
Submitted:
29 January 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Biological redox chemistry is traditionally described in terms of oxidant and reductant abundance, redox potential, and associated measures of oxidative stress. While informative, these scalar descriptors fail to explain why systems with comparable redox activity can exhibit profoundly different functional outcomes, or why the same pathology, across different biological contexts, may display opposing redox stress phenotypes. Here, we introduce redox coherence and redox decoherence as distinct emergent biological states arising from the integrity of a Redox Photonic Coupling System (RPCS). In this framework, redox chemistry is organized along two coupled axes - oxidative excitation and reductive assimilation - whose spatiotemporal synchronization within a nanodomain Coherence Interface (CI) determines whether redox-derived excitation is resolved into organized hydration shell architecture associated with adjacent biological substrates or dissipated through unstructured pathways.We define redox resilience as the capacity of the system to restore coherent resolution following perturbation, emphasizing recovery dynamics rather than static redox balance, and identify loss of this resilience as the defining feature of redox decoherence. Within this framework, oxidative and reductive stress are not primary causes but directional expressions of an underlying decoherent state, shaped by axis dominance and CI desynchronization.Distinct photonic outcomes are associated with these organizational states: Photonic Activation Quanta (PAQ) reflect coherent resolution and propagation of excitation, tightly coupled to organized water formation, whereas Decoherent Photon Emissions (DPE) mark dissipative resolution modes. The PAQ:DPE ratio thus provides a dynamic, state-sensitive readout of redox organization and recovery capacity, rather than a measure of oxidant or antioxidant burden alone.Together, this framework reframes redox biology as a state-dependent process governed by spatiotemporal organization, structured hydration and resilience, offering a unifying principle for interpreting redox physiology and pathology beyond redox magnitude.
Keywords:
redox coherence
; redox decoherence
; reodx homeostasis
; oxidative stress
; reductive stress
; redox biology
; systems biology
; redox signaling
1. Introduction
Redox reactions are fundamental to biological systems, underpinning metabolism, signaling, and stress responses. Conventional redox biology has largely characterized these processes through measures of oxidant burden, antioxidant capacity, and redox potential, often framing pathology as a consequence of excessive or insufficient redox activity [1,2,3]. While this scalar approach has been informative, it has proven insufficient to explain why biological systems with comparable redox activity can exhibit profoundly different functional outcomes. In particular, high levels of reactive oxygen species or metabolic flux may coexist with preserved physiological function in some contexts and with severe dysfunction in others [4,5], indicating that redox chemistry in living systems is governed not only by magnitude but by the manner in which it is organized.
Here, we propose that biological redox chemistry exists in at least two qualitatively distinct organizational states - redox coherence and redox decoherence - which emerge from the functional integrity of a Redox Photonic Coupling System (RPCS). In this framework, redox coherence arises when oxidative and reductive processes are dynamically paired, spatially constrained, temporally aligned and supported by structured hydration along two coupled axes: an oxidative excitation axis, exemplified by the controlled generation of highly reactive species such as the hydroxyl radical (•OH) [6,7], and a reductive assimilation axis, exemplified by glutathione (GSH)-dependent resolution of oxidative excitation [8,9,10,42]. When this coupling is preserved, redox reactions proceed coherently and give rise to structured photonic outcomes termed Photonic Activation Quanta (PAQ). Conversely, impairment or desynchronization of this coupling leads to redox decoherence, characterized by loss of pairing, energetic dissipation, molecular damage, and Decoherent Photon Emissions (DPE). Within this view, photonic emissions do not drive redox chemistry but arise as emergent signatures of its underlying organizational state. These axes represent functional roles rather than specific reactions and may be realized through different redox chemistries across cellular contexts.
Together, these observations point to a fundamental conceptual gap in redox biology: the absence of a state variable capable of describing how redox activity is organized in space and time [11,12,13]. Rather than asking how much redox chemistry is occurring, a biologically meaningful description must address whether redox reactions are coordinated, constrained, and recoverable following perturbation. This motivates a shift from scalar descriptors toward organizational redox states that capture the integrity of redox coupling itself.
1.1. Limitations of Scalar Redox Models
Scalar redox models reduce spatially localized and temporally dynamic redox processes to averaged values, obscuring whether oxidative reactions are appropriately paired with reductive counterparts, whether reactions remain spatially confined or become diffuse, and whether redox activity is productively channeled or dissipated through indiscriminate substrate oxidation [19,20,21].
Although useful for quantifying overall redox load, scalar descriptors have repeatedly failed to explain discordant biological outcomes observed under apparently similar redox conditions. Comparable levels of oxidants, antioxidants, or bulk redox potential may support physiological signaling in one context while producing extensive molecular damage in another. Consistent with this limitation, global antioxidant interventions often yield inconsistent or paradoxical effects, sometimes impairing essential cellular functions without preventing pathology [15,16]. Scalar models also fail to account for pathological states driven by excess or dysregulated reductive capacity, in which redox organization is destabilized despite physiological oxidant levels [17,18].
At their core, scalar models implicitly assume that redox reactions act uniformly and independently across space and time. By collapsing heterogeneous, dynamic redox activity into single values, these models obscure distinctions between fundamentally different organizational regimes and fail to capture the spatial, temporal, structured hydration availability and pairing constraints that ultimately determine biological outcome [19,20,21].
1.2. Redox Organization as a State Variable
The limitations of magnitude-based redox descriptors point to the need for qualitative organizational states. The relevant question is not how much redox chemistry is occurring, but how it is arranged: whether oxidative reactions are dynamically paired with appropriate reductive processes, whether reactions are spatially localized within nanodomains or allowed to diffuse, and whether redox activity is recoverable following perturbation [19,25].
We introduce redox coherence and redox decoherence as system-level organizational states that capture this dimension of redox behavior.
1.3. Overview of the Redox Photonic Coupling System (RPCS)
Redox coherence and redox decoherence do not arise from the properties of individual redox-active molecules but from the integrity of an underlying system that coordinates oxidative and reductive processes. We refer to this system as the Redox Photonic Coupling System (RPCS). The RPCS is not a discrete biochemical pathway or molecular complex but a functional redox architecture that governs how oxidative excitation and reductive assimilation are dynamically paired, spatially confined, and temporally aligned within living systems.
When intact, the RPCS supports coherent redox resolution by ensuring that physiological oxidant generation is matched with appropriate reductive capacity and structured hydration at spatiotemporally defined nanodomains [26,27]. When compromised, this coordination deteriorates, leading to redox decoherence even in the presence of abundant oxidants, reductants, or both.
In the sections that follow, we first formalize redox coherence and redox decoherence as emergent biological states (Section 2). We then describe the architecture of the Redox Photonic Coupling System (RPCS) and its organization along coupled oxidative and reductive axes (Section 3) and define the Coherence Interface as the nanodomain at which these axes converge and are resolved (Section 4). We next examine photonic outcomes as emergent readouts of redox organization, distinguishing Photonic Activation Quanta (PAQ) from Decoherent Photon Emissions (DPE) (Section 5). We then introduce redox resilience as a control property governing recovery and stability (Section 6), discuss redox decoherence as the organizational state that emerges when resilience is lost (Section 7) and lastly propose a series of testable predictions and experimental approaches, meant to probe the claims of this framework (Section 8).
2. Redox Coherence and Decoherence
Biological redox activity does not exist along a single continuum of excess or deficiency but instead occupies qualitatively distinct organizational regimes. In this section, we define redox coherence and redox decoherence as alternative system-level states that describe how oxidative and reductive processes are organized, paired, and constrained in space and time. These definitions establish the conceptual foundation for understanding redox behavior independently of redox magnitude and prepare the framework for introducing the architectural features that govern these states [33,34].
2.1. Redox Coherence
Redox coherence is a system-level organizational state in which oxidative and reductive reactions are dynamically paired, spatially constrained, temporally coordinated, and supported by local structured hydration, such that redox chemistry proceeds in an organized and non-dissipative manner. In coherent systems, oxidative excitation is matched by appropriately conditioned reductive capacity, enabling reactive intermediates to be resolved through ordered biochemical interactions rather than diffuse oxidation.
A defining feature of redox coherence is pairing. Oxidative excitations are coupled to compatible reductive counterparts through spatial proximity, temporal alignment, and functional compatibility, confining redox reactions to defined microenvironments and limiting energetic dissipation and collateral molecular damage [19,21,27,28].
Redox coherence does not imply low oxidant production. Coherent states may involve high redox flux provided that excitation remains effectively paired and channeled. Coherence therefore reflects the organization of redox activity rather than its magnitude and cannot be inferred from oxidant or antioxidant abundance alone [7,13,30].
Redox coherence is an emergent property of the system, arising from coordinated interactions among multiple redox processes rather than from any single molecule or pathway [33,34]. Coherence may be spatially heterogeneous, with localized disruptions occurring without immediate loss of global organization [19,21,27,29,38].
2.2. Redox Decoherence
Redox decoherence is a system-level organizational state in which oxidative and reductive processes fail to maintain structured redox resolution due to breakdown of the conditions governing pairing. This failure may arise from loss of spatial or temporal confinement, sustained dominance of either oxidative excitation or reductive assimilation, or disruption of local hydration. In decoherent states, oxidative excitation either escapes confined resolution or is suppressed under reductive dominance, resulting in diffuse, dissipative, and damaging redox outcomes.
Two convergent failure modes give rise to decoherence. First, disruption of structural or spatiotemporal constraints, including loss of locally organized hydration, impairs pairing and permits unconfined redox reactivity. Second, persistent dominance of either the oxidative excitation axis or the reductive assimilation axis destabilizes coherence even when spatial organization is preserved.
Redox decoherence is not defined by redox magnitude. It may arise under conditions of apparent redox homeostasis, where oxidant and reductant levels are balanced but organizational constraints required for coherent resolution are impaired [12,15,16]. Decoherence, like coherence, is an emergent and spatially distributed property, often manifesting locally before propagating system-wide [19,21,29,38].
Redox coherence and redox decoherence therefore represent alternative emergent organizational states of biological redox systems rather than properties of individual molecules or reactions [5,12,16]. Identical molecular components may support either state depending on their spatial arrangement, temporal coordination, pairing capacity, and hydrational continuity explaining why similar redox loads can yield divergent biological outcomes [6,13,19]. Redox coherence is distinct from redox homeostasis: quantitative balance does not ensure coherence, nor does elevated oxidant generation necessarily imply decoherence [1,11,35]. Instead, coherence reflects the organizational constraints imposed on redox chemistry and may be spatially heterogeneous, with localized disruptions arising without immediate loss of global organization [7,20,28].
