
Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
Human chromosome 2 (HSA2) originated from a telomere-to-telomere fusion event in thehuman lineage, supported by convergent cytogenetic and comparative-genomic signatures. The primaryunresolved questions are quantitative and empirical: how an (at least partially) underdominantrearrangement could establish under drift and realistic population structure, and whether fusion-proximalsequence behaved as a barrier to gene flow during later admixture with archaic hominins. Here, weintegrate (i) drift-aware Wright–Fisher simulations and a simple subdivided metapopulation model toquantify establishment probabilities under heterozygote fertility costs, including sensitivity to weaktransmission-ratio distortion (TRD; k>0.5); (ii) a tract-based assay of Neanderthal introgression at 2q13using a public IBDmix Vindija callset (hg19) benchmarked against a length-matched chromosome 2 null;and (iii) external evidence from recent T2T-CHM13 audits showing that reference completeness rescuessubstantial archaic sequence previously undetected in repeat-rich regions, constraining interpretations ofapparent 'introgression deserts' near pericentromeric sequence. Taken together, these results supportconservative, testable claims: establishment of an underdominant fusion is plausible under drift instructured populations and can be amplified by weak TRD, whereas introgression depletion at 2q13 inhg19-era callsets must be interpreted cautiously given callability vulnerabilities highlighted by T2T-basedremapping.
Keywords:
chromosome fusion
; human chromosome 2
; underdominance
; meiotic drive
; centromere competition
; archaic introgression
; paleogenomics
; T2T-CHM13
; Wright–Fisher simulation
; population structure
1. Introduction
Human chromosome 2 is a textbook example of a major karyotypic difference between humans (2n = 46) and other great apes (typically 2n = 48). Although the fusion origin of HSA2 is strongly supported, the evolutionary consequences of such a rearrangement are often discussed in ways that exceed the evidence. This manuscript treats the fusion as an established structural event and focuses on two questions that are explicitly testable: (i) the probability that a potentially underdominant fusion could establish under drift and population structure, and (ii) whether fusion-proximal sequence shows unusual reduction in archaic ancestry during later admixture, consistent with a barrier to gene flow. Throughout, executed results are distinguished from external audit evidence and from mechanistic or functional interpretations to avoid overclaiming.
2. Materials and Methods
2.1. Drift-Aware Establishment Simulations
We model a rare fusion allele under heterozygote fertility reduction (underdominance) using Wright–Fisher sampling with selection against heterozygotes (s_het = 0.02 as a conservative example). Transmission ratio distortion (TRD) is represented by parameter k, the probability that the fusion allele is transmitted from a heterozygote (k = 0.5 under Mendelian segregation). To evaluate the role of structure, we also simulate a subdivided metapopulation (D demes of size N_d) under an island model with migration rate m. Fixation probability is estimated from replicate simulations initiated from a single copy introduced into one deme.
2.2. Tract-Based Archaic Introgression Assay at 2q13 (hg19)
We quantify Neanderthal introgression in a conservative 2q13 core window (hg19 chr2:111–113 Mb) using a public IBDmix Vindija tract callset (hg19). For each individual, introgressed tracts are merged to non-overlapping intervals and the overlap with the target window is computed; the introgressed base-pair fraction is then averaged across individuals. A chromosome 2 length-matched null is constructed by sampling windows uniformly along chr2 while excluding 110–114 Mb to prevent overlap with the target region; depletion p-values are estimated by comparing the target statistic to the null distribution.
2.3. External Audit Evidence from Complete-Reference Remapping (T2T-CHM13)
Because 2q13 is repeat-rich and pericentromeric, hg19-era callsets can be sensitive to mappability and callability. We therefore interpret hg19-based tract results in light of recent T2T-CHM13 remapping analyses reporting higher mapping fractions near centromeres and recovery of introgressed sequence previously undetected in older references.
