherpesvirus-1 [2,3]. ILTV have very specific host range and predominantly affects galliform birds [4]. Although chickens are the most frequent hosts, pheasants, peafowl while, turkeys could be infected under experimental settings[5,6,7].
In poultry, the severe acute or epizootic form of ILT infection is characterized by significant sneezing, respiratory distress, expectoration of blood-mixed mucus, conjunctivitis, and severe hemorrhagic tracheitis along with high mortality reaching up to 70% (ranging from 5% - 70%), a milder form that typically ranges from 0.1% - 2% and is characterized by mild to severe conjunctivitis, sinusitis, and catarrhal tracheitis, as well as comparatively modest morbidity and infrequent fatality [8].
ILTV has an icosahedral shape with a hexagonal nucleocapsid that contains its double-stranded DNA genome. This nucleocapsid has 162 long and hollow capsomeres of 80–100 nm in diameter [9,10]. The ILTV genome is linear double-stranded DNA having a unique long (UL), unique short (US) and two inverted repeat (IR) sequences encoded by 150-155 kb nucleotides, with a GpC content of 48.2% [11,12,13]. Under electron microscopy, the ILTV virions resemble normal herpes virions, with outer envelopes made of glycoproteins and an icosahedral capsid with a DNA core enclosed in a tegument. The entire viral particle is between 200 to 350 nm in size, with the viral capsid diameter is about 100 nm [14].
The ILTV genome comprises eighty open reading frames (ORFs) distributed across three primary genomic regions: sixty-five ORFs are located within the unique long (UL) region, nine are situated in the unique short (US) region, and six are mapped within the inverted repeat (IR) segments [13]. Sixty-three of the eighty ORFs show similarities to the HSV-1 genome in terms of the location and structure of the in-silico predicted protein products.
The ILTV envelope contains eleven glycoproteins (UL27, UL44, US6, US8, US4, UL22, US7, US5, UL53, UL1, and UL10) which are encoded by highly conserved open reading frames (gB, gC, gD, gE, gG, gH, gI, gJ, gK, gL, and gM) respectively [15].
In contrast to HSV-1, the ILTV genome contains unique gene clusters. The first cluster, situated between UL45 and UL22, comprises five open reading frames designated ORF A–E. A second ILTV-specific cluster is located between UL1 and ICP4, encoding UL0 and a divergent UL-1 ortholog.[16].
Unlike other Alpha-herpes viruses, the ILTV genome lacks the UL16 gene and features an inverted conserved gene cluster in the UL region. Furthermore, UL47 is uniquely located in the US region between US3 and US4, representing a significant departure from the prototypical UL organization seen in HSV-1[17,18].
The deduced amino acid sequences of two ILTV-specific sections, UL0 and UL (-1), exhibit significant similarities, indicating a duplication event during virus evolution [19]. Defective ILTV that was unable to spread in permissive cells was produced by deleting the UL (-1) gene of ILTV and substituting it with the gene encoding green fluorescent protein (GFP) and the primary immediate promoter element of cytomegalovirus.
Accordingly, ILTV replication is significantly influenced by the UL (-1) gene [20]. Similar to other Alpha herpes viruses, the ILTV genome has two copies of OriS in the IR and terminal repeat (TR) sections, an OriL in the UL region, and three possible origins for DNA replication [13]. The properties of the ORFs differ from those of other alpha herpesviruses [21]. According to Kelly et al., 2009 [22], the tegument proteins aid in the capsid's movement into the cytoplasm and eventually the nucleus [22].
ILTV has a history of global spread than other well-known avian respiratory viral infections, yet it is still widely distributed. For many years, ILTV has been widely prevalent on almost every continent. Between 2000 and 2013, ILT outbreaks were documented in more than 100 countries [23]. Between 2007 and 2017, 88 cases of ILTV were confirmed in California alone in the United States [24], but most of these cases included mild clinical manifestations of ILTV infection However, ILTV continues to pose a serious risk to the global poultry industry. The virulence of the strain that is circulating in the area along with associated infection with other respiratory bacterial and viral pathogens significantly influences the morbidity and mortality of reported cases [25].
ILTV remains prevalent in backyard and commercial poultry systems in North America, which adds to the region's persistent health challenges. In Canada, ILTV infection is widespread in backyard flocks, and commercial businesses occasionally experience outbreaks [26] and comparable silent circulation patterns have been seen in other places, like Costa Rica, where significant ILTV seropositivity in unvaccinated backyard poultry raises the possibility that these flocks serve as neglected repositories maintaining regional transmission [27]. The persistence of circulating North American ILTV strains was highlighted by a field research conducted in North Alabama that showed continuous environmental ILTV contamination even after depopulation and cleaning [28]. Beyond North America, similar patterns have been observed in Latin America. For example, Brazil reported 15 ILTV outbreaks between 2012 and 2014, all of which were similar to the TCO vaccine strain, demonstrating the continued persistence of vaccination-derived ILTV after its emergence in 2002 [29]. Five ILTV isolates were retrieved from Alberta in Western Canada were found to be wild-type and unsimilar to live attenuated vaccines based on the genotyping analysis [30,31,32]. In Ontario, wild-type ILTV outbreaks do occur, but they are still less common than vaccine-associated outbreaks [33].
Molecular surveillance has emerged as a critical tool for monitoring viral pathogens across human and veterinary medicine. By analyzing viruses at the genomic level, researchers can map specific mutations that alter phenotypic or genotypic characteristics, potentially leading to the emergence of high-virulence variants. This high-resolution approach enables stakeholders and decision-makers to refine vaccination strategies and implement proactive interventions to ensure the robust protection of target hosts [34,35,36,37,38,39,40,41,42,43,44,45].
Genomic surveillance and epidemiological studies have highlighted the significant role of vaccine-derived strains in ILTV outbreaks across North America. Research in Western Canada demonstrated that ILTV isolates in British Columbia and Alberta are frequently genotyped using partial sequences of ORF a and ORF b, revealing that approximately 85% of these isolates are related to vaccine strains, specifically chicken embryo origin (CEO) revertant, while the remainder represent wild-type viruses [32]. Similarly, a surveillance study was conducted in the US in Maryland backyard flocks identified a high viral burden, characterized by a 26% PCR-based prevalence and a 77% seroprevalence. Notably, while global commercial outbreaks are commonly linked to CEO vaccines, molecular characterization of these Maryland backyard isolates indicated that eight out of ten ILTV-positive cases matched tissue culture origin (TCO) vaccines, suggesting distinct patterns of viral circulation and vaccine-derived drift in non-commercial poultry populations [46].
North American ILTV strains are classified into nine distinct genotypes (I–IX) based on comprehensive molecular characterization. Historically, these lineages were defined through PCR-RFLP targeting the ORF-b-TK intergenic region and the UL47, ICP4, gB, gG, and gM genes; however, recent advances in whole-genome and MinION sequencing have further refined these classifications. Under this framework, the United States standard challenge strain is assigned to genotype I, while genotypes II and III encompass tissue culture origin (TCO) vaccines and their associated virulent derivatives. Genotypes IV and V consist of chicken embryo origin (CEO) attenuated vaccines and related virulent revertant strains. Furthermore, genotype VI represents virulent, non-vaccine field strains circulating within commercial poultry operations, whereas genotypes VII–IX are predominantly composed of virulent isolates recovered from backyard flocks. [47].
Current molecular evidence indicates that a distinct subset of commercial ILTV isolates belongs to genotype VI, representing a lineage that is phylogenetically unrelated to established vaccinal strains. In contrast, the vast majority of isolates circulating in U.S. commercial poultry cluster within genotypes IV and V, which are characterized by their close genetic relationship to chicken embryo origin (CEO) vaccines [48]. Notably, genotypes V and VI exhibit heightened pathogenicity and superior replication kinetics compared to genotype IV and chicken embryo origin (CEO) vaccinal strains circulating in the United States. These enhanced growth characteristics suggest a higher level of viral fitness, contributing to the increased morbidity observed in outbreaks associated with these specific lineages [48]. Furthermore, genotype VI has been implicated in severe, rapidly disseminating ILT epidemics beyond North America, most notably in Brazil—underscoring its global epidemiological significance and potential for widespread geographic expansion [49]. Consistent with these observations, molecular characterization of ILTV isolates from British Columbia and Alberta between 2009 and 2018 identified genotype V as the predominant lineage. The concurrent detection of genotype VI and backyard-associated genotypes VII–IX further illustrates the sustained circulation and expanding genetic diversity of virulent, non-vaccinal ILTV strains within North America [32].
2. Materials and Methods:
2.1. Chicken Samples Collection and Processing
One hundred tracheal tissue samples were collected from twelve different chicken flocks and were kindly provided by the North Carolina Veterinary Diagnostic Laboratory Services, USA. The tissue samples were collected from 40 – 50 days old broiler infected chickens between October 2023 and November 2024. Processing of these tissue specimens was conducted as described in our previous study [50]. Simply, samples from each flock were treated individually and used for the preparation of 10 % tissue suspensions as previously described [50]. These tissue suspensions were stored at (-80oC) until further testing
2.2. The Total Viral DNA Isolation and Testing for the ILTV by Real-Time PCR
Viral genomic DNA was extracted from pooled tissue homogenates for each flock using the Quick-DNA/RNA Viral Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s protocol. The concentration and purity of the isolated DNA were assessed via spectrophotometry using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), with samples subsequently stored at (–20°C) for downstream analysis. Viral detection and quantification were performed via real-time PCR (qPCR) utilizing the PowerUp™ SYBR™ Green Master Mix (Applied Biosystems, Foster City, CA, USA) on a QuantStudio 3 Real-Time PCR System (Applied Biosystems). The nucleotide sequences of the primers used to amplify ILTV viral genome are: forward primer 5'-CCTTGCGTTTGAATTTTTCTGT-3' and reverse primer 5'-TTCGTGGGTTAGAGGTCTGT-3' targeting UL44 (gC) gene of ILTV was designed using primer3 online server [51]. Chicken GAPDH forward primer 5’-CACACAGAAGACGGTGGATG-3′ and reverse primer 5’-AACAGAGACATTGGGGGTTG-3′ [52] was used to normalized the ILTV viral genome expression following 2-^^CT values [53]
2.3. The Next Generation Sequencing (NGS)
For whole-genome sequencing (WGS) of the ILTV isolates, genomic DNA samples were submitted to Azenta Life Sciences (South Plainfield, NJ, USA). Library preparation and Next-Generation Sequencing (NGS) were performed according to previously established protocols [Insert Citation]. Briefly, the genomic architecture was resolved using [Illumina HiSeq/MiSeq] platforms, ensuring high-depth coverage for comprehensive de novo assembly or reference-based mapping [43,44]. Briefly, Next-Generation Sequencing (NGS) was conducted by constructing high-quality genomic DNA libraries followed by [Insert Platform, e.g., Illumina] sequencing. Raw sequence reads were processed for quality control and adapter removal using Trimmomatic (v0.36). The trimmed reads were subsequently mapped to the reference ILTV strain SA2 (GenBank Accession No. NC_006623) using [Insert Aligner, e.g., BWA or Bowtie2]. The final de novo assembled whole-genome sequences of the ILTV isolates were then deposited into the GenBank database.