3. Redox Photonic Coupling System (RPCS)
Biological redox coherence requires more than the presence of oxidants and reductants; it depends on an organizational architecture that governs how oxidative excitation is generated, delivered, and resolved. We define this architecture as the Redox Photonic Coupling System (RPCS) - a functional system that determines whether redox chemistry proceeds in a paired, constrained, and coherent manner or devolves into decoherent, dissipative behavior.In this section, we formalize the RPCS as a two-axis system, describe the molecular deliverables of each axis, and define the nanodomain-scale interface at which oxidative and reductive fluxes converge to determine redox state.
3.1. System Overview
The Redox Photonic Coupling System (RPCS) is a functional redox architecture rather than a discrete biochemical pathway or molecular complex. It describes the organizational condition under which oxidative and reductive processes are dynamically coordinated such that redox reactions remain paired, spatially constrained, and temporally aligned.
The RPCS is organized along two complementary and dynamically coupled axes:
- the Oxidative Excitation Axis (OEA), responsible for the controlled generation of oxidative electronic excitation, and
- the Reductive Assimilation Axis (RAA), responsible for capturing and resolving the oxidative excitation through reductive electronic supply.
Each axis is defined not only by its functional role but by a characteristic molecular deliverable. The principal deliverable of the OEA is the hydroxyl radical (•OH), representing the maximal expression of oxidative excitation in biological systems. The principal deliverable of the RAA is reduced glutathione (GSH), which functions as the dominant intracellular carrier of reductive potential and the primary molecular agent for excitation assimilation [41,44].
Coupling between these axes occurs at a Coherence Interface (CI) - a nanodomain-scale, spatially and temporally constrained functional zone in which oxidative excitation generated along the OEA encounters sufficient, appropriately positioned reductive capacity from the RAA. Redox coherence is preserved when CI integrity is maintained; redox decoherence emerges when it is lost.
The RPCS should be understood as a necessary condition for redox coherence, not as a determinant of specific downstream biological functions. Its integrity governs whether coherent redox organization is possible, without prescribing how redox chemistry is ultimately deployed in metabolism, signaling, or adaptation.
Although the RPCS is present in multiple cellular compartments, including mitochondria, cytosol, membranes, and organelle-associated microenvironments, the present work focuses on the mitochondrial RPCS, where spatial organization, redox intermediates, and excitation-resolution coupling are best characterized [7,31,39]. This focus is illustrative rather than exclusive.
Figure 1.
Functional architecture of the Redox Photonic Coupling System (RPCS). This schematic illustrates the RPCS as a functional architecture coordinating two coupled redox fluxes: the Oxidative Excitation Axis (OEA), which delivers localized oxidative excitation as hydroxyl radical (•OH), and the Reductive Assimilation Axis (RAA), which delivers energetically competent reductive capacity as glutathione (GSH). These axes converge at the Coherence Interface (CI), depicted here as a nanodomain control interface specifying the local conditions under which redox-derived excitation is coherently resolved or dissipated.
Figure 1.
Functional architecture of the Redox Photonic Coupling System (RPCS). This schematic illustrates the RPCS as a functional architecture coordinating two coupled redox fluxes: the Oxidative Excitation Axis (OEA), which delivers localized oxidative excitation as hydroxyl radical (•OH), and the Reductive Assimilation Axis (RAA), which delivers energetically competent reductive capacity as glutathione (GSH). These axes converge at the Coherence Interface (CI), depicted here as a nanodomain control interface specifying the local conditions under which redox-derived excitation is coherently resolved or dissipated.
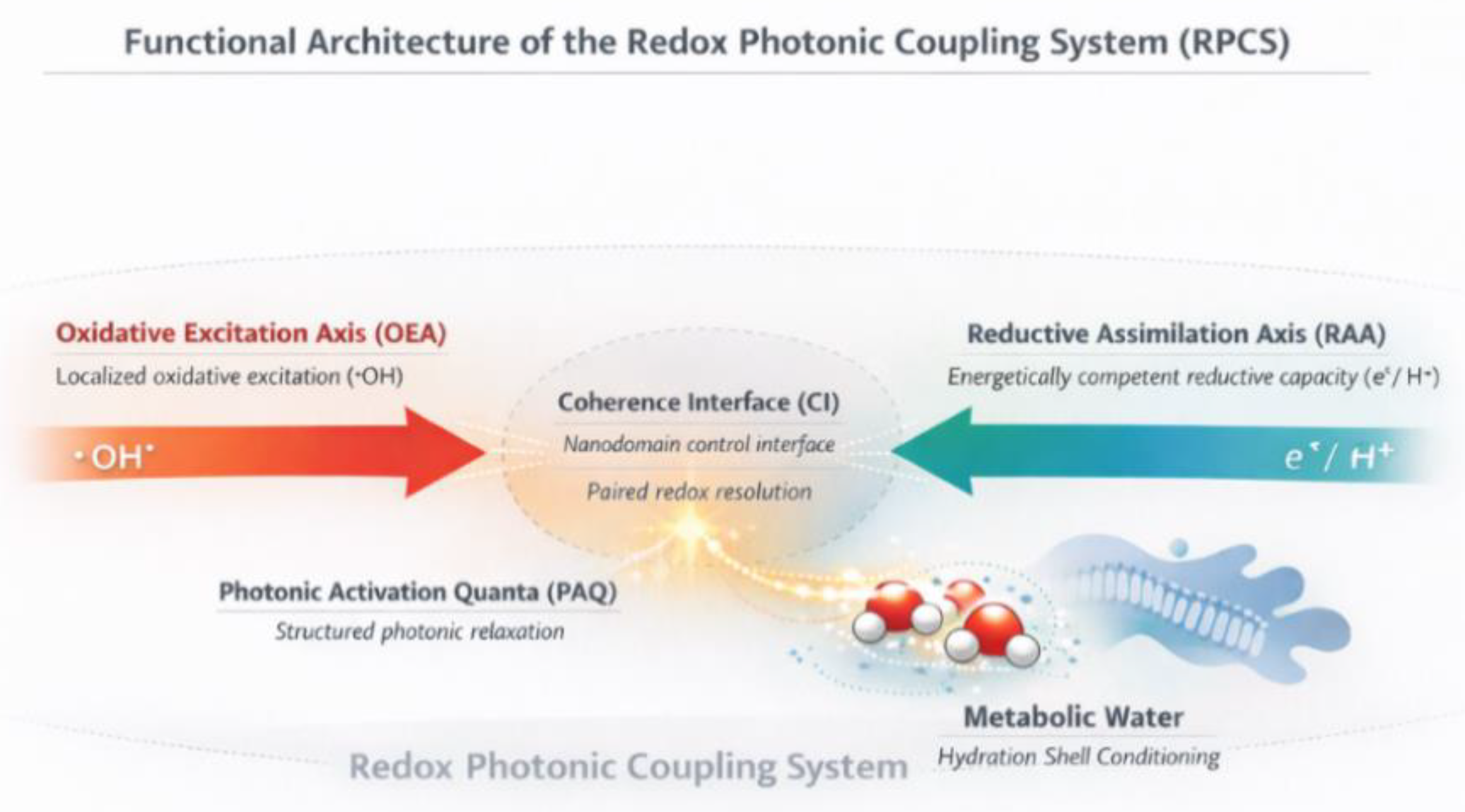
3.2. The Oxidative Excitation Axis (OEA)
The Oxidative Excitation Axis (OEA) represents the component of the RPCS responsible for the ordered generation of oxidative electronic excitation. In mitochondria, this axis is initiated within the electron transport chain (ETC), primarily at Complex I (NADH ubiquinone oxidoreductase) and Complex III (ubiquinol cytochrome c oxidoreductase), which serve as physiologically regulated entry points for excitation [39,40]. Superoxide anion generation is not limited to the mitochondrial electron transport chain (ETC) components, other sources being nicotinamide adenine dinucleotide phosphate oxidases (NOX) expressed in cell membranes, cytoplasmic xanthine oxidase (XO), and flavin-centered oxidases in peroxisomes and other cellular compartments [51]. For purposes of mechanistic exemplification, the present work describes the oxidative excitation axis initiated at the electron transport chain (ETC).
Step I - Superoxide Generation
At Complex I, reduced flavin mononucleotide (FMNH2) and iron–sulfur centers donate single electrons to molecular oxygen. At Complex III, semiquinone intermediates formed during the Q cycle similarly transfer single electrons to oxygen. In both cases, the result is superoxide anion (O2•−) formation within membrane-associated nanodomains.
Within the RPCS framework, superoxide generation is interpreted not as an accidental electron leak but as a physiological initiation of oxidative excitation, spatially confined and functionally integrated [39,40].
Reaction:
O2 + e− → O2•−
Step II - Hydrogen Peroxide Formation
Superoxide is rapidly converted to hydrogen peroxide (H2O2) by mitochondrial superoxide dismutase (SOD2). Hydrogen peroxide functions as a stabilized excitation intermediate, preserving redox potential while extending the spatial and temporal window for controlled processing [38,40].
Reaction:
2 O2•− + 2 H+ → H2O2 + O2
Step III — Hydroxyl Radical Formation
Within redox-active nanodomains containing transition metals, hydrogen peroxide undergoes Fenton-type reactions, yielding the hydroxyl radical (•OH) - the terminal deliverable of the OEA.
The hydroxyl radical possesses a singly occupied molecular orbital (SOMO) with an energetically accessible lowest unoccupied molecular orbital (LUMO), conferring extreme reactivity and imposing the strictest requirement for immediate reductive pairing [36]. The biological role of hydroxyl radical (•OH) within a coherent system is therefore not diffusion or accumulation, but ultra-localized, transient participation in paired redox events. When pairing fails, the same SOMO/LUMO-driven reactivity underlies indiscriminate substrate oxidation and the transition to redox decoherence.
Reaction:
H2O2 + Fe2+ → •OH + OH− + Fe3+
This reaction represents the culmination of oxidative excitation along the OEA and produces the principal deliverable of the oxidative excitation axis. This progression reflects a structured physiological excitation sequence, not a pathological escalation. Whether excitation is resolved coherently or results in damage depends entirely on downstream assimilation capacity.
Figure 2.
Oxidative Excitation Axis (OEA). Ordered generation of oxidative electronic excitation originating at mitochondrial ETC Complexes I and III, progressing through superoxide anion generation, hydrogen peroxide formation as stabilized intermediate, and culminating in hydroxyl radical formation via the Fenton reaction as the terminal excitation deliverable.
Figure 2.
Oxidative Excitation Axis (OEA). Ordered generation of oxidative electronic excitation originating at mitochondrial ETC Complexes I and III, progressing through superoxide anion generation, hydrogen peroxide formation as stabilized intermediate, and culminating in hydroxyl radical formation via the Fenton reaction as the terminal excitation deliverable.