3. Results
3.1. Cytogenetic Evidence and Operational Locus Definition
HSA2 exhibits fusion-consistent signatures used in cytogenetics and comparative genomics. For quantitative tests we define a conservative 2q13 core window (111–113 Mb in hg19), with an extended 110–114 Mb window for sensitivity analyses.
Figure 1.
HSA2 fusion schematic and operational locus definition (schematic).

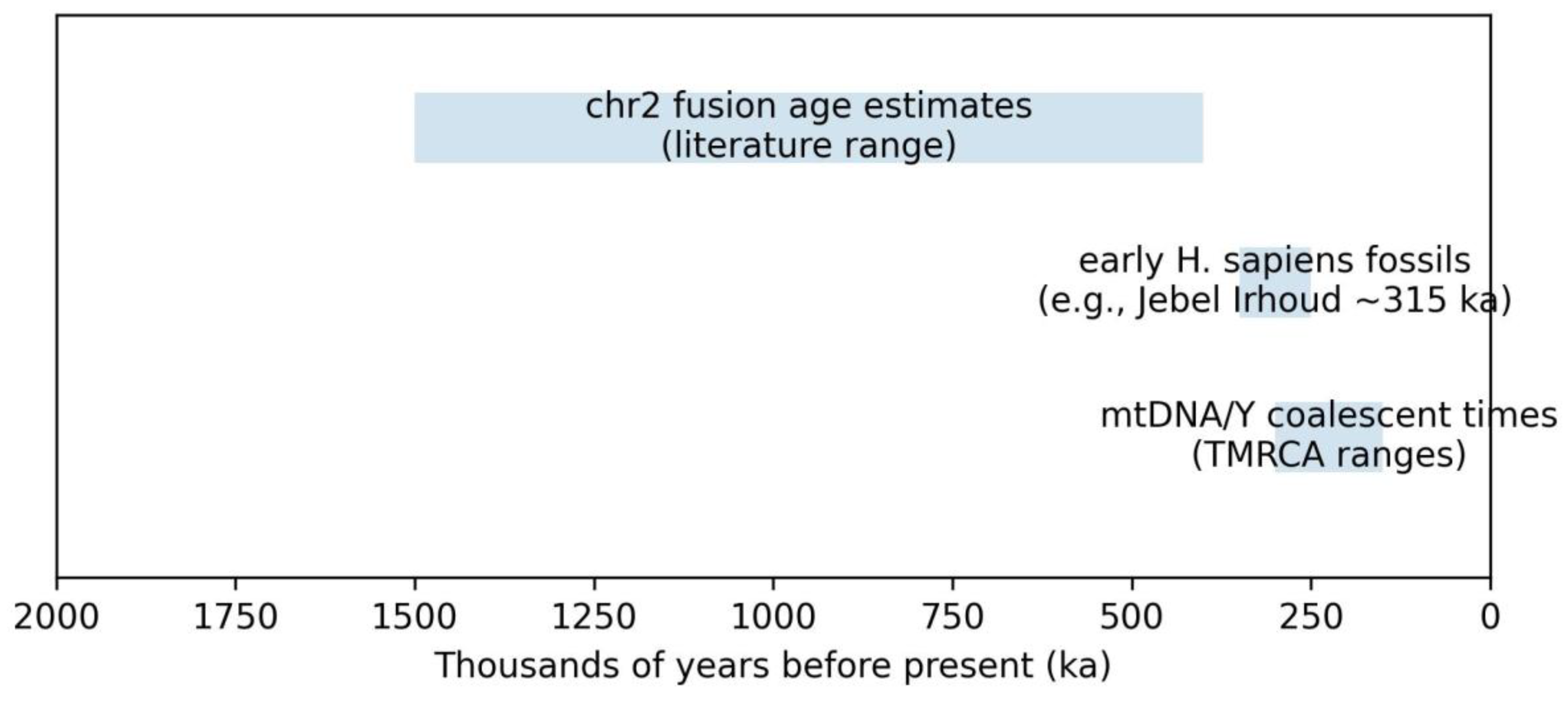
3.2. Distinct Clocks: Fusion Age, Fossil Minima, and Coalescent Times
Fusion event ages, fossil ages, and lineage coalescent times quantify distinct processes and should not be conflated. Recognizing this distinction prevents narratives that either over-attribute sapiens-specific speciation to the fusion or dismiss the fusion as evolutionarily irrelevant without quantitative evaluation.