2.4. Multiple Sequence Alignment (MSA) and Phylogenetic Analysis
Multiple sequence alignment (MSA) and pairwise identity analysis of the three ILTV isolates (ILTV-B1, ILTV-B3, and ILTV-B4) were conducted using Geneious Prime (v2024.11.0.2). Representative ILTV sequences encompassing genotypes I through IX were retrieved from the NCBI GenBank database to serve as reference strains for comparative analysis. Phylogenetic reconstruction was performed using the Maximum Likelihood (ML) method within MEGA 11 software, with nodal support assessed via a bootstrap analysis of 1,000 replicates. The evolutionary model was selected based on the lowest Bayesian Information Criterion (BIC) score to ensure optimal tree topology [54].
2.5. Comparative Divergence Analysis of the Key ILTV Proteins
A comparative divergence analysis was conducted for the UL27 (gB) and US5 (gJ) genes of isolates ILTV-B1, ILTV-B3, and ILTV-B4 against the SA2 and USA reference strains. Multiple sequence alignments (MSA) to determine nucleotide and amino acid identity were performed using Geneious Prime (v2024.11.0.2). This analysis focused on identifying conserved domains and site-specific variations within these critical glycoproteins to assess evolutionary distance and potential phenotypic shifts relative to established reference lineages.
2.6. In Silico Structural Modeling of Some Key ILTV Proteins
The tertiary protein structures of the UL27 (gB) and US5 (gJ) glycoproteins were predicted using the AlphaFold 3 server to assess the impact of observed mutations on protein folding. This deep-learning-based modeling utilized the isolates' deduced amino acid sequences to generate high-resolution structural predictions. Model reliability was rigorously evaluated using the predicted Local Distance Difference Test (pLDDT) scores and Predicted Alignment Error (PAE) matrices. The resulting three-dimensional coordinates were then superimposed onto reference structures to identify potential conformational shifts or alterations in neutralizing epitopes induced by site-specific substitutions [55].
2.7. Statistical Analysis
All data collected in this study were subjected to a one-way analysis of variance (ANOVA) to determine statistically significant differences between groups. Post-hoc comparisons were conducted using [Tukey’s] multiple comparisons test where applicable. Statistical significance was defined as p < 0.05, with significant results denoted by an asterisk (*). All computational analyses and figure generations were performed using GraphPad Prism (v9.0.0, GraphPad Software, San Diego, CA, USA).
3. Results:
3.1. Molecular Detection and Selection of ILTV Isolates
The presence of ILTV within tracheal tissue specimens was confirmed using a highly sensitive quantitative real-time PCR (qPCR) assay. Among the screened populations, three specific chicken flocks exhibited high viral loads, as evidenced by significantly low cycle threshold (Ct) values. These isolates designated as (ILTV-B1, ILTV-B3, and ILTV-B4) were selected for downstream genomic characterization based on their high viral genome copy numbers. The diagnostic profiles and relative quantification for these representative samples are detailed in Figure 1.
3.2. The Genome Structure and Organization of ILTV Isolates Reported in This Study
The complete genome sequences of isolates ILTV-B1, ILTV-B3, and ILTV-B4 were successfully assembled and deposited into the NCBI GenBank database under accession numbers PX492157, PX496590, and PX522223, respectively. The genome of ILTV-B1 spans 152,975 bp and exhibits 99.997% nucleotide identity with the ILTV SA2 reference strain (Figure 2, Table 1). In contrast, both ILTV-B3 and ILTV-B4 possess a slightly larger genome size of 152,978 bp. The ILTV-B3 genome sequence shares 99.98% homology with the SA2 strain; however, a notable genomic variation was identified: a three-nucleotide insertion (ACC) at position 1,673. This insertion results in an additional proline residue at position 559 within the ORF F gene product relative to the SA2 reference. ILTV-B4 demonstrated 99.9% sequence identity with the SA2 strain. Comprehensive genomic annotations and the structural organization of all three isolates are detailed (Supplementary Table 1).
3.3. Phylogenetic Analysis Based on the Full-Length Genome Sequences of ILTV-B1, B3, and B4
A phylogenetic tree was reconstructed using the whole-genome sequences of 52 ILTV isolates, representing a global distribution of genotypes I through IX, to contextualize ILTV-B1, ILTV-B3, and ILTV-B4. Phylogenetic topology revealed that the three isolates from this study clustered monophyletically with the Australian vaccinal strains A20 and SA2 (Figure 3). This clade also included North American strains S2.816 and 6.48.88, which were isolated in 2017. Despite the proximity to certain vaccinal lineages, the overall genomic characterization and phylogenetic placement categorize ILTV-B1, ILTV-B3, and ILTV-B4 within the virulent field-type lineages associated with genotypes VI–IX (Figure 3).
3.4. Phylogenetic Analysis of Some Key Genes of ILTV-B1, B3, and B4 Isolates
Phylogenetic reconstruction of five key marker genes (ICP4, UL23 (TK), UL27 (gB), US4 (gG), and US5 (gJ) was conducted to evaluate the evolutionary relationships of isolates ILTV-B1, -B3, and -B4 against globally distributed strains. Analysis of the ICP4, UL23 (TK), and US4 (gG) loci consistently placed the three study isolates within a cluster comprising Australian vaccinal strains (SA2 and A20) and North American field strains (S2.816 and 6.48.88) (Figure 4A, 4B, and 4C). Similarly, the US5 (gJ) gene showed high homology with SA2, A20, and the Chinese isolate WG (Figure 4E). In contrast, the UL27 (gB) gene exhibited a distinct and discordant clustering pattern compared to the whole-genome analysis (Figure 4D). Specifically, while ILTV-B1 remained clustered with the SA2 strain, ILTV-B3 grouped with the classical Australian isolates V1-99 and CSW-1 (isolated in 1999 and 1970, respectively). Furthermore, ILTV-B4 clustered independently with the USDA reference strain and the Italian isolate 757/11. These findings suggest a higher degree of genetic heterogeneity or potential recombination events within the UL27 locus among these isolates.
3.5. Mapping the Notable Mutations Within the UL27 (gB) Genes of ILTV-(B1, B3, and B4) Isolates Reported in This Study
The phylogenetic analysis of UL27 (gB) gene shows different clustering patterns when compared to the complete genome and other key genes results. Therefore, we investigated to identify the mutations in UL27 gene. Multiple sequence alignment of UL27 (gB) gene of ILTV-B1, ILTV-B3, ILTV-B4, SA2, and USA reference isolates were performed. Results showed four notable variations in UL27 (gB) gene (Figure 5A – 5J). The first substitution was observed at position 44 of UL27, where SA2 encodes histidine (Figure 5B), while the USDA reference isolate encodes arginine at the same position. The ILTV-B1 and ILTV-B3 encode histidine at position 44 (Figure 5B), while ILTV-B4 encodes arginine at the same position (Figure 5C). At position 551 of UL27 gene SA2 and ILTV-B1 encode valine (Figure 5D), while the ILTV-B3 and ILTV-B4 encodes methionine at same position (Figure 5E). At position 799 ILTV-B1 isolates encode serine instead of proline (Figure 5F, 5G). Similarly, at position 805 ILTV-B1 isolate encodes arginine instead of lysine (Figure 5H, 5I).
3.6. Mutations and Substitution Analysis Within the US5 (gJ) Gene of ILTV- Isolates Reported in This Study
Multiple sequence alignment (MSA) of the US5 (gJ) gene was conducted for isolates ILTV-B1, B3, and -B4 in comparison with the SA2 reference strain. The analysis identified four unique non-synonymous mutations exclusive to the ILTV-B4 isolate (Figure 6A–6C). At nucleotide position 494, ILTV-B4 possesses an alanine residue, whereas SA2, ILTV-B1, and ILTV-B3 encode valine (V494A; Figure 6D–6E). At position 647, ILTV-B4 encodes threonine, contrasting with the isoleucine found in the other three isolates (I647T; Figure 6F–6G). A third substitution was identified at position 694, where ILTV-B4 substitutes alanine for threonine (A694T; Figure 6H–6I). Finally, a notable transition at position 887 results in a phenylalanine-to-leucine substitution (F887L) unique to the ILTV-B4 isolate (Figure 6J–6K).
3.7. Mutations and Substitution Analysis Within the Structural Proteins of ILTV-B1, ILTV-B3, ILTV-B4 Compared to the ILTV Vaccine Strains
Comparative multiple sequence alignment (MSA) revealed a distinct landscape of amino acid substitutions within the ILTV-B1, -B3, and -B4 isolates relative to widely utilized vaccine strains of North American (LT-Blen, Laryngo Vacc, and US-IVAX), Australian (Serva/SA2), and European (Nobilis Laringovac®) origins (Table 2). These non-synonymous mutations were distributed across 14 distinct viral proteins, with heavy clustering within the structural envelope glycoproteins. Specifically, significant polypeptide alterations were identified in gH (UL22), gB (UL27), gD (US6), and gG (US4). Beyond the structural components, the Thymidine Kinase (UL23) and the major transcriptional regulator ICP4 exhibited substantial variability, with the latter harboring seven substitutions across its coding sequence. The spatial distribution and frequency of these mutations underscore a significant genetic divergence between the field isolates and the attenuated vaccine strains. While the sequenced isolates maintain a high degree of homology with the Australian SA2 strain, they demonstrate a pronounced evolutionary distance from the North American vaccinal lineages (Figure 3).