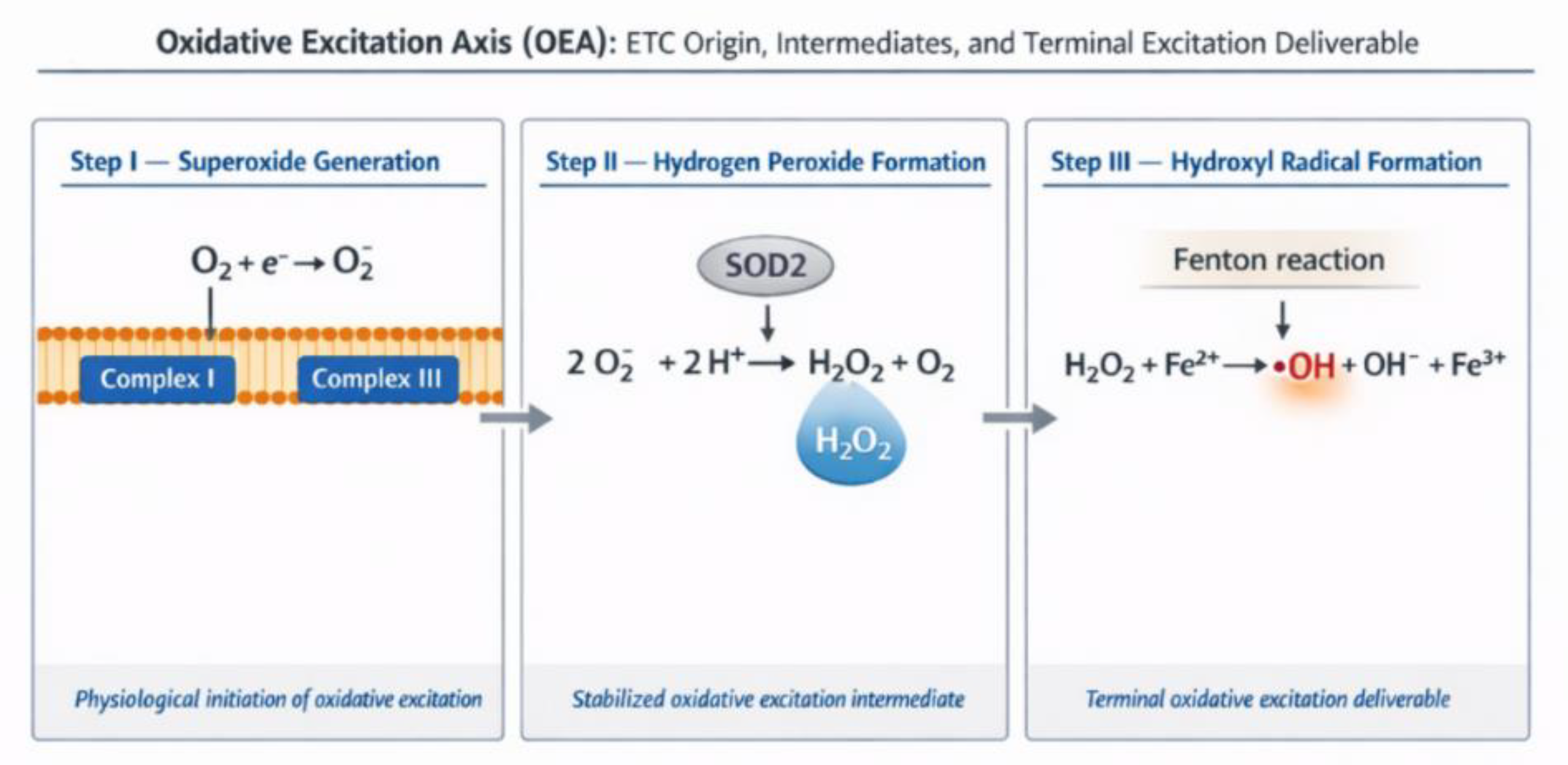
3.3. The Reductive Assimilation Axis (RAA)
The Reductive Assimilation Axis (RAA) represents the component of the Redox Photonic Coupling System responsible for assimilating oxidative excitation generated along the Oxidative Excitation Axis. Its deliverables are reducing electrons and hydrogen donors, exemplified by reduced glutathione (GSH), whose functional efficacy depends on continuous recycling from oxidized glutathione (GSSG) and on the energetic state of the electrons they carry. While de novo glutathione synthesis establishes basal reductive capacity, the RAA primarily concerns the dynamic regeneration and energetic conditioning of glutathione, enabling repeated and coherent redox resolution at active Coherence Interfaces [42,43,44].
From an electronic structure perspective, GSH carries a high-energy electron in its highest occupied molecular orbital (HOMO), localized on the thiolate sulfur of its cysteine residue. This HOMO-confined electron endows GSH with strong nucleophilic and reducing capacity, enabling rapid and selective electron donation to highly reactive oxidative species, including the hydroxyl radical [37]. Within the RPCS, GSH therefore functions not merely as an antioxidant buffer, but as a vector for delivering reductive electronic energy precisely where oxidative excitation must be assimilated.
The energetic state of the GSH HOMO is not intrinsic or static. It is continuously regenerated by nicotinamide adenine dinucleotide phosphate in its reduced form (NADPH) through the action of glutathione reductase (GSR), which catalyzes the reduction of oxidized glutathione (GSSG) back to GSH [45].
This reaction constitutes the core biochemical operation of the RAA:
GSSG + NADPH + H+ → 2 GSH + NADP+
Through this process, NADPH supplies paired high-energy reducing equivalents via hydride ion (H−) transfer, preserving electron pairing and energetic integrity during glutathione recycling and restoring the high-energy electronic configuration of the thiolate sulfur in GSH; we refer to this process here as electronic conditioning. This mode of electron delivery maintains energetic consistency within the reductive assimilation pathway, ensuring that regenerated GSH retains full reductive competence for subsequent redox engagement. Accordingly, the functional capacity of the RAA depends directly on NADPH availability, turnover, and regeneration efficiency.
Figure 3.
Reductive Assimilation Axis (RAA). The Reductive Assimilation Axis (RAA) regenerates and energetically conditions glutathione (GSH) through NADPH-dependent recycling across cellular compartments, preparing reductive capacity upstream of Coherence Interface engagement.
Figure 3.
Reductive Assimilation Axis (RAA). The Reductive Assimilation Axis (RAA) regenerates and energetically conditions glutathione (GSH) through NADPH-dependent recycling across cellular compartments, preparing reductive capacity upstream of Coherence Interface engagement.
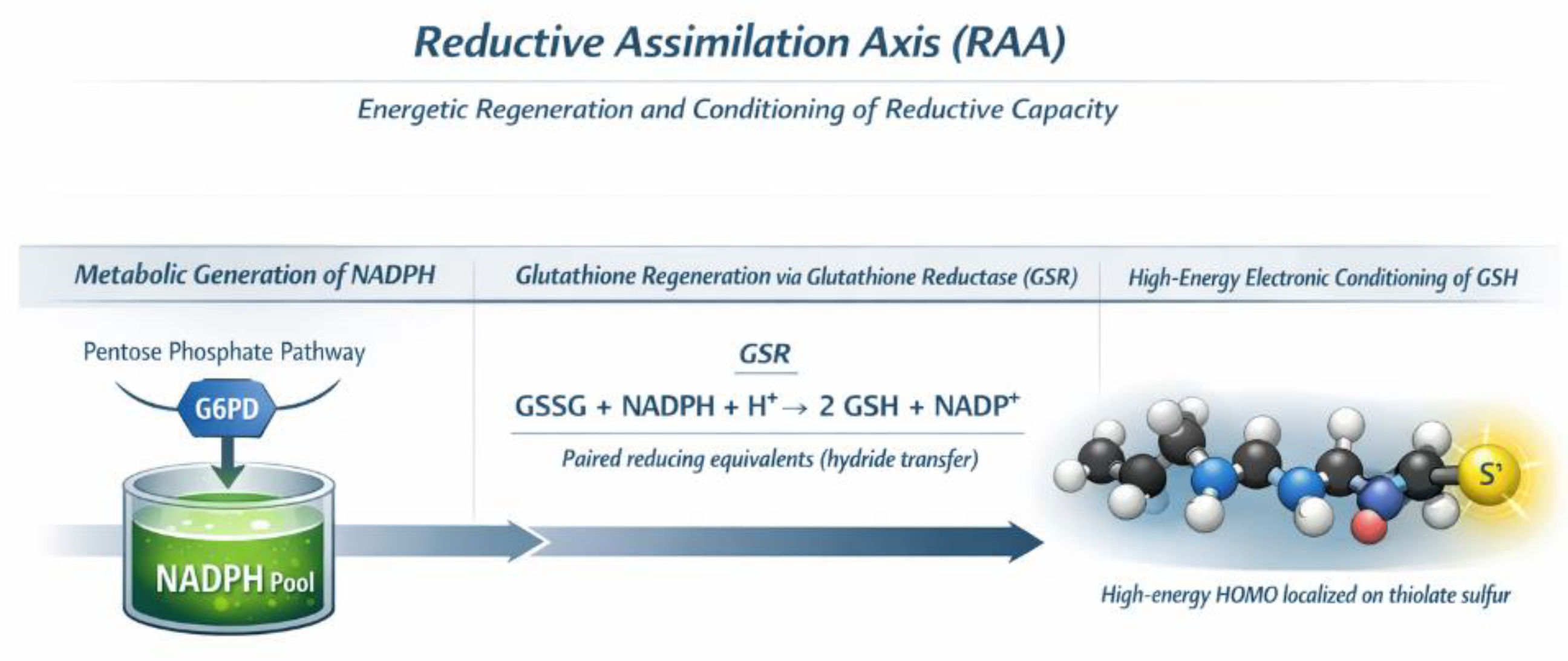
In mammalian cells, a major source of cytosolic NADPH is the pentose phosphate pathway (PPP), whose flux is governed by glucose-6-phosphate dehydrogenase (G6PD). This cytosolic NADPH pool supports glutathione recycling and redox control at Coherence Interfaces located in the cytosol, plasma membrane, endoplasmic reticulum, and peroxisomes -compartments that host multiple oxidative excitation sites, including NADPH oxidases and metal-dependent reactions [51].
NADPH itself does not freely cross mitochondrial membranes [52]. Instead, reducing equivalents generated in the cytosol are transferred to mitochondria via redox shuttle systems, while mitochondria also possess intrinsic NADPH-generating pathways, including NADP+-dependent isocitrate dehydrogenase (IDH2), malic enzyme (ME3), and nicotinamide nucleotide transhydrogenase (NNT) [53,54]. Together, these mechanisms sustain compartment-specific NADPH pools and enable local glutathione recycling within mitochondrial Coherence Interfaces.
