
Submitted:
27 January 2026
Posted:
28 January 2026
You are already at the latest version
Abstract
This study addresses the challenge of consistently transferring atomistic parameters of the C–C bond into phenomenological material characteristics within framework of continuum mechanics. Particular attention is given to determining the effective transverse diameter of the covalent C–C bond in carbon nanostructures. The dependence of this diameter on the Poisson’s ratio ν is examined, and the influence of the interatomic stiffness constants k_r,k_θ,and k_τ is systematically analyzed. Classical representative-volume models of the C–C bond based on the Euler–Bernoulli beam hypothesis violate thermodynamic stability conditions and lead to nonphysical Poisson’s ratio values exceeding 0.5, due to the neglect of shear deformation effects. To overcome this limitation, an approach based on Timoshenko beam theory is proposed, accounting for both bending and shear deformations. This approach enables estimation of energetically equivalent states between the phenomenological representative volume and the corresponding atomistic C–C bond model. As a result, a sixth-order algebraic equation is derived linking the effective bond diameter, the Poisson’s ratio, and the molecular mechanics force constants. Analysis of this equation reveals a narrow range of effective bond diameters and Poisson’s ratios for which thermodynamic stability conditions are satisfied. Within this range, physically consistent macroscopic material parameters can be directly expressed in terms of atomistic force constants.
Keywords:
carbon
; covalent bond
; molecular mechanics
; structural mechanics
; nanotubes
; Poisson’s ratio
; effective bond diameter
1. Introduction
Carbon nanostructures, such as nanotubes and graphene, possess unique mechanical properties — high stiffness, strength, and resistance to deformation. Their behavior at the nanoscale is typically described using molecular dynamics (MD) methods, which allow the explicit consideration of atomic interactions through empirical potentials, such as Morse or Lennard-Jones.
However, the direct application of MD models for describing large-scale systems is associated with significant computational costs, due to the need to simulate a representative volume of material containing a relatively small number of molecules or atoms, as well as the limitation to modeling processes over very short time intervals (from picoseconds to nanoseconds) [1]. This significantly complicates the macroscopic modeling of nanomodified materials and structures derived from them, and in some cases renders such modeling impossible.
In this context, there is a need to develop specialized multiscale models that allow for a consistent transition from atomistic mechanics parameters (bond stiffness under tension, bending, and torsion, potential parameters) to phenomenological parameters of solid mechanics (elastic modulus and Poisson’s ratio, shear modulus, plasticity, etc.). Such a transition requires the development of specialized models in which an individual chemical bond is interpreted as a mechanical element with angular, radial, and torsional stiffness. In this regard, the question arises regarding the correct choice of geometric parameters, in particular the effective diameter of the C–C chemical bond, which allows the reduction of the interatomic interaction Equations to the classical Equations of continuum mechanics.
The main difficulty lies in the fact that the formulas of structural mechanics include the Poisson’s ratio, whereas at the level of an individual chemical bond this parameter is not explicitly defined. As a result, traditional methods for determining the energetically equivalent mechanical properties of an effective rod depend on an arbitrarily chosen value of the Poisson’s ratio ν. Moreover, the direct application of classical beam theory Equations to carbon nanostructures frequently results in nonphysical values of the Poisson’s ratio, exceeding the upper limit of ν = 0.5 permitted for isotropic continuum materials.
Thus, a contradiction arises when directly transferring the molecular dynamics bond stiffnesses of the C–C bond into the classical beam theory Equations. From the well-known relations that energetically link the force constant of molecular mechanics with the physico-mechanical characteristics of a rod with a circular cross-section of diameter d within the straight-normal (Euler–Bernoulli) hypothesis [2] (, , and ), it follows that . After substituting the known values for the C–C covalent bond, we obtain , , and . At the same time, if the Poisson’s ratio ν is determined from the known relations for an isotropic body, as , we obtain , which is unphysical, since it violates the thermodynamic stability conditions for the continuum mechanics model of isotropic bodies (volumetric compression leads to a negative increment energy).
Thus, the use of classical formulas for tension, bending, and torsion of a beam to determine the effective physical parameters of the C–C covalent bond can lead to nonphysical values of the Poisson’s ratio, thereby highlighting the need to address the problem of a consistent and physically justified coupling between atomistic and continuum models.