4. Discussion
The ILTV; Gallid alphaherpesvirus 1 is a prominent member of the Alphaherpesvirinae subfamily within the Ortho-Herpesviridae family. Consistent with other Alpha Herpesviruses, ILTV is characterized by a relatively low nucleotide substitution rate compared to many other DNA viruses. This evolutionary stability is primarily attributed to the high-fidelity proofreading activity of the viral DNA polymerase, which minimizes the accumulation of spontaneous mutations during genome replication [56].
LTV possesses a linear, double-stranded DNA genome of approximately 150 kb with a mean G+C content of 48%. While the virus is categorized into several distinct genotypic groups based on global epidemiological data, localized variations continue to emerge. In the present study, three field isolates designated ILTV-B1, ILTV-B3, and ILTV-B4 were recovered from ILTV-positive poultry flocks in North Carolina, USA. To elucidate their genetic architecture and evolutionary standing, these isolates were subjected to high-depth whole-genome sequencing (WGS).
A phylogenetic tree was produced by multiple sequence alignment of the whole genome sequences [57]. The clusters of various ILTV strains are displayed in Figure 3. Nine genotypic clusters been developed based on sequence analysis [32].
The global population of ILTV is categorized into distinct genotypic clusters based on their evolutionary origin. Clusters I, II, and III are primarily composed of Tissue Culture Origin (TCO) vaccine strains, while Chicken Embryo Origin (CEO) vaccines and their immediate derivatives constitute Cluster IV. Additionally, Cluster V is characterized by CEO-related field strains. In contrast, virulent wild-type ILTV isolates are typically sequestered within genotypic clusters VI through IX, which are genetically divergent from standard North American vaccinal lineages. The isolates characterized in this study ILTV-B1, -B3, and -B4 map to these latter clusters (VI–IX). Interestingly, despite their wild-type classification, they exhibit the highest degree of genetic homology with the Australian vaccine strains SA2 and A20, suggesting a unique evolutionary trajectory or historical introduction.
The American vaccines, which are frequently used to prevent and control ILTV infections worldwide, belonged to CEO Vaccine clusters, while the sequenced isolates belonged to the wild-type genotypic clusters. The vaccine strain itself can revert to virulence after bird-to-bird transmissions, potentially causing outbreaks.
While American Chicken Embryo Origin (CEO) vaccine strains are widely utilized for the global control of ILTV, they are phylogenetically restricted to specific vaccinal clusters. In contrast, the isolates characterized in this study ILTV-B1, -B3, and -B4—align with virulent wild-type genotypic clusters [58]. A critical concern in ILTV management is the inherent instability of live-attenuated CEO vaccines; these strains maintain the capacity to recover pathogenicity through successive bird-to-bird passages. This process, known as reversion to virulence, often leads to the emergence of "vaccine-derived" outbreaks that can mirror the clinical severity of wild-type infections. The clear genotypic separation between the sequenced isolates and these American CEO clusters suggests that the current North Carolina outbreak may be driven by distinct field-type lineages rather than simple vaccine reversion.
A comprehensive sequence analysis was conducted by aligning the ILTV-B1, -B3, and -B4 isolates against a panel of internationally utilized vaccinal strains. This comparative framework included North American Chicken Embryo Origin (CEO) and Tissue Culture Origin (TCO) strains (LT-Blen, Laryngo Vac, and US-IVAX), as well as European and Australian reference lineages such as the Serva and Nobilis strains.
Detailed sequence analysis of the Unique Long (UL) region identified a significant density of both synonymous and non-synonymous mutations across the structural and regulatory loci. The sequenced ILTV isolates (ILTV-B1, -B3, and -B4) exhibited a high degree of genetic heterogeneity, characterized by more than nine distinct nucleotide variants within conserved regions. The genes (gC, UL3, UL4, UL11, UL12, UL13, UL14, UL20, UL24, UL25, UL26.5, UL31, UL32, UL33, UL34, UL35, UL40, UL45, UL 49, and UL 51) may not be involved in ILTV virulence because the amino acid sequences of these genes differ between strains [59].
The ICP4 gene regulates viral transcription, which is necessary to induce infection [60,61]. The protein's potentially flexible region enables it to effectively interact with a variety of transcription factors [62]. Stress factors and latency infection periods affect ICP4 expression levels. As a result, ICP4 and latent viral reactivation are closely linked [63].
Comparative genomic analysis of the ILTV-B1, -B3, and -B4 isolates against North American and European vaccinal lineages identified seven distinct non-synonymous mutations at nucleotide positions 815, 2240, 2523, 2531, 3426, 3909, and 4302. These substitutions represent significant points of divergence between the field isolates and attenuated reference strains.
Glycoproteins are linked to host range diversity and are crucial for virus attachment to and penetration of host cells [64]. Three mutations were found at amino acid positions 131, 1651, and 1931 on the gB protein, as indicated in Table 2. Furthermore, compared to American and European vaccinal strains, the gD protein had five amino acids mutations at positions 61, 580, 592, 644, and 752, and the gL protein had one mutation at aa position 377, indicating the significance of these amino acids on the gB, gD, and gL proteins in determining the cell tropism of ILTVs [59].
The connection between the virus and the host depends on the glycoproteins gB [65,66], gC [67,68], gD [69,70], and gH/gL [71,72,73]. Mutations in these genes can change the structure and function of the relevant protein, which can impact or prevent the virus from interacting with the receptors on the host cells [57,74]. In this study we found mutations at amino acid position 44, 551, 799, and 805 of gB glycoprotein of ILTV-B1, ILTV-B3, and ILTV-B4 when compared to SA2 and USDA reference isolate (Figure 5A – 5J).
The translated protein functional activity may be impacted by this substitution. Another unique gene of ILTV is the ORF B gene [75]. The importance of this protein in viral replication in vitro has been evaluated in earlier research using an ORF B deletion mutant of ILTV [75]. When reproduced in immortalized chicken hepatoma cells, the ORF-B deletion mutant produced viral plaques that were comparable in size to the wild type virus but replicated to lower titers [75].
In this study, in gC and gM, no variation was observed at the amino acid level. Strong evolutionary conservation of these envelope proteins is indicated by the lack of amino acid variation in the glycoproteins gC and gM among the examined ILTV isolates, underscoring their functional significance in the viral life cycle. While not necessary for viral replication in vitro, gC facilitates effective virus attachment, entrance, cell-to-cell transmission, and immunogenicity. Its conservation indicates significant selection pressure to preserve important virus–host interactions [4,76]. Like this, gM is a highly conserved herpesvirus envelope protein that plays a role in virion assembly, secondary envelopment, intracellular trafficking, and virus egress, frequently through the formation of a gM/gN complex. In the Alpha herpesviruses, even slight changes in gM have been demonstrated to hinder virion morphogenesis and viral spread [77].
Compared to other viral envelope glycoproteins, the ILTV gG exhibits a comparatively high degree of amino acid polymorphism, which is consistent with its non-essential but biologically significant function in virus host interactions. Eight significant point mutations at amino acid positions 133, 173, 292, 344, 387, 825, 850, and 872 were found in the current investigation, suggesting continued genetic diversification within the gG coding area. Instead of being involved in structural processes like virion assembly, ILTV gG is a secreted and envelope-associated protein that has been linked to immunological regulation and virulence, which may allow for better tolerance for sequence variation [4,76]. Several functional areas may be under selective pressure, either due to host immunological responses or adaptability to various epidemiological circumstances, according to the distribution of mutations found throughout the gG sequence in this study [78].
A single mutation was found in gE at position 629; gE and gI are necessary for effective cell-to-cell transmission and pathogenicity in ILTV, and the little change is in line with its crucial function in viral dissemination [4,78]. Five-point mutations were found in gI at positions 115, 533, 555, 631, and 670. Since gI interacts with gE to promote neurotropism and cell-to-cell dissemination, these mutations may be responsible for strain-specific variations in tissue tropism or virulence [78]. ILTV glycoprotein sequence analysis showed little variation in gH, gJ, and gK. Four-point mutations were found in gH at locations 1202, 1696, 1703, and 1813. Glycoprotein J showed several alterations, such as insertions/deletions in 1962 and 1991 and point mutations at 23, 765, 903, 1276, and 1481. One mutation was found in glycoprotein K at location 376 (Table 2).
ILTV has three origins of viral DNA replication, like many other Alpha herpesviruses. with one copy of OriL in the UL region and two copies of OriS in the IRS and TRS regions [79]. Although deletions of oriL or both copies of oriS had no influence on viral replication in vitro, the PCR product of this region was sequenced in our investigation to ensure the sequence accuracy [80,81]. Since a deletion has been identified in the OriS of virulent strains LJS09 and USDA but not in those of other virulent or attenuated vaccine strains, the results of the OriS sequences may support these earlier findings.
The UL15 sequence analysis identified several amino acid changes at positions 403, 660, 1360, 2366, and 2794 (Table 2). A highly conserved terminase packaging subunit is encoded by homologues of the ILTV UL15 gene in other herpesviruses [82]. It has been determined that twenty highly conserved UL15 sites are necessary for cleavage and packing of DNA [82]. The attenuated phenotype of A20 ILTV may potentially be caused by an interaction between the two mutant gene products (UL15 and ORF B). This is especially pertinent in light of earlier research on herpes simplex virus 1, which showed that UL15 interacts with several other herpesvirus proteins, such as UL28 and UL33 [83].