Thus, the RAA does not correspond to a single biochemical pathway, but to a distributed energetic supply network that translates metabolic reducing power into spatially and temporally available reductive capacity. When NADPH production, reducing-equivalent transfer, glutathione recycling, or nanodomain delivery are compromised within a given compartment, the RAA fails to meet the energetic demands imposed by oxidative excitation, predisposing the system to redox decoherence.
The high-energy electrons carried by NADPH, and subsequently by the highest occupied molecular orbital of glutathione, originate from carbohydrate metabolism ultimately rooted in solar energy captured through photosynthetic carbon fixation. In this sense, the Reductive Assimilation Axis represents the downstream endpoint of a long biological energy-transduction chain, linking solar-derived chemical energy to reductive electronic power and, ultimately, to paired assimilation of oxidative excitation. When the Redox Photonic Coupling System is intact, this reducing power is delivered to nanodomain-scale Coherence Interfaces, where it supports organized redox chemistry and coherent resolution rather than dissipative outcome.
3.4. Redox Coupling at the Coherence Interface
The Coherence Interface (CI) is a control interface within the Redox Photonic Coupling System at which the Oxidative Excitation Axis and the Reductive Assimilation Axis converge. It is a spatiotemporally confined nanodomain, defined by nanometer-scale spatial confinement, temporal synchronization, stoichiometric adequacy and hydrational continuity, where oxidative excitation is either productively assimilated or allowed to devolve into redox decoherence.
At the CI, the hydroxyl radical (•OH) generated along the Oxidative Excitation Axis encounters glutathione (GSH) delivered by the Reductive Assimilation Axis. This encounter constitutes the decisive redox pairing event of the RPCS. When pairing occurs within an intact CI, oxidative excitation is neutralized through a tightly constrained electron and hydrogen-transfer process rather than through diffuse substrate attack.
The core chemical outcome of this pairing can be summarized schematically as:
2 •OH + 2 GSH → GSSG + 2 H2O + 2 hν (ultraweak photon emission)
In this reaction, two molecules of glutathione neutralize two hydroxyl radicals through hydrogen atoms transfer from the cysteine thiol, a process enabled by the high-energy electronic state of the glutathione highest occupied molecular orbital (HOMO). The donated hydrogen atoms complete radicals neutralization, converting reduced glutathione to oxidized glutathione (GSSG), generating two molecules of water, and resolving the associated oxidative electronic excitation. To first approximation, this redox event can be described as a discrete orbital-level coupling process, in which electrons originating from the glutathione HOMO resolve pre-existing radical electronic states established during hydroxyl radical formation [36,37].
Within this framework, energy released during redox pairing is not treated as nonspecific dissipation, but as quantized electronic relaxation arising from orbital-level coupling between high-energy electrons supplied by the reductive axis and radical electronic states generated along the oxidative axis. The resulting ultraweak photon emission (UPE) thus reflects a structured electronic transition intrinsic to successful redox pairing, rather than a byproduct of random molecular damage. Orbital-level analyses of hydroxyl radical reactions in aqueous environments have demonstrated that reactivity is governed by pre-existing frontier molecular orbitals rather than by random energy dissipation [36]. By explicitly framing redox coupling in orbital-level terms, this work links chemical reactivity, photonic emission, and system organization within a unified electronic framework [47,48,49,50].
Figure 4.
The Coherence Interface (CI): Orbital-coupled H-atom transfer and photonic relaxation. The Coherence Interface (CI) is shown as a nanodomain-scale electronic coupling site where oxidative excitation is resolved through frontier-orbital alignment rather than bulk antioxidant chemistry. Energetic alignment between a sulfur-centered donor state of reduced glutathione (GSH) and the singly occupied molecular orbital (SOMO) of the hydroxyl radical (•OH) enables a tightly coupled hydrogen-atom transfer (H), forming metabolic water in situ. Electronic reorganization during this transfer undergoes quantized relaxation, giving rise to a discrete Photonic Activation Quantum (PAQ), which is immediately captured by the newly formed water, preserving local energetic coherence.
Figure 4.
The Coherence Interface (CI): Orbital-coupled H-atom transfer and photonic relaxation. The Coherence Interface (CI) is shown as a nanodomain-scale electronic coupling site where oxidative excitation is resolved through frontier-orbital alignment rather than bulk antioxidant chemistry. Energetic alignment between a sulfur-centered donor state of reduced glutathione (GSH) and the singly occupied molecular orbital (SOMO) of the hydroxyl radical (•OH) enables a tightly coupled hydrogen-atom transfer (H), forming metabolic water in situ. Electronic reorganization during this transfer undergoes quantized relaxation, giving rise to a discrete Photonic Activation Quantum (PAQ), which is immediately captured by the newly formed water, preserving local energetic coherence.
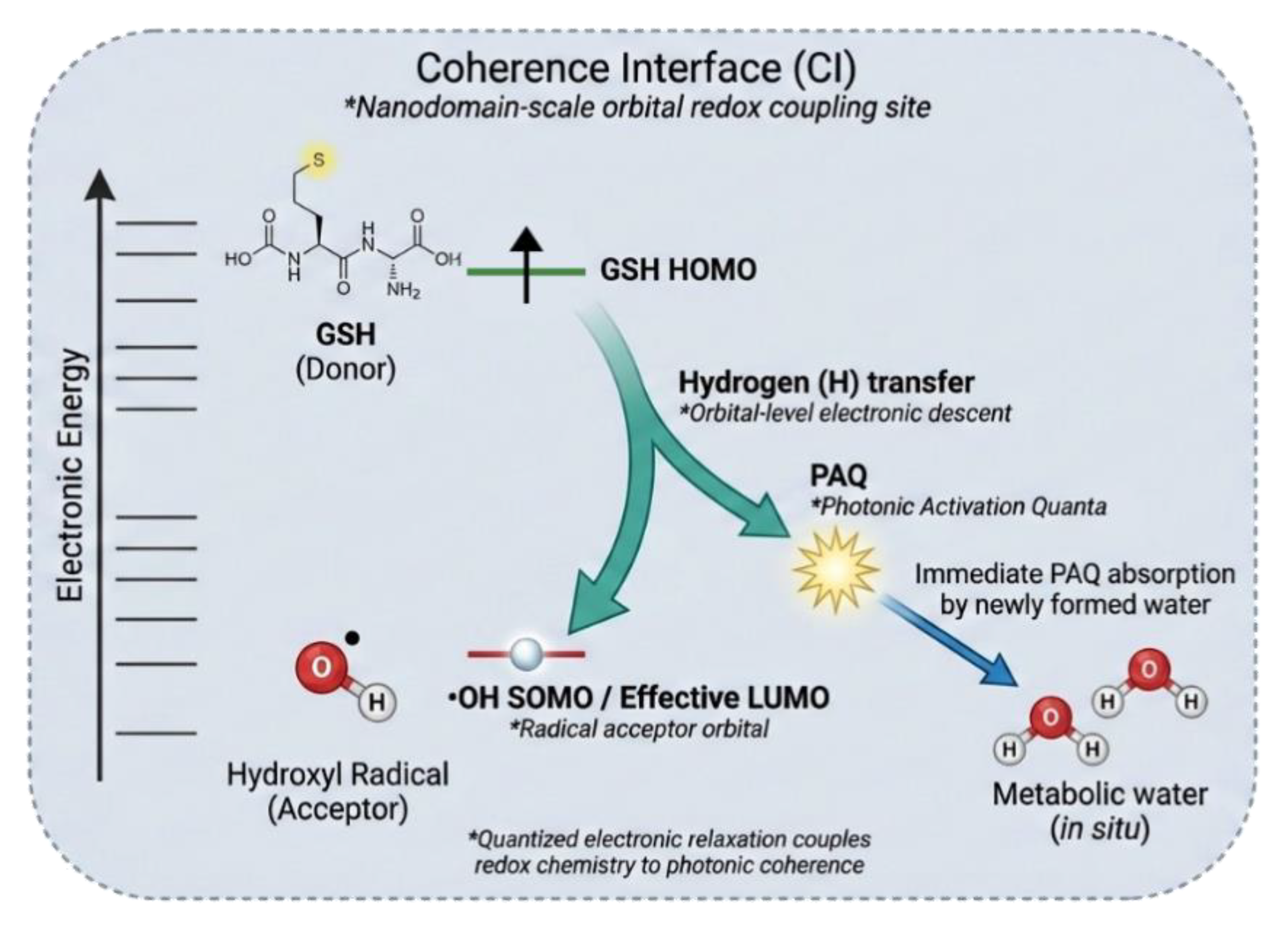
Photonic emissions arising from successful redox pairing are defined here as Photonic Activation Quanta (PAQ) - coherent photons with defined spatial and temporal properties generated during quantized electronic relaxation. We propose that these PAQs do not propagate freely but are captured within the local hydration environment formed in situ on the timescale of redox pairing between hydroxyl radical and glutathione, owing to their nanometer-scale proximity and temporal synchrony with the redox event [50].
The redox pairing reaction at the Coherence Interface thus simultaneously generates oxidized glutathione (GSSG), metabolic water, and PAQs. Newly formed water molecules act as primary absorbers of PAQs, capturing photonic coherence at the time of their formation. This absorption couples electronic coherence directly into the hydrogen-bonding network of water, imparting organizational properties that distinguish this water from bulk or diffusely generated metabolic water.
We further propose that these PAQ-conditioned water molecules subsequently participate in the hydration shells of nearby proteins, membranes, and macromolecular assemblies, locally stabilizing coherent organization within adjacent hydration environments. Through this mechanism, redox coherence is locally embedded within hydration layers adjacent to the Coherence Interface, influencing molecular organization and dynamics within those immediate environments. While the detailed physical properties and biological consequences of such hydration effects are beyond the scope of the present work, this process provides a mechanistic connection between redox coupling, photonic coherence, and structured hydration.”
Accordingly, within this framework, PAQs function as local markers of coherent energy resolution whose biological effects are mediated through water rather than as freely propagating signals. Redox coherence is thus sustained by a coupled triad of paired redox chemistry, coherent photonic generation, and hydration-shell organization, whereas loss of Coherence Interface integrity disrupts all three simultaneously, leading to decoherent photon emission, unstructured water formation, and redox decoherence.
4. The Coherence Interface as a Dynamic Control Domain
The Coherence Interface (CI) functions as the central dynamic control domain of the Redox Photonic Coupling System, governing whether biological redox activity manifests as coherence or decoherence. As established in Section 3.4, the CI is the site at which oxidative excitation and reductive assimilation are resolved through paired redox chemistry, coherent photonic generation, and localized metabolic water formation. Notably, the Coherence Interface does not generate oxidative or reductive flux, does not initiate redox reactions, and does not function as an independent system component; it specifies the local conditions under which redox-derived excitation is resolved coherently or dissipatively. The present section shifts focus from the chemical identity of the CI to its operational integrity, failure modes, and role in redox state transitions [19,21,22].