In this work, we show that there exists a specific range of variation of the effective C–C bond diameter within which the conditions of thermodynamic stability are satisfied when transitioning from molecular-mechanics Equations to phenomenological Equations of solid mechanics. This eliminates the problem of obtaining nonphysical values of the Poisson’s ratio and allows the mean diameter within this range to be considered as a universal bond characteristic, ensuring a consistent transition from the atomistic description of the C–C bond to its continuum analogue.
2. Materials and Methods
2.1. State of the Art in Modeling the Effective Mechanical C–C Bond Properties
The carbon–carbon covalent (C–C) bond is a key element in the structure of carbon materials. Its electron density determines the spatial configuration, which in turn governs the mechanical and physical properties of nanostructures, including graphene, carbon nanotubes, and diamond-like materials. Research aimed at determining the macroscopic characteristics of C–C bonds from molecular mechanics parameters began to develop actively in the late 1980s and received particular attention after the discovery of carbon nanotubes [3]. The main focus was on establishing the relationship between empirical interatomic potentials and effective elastic constants.
To date, several key research directions have emerged in the literature, such as:
The determination of macroscopic mechanical properties of carbon nanostructures, such as carbon nanotubes and graphene, is directly related to the accurate description of the properties and geometric parameters of the C–C covalent bond. To date, one of the key challenges remains the transition from atomistic molecular mechanics parameters (bond stiffnesses, potential functions) to macroscopic continuum mechanics parameters (Young’s modulus, Poisson’s ratio, shear modulus).
However, the literature shows significant discrepancies in the values of the so-called “effective diameter” of the C–C bond, which lead to substantial variations in the calculated effective elastic modulus and Poisson’s ratio of representative C–C bond volumes. Early works [5,6,27], laid the foundation for the theory of interatomic interaction potentials describing atomic interactions.
Based on these studies, the REBO (Reactive Empirical Bond-Order) potentials [7] and their AIREBO [9] modifications were developed, which are widely used in molecular dynamics simulations [8]. These models allow the determination of microscopic bond stiffnesses; however, transitioning to macroscopic characteristics requires the introduction of additional geometric parameters, including the effective bond diameter. In particular, the effective values of Young’s modulus calculated based on REBO and AIREBO potential range from 0.9 to 1.3 TPa [28], while the Poisson’s ratios range from 0.15 to 0.45. At the same time, the use of structural-mechanical analogies sometimes leads to nonphysical values of ν > 0.5 [29,30,31].
Recent studies [30,32] show that when choosing an effective diameter d for the C–C bond in the range of 0.04 nm to 0.10 nm, significant discrepancies can arise in the calculation of the Poisson’s ratio ν. For example, when using d0.1 nm, the values of ν can exceed 0.5, which is unphysical from the perspective of structural mechanics, as it violates the condition of thermodynamic stability. With a more precise consideration of molecular-mechanical parameters and a refined value range of d ≈ 0.076 nm – 0.09 nm, the resulting values of ν return to the physically permissible range (0 < ν < 0.5).
Studies addressing this problem have shown that different potentials (REBO, AIREBO, TERSOFF, REAXFF) [4] yield significantly varying values of elastic moduli if the bond diameter correction is not taken into account [23]. Particular attention in the literature is paid to the fact that standard beam and bar theory Equations, when directly applied at the level of a covalent bond, can lead to unphysical results if a diameter value of around 0.1 nm is used without correction. Therefore, the commonly accepted bond diameter d = 0.147 nm should not be used without appropriate corrections [17,30].
Thus, the correct choice of the effective C–C bond diameter is central to achieving a consistent transition from molecular to structural mechanics.
2.2. Selection of calculation schemes for C-C bond
The method proposed in this work for determining the physics-mechanical characteristics of a C–C covalent bond between two carbon atoms consists of the following steps:
- Determination of the effective C–C bond diameter, taking shear deformation into account, using a refined Timoshenko beam model;
- Determination of the effective Poisson’s ratio;
- Determination of the effective Young’s modulus based on the Morse potential;
- Determination of the effective shear modulus of the C–C bond.