The tertiary structures of the UL27 (gB) and US5 (gJ) glycoproteins were modeled using AlphaFold3 to assess the potential impact of identified sequence variations. Structural mapping of these non-synonymous mutations onto the high-confidence models revealed that polymorphisms are strategically distributed across critical functional domains. In gB, several amino acid substitutions were localized within key antigenic epitopes, as well as the membrane fusion and receptor-binding domains. These structural alterations suggest potential modifications in viral entry kinetics and altered immunogenicity, which may facilitate escape from neutralizing antibodies. Conversely, mutations within gJ were predominantly identified within surface-exposed loops and regulatory motifs, potentially modulating host-virus interface dynamics and enhancing immune evasion strategies. Collectively, these structural insights indicate that ILTV glycoproteins are undergoing continuous genetic diversification. Such molecular evolution carries significant implications for viral fitness, clin
To categorize ILTV strains, more accurate diagnostic techniques will be developed using genome sequence comparisons. The thorough comparisons of ILTV strains shed light on the functional gene families that may affect the attenuation, pathogenesis, virulence, and life cycle of the ILTV virus. Both the examination of sequence variations and the prediction of mutational impacts on the functions of viral proteins are made possible by the viability of complete genome sequencing utilizing NGS techniques.
The ILTV isolates whole genome sequencing showed that they were genetically similar to Australian field strains, but that they differed significantly at the amino acid level in several glycoproteins when compared to commonly used American vaccine strains. These variations indicate that strain-specific vaccines could be required to offer the best defense against circulating ILTV strains and highlight the possible limitations of cross-regional vaccine efficacy. The sequence and structural variants that have been found highlight the significance of continuous genomic surveillance and comparative analysis to better understand the genetic determinants of ILTV pathogenicity and immune evasion, improve disease control, and influence vaccine design.
Targeted mutagenesis of the ILTV genome would be required to conclusively identify if one or both non-synonymous SNPs found in this study are crucial for the attenuation of A20 ILTV. Since CEO-vaccine-related isolates of ILTV have often been found to be the cause of disease, this is essential for researching the epidemiology of ILTV during disease outbreaks [60,61].
Acknowledgments
This study was supported by Long Island University (LIU) (Grant No. 40262), with financing provided by Helaine Lerner and Joan Rechnitz, as part of the Heilbrunn Family College of Veterinary Medicine Research and Scholarship Fund (Research Project #1) at LIU.
References
- Cover, M.S., The early history of infectious laryngotracheitis. Avian Dis, 1996. 40(3): p. 494-500. [CrossRef]
- Davison, A.J., et al., The order Herpesvirales. Arch Virol, 2009. 154(1): p. 171-7. [CrossRef]
- Mettenleiter, T.C., B.G. Klupp, and H. Granzow, Herpesvirus assembly: an update. Virus Res, 2009. 143(2): p. 222-34. [CrossRef]
- Fuchs, W., et al., Molecular biology of avian infectious laryngotracheitis virus. Vet Res, 2007. 38(2): p. 261-79. [CrossRef]
- Garcia, M., Current and future vaccines and vaccination strategies against infectious laryngotracheitis (ILT) respiratory disease of poultry. Vet Microbiol, 2017. 206: p. 157-162. [CrossRef]
- Crawshaw, G.J. and B.R. Boycott, Infectious laryngotracheitis in peafowl and pheasants. Avian Dis, 1982. 26(2): p. 397-401. [CrossRef]
- Winterfield, R.W. and I.G. So, Susceptibility of turkeys to infectious laryngotracheitis. Avian Dis, 1968. 12(1): p. 191-202. [CrossRef]
- Ou, S.C. and J.J. Giambrone, Infectious laryngotracheitis virus in chickens. World J Virol, 2012. 1(5): p. 142-9. [CrossRef]
- Watrach, A.M., L.E. Hanson, and M.A. Watrach, The Structure of Infectious Laryngotracheitis Virus. Virology, 1963. 21: p. 601-8. [CrossRef]
- Mo, J. and J. Mo, Infectious Laryngotracheitis Virus and Avian Metapneumovirus: A Comprehensive Review. Pathogens, 2025. 14(1). [CrossRef]
- McGeoch, D.J., A. Dolan, and A.C. Ralph, Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol, 2000. 74(22): p. 10401-6. [CrossRef]
- Morales Ruiz, S., et al., Full-Genome Sequence of Infectious Laryngotracheitis Virus (Gallid Alphaherpesvirus 1) Strain VFAR-043, Isolated in Peru. Genome Announc, 2018. 6(10). [CrossRef]
- Lee, S.W., et al., First complete genome sequence of infectious laryngotracheitis virus. BMC Genomics, 2011. 12: p. 197. [CrossRef]
- Granzow, H., et al., Egress of alphaherpesviruses: comparative ultrastructural study. J Virol, 2001. 75(8): p. 3675-84. [CrossRef]
- Piccirillo, A., et al., Full Genome Sequence-Based Comparative Study of Wild-Type and Vaccine Strains of Infectious Laryngotracheitis Virus from Italy. PLoS One, 2016. 11(2): p. e0149529. [CrossRef]
- Fuchs, W. and T.C. Mettenleiter, DNA sequence and transcriptional analysis of the UL1 to UL5 gene cluster of infectious laryngotracheitis virus. J Gen Virol, 1996. 77 ( Pt 9): p. 2221-9. [CrossRef]
- Wild, M.A., S. Cook, and M. Cochran, A genomic map of infectious laryngotracheitis virus and the sequence and organization of genes present in the unique short and flanking regions. Virus Genes, 1996. 12(2): p. 107-16. [CrossRef]
- McGeoch, D.J., et al., The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol, 1988. 69 ( Pt 7): p. 1531-74. [CrossRef]
- Thureen, D.R. and C.L. Keeler, Jr., Psittacid herpesvirus 1 and infectious laryngotracheitis virus: Comparative genome sequence analysis of two avian alphaherpesviruses. J Virol, 2006. 80(16): p. 7863-72. [CrossRef]
- Nadimpalli, M., et al., Impairment of infectious laryngotracheitis virus replication by deletion of the UL[-1] gene. Arch Virol, 2017. 162(6): p. 1541-1548. [CrossRef]
- McGeoch, D.J., F.J. Rixon, and A.J. Davison, Topics in herpesvirus genomics and evolution. Virus Res, 2006. 117(1): p. 90-104. [CrossRef]
- Kelly, B.J., et al., Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res, 2009. 145(2): p. 173-86. [CrossRef]
- Menendez, K.R., et al., Molecular epidemiology of infectious laryngotracheitis: a review. Avian Pathol, 2014. 43(2): p. 108-17. [CrossRef]
- Blakey, J., et al., Retrospective analysis of infectious laryngotracheitis in backyard chicken flocks in California, 2007-2017, and determination of strain origin by partial ICP4 sequencing. J Vet Diagn Invest, 2019. 31(3): p. 350-358. [CrossRef]
- Devlin, J.M., et al., Glycoprotein G is a virulence factor in infectious laryngotracheitis virus. J Gen Virol, 2006. 87(Pt 10): p. 2839-2847. [CrossRef]
- Perez Contreras, A., et al., Analysis of Whole-Genome Sequences of Infectious laryngotracheitis Virus Isolates from Poultry Flocks in Canada: Evidence of Recombination. Viruses, 2020. 12(11). [CrossRef]
- Hernandez-Divers, S.M., et al., Backyard chicken flocks pose a disease risk for neotropic birds in Costa Rica. Avian Dis, 2008. 52(4): p. 558-66. [CrossRef]
- Dormitorio, T.V., J.J. Giambrone, and K.S. Macklin, Detection and isolation of infectious laryngotracheitis virus on a broiler farm after a disease outbreak. Avian Dis, 2013. 57(4): p. 803-7. [CrossRef]
- Parra, S.H., et al., Persistence of the tissue culture origin vaccine for infectious laryngotracheitis virus in commercial chicken flocks in Brazil. Poult Sci, 2015. 94(11): p. 2608-15. [CrossRef]
- Chacon, J.L., et al., Persistence and spreading of field and vaccine strains of infectious laryngotracheitis virus (ILTV) in vaccinated and unvaccinated geographic regions, in Brazil. Trop Anim Health Prod, 2015. 47(6): p. 1101-8. [CrossRef]
- Garcia, M. and G. Zavala, Commercial Vaccines and Vaccination Strategies Against Infectious Laryngotracheitis: What We Have Learned and Knowledge Gaps That Remain. Avian Dis, 2019. 63(2): p. 325-334. [CrossRef]
- Barboza-Solis, C., et al., Genotyping of Infectious Laryngotracheitis Virus (ILTV) Isolates from Western Canadian Provinces of Alberta and British Columbia Based on Partial Open Reading Frame (ORF) a and b. Animals (Basel), 2020. 10(9). [CrossRef]
- Ojkic, D., et al., Characterization of field isolates of infectious laryngotracheitis virus from Ontario. Avian Pathol, 2006. 35(4): p. 286-92. [CrossRef]
- Al-Mubarak, A.I.A., et al., Detection of Avian Orthoavulavirus-1 genotypes VI.2.1 and VII.1.1 with neuro-viscerotropic tropism in some backyard pigeons (Columbidae) in Eastern Saudi Arabia. Front Vet Sci, 2024. 11: p. 1352636. [CrossRef]
- Al-Mubarak, A.I.A., et al., Avian encephalomyelitis virus in backyard chickens. Vet World, 2023. 16(9): p. 1866-1870. [CrossRef]
- Alsultan, M.A., M.A. Alhammadi, and M.G. Hemida, Infectious bronchitis virus from chickens in Al-Hasa, Saudi Arabia 2015-2016. Vet World, 2019. 12(3): p. 424-433. [CrossRef]
- Ba Abduallah, M.M. and M.G. Hemida, Comparative analysis of the genome structure and organization of the Middle East respiratory syndrome coronavirus (MERS-CoV) 2012 to 2019 revealing evidence for virus strain barcoding, zoonotic transmission, and selection pressure. Rev Med Virol, 2021. 31(1): p. 1-12. [CrossRef]
- Hemida, M.G., The next-generation coronavirus diagnostic techniques with particular emphasis on the SARS-CoV-2. J Med Virol, 2021. 93(7): p. 4219-4241. [CrossRef]
- Hemida, M.G., et al., MERS coronavirus in dromedary camel herd, Saudi Arabia. Emerg Infect Dis, 2014. 20(7): p. 1231-4. [CrossRef]
- Hemida, M.G., et al., Phylogenetic Analysis of MERS-CoV in a Camel Abattoir, Saudi Arabia, 2016-2018. Emerg Infect Dis, 2020. 26(12): p. 3089-3091. [CrossRef]
- Hemida, M.G., et al., Foot-and-mouth disease virus O/ME-SA/Ind 2001 lineage outbreak in vaccinated Holstein Friesian cattle in Saudi Arabia in 2016. Vet Q, 2018. 38(1): p. 88-98. [CrossRef]
- Malik, Y.S., et al., SARS-CoV-2 Spike Protein Extrapolation for COVID Diagnosis and Vaccine Development. Front Mol Biosci, 2021. 8: p. 607886. [CrossRef]
- Shah, A.U., et al., Comparative Genome Sequencing Analysis of Some Novel Feline Infectious Peritonitis Viruses Isolated from Some Feral Cats in Long Island. Viruses, 2025. 17(2). [CrossRef]
- Shah, A.U., P. Gauger, and M.G. Hemida, Isolation and molecular characterization of an enteric isolate of the genotype-Ia bovine coronavirus with notable mutations in the receptor binding domain of the spike glycoprotein. Virology, 2025. 603: p. 110313. [CrossRef]
- Singh, K., Y.S. Malik, and M.G. Hemida, Comprehensive Evolutionary and Structural Analysis of the H5N1 Clade 2.4.3.4b Influenza a Virus Based on the Sequences and Data Mining of the Hemagglutinin, Nucleoprotein and Neuraminidase Genes Across Multiple Hosts. Pathogens, 2025. 14(9). [CrossRef]
- Madsen, J.M., et al., Evaluation of Maryland backyard flocks and biosecurity practices. Avian Dis, 2013. 57(2): p. 233-7. [CrossRef]
- Spatz, S.J., et al., MinION sequencing to genotype US strains of infectious laryngotracheitis virus. Avian Pathol, 2019. 48(3): p. 255-269. [CrossRef]
- Oldoni, I., et al., Pathogenicity and growth characteristics of selected infectious laryngotracheitis virus strains from the United States. Avian Pathol, 2009. 38(1): p. 47-53. [CrossRef]
- Chacon, J.L., et al., Molecular Characterization of the Infectious Laryngotracheitis Virus (ILTV) Involved in Poultry Outbreaks Reveals the Virus Origin and Estimated Spreading Route. Viruses, 2025. 17(2). [CrossRef]
- Shah, A.U., et al., Some novel field isolates belonging to lineage-1 of the genotype GI-avian infectious bronchitis virus (AIBV) show strong evidence of recombination with field/vaccinal strains. Infect Genet Evol, 2025. 129: p. 105723. [CrossRef]
- Rozen, S. and H. Skaletsky, Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol, 2000. 132: p. 365-86.