Rather than representing a static structural feature, the CI is inherently responsive, continuously responding to fluctuations in oxidative excitation, reductive supply, metabolic flux, and nanodomain organization. Its integrity determines whether HOMO–LUMO coupling can be sustained, whether Photonic Activation Quanta (PAQ) are generated and absorbed locally, and whether the metabolic water produced is incorporated into hydration shells [36,37]. In this sense, the CI acts as a state-determining control domain, separating organized redox function from dissipative redox damage.
Redox coherence and redox decoherence are therefore not gradual outcomes of increasing oxidative burden, but distinct system states that emerge from the preservation or collapse of CI integrity. Understanding the CI as a dynamic control surface allows redox pathology to be reframed as a failure of coupling conditions rather than as a simple excess of reactive species or reductive capacity [5,32].
4.1. Operational Definition of Coherence Interface Integrity
The integrity of the Coherence Interface (CI) can be defined operationally by the system’s capacity to sustain productive redox pairing under physiological conditions. CI integrity is not determined by the presence of specific molecules alone, but by whether a set of necessary dynamical conditions is simultaneously satisfied within nanodomain-scale environments.
For the CI to remain intact, all four of the following conditions must be met:
I. Nanospatial Confinement
Oxidative excitation, particularly in the form of hydroxyl radicals, must remain within a spatial radius that permits immediate interaction with reduced glutathione. Nanodomain-scale confinement ensures that oxidative species do not escape the pairing zone. Loss of nanospatial confinement allows oxidative excitation to diffuse beyond the reach of reductive assimilation, precluding HOMO–LUMO coupling and favoring indiscriminate substrate oxidation.
II. Temporal Synchronization
The availability of reduced glutathione must be synchronized with the timescale of oxidative excitation. Even when reductive capacity is sufficient in bulk, delays in the local delivery or regeneration of GSH at the nanodomain scale disrupt pairing. Temporal desynchronization prevents coherent electronic relaxation and abolishes the coupled formation of PAQs and metabolic water.
III. Energetic Adequacy
Reduced glutathione must carry sufficient electronic energy in its HOMO to neutralize the oxidative excitation of hydroxyl radical. This energetic adequacy depends on sustained NADPH regeneration through the pentose phosphate pathway and the other mitochondrial specific pathways and efficient glutathione reductase activity. Depletion of NADPH, impairment of reductive recycling, or limitation of metabolic flux lowers the effective reducing power of the RAA and destabilizes the CI.
IV. Hydrational Continuity
Successful redox pairing must be accompanied by the integration of the metabolic water into contiguous hydration shell networks surrounding nearby macromolecules. Hydrational continuity refers to the system’s capacity to embed this newly formed water into structured hydrogen-bonded layers, enabling stabilization of the nanodomain and transfer of coherence beyond the immediate redox pairing site. Through this integration, photonic coherence associated with PAQ is transmitted via hydration shells to adjacent proteins, membranes, and macromolecular assemblies, reinforcing organized redox activity across spatial scales. Disruption of hydration shell formation or persistence prevents stabilization and propagation of coherence, feeds back negatively on Coherence Interface integrity, and compromises sustained redox organization [19,21,28,29].
When all four conditions are satisfied, the CI supports HOMO-LUMO coupling, PAQ generation and absorption, and the emergence of a directed flux of metabolic water, maintaining redox coherence as an emergent system state. Failure of any single condition destabilizes the CI and initiates the transition toward redox decoherence. Notably, such transitions may occur without measurable changes in bulk oxidant or antioxidant levels, highlighting the limitations of scalar redox descriptors [5,12,30].
4.2. Failure Modes of the Coherence Interface
Failure of the Coherence Interface (CI) occurs when the convergence of oxidative excitation flux and reductive electron flux can no longer be resolved into a unified flux of metabolic water. Such failure does not require an increase in oxidant production or a depletion of antioxidants per se; rather, it reflects disruption of one or more of the dynamical conditions that sustain CI integrity. CI failure therefore represents a loss of coupling, not simply a change in redox magnitude [5,9,32].
The principal failure modes of the CI can be classified according to which integrity condition is violated.
I. Nanospatial Failure (Loss of Confinement)
Nanospatial failure may occur through membrane disruption, loss of macromolecular organization, altered lipid composition, or breakdown of protein assemblies that normally define nanodomain boundaries. When nanospatial failure arises, oxidative excitation escapes the nanodomain in which reductive assimilation is available. In this mode, reactive intermediates and hydroxyl radicals diffuse beyond the effective pairing radius of reduced glutathione before HOMO–LUMO coupling can occur. As a result, oxidative excitation is resolved through nonspecific substrate oxidation rather than through paired redox chemistry.
II. Temporal Failure (Desynchronization of Fluxes)
Temporal failure occurs when the timing of oxidative excitation and reductive availability becomes misaligned. Even when sufficient reductive capacity exists in bulk, delays in the delivery or regeneration of reduced glutathione at the CI prevent immediate pairing with hydroxyl radicals. Temporal desynchronization abolishes coherent electronic relaxation, suppresses PAQ formation and absorption, and diverts excitation into unpaired reactions. This failure mode highlights that redox coherence depends on when reductive power arrives, not only on how much is present.
III. Energetic Failure (Insufficient Reductive Potential)
Energetic failure reflects inadequate electronic energy within the reductive flux, such that reduced glutathione lacks sufficient HOMO energy to neutralize oxidative excitation. This condition arises when NADPH regeneration is compromised, glutathione reductase activity is impaired, or pentose phosphate pathway flux is insufficient. In this mode, pairing attempts may occur but fail to complete effective HOMO–LUMO coupling, resulting in partial neutralization, aberrant photon emission, or diversion of excitation into decoherent pathways.
IV. Hydrational Failure (Loss of Structured Hydration Continuity)
Hydrational failure occurs when water generated at the CI cannot be retained or incorporated into structured hydration shells. Disruption of hydration continuity destabilizes nanodomain architecture and feeds back negatively on spatial confinement and temporal synchronization. In this mode, even successful redox pairing events fail to reinforce CI structure, leading to progressive loss of coherence. Hydrational failure therefore represents a secondary route to CI collapse that can become self-reinforcing under sustained perturbation. [19,21,27,28].
These failure modes are not mutually exclusive; CI breakdown often involves coupled violations, in which an initial perturbation in one condition propagates to others. For example, energetic failure may lead to incomplete pairing, reducing coherent water formation and precipitating hydrational failure, which in turn exacerbates nanospatial diffusion [33,34].
Importantly, all CI failure modes share a common outcome: the inability to resolve converging redox fluxes into coherent water and structured energy outputs. When this resolution fails, metabolic water production becomes diffuse and ultraweak photon emissions lose structure, giving way to decoherent photon emissions. This transition marks the emergence of redox decoherence as a system-level state [47,48,49,50].
By framing CI breakdown in terms of discrete failure modes rather than global oxidative stress, this model explains how biological systems can enter decoherent redox regimes under conditions of unchanged or even elevated antioxidant capacity. CI failure thus provides a mechanistic bridge between microscopic redox disorganization and macroscopic functional decline.
4.3. Transition from Redox Coherence to Redox Decoherence
The transition from redox coherence to redox decoherence represents a qualitative change in system state rather than a gradual escalation of oxidative burden. Within the framework of the Redox Photonic Coupling System, this transition is governed by the loss of Coherence Interface (CI) integrity, which marks the failure to resolve converging redox fluxes into metabolic water and structured photonic outputs.
In a redox-coherent state, oxidative excitation flux and reductive electron flux converge at the CI and are transduced into a unified flux of coherent water, accompanied by the generation and immediate absorption of Photonic Activation Quanta (PAQ). This coupled resolution stabilizes nanodomain organization, sustains hydration-shell structure, and maintains dynamic alignment between oxidative and reductive processes. Redox coherence is therefore self-reinforcing: successful pairing events actively preserve the conditions required for subsequent pairing [19,21].
Redox decoherence emerges when CI failure modes described in Section 4.2 disrupt this resolution. Violation of any CI integrity conditions progressively impairs effective HOMO–LUMO coupling between oxidative excitation and reductive capacity. As a result, PAQ generation collapses and is replaced by Decoherent Photon Emissions (DPE), while metabolic water formation becomes spatially diffuse and structurally unorganized. The system thus transitions from a state characterized by structured hydration-mediated organization to one dominated by dissipative redox chemistry.
Once initiated, redox decoherence tends to propagate. Diffuse water formation fails to reinforce hydration shells, destabilizing nanodomain architecture and further impairing CI confinement. DPE-dominated energy release lacks the spatial and temporal structure necessary to restore alignment between fluxes. These effects form a positive feedback loop in which loss of coherence begets further decoherence, even in the absence of increased oxidative input [33,34,35].
The coherence–decoherence transition can therefore be understood as a state boundary defined by CI functionality. On one side of this boundary, redox chemistry is organized, paired, and coherence-generating; on the other, redox activity persists but is unpaired, dissipative, and damaging. Recognizing this transition as a system-level state change provides a unifying explanation for disparate redox phenomena and sets the stage for distinguishing coherent and decoherent photonic outcomes in the following section.
5. Photonic Outcomes of Redox Organization
Photonic emissions are among the most sensitive manifestations of biological redox activity, yet their physiological meaning has remained poorly defined. Traditionally observed as ultraweak photon emission, these signals have often been interpreted as nonspecific byproducts of oxidative reactions or molecular damage. Within the framework developed here, photonic emissions are instead understood as state-dependent outcomes of redox organization, reflecting whether oxidative and reductive fluxes are coherently coupled at the Coherence Interface [47,48,55,56].
When oxidative excitation and reductive electron flux converge productively at an intact Coherence Interface, electronic excitation is resolved through orbital-level coupling, coherent photonic relaxation, and localized metabolic water formation. Under these conditions, photonic output is structured, spatially confined, and temporally synchronized, reflecting organized redox resolution. When Coherence Interface integrity is compromised, excitation is instead released through diffuse, unstructured photonic emissions associated with failed pairing and decoherent redox chemistry.
We therefore distinguish two qualitatively distinct photonic regimes: Photonic Activation Quanta (PAQ), which arise from redox coherence, and Decoherent Photon Emissions (DPE), which accompany redox decoherence. These regimes do not represent different intensities of the same phenomenon, but distinct modes of energy resolution corresponding to different organizational states of the redox system. Photonic emissions thus provide a direct, noninvasive readout of redox state-space, linking electronic structure, flux coupling, and hydration organization.