Figure 2.
Distinct clocks relevant to HSA2 interpretation: fusion event age estimates, fossil minima, and coalescent times.
Figure 2.
Distinct clocks relevant to HSA2 interpretation: fusion event age estimates, fossil minima, and coalescent times.
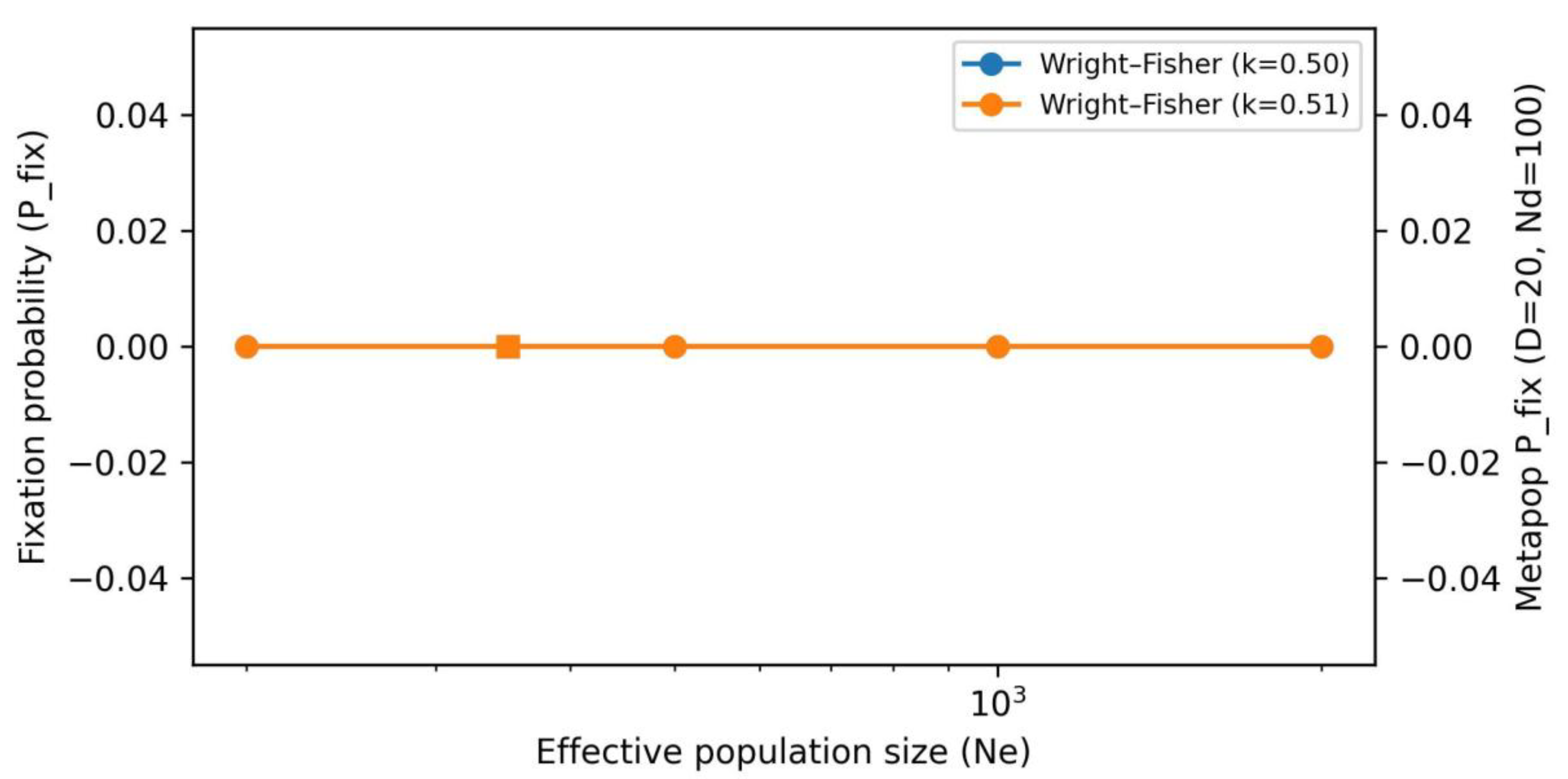
3.3. Establishment Probability Under Underdominance Is Sensitive To Structure and Weak TRD
Under a modest heterozygote fertility cost, fixation probability is low under panmixia but increases with population subdivision, consistent with drift amplification in demes. In sensitivity analyses, weak TRD (k = 0.51) can counteract modest underdominance, increasing establishment probability. Here k is treated as a sensitivity parameter unless independently supported by cytological or molecular evidence of segregation distortion.
Figure 3.
Fixation probability under Wright–Fisher versus a subdivided metapopulation (illustrative parameterization).
Figure 3.
Fixation probability under Wright–Fisher versus a subdivided metapopulation (illustrative parameterization).
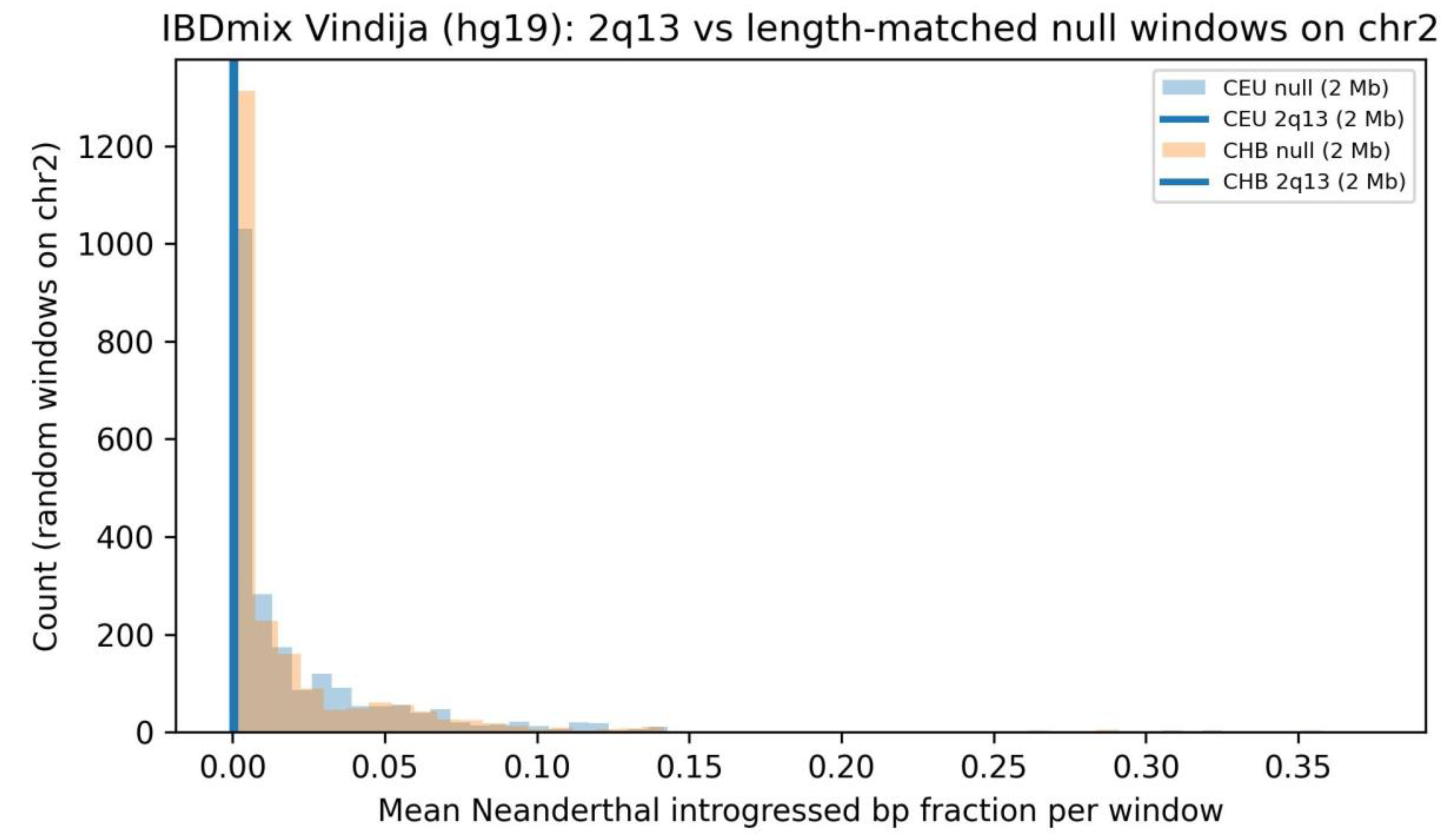
3.4. Introgression Signal at 2q13 in hg19-Era Tract Callsets Is Lower-Tail but Technically Vulnerable