- Shah, A.U., et al., From nasal to basal: single-cell sequencing of the bursa of Fabricius highlights the IBDV infection mechanism in chickens. Cell Biosci, 2021. 11(1): p. 212. [CrossRef]
- Livak, K.J. and T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402-8. [CrossRef]
- Kumar, S., et al., MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol Biol Evol, 2024. 41(12). [CrossRef]
- Abramson, J., et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024. 630(8016): p. 493-500. [CrossRef]
- Thiry, E., et al., Recombination in alphaherpesviruses. Rev Med Virol, 2005. 15(2): p. 89-103. [CrossRef]
- Uchida, H., et al., A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J Virol, 2010. 84(23): p. 12200-9. [CrossRef]
- Dufour-Zavala, L., Epizootiology of infectious laryngotracheitis and presentation of an industry control program. Avian Dis, 2008. 52(1): p. 1-7. [CrossRef]
- Kong, C., et al., Complete genome sequence of the first Chinese virulent infectious laryngotracheitis virus. PLoS One, 2013. 8(7): p. e70154. [CrossRef]
- Godowski, P.J. and D.M. Knipe, Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc Natl Acad Sci U S A, 1986. 83(2): p. 256-60. [CrossRef]
- Watson, R.J. and J.B. Clements, A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature, 1980. 285(5763): p. 329-30. [CrossRef]
- Wagner, L.M., et al., The N terminus and C terminus of herpes simplex virus 1 ICP4 cooperate to activate viral gene expression. J Virol, 2012. 86(12): p. 6862-74. [CrossRef]
- Huang, W., et al., The influence of stress factors on the reactivation of latent herpes simplex virus type 1 in infected mice. Cell Biochem Biophys, 2011. 61(1): p. 115-22. [CrossRef]
- Wang, J., et al., Complete genome sequence of virulent duck enteritis virus (DEV) strain 2085 and comparison with genome sequences of virulent and attenuated DEV strains. Virus Res, 2011. 160(1-2): p. 316-25. [CrossRef]
- Williams, R.K. and S.E. Straus, Specificity and affinity of binding of herpes simplex virus type 2 glycoprotein B to glycosaminoglycans. J Virol, 1997. 71(2): p. 1375-80. [CrossRef]
- Bender, F.C., et al., Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol, 2005. 79(18): p. 11588-97. [CrossRef]
- Mardberg, K., et al., Basic amino acids as modulators of an O-linked glycosylation signal of the herpes simplex virus type 1 glycoprotein gC: functional roles in viral infectivity. Glycobiology, 2004. 14(7): p. 571-81. [CrossRef]
- Adamiak, B., et al., Human antibodies to herpes simplex virus type 1 glycoprotein C are neutralizing and target the heparan sulfate-binding domain. Virology, 2010. 400(2): p. 197-206. [CrossRef]
- Whitbeck, J.C., et al., Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol, 1997. 71(8): p. 6083-93. [CrossRef]
- Whalley, J.M., et al., Host cell tropism of equine herpesviruses: glycoprotein D of EHV-1 enables EHV-4 to infect a non-permissive cell line. Arch Virol, 2007. 152(4): p. 717-25. [CrossRef]
- Westra, D.F., et al., Glycoprotein H of herpes simplex virus type 1 requires glycoprotein L for transport to the surfaces of insect cells. J Virol, 1997. 71(3): p. 2285-91. [CrossRef]
- Chen, J., T.S. Jardetzky, and R. Longnecker, The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J Virol, 2013. 87(7): p. 3620-7. [CrossRef]
- Heldwein, E.E. and C. Krummenacher, Entry of herpesviruses into mammalian cells. Cell Mol Life Sci, 2008. 65(11): p. 1653-68. [CrossRef]
- Uchida, H., et al., Novel mutations in gB and gH circumvent the requirement for known gD Receptors in herpes simplex virus 1 entry and cell-to-cell spread. J Virol, 2013. 87(3): p. 1430-42. [CrossRef]
- Veits, J., T.C. Mettenleiter, and W. Fuchs, Five unique open reading frames of infectious laryngotracheitis virus are expressed during infection but are dispensable for virus replication in cell culture. J Gen Virol, 2003. 84(Pt 6): p. 1415-1425. [CrossRef]
- Davison, A.J., Herpesvirus systematics. Vet Microbiol, 2010. 143(1): p. 52-69. [CrossRef]
- Li, C., et al., The Roles of Envelope Glycoprotein M in the Life Cycle of Some Alphaherpesviruses. Front Microbiol, 2021. 12: p. 631523. [CrossRef]
- García, M. and S. Spatz, Infectious Laryngotracheitis, in Diseases of Poultry. 2020. p. 189-209.
- Ziemann, K., T.C. Mettenleiter, and W. Fuchs, Gene arrangement within the unique long genome region of infectious laryngotracheitis virus is distinct from that of other alphaherpesviruses. J Virol, 1998. 72(1): p. 847-52. [CrossRef]
- Igarashi, K., et al., Construction and properties of a recombinant herpes simplex virus 1 lacking both S-component origins of DNA synthesis. J Virol, 1993. 67(4): p. 2123-32. [CrossRef]
- Polvino-Bodnar, M., P.K. Orberg, and P.A. Schaffer, Herpes simplex virus type 1 oriL is not required for virus replication or for the establishment and reactivation of latent infection in mice. J Virol, 1987. 61(11): p. 3528-35. [CrossRef]
- Przech, A.J., D. Yu, and S.K. Weller, Point mutations in exon I of the herpes simplex virus putative terminase subunit, UL15, indicate that the most conserved residues are essential for cleavage and packaging. J Virol, 2003. 77(17): p. 9613-21. [CrossRef]
- Higgs, M.R., V.G. Preston, and N.D. Stow, The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J Gen Virol, 2008. 89(Pt 7): p. 1709-1715. [CrossRef]
Figure 1.
ILTV viral load quantification by PCR. Tissue samples from four infected chicken flocks (B1 – B4) were tested for ILTV viral load. Tissue samples from each flock were pooled together. Chicken tissue samples negative for ILTV were used as control.
Figure 1.
ILTV viral load quantification by PCR. Tissue samples from four infected chicken flocks (B1 – B4) were tested for ILTV viral load. Tissue samples from each flock were pooled together. Chicken tissue samples negative for ILTV were used as control.