5.1. Photonic Activation Quanta (PAQ)
Photonic Activation Quanta (PAQ) are defined as coherent photonic entities generated during successful redox coupling at the Coherence Interface. PAQs arise from quantized electronic relaxation events associated with HOMO-LUMO transitions that occur when oxidative excitation carried by hydroxyl radicals is productively paired with reductive electrons delivered by reduced glutathione. In contrast to diffuse ultraweak photon emissions, PAQs possess defined spatial and temporal properties, reflecting their origin within nanodomain-confined, synchronized redox events. In this regime, HOMO-LUMO coupling proceeds with sufficient coherence that the resulting photonic emission is matched to the acceptance modes of newly formed metabolic water, enabling immediate absorption and localized retention.
Within the Redox Photonic Coupling System, PAQs are not passive byproducts of chemistry but active coherence vectors. They emerge only when the conditions required for Coherence Interface integrity-nanospatial confinement, temporal synchronization, energetic adequacy, and hydrational continuity- are simultaneously satisfied and therefore constitute a direct manifestation of redox coherence as a system state. The absence of PAQ generation signals failure of these conditions and marks the transition toward redox decoherence.
A defining feature of PAQs is their immediate absorption by metabolic water molecules formed in situ during hydroxyl radical neutralization. Owing to nanometer-scale proximity and temporal synchrony with redox pairing, newly generated water molecules act as primary absorbers of PAQs, coupling photonic coherence directly into the hydrogen-bonding network of water. Through subsequent incorporation into hydration shells of nearby proteins, membranes, and macromolecular assemblies, PAQ-conditioned water propagates coherence beyond the immediate site of redox coupling.
Taken together, PAQs define the coherent photonic regime of biological redox chemistry. Their generation, absorption, and downstream effects distinguish redox coherence from redox decoherence and provide a principled basis for differentiating organized redox physiology from dissipative redox pathology.
5.2. Decoherent Photon Emissions (DPE)
Decoherent Photon Emissions (DPE) arise when one or more integrity conditions of the Coherence Interface (CI) are not satisfied, preventing the successful resolution of redox-derived excitation into stable, propagating metabolic water network. In contrast to Photonic Activation Quanta (PAQ), DPE do not signify a simple absence of redox pairing or HOMO-LUMO coupling per se. Rather, they reflect condition-specific failures in the stabilization, coordination, or propagation of coherence following photonic emission.
Because CI integrity depends on multiple jointly necessary conditions, the physical meaning and biological consequences of DPE depend on which condition is violated.
When nanospatial confinement is lost, oxidative excitation escapes the effective pairing radius of reduced glutathione. Hydroxyl radicals then react indiscriminately with nearby molecular substrates rather than being resolved within the CI. Under these conditions, photonic emissions arise from fragmented electronic relaxation during random substrate oxidation, producing DPE that are spatially diffuse and temporally unsynchronized. This mode of failure is typically associated with widespread oxidative damage, absence of metabolic water formation at the CI, and dissipation of energy through unstructured photon emission and heat.
When temporal synchronization between oxidative excitation and reductive availability is disrupted, redox pairing is delayed, fragmented, or diverted into alternative relaxation pathways. In such cases, electronic excitation may relax before or outside the pairing event, producing diffuse photonic emissions that are not synchronized with metabolic water formation; these emissions constitute Decoherent Photon Emissions.
Energetic adequacy failure occurs when reductive carriers are present but lack the appropriate electronic energy state required for coherent redox resolution. In this condition, reduced glutathione may be chemically reduced and abundant yet insufficiently energized to support effective orbital-level coupling with oxidative excitation. Within the Redox Photonic Coupling System, energetic adequacy depends not on glutathione concentration alone but on the energetic provenance of the electrons it carries, which under physiological conditions are supplied via NADPH. When this upstream energetic chain is compromised, glutathione may retain reducing capacity while lacking the energetic load required for structured excitation resolution. This failure mode may also arise when reductive capacity is supplied exogenously, without restoration of endogenous NADPH-generating metabolism, or when upstream substrates fail to sustain effective energetic loading. Under these conditions, redox pairing and nominal HOMO-LUMO coupling may still occur, but the resulting relaxation lacks sufficient coherence to stabilize and propagate organized outcomes.
When hydrational continuity is compromised, photon absorption by newly formed water molecules may still occur, but coherence cannot be stabilized or propagated beyond the immediate absorption event. The surrounding molecular environment lacks a sufficiently structured hydrogen-bond network to embed and transmit the excitation. As a result, absorbed energy rapidly thermalizes rather than reinforcing nanodomain organization, and DPE dominate the observable photonic output despite local photon absorption.
In biological systems, violations of CI integrity are often partial, transient, or spatially heterogeneous across nanodomains. Under such boundary conditions, PAQ and DPE may coexist, with their relative prevalence reflecting the degree and nature of CI impairment. In this regime, the PAQ:DPE ratio functions as a continuous, state-sensitive measure of redox organization, indicating whether the system is biased toward coherence preservation or toward progressive decoherence.
5.3. Photonic Activation Quanta and Decoherent Photon Emissions as Coordinates of Redox State
Photonic Activation Quanta (PAQ) and Decoherent Photon Emissions (DPE) define two qualitatively distinct modes of photonic energy resolution in biological redox chemistry that arise from differences in organizational integrity at the Coherence Interface (CI), rather than from differences in redox magnitude. Together, they define a photonic state-space reflecting the degree to which oxidative and reductive fluxes are coherently coupled and resolved.
Physiologically, optimal redox function corresponds to PAQ dominance. Transient DPE generation may accompany acute perturbations, but sustained DPE dominance indicates loss of CI integrity and drift toward redox decoherence. Redox resilience is therefore defined as the capacity to rapidly re-establish PAQ-mediated resolution following transient DPE emergence. The PAQ:DPE ratio thus provides both a snapshot of redox organization and a dynamic measure of proximity to, and recoverability from, decoherent states.
6. Redox Resilience as a Biological Control Property
Biological systems are continuously exposed to fluctuations in redox demand arising from metabolism, signaling, and environmental stressors [1]. Under physiological conditions, such perturbations do not destabilize redox organization but are actively absorbed and resolved through redox resilience - the capacity of the system to rapidly restore PAQ-dominant resolution following transient challenges. In this regime, temporary excursions into decoherent photonic output may occur, but they do not persist, and Coherence Interface integrity is promptly re-established.
Redox resilience therefore represents a control property of biological redox systems, reflecting their ability to maintain organized energy resolution, coherent water formation, and hydration-mediated structure despite dynamic stress. Rather than being defined by static redox balance, resilience is expressed through recovery dynamics, determining whether perturbations are contained or allowed to propagate into sustained organizational failure.
In this section, we describe the general features of redox resilience and the conditions that support rapid restoration of PAQ-dominant states, setting the stage for understanding how loss of resilience leads to redox decoherence.
6.1. General Features of Redox Resilience
Redox resilience is the capacity of a biological system to maintain and rapidly restore organized redox resolution in the face of transient perturbations. It is expressed through specific, identifiable system properties rather than through static redox balance. The principal features of redox resilience are as follows:
I. Rapid Recovery of PAQ-Dominant Resolution
A defining feature of redox resilience is the ability to rapidly re-establish PAQ-dominant photonic output following transient DPE generation. Brief DPE emergence reflects momentary challenges to one or more Coherence Interface (CI) integrity conditions and does not, by itself, signify loss of organizational control. Resilience is expressed by the system’s capacity to terminate DPE-generating modes and restore coherent photonic resolution before decoherent processes propagate.
II. Energetically Efficient Resolution of Excitation
Resilient redox systems preferentially resolve electronic excitation through metabolic water formation and structured photonic relaxation, rather than through non-radiative thermal dissipation [50]. By embedding excitation into hydration-mediated organization, resilient states minimize energy loss as heat and preserve hydration shell architecture. This energetic efficiency supports sustained redox signaling and metabolic function without progressive structural erosion.
III. Structural Robustness of the Coherence Interface
Redox resilience depends on the structural robustness of the Coherence Interface under fluctuating redox demand. Nanospatial confinement, temporal synchronization, energetic adequacy, and hydrational continuity must remain sufficiently intact to permit rapid re-coupling following perturbation. Temporary stress to individual conditions may occur, but resilience reflects the system’s capacity to reconstitute CI integrity before decoherence becomes self-amplifying.
IV. Independence from Antioxidant Abundance
Redox resilience is independent of absolute antioxidant levels [12,16]. A system may possess substantial antioxidant capacity yet exhibit poor resilience if CI integrity is not rapidly restored following perturbation. Conversely, resilient systems tolerate transient oxidative excursions without accumulating damage, demonstrating that recovery dynamics and organizational control, not redox magnitude, govern stability.
Together, these features define redox resilience as the capacity of a biological system to remain organized, energy-efficient, and functionally stable despite ongoing perturbation. Transient deviations from coherence are absorbed and corrected, allowing PAQ-dominant resolution and coherent water organization to be re-established without lasting disruption. Viewed in this light, resilience emerges as the central control property that preserves physiological redox organization and delays the onset of sustained decoherence.
7. Redox Decoherence as Loss of Redox Resilience
Redox decoherence represents the organizational state that emerges when redox resilience is lost. Rather than reflecting transient deviations from optimal redox resolution, decoherence arises when recovery fails, when excursions into dissipative modes persist and progressively reshape system behavior. Under these conditions, redox activity continues, but its outcomes shift away from organized energy resolution toward energetic inefficiency, structural erosion, and loss of functional stability.
Importantly, redox decoherence does not require extreme oxidant excess, antioxidant depletion, or catastrophic metabolic failure. Instead, it emerges when the system can no longer reliably re-establish PAQ-dominant resolution and coherent water organization following perturbation. Loss of redox resilience therefore transforms redox chemistry from an adaptive, self-correcting process into a source of cumulative disorder [1,5].
7.1. General Features of Redox Decoherence
Redox decoherence is characterized by a reproducible set of organizational features that reflect sustained loss of recovery capacity rather than acute redox stress. Once redox resilience is compromised, these features become interdependent and self-reinforcing.
A defining hallmark of decoherence is persistent dominance of Decoherent Photon Emissions (DPE). In contrast to resilient systems, where DPE emergence is brief and reversible, decoherent systems fail to restore PAQ-dominant photonic output after perturbation. Persistent DPE dominance is consistent with impaired recovery at the Coherence Interface rather than ongoing acute stress.
As recovery capacity erodes, redox-derived excitation is increasingly resolved through unstructured photon emission and non-radiative dissipation as heat, rather than through coherent water-mediated organization. This shift marks a transition toward dissipative energy resolution, reducing energetic efficiency and further weakening the mechanisms required for recovery.