In the IBDmix Vindija hg19 tract representation, 2q13 falls in the lower tail of introgressed fraction relative to a chr2 length-matched null for CEU and CHB. However, because 2q13 is repeat-rich and pericentromeric, callability differences can generate apparent depletion. Accordingly, this signal is treated as a motivation for stricter callability-aware reanalysis rather than as a standalone proof of a reproductive barrier.
Figure 4.
IBDmix Vindija (hg19): 2q13 window versus a length-matched chr2 null distribution (2 Mb).
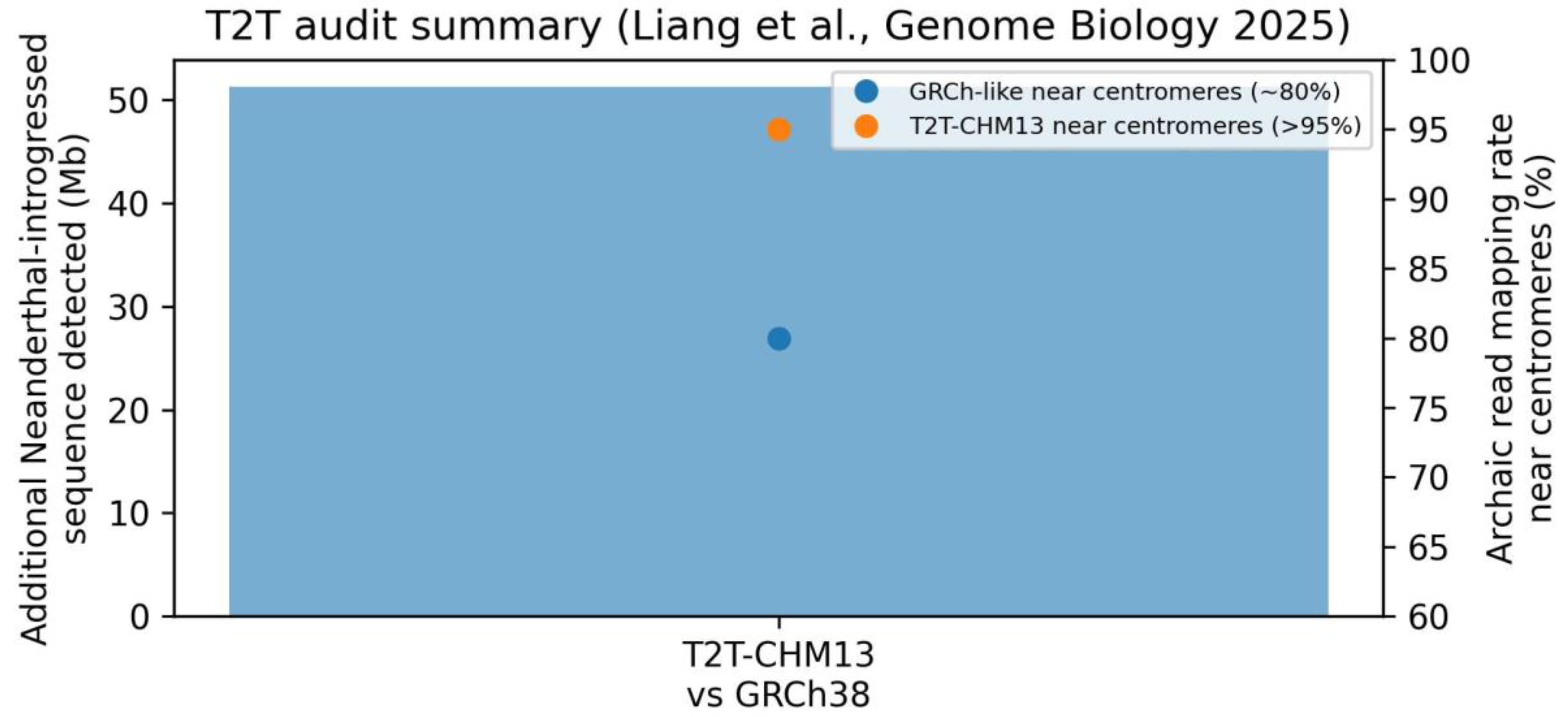
3.5. T2T-CHM13 Remapping Constrains the Interpretation of Putative Deserts
Recent T2T-CHM13 audits report substantial improvements in mapping near centromeres and recovery of archaic sequence previously missed under older references, demonstrating that reference completeness can materially alter inferred introgression landscapes in repeat-rich regions. This external evidence supports a conservative interpretation of hg19-era depletion at 2q13: the signal may reflect callability artifacts as much as selection against introgression.