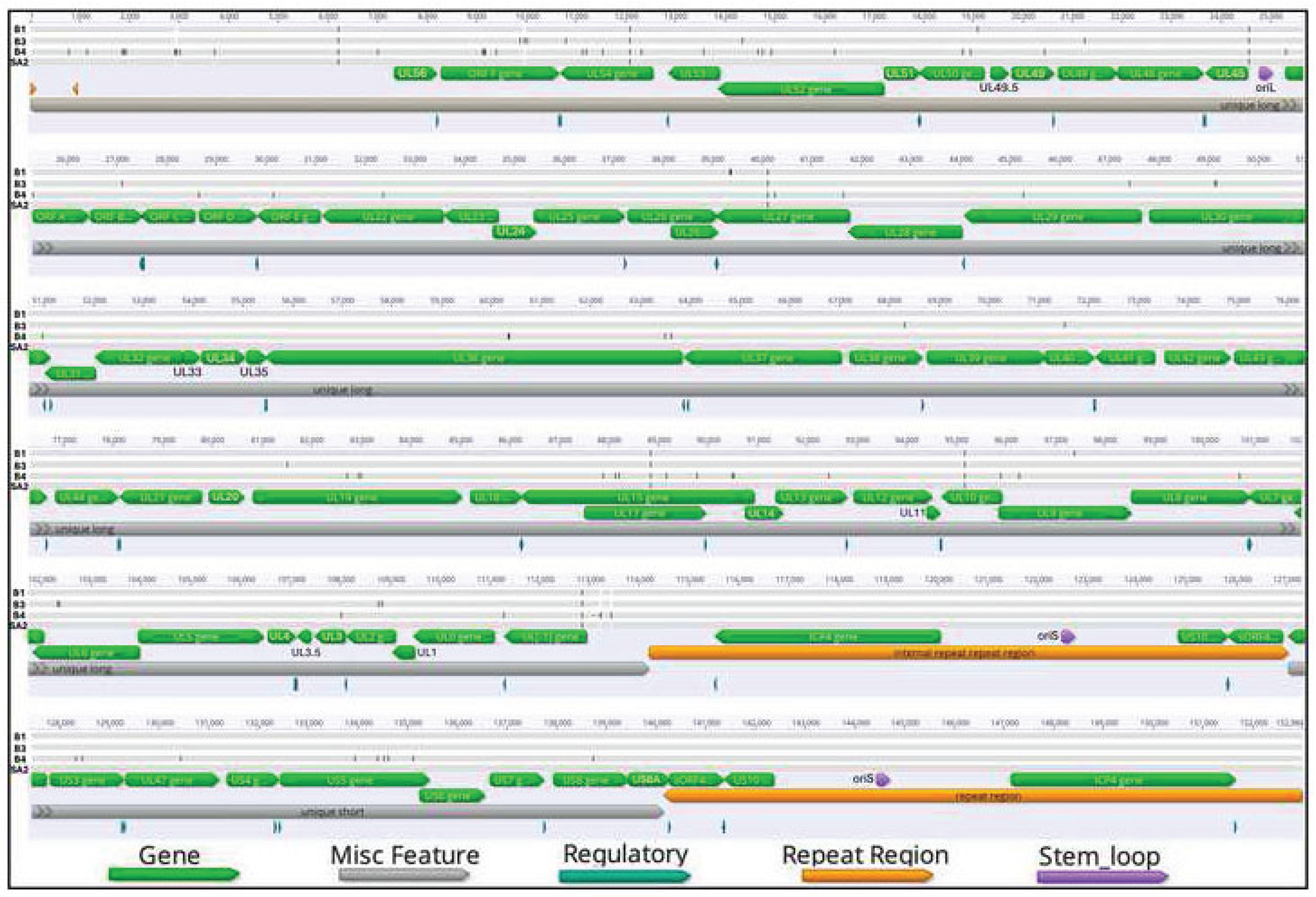
Figure 2.
Complete genome alignment of ILTV-B1, ILTV-B3, ILTV-B4, and SA2 isolates. The black lines indicate nucleotides differences. Dashes indicates gaps in sequences. The alignment was performed using Geneious V.9. The locations and sizes of predicted ORFs were annotated using Geneious. The green lines indicate genes, gray lines indicate miscellaneous features, blue lines indicate regulatory regions, orange lines indicate repeat regions, purple line indicates stem loop regions.
Figure 2.
Complete genome alignment of ILTV-B1, ILTV-B3, ILTV-B4, and SA2 isolates. The black lines indicate nucleotides differences. Dashes indicates gaps in sequences. The alignment was performed using Geneious V.9. The locations and sizes of predicted ORFs were annotated using Geneious. The green lines indicate genes, gray lines indicate miscellaneous features, blue lines indicate regulatory regions, orange lines indicate repeat regions, purple line indicates stem loop regions.
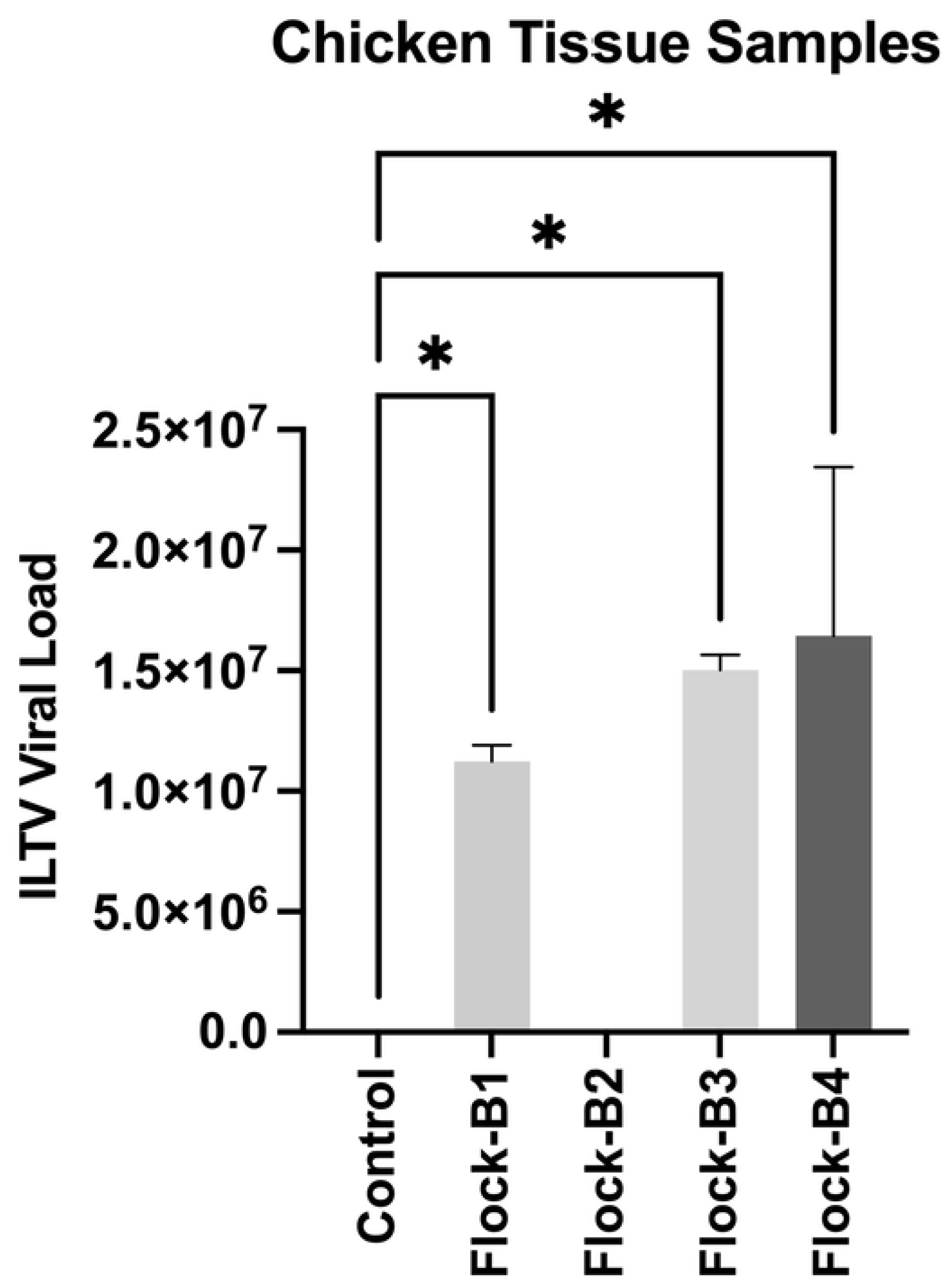
Figure 3.
Phylogenetic analysis of the complete genome of sequences of 52 ILTV isolates from different geographical locations. The maximum log likelihood of the tree was -256,708.88. The percentage of replicate trees associated with taxa clustered together (1,000 replicates) is shown below at each branch. The complete genome sequences of ILTV-B1, ILTV-B3, and ILTV-B4 isolates identified in this study are shown in red boxes. The genotypes and groups of isolates shown at the right side of the tree are separated based on genotype clustering in the phylogenetic tree. The tree was generated using MEGA12 software.
Figure 3.
Phylogenetic analysis of the complete genome of sequences of 52 ILTV isolates from different geographical locations. The maximum log likelihood of the tree was -256,708.88. The percentage of replicate trees associated with taxa clustered together (1,000 replicates) is shown below at each branch. The complete genome sequences of ILTV-B1, ILTV-B3, and ILTV-B4 isolates identified in this study are shown in red boxes. The genotypes and groups of isolates shown at the right side of the tree are separated based on genotype clustering in the phylogenetic tree. The tree was generated using MEGA12 software.

Figure 4.
Phylogenetic analysis of the ILTV genes sequences from 38 different isolates from different geographical locations. (A) Phylogenetic tree showing multiple sequence alignment of ICP4 gene with a maximum log likelihood of -7,055.90. (B) Phylogenetic tree showing multiple sequence alignment of UL23 (TK) gene with a maximum log likelihood of -1,666.27. (C) Phylogenetic tree showing multiple sequence alignment of US4 (gG) gene with maximum log likelihood of -1,667.19. (D) Phylogenetic tree showing multiple sequence alignment of UL27 (gB) gene with maximum log likelihood of -3,912.94. (E) Phylogenetic tree showing multiple sequence alignment of US5 (gJ) gene with maximum log likelihood of -4,621.84. The percentage of replicates of all five trees were associated with taxa clustered together (1,000 replicates) and is shown below at each branch. The gene sequences of ILTV-B1, ILTV-B3, and ILTV-B4 isolates identified in this study are shown in red boxes. The trees were generated using MEGA12 software.
Figure 4.