Loss of redox resilience is also accompanied by progressive destabilization of hydration-shell architecture, which serves as the physical substrate for coherence propagation. Even when redox pairing and photon absorption occur, absorbed excitation cannot be stabilized or transmitted if hydration structure is impaired. This directly limits the system’s capacity to regain coherence following perturbation.
These processes form a self-amplifying loop. Disrupted hydration impairs nanospatial confinement, temporal synchronization, and energetic coordination at the Coherence Interface, further reducing resilience. Each failed recovery episode increases the likelihood of subsequent DPE dominance, progressively locking the system into a decoherent regime with diminishing probability of spontaneous restoration.
Viewed in this light, oxidative and reductive stress are not primary causal states but directional expressions of an underlying decoherent redox organization. When oxidative excitation dominates under impaired Coherence Interface integrity, excitation is dissipated through diffuse photonic emission, heat, and substrate oxidation, producing oxidative stress phenotypes [1,11,14,35]. Conversely, when reductive capacity dominates while coupling discipline is compromised, excitation dynamics are progressively suppressed despite preserved reductive resources, producing reductive stress phenotypes [17,18,32]. Although mechanistically opposite, both outcomes arise from the same failure of organized redox resolution and reflect surface manifestations of redox decoherence rather than independent pathological drivers [5,12,16].
7.2. Conceptual and Translational Implications of Redox Decoherence
Framing redox decoherence as an underlying organizational state rather than as a consequence of oxidant and reductant imbalances has several important implications.
First, it reframes redox pathology as a failure of coupling, synchronization, and recovery, rather than as excess or deficiency along a single redox axis. Oxidative and reductive stress become directional phenotypes of decoherence, shaped by which axis dominates and which Coherence Interface conditions are violated. This perspective resolves long-standing inconsistencies in literature where similar redox burdens produce divergent functional outcomes.
Second, it explains redox paradoxes in which biological dysfunction occurs despite low oxidant burden or high reductive capacity. It clarifies why antioxidant or pro-reductive interventions often fail when Coherence Interface integrity and recovery dynamics are not restored, allowing redox activity to remain high even as biological organization deteriorates [12,15,18].
Third, it positions redox organization as a dynamic and recoverable state variable, governed by recovery kinetics rather than static measurements. In this framework, recurrence, persistence, and restoration of coherent coupling, rather than instantaneous redox levels, determine whether systems remain resilient or drift toward decoherence. The PAQ:DPE ratio therefore emerges as a dynamic indicator of redox state and resilience.
Finally, this framework underscores the limitations of scalar redox biomarkers used in isolation and motivates measurement strategies sensitive to organizational state, coupling integrity, and photonic resolution mode [32]. While the upstream drivers that bias Coherence Interface desynchronization-genetic, metabolic, environmental, infectious, or age-related- are not addressed here, the present work establishes a unifying organizational principle within which such drivers can be investigated without conflating cause and manifestation.
8. Testable Predictions and Experimental Approaches
Although the Redox Photonic Coupling System (RPCS) framework is primarily theoretical, it is explicitly designed to be empirically falsifiable through measurable proxies of redox organization, photonic output, and recovery dynamics. This section outlines testable predictions derived from the concepts of redox coherence, decoherence, and resilience, using established experimental approaches, such as localized ROS imaging, ultraweak photon emission (UPE) detection, and hydration shell analysis, to probe the framework’s core claims. These predictions aim to distinguish coherent from decoherent redox states without requiring direct measurement of Photonic Activation Quanta (PAQ) or Decoherent Photon Emissions (DPE), which remain technically inaccessible for now.
8.1. Predictions Related to Redox Coherence and Decoherence
Hypothesis 1.
Under experimentally matched oxidative excitation loads, redox-coherent systems will preserve cellular function and exhibit structured photonic output, whereas redox-decoherent systems will display diffuse photonic emissions and molecular damage.
Rationale: Redox coherence is independent of magnitude; coherent systems channel excitation into organized outcomes, while decoherent systems dissipate it.
Testable prediction: In resilient cells (e.g., young or stress-adapted fibroblasts), transient H2O2 elevations induced by mitochondrial stressors will increase structured photonic emission proxies without inducing lipid or DNA damage. In decoherent cells (e.g., senescent or neurodegenerative models), comparable excitation will shift photonic output toward diffuse patterns accompanied by oxidative damage markers.
Methods: Combine compartment-specific ROS probes [29] with UPE detection [47,48] and correlate photonic structure with functional readouts such as mitochondrial membrane potential and cell viability.
Hypothesis 2.
Disruption of Coherence Interface (CI) integrity will convert PAQ-dominant regimes to DPE-dominant regimes without requiring changes in bulk oxidant or antioxidant levels.
Rationale: CI failure decouples oxidative and reductive fluxes even when scalar redox metrics remain balanced.
Testable prediction: Perturbations that increase glutathione recycling demand or impair reductive regeneration will shift photonic output toward DPE proxies and destabilize hydration-dependent organization, despite preserved global redox potential.
8.2. Predictions Related to Redox Resilience
Hypothesis 3.
Redox-resilient systems will rapidly re-establish coherent photonic output following perturbation, whereas non-resilient systems will exhibit persistent decoherent emissions.
Rationale: Resilience is defined by recovery kinetics rather than static redox balance.
Testable prediction: In preconditioned or hormetic models [4], acute oxidative challenges will produce transient decoherent signatures followed by rapid restoration of structured photonic output and function. In NADPH or GSH compromised systems, the same challenges will result in sustained DPE dominance and progressive dysfunction.
8.3. Predictions Linking Photonic Output and Hydration Organization
Hypothesis 4.
Coherent redox resolution is predicted to enhance structured hydration environments, enabling organizational effects that extend beyond the immediate redox pairing site.
Rationale: Structured photonic relaxation is proposed to stabilize hydration shell organization, linking redox coherence to macromolecular dynamics.
Testable prediction: In coherent regimes, redox pairing will correlate with enhanced hydration shell stability and improved enzymatic or membrane function, whereas decoherent regimes will show diminished hydration organization and reduced functional efficiency.
Methods: Combine UPE measurements [47,48] with terahertz time-domain spectroscopy or dielectric relaxation spectroscopy to quantify water dynamics around proteins and membranes [21,28] and functional assays of redox-sensitive proteins.
Together, these predictions provide a falsifiable experimental roadmap for evaluating redox coherence, decoherence, and resilience using existing tools. Positive or negative outcomes can directly support, refine, or challenge specific elements of the framework, enabling redox biology to be evaluated as an organizational and state-dependent process rather than solely through scalar redox measures.
Conclusions
In this work, we present a framework that redefines biological redox chemistry at the level of system organization, introducing coherence, coupling discipline, and recovery capacity as primary descriptors rather than redox magnitude alone. We identify redox coherence and redox decoherence as emergent system states arising from the integrity of a Redox Photonic Coupling System (RPCS), in which oxidative excitation and reductive assimilation axes must remain spatiotemporally synchronized and supported by structured hydration at a Coherence Interface to sustain organized redox resolution.
Within this framework, redox resilience emerges as a system-level control property of physiological redox activity, determining whether perturbations resolve coherently or progress toward persistent organizational degradation. Erosion of redox resilience gives rise to redox decoherence, an underlying organizational state from which oxidative- or reductive-stress phenotypes may emerge depending on axis dominance and the specific mode of Coherence Interface desynchronization. Oxidative and reductive stress are therefore repositioned as context-dependent expressions of decoherence rather than as primary causal drivers.
Photonic behavior functions as an integrated readout of redox organization rather than an independent outcome. Coherent redox resolution is accompanied by structured photonic relaxation embedded within hydration-mediated architecture, whereas impaired synchronization or recovery is associated with dissipative photonic release. Accordingly, the relative prevalence of Photonic Activation Quanta (PAQ) versus Decoherent Photon Emissions (DPE) reflects the system’s organizational state and resilience, not its absolute oxidant or reductant burden.
Although direct measurement of Photonic Activation Quanta (PAQ) and Decoherent Photon Emissions (DPE) is not yet feasible, the framework advances testable predictions by linking established experimental proxies of redox organization to underlying state and recovery dynamics.
By framing redox biology as a state-dependent process governed by spatiotemporal organization and recovery dynamics, this work offers a unifying principle for interpreting redox physiology and pathology across diverse biological contexts. While the upstream determinants that bias systems toward loss of coherence are beyond the scope of this study, the framework establishes a common organizational language within which heterogeneous mechanisms can be examined without confusing primary organization with downstream manifestation. Together, these concepts provide a new foundation for future experimental and theoretical efforts aimed at understanding how redox organization is maintained, lost, and potentially restored.
Authorship and Terminology Declaration
Conceptual Authorship
The conceptual framework presented in this manuscript, including the definitions of Redox Coherence, Redox Decoherence, Redox Resilience, the Redox Photonic Coupling System (RPCS), the Oxidative Excitation Axis, the Reductive Assimilation Axis, the Coherence Interface, Photonic Activation Quanta (PAQ), Decoherent Photon Emissions (DPE), PAQ;DPE ratio and Hydrational Continuity was developed by the author as an original theoretical construct. These concepts do not represent reinterpretations or rebranding of existing models, but a novel organizational framework intended to unify disparate observations in redox biology under a state-dependent, systems-level perspective.
While individual molecular components referenced in this work (e.g., reactive oxygen species, glutathione, NADPH, hydration shells, photon emission) are well established in the literature, their integration into a coherent organizational model centered on coupling discipline, photonic outcomes, and recovery dynamics is original to this manuscript.
Terminology and Definitions
Several terms introduced herein are newly defined or used in a formally novel way. These include, but are not limited to:
- Redox Coherence: An emergent organizational state of biological redox activity in which oxidative and reductive processes are dynamically coupled, spatially constrained, temporally synchronized, and maintained across a continued hydration network, enabling paired redox resolution and organized energy handling.
- Redox Decoherence: An emergent organizational state of biological redox activity defined by loss of organizational constraints, including oxidative-reductive coupling, spatial confinement, temporal synchronization, and hydrational continuity, with redox energy resolved primarily through dissipation and increased damage to biological substrates.
- Redox Photonic Coupling System (RPCS): A distributed redox architecture that coordinates the generation, delivery, and spatiotemporal synchronization of oxidative excitation and reductive assimilation fluxes across biological systems, supported by continuous hydration networks that enable coherent energy resolution.
- Oxidative Excitation Axis (OEA): A functional redox axis within the Redox Photonic Coupling System responsible for delivering localized oxidative excitation to the Coherence Interface. Its deliverables are electronically excited oxidative species, exemplified by hydroxyl radicals, that carry excitation requiring immediate paired resolution.