Let us consider each of these points separately.
Accounting for shear deformation according to Timoshenko in modeling the C–C covalent bond as a beam model is necessary due to the fundamental difference between the assumptions of classical Euler–Bernoulli bending theory and the physical reality of interatomic interactions at the nanoscale.
Classical Euler–Bernoulli beam theory is based on two key assumptions:
- Plane section hypothesis: It is assumed that the beam’s cross-sections remain plane and perpendicular to the neutral axis during bending.
- Neglect of transverse shear: This model assumes infinite material stiffness with respect to transverse shear ().
These assumptions work well only for “very long” and “very thin” beams, where the primary contribution to deformation comes from pure bending, and shear deformation is negligibly small. In the case of modeling an individual C–C covalent bond as a beam in a continuum model (for example, to calculate the elastic properties of carbon nanotubes), the C–C bond has a length of approximately 0.142 nm, and the effective “bond diameter,” determined according to the plane section hypothesis (), is not negligibly small compared to the bond length.
In this context, the C–C bond, according to the classification of beam theories, can be considered a geometrically “short” or “thick” beam. Therefore, the use of Euler–Bernoulli theory in this case leads to an artificially overestimated bond diameter and an underestimated bond elastic modulus as result of compensating for the neglected shear deformation. In addition, when real atomic structures (e.g. molecules or nanotubes) bend or vibrate, the angle between bonds can change significantly, which further necessitates to accounting for transverse shear deformation in the continuum model.
Therefore, in this work, the Timoshenko model is adopted as the fundamental hypothesis for the transition from the atomic model to the continuum, whose advantages can be summarized as follows:
- Timoshenko theory, by introducing the shear angle as an independent parameter, removes the assumption of cross-section perpendicularity to the neutral axis and accounts for both bending deformation and transverse shear deformation independently.
- The total potential deformation energy in the Timoshenko model includes not only the bending energy but also the shear energy. In this case, the Timoshenko model provides a more physically adequate representation of the behavior of continuum bond models when simulating the actual behavior of atoms and bonds, especially when analyzing local bending and high-frequency vibrations (when the wavelengths are comparable to interatomic distances).
- Furthermore, accounting for shear strain plays a significant role in the analysis of high-frequency vibrations (for example, when determining the natural modes and frequencies of vibrations of nanostructures).
In addition, the Timoshenko model allows for a more adequate description of local stiffness variations caused by defects (e.g., Stone–Wales defects) or changes in the orientation of C–C bonds, which is important for a more accurate determination of the strength characteristics of nanostructures.
Thus, accounting for shear according to Timoshenko is not strictly necessary for determining the intrinsic diameter of the C–C bond, but is required for the correct application of continuum models when calculating the mechanical and dynamic properties of systems composed of these bonds (e.g., carbon nanotubes), where classical simplified Euler–Bernoulli theories produce significant errors due to the considerable influence of transverse shear and can lead to unphysical results when obtaining effective physics-mechanical and geometric parameters.
2.3. C-C bond shear effect
The simplest method for accounting for the shear effect was proposed in [33], where a shear coefficient was introduced into the bending Equations of the Timoshenko beam.
There are several methods for determining the shear coefficient . In general, it should satisfy the following condition:
where is the transverse shear stress acting in the beam, is the cross-sectional area of the beam, is the shear modulus of the beam material, and is the rotation angle of the cross-section normal during deformation.
According to [34], the shear coefficient can be expressed by an Equation depending on the Poisson’s ratio and the geometric parameters of the beam’s cross-section as follows:
where is a harmonic function depending on the coordinates of the beam’s cross-section and the Poisson’s ratio [34], is the moment of inertia of the cross-section about the -axis, and is the moment of inertia of the cross-section about the -axis.
For a solid circular cross-section beam, the shear coefficient is equal to
and for a solid rectangular cross-section, it is equal to
In this case, it does not depend on the ratio of the beam’s width to its height [34].
The total steric potential energy of a diatomic C–C system is associated with the spatial arrangement of the atoms. For small linear elastic deformations, neglecting electrostatic interactions and the energy associated with nonbonded van der Waals forces, it can be expressed as [2,11]:
where:
— energy associated with bond tension;
— energy associated with bond angle change during bending;
— energy accounting for total torsion under the dihedral angle and out-of-plane torsion energy .