Phylogenetic analysis of the ILTV genes sequences from 38 different isolates from different geographical locations. (A) Phylogenetic tree showing multiple sequence alignment of ICP4 gene with a maximum log likelihood of -7,055.90. (B) Phylogenetic tree showing multiple sequence alignment of UL23 (TK) gene with a maximum log likelihood of -1,666.27. (C) Phylogenetic tree showing multiple sequence alignment of US4 (gG) gene with maximum log likelihood of -1,667.19. (D) Phylogenetic tree showing multiple sequence alignment of UL27 (gB) gene with maximum log likelihood of -3,912.94. (E) Phylogenetic tree showing multiple sequence alignment of US5 (gJ) gene with maximum log likelihood of -4,621.84. The percentage of replicates of all five trees were associated with taxa clustered together (1,000 replicates) and is shown below at each branch. The gene sequences of ILTV-B1, ILTV-B3, and ILTV-B4 isolates identified in this study are shown in red boxes. The trees were generated using MEGA12 software.
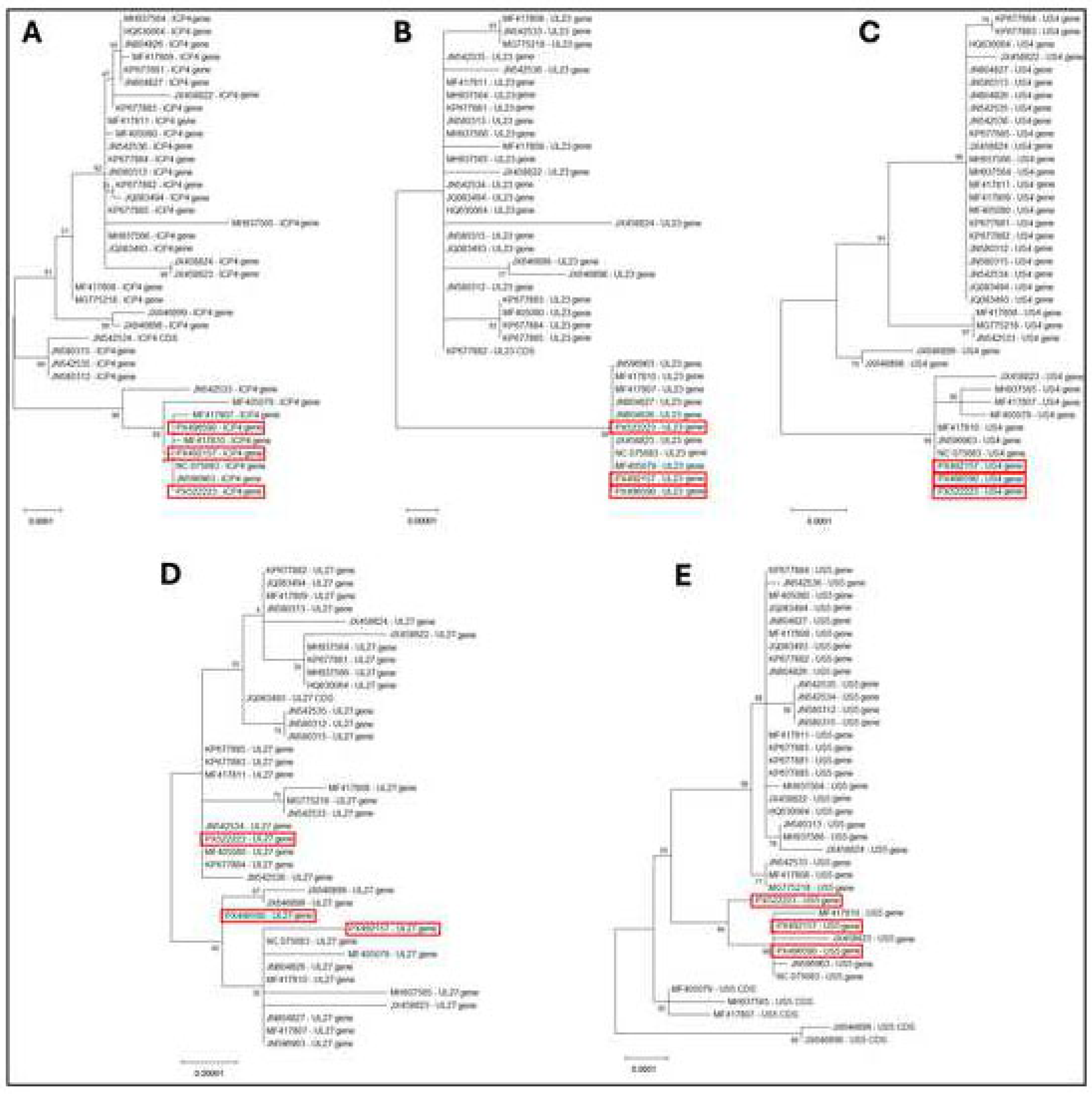
Figure 5.
Divergence analysis of the UL27 (gB) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate USDA and SA2. (A) Multiple sequence alignment of the UL27 (gB) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate USDA and SA2, showing mismatches of nucleotide and amino acids at four different sites. The multiple sequence alignment was performed by using SnapGene V6.0.2 software. (B) Protein structure of UL27 (gB) gene of reference isolate SA2 showing histidine (His) at 44 positions, (C) which substitutes to arginine (Arg) at the same position in ILTV-B1 isolate. (D) Valine (Val) at position 551 of SA2 isolate substitutes to (E) methionine (Met) at the same position in ILTV-B1 isolate. (F) Proline (Pro) at position 799 of the SA2 isolate substitutes to (G) serine (Ser) at the same position in ILTV-B1 isolate. (H) Lysine (Lys) at position 805 of the SA2 isolate substitutes to (I) arginine (Arg) at the same position in ILTV-B1 isolate. The amino acid locations are indicated by a red arrowhead. (J) Protein structure prediction of UL27 (gB) gene of ILTV-B1 isolate. The sites of mismatches are highlighted in red boxes. The protein structure was designed using AlphaFold3 server.
Figure 5.
Divergence analysis of the UL27 (gB) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate USDA and SA2. (A) Multiple sequence alignment of the UL27 (gB) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate USDA and SA2, showing mismatches of nucleotide and amino acids at four different sites. The multiple sequence alignment was performed by using SnapGene V6.0.2 software. (B) Protein structure of UL27 (gB) gene of reference isolate SA2 showing histidine (His) at 44 positions, (C) which substitutes to arginine (Arg) at the same position in ILTV-B1 isolate. (D) Valine (Val) at position 551 of SA2 isolate substitutes to (E) methionine (Met) at the same position in ILTV-B1 isolate. (F) Proline (Pro) at position 799 of the SA2 isolate substitutes to (G) serine (Ser) at the same position in ILTV-B1 isolate. (H) Lysine (Lys) at position 805 of the SA2 isolate substitutes to (I) arginine (Arg) at the same position in ILTV-B1 isolate. The amino acid locations are indicated by a red arrowhead. (J) Protein structure prediction of UL27 (gB) gene of ILTV-B1 isolate. The sites of mismatches are highlighted in red boxes. The protein structure was designed using AlphaFold3 server.
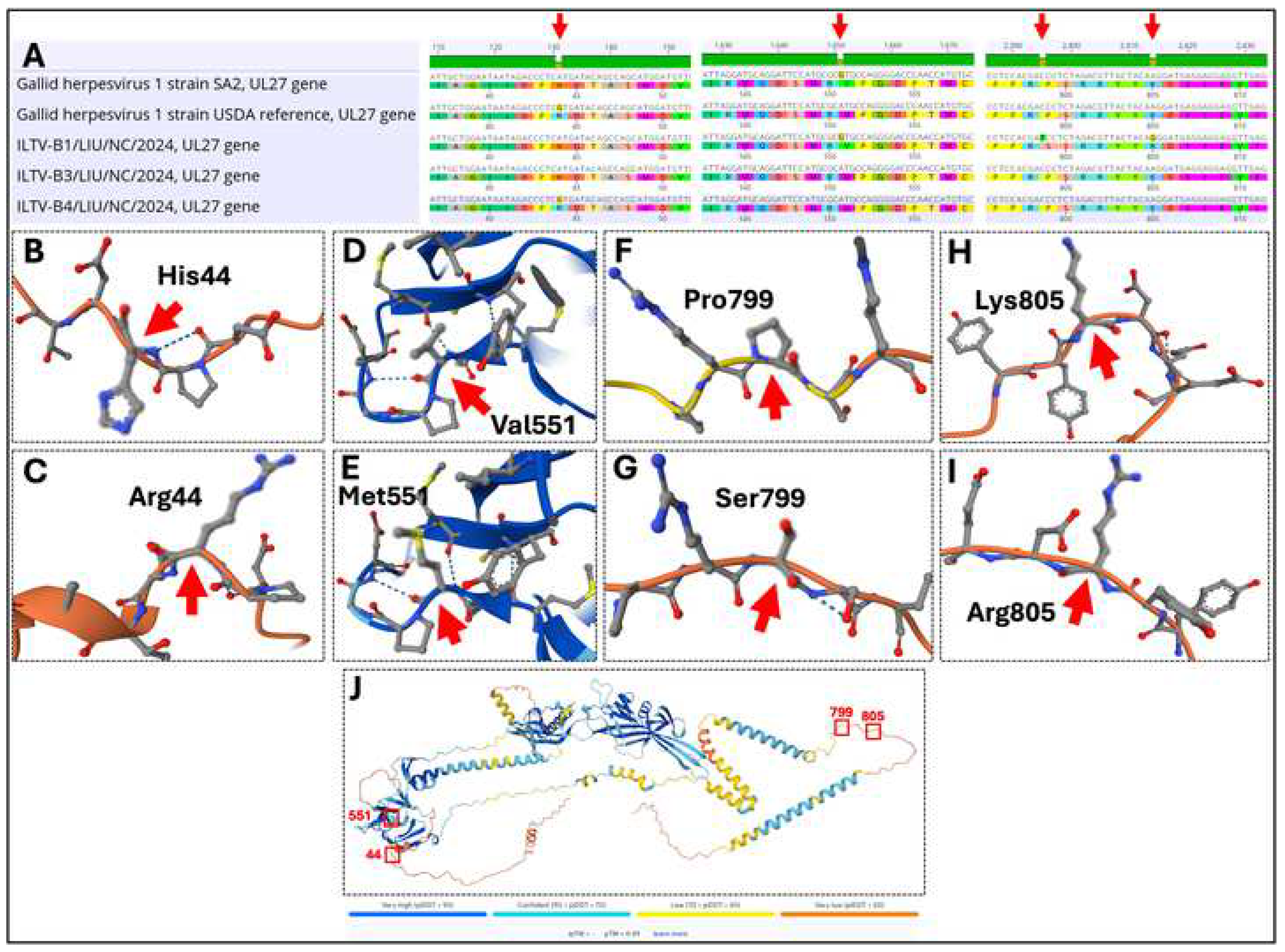
Figure 6.