Figure 5.
External audit evidence: reference completeness rescues introgressed sequence and improves mapping near centromeres (summary).
Figure 5.
External audit evidence: reference completeness rescues introgressed sequence and improves mapping near centromeres (summary).
4. Discussion
These results support bounded conclusions. First, establishment of an underdominant fusion is plausible under drift, especially in structured populations, and weak TRD can enlarge the plausible parameter space. Second, tract-based introgression depletion at 2q13 in hg19-era callsets is not sufficient to conclude a fusion-mediated barrier because the region’s sequence architecture makes it vulnerable to callability artifacts. Third, T2T-CHM13 remapping results provide an external constraint that motivates re-evaluation of 'deserts' in repeat-rich regions under complete references. Mechanistically, centromere competition in asymmetric female meiosis provides a plausible biological interpretation for weak TRD, but it remains an empirical question whether HSA2 exhibited measurable segregation distortion. Functionally, any claim that the fusion reorganized 3D genome topology and altered expression should be framed as falsifiable until supported by comparative Hi-C/Micro-C and transcriptomic analyses robust to paralog mapping.
4.1. Reference Completeness as an Evidentiary Constraint at 2q13
A central interpretive constraint is that the tract assay in this manuscript is computed on an hg19-based representation. This is not merely a historical artifact: the 2q13 region is enriched for segmental duplications and telomeric-like repeats, and older references can compress, misplace, or omit difficult sequence. Complete-reference resources such as T2T-CHM13 therefore change the epistemic status of depletion claims in repeat-rich contexts. Accordingly, the strongest form of the introgression argument is not a literature comparison but a like-for-like reanalysis in which the same archaic and modern reads are mapped to both GRCh/hg19-era references and T2T-CHM13 with matched filters and callability masks. Under that framework, a genuine biological barrier would be supported only if reduced archaic ancestry at 2q13 persists after controlling for callable sites under the complete reference.