The force constants , and represent the bond’s resistance to tension, bending, and torsion, respectively. , correspond to the increments of longitudinal displacement under tension, angle changes during bending, and bond torsion, respectively. A schematic representation of the interatomic interactions is shown in Figure 1(a).
Following the procedure described in [2], we write the expressions for determining the potential energy of the beam in the cases of tension, bending, and torsion (Figure 1(b)):
where — Young’s modulus; — shear modulus; — cross-sectional area; — beam length; — moment of inertia; — polar moment of inertia of a circular cross-section beam; ∆L, α, and ∆β — increment of length, bending angle, and increment of torsion angle, respectively; — effective bond beam diameter, nm.
By comparing the molecular mechanics Equations with Equations (6)–(8) of structural mechanics, we obtain the well-known relations [2]:
from which it follows that
According to [37], the bending stiffness of the effective representative beam volume, accounting for the correction for the shear coefficient K in Equation (3), is given by [33]:
Taking into account relations (10), we obtain the expression for the shear deformation constant :
By equating the bending stiffness of the effective representative bond volume (11) to the molecular mechanics force constant for bond bending (), and substituting (14) and (10) into (11), we obtain the following relation, which establishes the connection between the effective diameter and the Poisson’s ratio through the molecular mechanics force constants:
After combining like terms in (15), we obtain a sixth-order algebraic Equation containing nonzero coefficients only for even powers of , or, in other words, a bicubic Equation with respect to the sought diameter , namely:
Similarly, we obtain the Equation for calculating the Poisson’s ratio :
In [33], based on the same methodology, an expression for was obtained, similar to expression (15), namely:
Equation (16), after a change of variables, is reduced to an algebraic cubic Equation. Solving it using Cardano’s method with the force constants corresponding to the C–C bond [2] (), we obtain the curve of the C–C bond diameter as a function of the Poisson’s ratio , shown in Figure 2.
Comparison of the obtained results with the data presented in [33] demonstrates their close agreement (Figure 2).
on the Poisson’s ratio .
3. Results
The results of the function in Equation (16), with the Poisson’s ratio varied in the range from 0 to 0.5, which corresponds to physically admissible values of this parameter for continuum materials, are shown in Figure 3 and Figure 4.
for different values of Poisson’s ratio in the range from 0 to 0.5.
depending on the Poisson’s ratio in the range from 0 to 0.5.
As can be seen from the graphs in Figure 3 and Figure 4, for Poisson’s ratio values within this range, the C–C bond diameter varies from 0.08393 nm at to 0.08474 nm at . The mean C–C bond diameter corresponds approximately to 0.0844 nm at a Poisson’s ratio of .
The obtained numerical range of for the C-C bond indicates that, when the Poisson’s ratio is confined within physically admissible limits for continuum media, the effective diameter of the C–C bond remains nearly constant and exhibits only a weak dependence on ν. From the standpoint of structural mechanics, such behavior is expected, because Poisson’s ratio primarily affects the redistribution of transverse shear stiffness, rather than the longitudinal stiffness of the representative volume.
In this context, the obtained diameter can be interpreted as the lower-bound estimate of the effective transverse cross-section of the bond, which ensures equivalence between the molecular bending stiffness and the bending stiffness of a representative continuum element modeled within the Timoshenko framework. In contrast to the widely accepted nominal diameter of = 0.147 nm, which is directly associated with the equilibrium spatial configuration of atomic orbitals, the present estimate represents a mechanically consistent characteristic derived strictly from energy-equivalence considerations. The presented correspondence between continuum and atomistic descriptions provides a fundamentally justified measure of the bond diameter, which prevents overestimation of flexibility under bending and avoids unphysical Poisson’s ratio values (e.g., ν > 0.5) that often arise when using simplified Euler–Bernoulli formulas.