Divergence analysis of US5 (gJ) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference strain SA2. (A) Multiple sequence alignment of the complete US5 (gJ) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate SA2. (B) Multiple sequence alignment showing mismatches of nucleotide and amino acids at four different sites of ILTV-B4 isolate. The multiple sequence alignment was performed by using SnapGene V6.0.2 software. (C) Protein structure prediction of US5 (gJ) gene of ILTV-B4 isolate. The sites of mismatches are highlighted in red boxes. (D) Protein structure of US5 (gJ) gene of reference isolate SA2 showing valine (Val) at 494 positions, (E) which substitute to alanine (Ala) at the same position in ILTV-B4 isolate. (F) Isoleucine (Ile) at position 647 of SA2 isolate substitutes to (G) threonine (Thr) at the same position in ILTV-B4 isolate. (H) Alanine (Ala) at position 694 of the SA2 isolate substitutes to (I) threonine (Thr) at the same position in ILTV-B4 isolate. (J) Phenylalanine (Phe) at position 887 of the SA2 isolate substitutes to (K) leucine (Leu) at the same position in ILTV-B4 isolate. The amino acid locations are indicated by a red arrowhead. The protein structure was designed using AlphaFold3 server.
Figure 6.
Divergence analysis of US5 (gJ) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference strain SA2. (A) Multiple sequence alignment of the complete US5 (gJ) gene of ILTV-B1, ILTV-B3, and ILTV-B4 with reference isolate SA2. (B) Multiple sequence alignment showing mismatches of nucleotide and amino acids at four different sites of ILTV-B4 isolate. The multiple sequence alignment was performed by using SnapGene V6.0.2 software. (C) Protein structure prediction of US5 (gJ) gene of ILTV-B4 isolate. The sites of mismatches are highlighted in red boxes. (D) Protein structure of US5 (gJ) gene of reference isolate SA2 showing valine (Val) at 494 positions, (E) which substitute to alanine (Ala) at the same position in ILTV-B4 isolate. (F) Isoleucine (Ile) at position 647 of SA2 isolate substitutes to (G) threonine (Thr) at the same position in ILTV-B4 isolate. (H) Alanine (Ala) at position 694 of the SA2 isolate substitutes to (I) threonine (Thr) at the same position in ILTV-B4 isolate. (J) Phenylalanine (Phe) at position 887 of the SA2 isolate substitutes to (K) leucine (Leu) at the same position in ILTV-B4 isolate. The amino acid locations are indicated by a red arrowhead. The protein structure was designed using AlphaFold3 server.
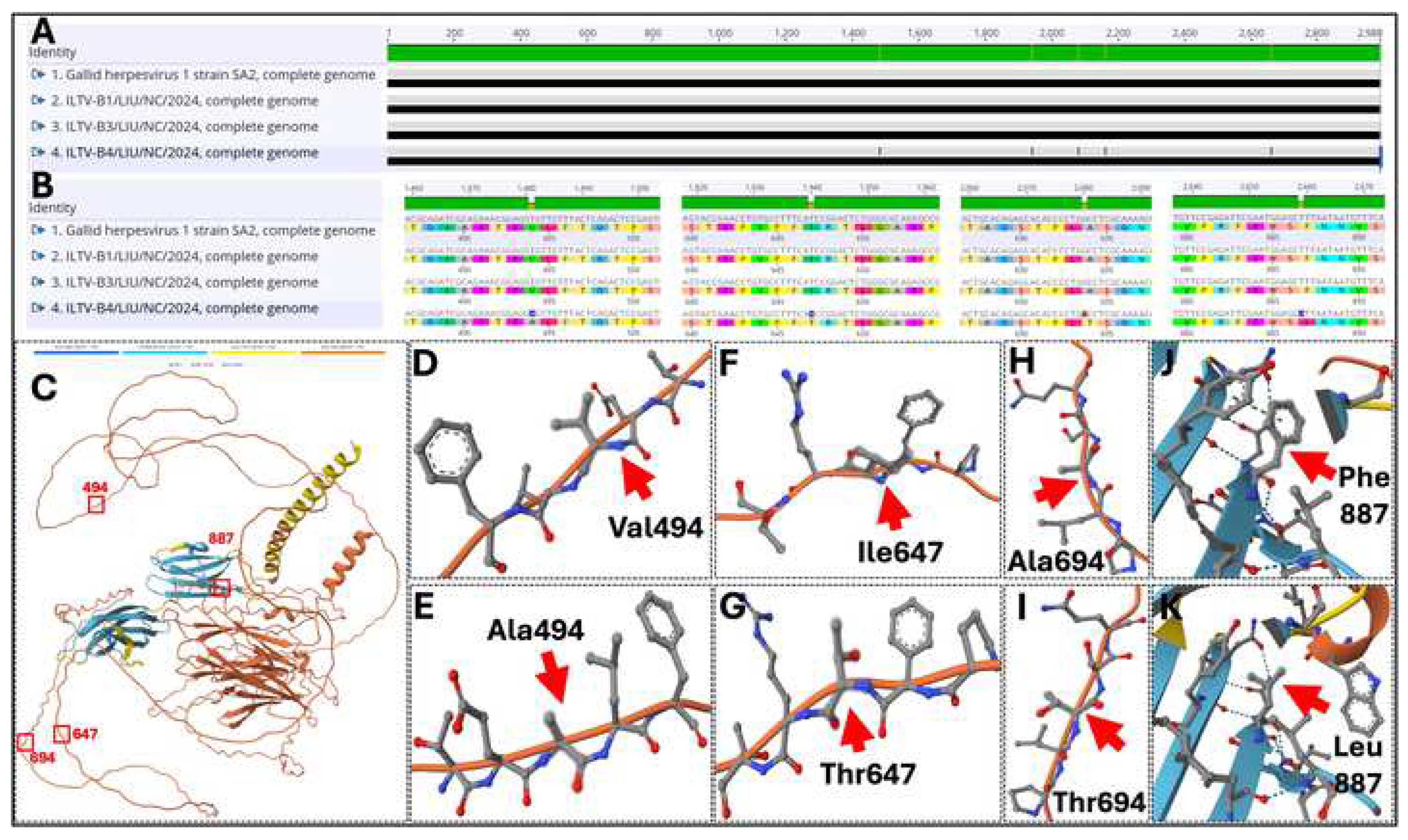

Table 1.
Comparative genomic features and GenBank accession numbers of Infectious Laryngotracheitis Virus (ILTV) isolates B1, B3, and B4.
Table 1.
Comparative genomic features and GenBank accession numbers of Infectious Laryngotracheitis Virus (ILTV) isolates B1, B3, and B4.
| Isolate Name | Accession No. | Genome length (bp) | UL (bp) | IR (bp) | US (bp) | TR (bp) | G+C content |
|---|---|---|---|---|---|---|---|
| ILTV-B1/LIU/NC/2024 | PX492157 | 152,975 | 114,179 | 12,835 | 13,126 | 12,835 | 48.1% |
| ILTV-B3/LIU/NC/2024 | PX496590 | 152,978 | 114,182 | 12,835 | 13,126 | 12,835 | 48.1% |
| ILTV-B4/LIU/NC/2024 | PX522223 | 152,978 | 114,182 | 12,835 | 13,126 | 12,835 | 48.1% |
Table 2.
Comparative analysis of amino acid substitutions in ILTV-B1, -B3, and -B4 isolates relative to global vaccinal reference strains (American, Australian, and European lineages).
Table 2.
Comparative analysis of amino acid substitutions in ILTV-B1, -B3, and -B4 isolates relative to global vaccinal reference strains (American, Australian, and European lineages).
| Gene | Protein | Position | Amino Acid Substitution |
|---|---|---|---|
| UL22 | Envelope glycoprotein H | 1202 | H → R/H |
| 1696 | I → V | ||
| 1703 | L → S | ||
| 1813 | N → H | ||
| UL23 | Thymidine kinase | 755 | T → M |
| UL27 | Envelope glycoprotein B | 131 | R → H |
| 1651 | M → V/M | ||
| 1931 | I → T | ||
| 2395 | P → P/S | ||
| 2414 | K → R/K | ||
| UL1 | Envelope glycoprotein L | 377 | P → Q |
| UL53 | Envelope glycoprotein K | 646 | V → I |
| UL49.5 | Envelope glycoprotein N | 46 | V → M |
| 118 | Y → H | ||
| US4 | Envelope glycoprotein G | 133 | I → L |
| 173 | V → G | ||
| 292 | H → N | ||
| 344 | V → G | ||
| 353 | A → V | ||
| 387 | Q → H | ||
| 850 | F → L | ||
| 872 | Q → R | ||
| US5 | Envelope glycoprotein J | 23 | R → H |
| 903 | M → I | ||
| 1276 | T → A | ||
| 1481 | A → V | ||
| US6 | Envelope glycoprotein D | 61 | K → E |
| 580 | V → L | ||
| 592 | P → S | ||
| 644 | T → N | ||
| 752 | Q → R | ||
| US7 | Envelope glycoprotein I | 115 | I → V |
| 533 | A → D | ||
| 555 | L → F | ||
| 631 | A → T | ||
| 670 | V → I | ||
| US8 | Envelope glycoprotein E | 629 | K → R |
| ICP4 | Transcriptional regulator | 815 | A → T |
| 2240 | N → D | ||
| 2523 | L → P | ||
| 2531 | S → P | ||
| 3426 | V → G | ||
| 3909 | L → P | ||
| 4302 | P → L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