4.2. Beyond an Illustrative Island Model: Demographic Realism and Time-varying Structure
The establishment simulations intentionally begin with an analytically transparent 'toy' metapopulation, which clarifies how drift and subdivision can assist fixation under underdominance. However, Pleistocene hominin demography was neither static nor well approximated by identical, permanently connected demes. Effective sizes likely varied over time, connectivity was heterogeneous, and gene flow among lineages changed with geography and climate. These complexities matter because the probability of establishment is sensitive to the distribution of short-term N_e, the duration of local isolation, and the tempo of subsequent mixing. Therefore, any strong claim about plausibility should ultimately be framed against demographic models that allow time-varying structure (e.g., changing migration matrices, episodic bottlenecks and expansions, and archaic-modern admixture pulses). Within this manuscript we treat the island model as a conservative demonstration of principle rather than a reconstruction of hominin history.
4.3. Interpreting k: From Sensitivity Parameter to Biological Quantity
The TRD parameter k is used here to quantify how small deviations from Mendelian segregation could amplify establishment probability under underdominance. In the current manuscript, k is not asserted as an observed property of HSA2; it is a sensitivity axis. Nevertheless, the parameter is not free-floating: centromere competition in asymmetric female meiosis provides a well-defined biological pathway by which weak TRD can arise, and it motivates measurable molecular proxies (e.g., centromeric chromatin and kinetochore assembly metrics) that could, in principle, constrain k for a given rearrangement. This connection is important because it moves the model from 'fit by convenience' to 'parameter linked to observable cell biology', even while acknowledging that the relevant measurements are not yet reported for HSA2 specifically.
5. Conclusions
Human chromosome 2 fusion is a secure structural event, but its evolutionary significance must be evaluated with quantitative population-genetic models and callability-aware paleogenomic analyses. Under modest underdominance, establishment can be drift-assisted in structured populations and may be amplified by weak TRD; meanwhile, apparent introgression depletion at 2q13 in hg19-era tract callsets is best interpreted cautiously given T2T-era evidence that reference completeness rescues introgressed sequence in difficult genomic regions.
Algorithm and Code Availability
We provide a reproducible realignment workflow for mapping raw archaic-hominin reads onto the complete T2T-CHM13 reference and extracting callability-aware summaries over user-defined regions. The workflow is implemented as a Snakemake pipeline using widely adopted tools (bwa/samtools/bedtools) with ancient-DNA-appropriate alignment parameters and strict post-alignment filtering (duplicate removal and MAPQ thresholds). Per-sample outputs include a filtered BAM, base-level depth tracks, callable-site masks, and a region-level summary table (mean depth and callable fraction). This workflow enables direct T2T-based reanalysis of fusion-proximal sequence under matched filters, providing an empirical constraint on introgression 'desert' interpretations in repeat-rich regions.
References
- Ijdo, JW; Baldini, A; Ward, DC; Reeders, ST; Wells, RA. Origin of human chromosome 2: an ancestral telomere–telomere fusion; Proc Natl Acad Sci USA, 1991. [Google Scholar]
- Chmátal, L; Gabriel, SI; Mitsainas, GP; et al. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution. Curr Biol. 2014, 24, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S; Koren, S; Rhie, A; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Liang, S-A; et al. A refined analysis of Neanderthal-introgressed sequences in modern humans with a complete reference genome. Genome Biol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K; et al. The complete genome sequence of a Neanderthal from the Altai Mountains; Nature, 2014. [Google Scholar]
- Prüfer, K; et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 2017. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