- Reductive Assimilation Axis (RAA): A functional redox axis within the Redox Photonic Coupling System responsible for delivering energetically competent reductive capacity to the Coherence Interface. Its deliverables are reducing electrons and hydrogen donors, exemplified by reduced glutathione, capable of resolving oxidative excitation through paired, orbital-level coupling.
- Coherence Interface (CI): A nanodomain control interface within the Redox Photonic Coupling System where oxidative and reductive fluxes converge and where redox-derived excitation is resolved coherently or dissipatively according to local coupling conditions.
- Photonic Activation Quanta (PAQ): Structured photonic outputs generated during coherent redox resolution at the Coherence Interface, reflecting successful orbital-level coupling and subsequent transfer of excitation into organized, hydration-mediated structures.
- Decoherent Photon Emissions (DPE): Diffuse photonic outputs whose energy is dissipated rather than embedded into coherent water-mediated organization, arising when redox-derived excitation is not coherently resolved and reflecting failures in coupling, stabilization, or propagation of excitation following redox reactions.
- PAQ:DPE ratio: A relative measure expressing the balance between photonic signatures associated with coherent redox resolution (PAQ) and those associated with dissipative redox resolution (DPE), used as an organizational indicator of redox state.
- Redox Resilience: The capacity of a biological system to absorb perturbations and re-establish coherent redox resolution, restoring organized coupling at the Coherence Interface rather than allowing persistent dissipative behavior.
- Hydrational Continuity: The ability of the metabolic water formed during redox coupling to integrate into contiguous hydration shell networks, enabling redox-derived signals to propagate into adjacent proteins, membranes, and macromolecular assemblies through structured hydrogen-bond networks.
These terms are explicitly defined within the manuscript and should be interpreted according to those definitions. Their usage reflects intentional conceptual development rather than stylistic variation.
Acknowledgments
The author gratefully acknowledges his wife, Michaela Neatu, for her patience, intellectual openness, and sustained support throughout the development of this work. As an independent researcher, the author relied on continuous dialogue to test and refine emerging concepts, and her engagement, questions, and feedback were instrumental in clarifying assumptions, strengthening the framework, and improving communication. This iterative exchange played a significant role in advancing the ideas presented here.
Use of Language-Assistance Tools
Language-assistance tools were used for editing and clarity. All scientific content and interpretations are the responsibility of the author.
References
- Sies, H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Halliwell, B; Gutteridge, JMC. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, 2015. [Google Scholar]
- Forman, HJ; Maiorino, M; Ursini, F. Signaling functions of reactive oxygen species. Am J Physiol Cell Physiol. 2010, 298(2), C407–C429. [Google Scholar] [CrossRef]
- Ristow, M; Schmeisser, K. Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Nat Rev Mol Cell Biol. 2014, 15(5), 365–371. [Google Scholar] [CrossRef]
- Jones, DP. Redefining oxidative stress. Antioxid Redox Signal. 2006, 8(9-10), 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008, 4(5), 278–286. [Google Scholar] [CrossRef] [PubMed]
- Murphy, MP. How mitochondria produce reactive oxygen species. Biochem J 2009, 417(1), 1–13. [Google Scholar] [CrossRef] [PubMed]
- Meister, A; Anderson, ME. Glutathione. Annu Rev Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Jones, DP. Redox sensing and signaling by hydrogen peroxide in mammalian cells. Free Radic Biol Med. 2010, 48(5), 507–515. [Google Scholar] [CrossRef]
- Lu, SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009, 30(1-2), 42–59. [Google Scholar] [CrossRef]
- Sohal, RS; Forster, MJ. Caloric restriction and the aging process: a critique. Free Radic Biol Med. 2014, 73, 366–382. [Google Scholar] [CrossRef]
- Poljsak, B; Šuput, D; Milisav, I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Sies, H; Jones, DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020, 21(7), 363–383. [Google Scholar] [CrossRef]
- Betteridge, DJ. What is oxidative stress? Metabolism 2000, 49((2) Suppl 1, 3–8. [Google Scholar] [CrossRef]
- Bjelakovic, G; Nikolova, D; Gluud, LL; Simonetti, RG; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 2007, 297(8), 842–857. [Google Scholar] [CrossRef]
- Ristow, M; Zarse, K; Oberbach, A; et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 2009, 106(21), 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, NS; Connell, P; Christians, ES; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007, 130(3), 427–439. [Google Scholar] [CrossRef] [PubMed]
- Brewer, AC; Mustafi, SB; Murray, TV; Rajasekaran, NS; Benjamin, IJ. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid Redox Signal. 2013, 18(9), 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Jones, DP; Go, YM. Redox compartmentalization in eukaryotic cells. Antioxid Redox Signal. 2010, 12(10), 1273–1290. [Google Scholar] [CrossRef]
- Sies, H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014, 289(13), 8735–8741. [Google Scholar] [CrossRef]
- Chen, YR; Zweier, JL. Mitochondrial ROS microdomains in cardiac mitochondria. Circ Res. 2014, 114(3), 524–537. [Google Scholar] [CrossRef]
- Winterbourn, CC; Hampton, MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008, 45(5), 549–561. [Google Scholar] [CrossRef]
- Go, YM; Jones, DP. Redox theory of aging: implications for health and disease. Antioxid Redox Signal. 2017, 27(6), 359–379. [Google Scholar] [CrossRef] [PubMed]
- Holmström, KM; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signaling. Nat Rev Mol Cell Biol. 2014, 15(6), 411–421. [Google Scholar] [CrossRef] [PubMed]
- Scialò, F; Fernández-Ayala, DJ; Sanz, A. Control of mitochondrial ROS production in health and disease. Trends Endocrinol Metab. 2017, 28(6), 449–465. [Google Scholar] [CrossRef]
- Cogliati, S; Enriquez, JA; Scorrano, L. Mitochondrial cristae: where beauty meets functionality. Cell. 2016, 166(2), 430–444. [Google Scholar] [CrossRef]
- Antunes, F; Cadenas, E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000, 475(2), 121–126. [Google Scholar] [CrossRef]
- Go, YM; Jones, DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 2008, 1780(11), 1273–1290. [Google Scholar] [CrossRef]
- Morgan, B; Van Laer, K; Owusu, TN; et al. Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat Chem Biol. 2016, 12(6), 437–443. [Google Scholar] [CrossRef]
- Murphy, MP; Holmgren, A; Larsson, NG; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13(4), 361–366. [Google Scholar] [CrossRef]
- Kirkinezos, IG; Moraes, CT. Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol. 2001, 12(6), 449–457. [Google Scholar] [CrossRef]
- Jones, DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008, 295(4), C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Systems biology: a brief overview. Science 2002, 295(5560), 1662–1664. [Google Scholar] [CrossRef]
- Noble, D. A theory of biological relativity: no privileged level of causation. Interface Focus. 2012, 2(1), 55–64. [Google Scholar] [CrossRef] [PubMed]
- Balaban, RS; Nemoto, S; Finkel, T. Mitochondria, oxidants, and aging. Cell. 2005, 120(4), 483–495. [Google Scholar] [CrossRef] [PubMed]
- Jayathilaka, P; Pathiraja, G; Bandara, A; Subasinghe, N; Nanayakkara, N. Theoretical study of phenol and hydroxyl radical reaction mechanism in aqueous medium by the DFT/B3LYP/6-31+G(d,p)/CPCM model. Can J Chem. 2014, 92(9), 809–813. [Google Scholar] [CrossRef]
- Aguiar, ASN; et al. New insights on glutathione’s supramolecular arrangements and electronic structure. Molecules 2022, 27(22), 7958. [Google Scholar] [CrossRef]
- Chance, B; Sies, H; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979, 59(3), 527–605. [Google Scholar] [CrossRef]
- Quinlan, CL; Perevoshchikova, IV; Hey-Mogensen, M; Orr, AL; Brand, MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Winterbourn, CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008, 4(5), 278–286. [Google Scholar] [CrossRef]
- Buettner, GR; Jurkiewicz, BA. Chemistry and biochemistry of ascorbic acid. In Antioxidants in Disease Mechanisms and Therapy. Advances in Pharmacology; Sies, H, Ed.; Academic Press, 1996; Vol 38, pp. 91–115. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J Biol Chem. 1988, 263(33), 17205–17208. [Google Scholar] [CrossRef]
- Pollak, N; Dölle, C; Ziegler, M. The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochem J. 2007, 402(2), 205–218. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A; Prigione, A; Cramer, T; et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015, 90(3), 927–963. [Google Scholar] [CrossRef] [PubMed]
- Niggli, HJ. Ultraweak photons emitted by cells: biophotons. J Photochem Photobiol B 1992, 14(1-2), 144–146. [Google Scholar] [CrossRef] [PubMed]
- Popp, FA; Li, KH; Gu, Q (Eds.) Recent Advances in Biophoton Research and Its Applications; World Scientific: Singapore, 1992. [Google Scholar]
- Van Wijk, R; Van Aken, JM. Photon emission from living systems. In Recent Advances in Biophoton Research and Its Applications; Popp, FA, Li, KH, Gu, Q, Eds.; World Scientific: Singapore, 1992; pp. 95–113. [Google Scholar]
- Rastogi, A; Pospíšil, P. Ultra-weak photon emission as a non-invasive tool for monitoring of oxidative processes and stress tolerance in plants. J Photochem Photobiol B 2011, 105(1), 1–7. [Google Scholar] [CrossRef]
- Milonni, PW; Bochove, EJ; Cook, RJ. Quantum theory of spontaneous emission and excitation near a phase-conjugating mirror. J Opt Soc Am B 1989, 6(10), 1932–1941. [Google Scholar] [CrossRef]
- Lakowicz, JR. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Bianchini, BHS; Martelossi-Cebinelli, G; Carneiro, JA; et al. Superoxide anion generation, its pathological cellular and molecular roles and pharmacological targeting in inflammatory pain: lessons from the potassium superoxide model. Future Pharmacol. 2025, 5(4), 60. [Google Scholar] [CrossRef]
- Pollak, N; Dölle, C; Ziegler, M. The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochem J. 2007, 402(2), 205–218. [Google Scholar] [CrossRef]
- Jo, SH; Son, MH; Koh, HJ; et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001, 276(19), 16168–16176. [Google Scholar] [CrossRef]
- Nesci, S; Trombetti, F; Pagliarani, A. Nicotinamide nucleotide transhydrogenase as a sensor of mitochondrial biology. Trends Cell Biol. 2020, 30(1), 1–3. [Google Scholar] [CrossRef]
- Cifra, M; Pospíšil, P. Ultra-weak photon emission -a brief review. Frontiers in Physiology 2024, 15, 1348915. [Google Scholar] [CrossRef]
- Ren, Z; Yi, J; et al. The application and trend of ultra-weak photon emission in biology and medicine. Front Chem. 2023, 11, 1140128. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