Thus, for engineering calculations of nanostructures, such as carbon nanotubes, it is recommended to use the effective C–C bond diameter , which, according to Equation (17), corresponds to a Poisson’s ratio of . This value of may therefore serve as a reference geometric parameter in subsequent derivations of homogenized physical and mechanical characteristics for periodic carbon-containing nanostructures (CNT arrays, graphene sheets, graphite laminates) under both static deformation and dynamic loading, including the propagation of waves at characteristic nanoscale lengths.
Let us consider the physical meaning of these results.
As demonstrated in [21], most chemical bonding concepts can be adequately described using the Electron Localization Function (ELF). The distribution of electron density is a fundamental characteristic of a molecule, determining its physical and chemical properties. The effective overlap region of the C–C σ-bond can be evaluated either by performing Density Functional Theory (DFT) calculations [20] or from molecular visualization moiré patterns [21].
The work [21] presents studies on the ELF distribution for various molecules and notes that the central positions of C atoms within molecular structures act as invariant attractors. This indicates that the topology of structurally invariant attractors (the centroids of structural elements) is always preserved regardless of the chemical correlation between neighboring groups (changes in bond angles or unit cell dimensions), as long as the number of primary neighbors around each invariant attractor remains unchanged [21]. Based on this, assuming that the distance between two carbon atoms is 0.142 nm, and by appropriately scaling the data shown in Figure 5, the effective diameter of the C–C bond’s electron density cross-section is found to vary in the range of approximately 0.08 - 0.09 nm. These estimates of the effective transverse width of the C–C σ-bond overlap are in good agreement with the results obtained from DFT calculations, which lie in the range of approximately 0.07 -
0.09 nm [20], as well as with the results presented above.
molecule (extended Hückel method) [21].
ELF calculations and experimental data indicate that the electron density in the C–C σ-bond region is highly concentrated around the bond axis, and the “characteristic bond width,” where the density remains high—e.g., at least 95% of the maximum—is approximately 0.1 nm in diameter. These findings are corroborated by numerous studies on ELF analysis for graphene and diamond [22].
To describe the transverse distribution of electron density, a smooth normal distribution is used, which adequately captures the symmetry and shape of the σ-bond electron cloud. The adopted model allows for the analytical calculation of electron density fractions as a function of the distance from the bond axis.
Assuming a Gaussian distribution for the function along the radius perpendicular to the C–C bond axis, we can write:
where is the distance from the bond axis (transverse direction) in nm; describes the maximum density at the center, and nm is the effective width parameter of the distribution.
Based on Equation (19), a normalized curve of the Gaussian electron density distribution (ELF), , along the radius (perpendicular to the bond axis) is obtained, as shown in Figure 6.
To estimate the radius of the region containing a specified percentage of the electron density, an integral calculation of the cumulative density is performed, assuming a cylindrical geometry for the bond. Based on the chosen profile, the radii corresponding to 20%, 50%, and 90% of the cumulative electron density are determined. These radii uniquely define the transverse size of the region of maximum electron localization. A 2D map of the electron density distribution in the transverse section of the C–C bond, corresponding to the Gaussian distribution given by Equation (19), is shown in Figure 7. Calculations show that the transverse radius of the region containing from 90% to 99% of the electron density increases from 0.04 nm to 0.05 nm. The difference between these cumulative density levels reflects the exponential decay of electron density along the transverse radius of the bond.
Determining the transverse diameter of the bond’s electron cloud is crucial for refining the distribution of mechanical loads in nanostructures, as the thickness of the electron density influences the calculation of bending and torsional stiffnesses when these values are incorporated into multiscale models for an accurate description of the mechanical behavior of carbon materials. Analysis of the electron density distribution curve indicates that the effective radius of the C–C bond lies within the range , while the corresponding effective diameter falls within .
The obtained value of the average C–C bond diameter, according to Equation (16), , is fully consistent with these data. The deviation from the average diameter ranges from –0.514% to 0.43%, which lies within the accuracy limits of the molecular mechanics force constants , and the adopted shear coefficient .
Given the known average value of the C–C bond diameter, the effective Young’s modulus of the C–C bond can be determined based on the generalized Morse potential , which represents a semi-empirical approximation of the true interatomic interaction potential and is expressed as [14]:
where is the specific potential energy of the bond, is the bond dissociation energy between the atoms, is a parameter defining the curvature of the potential well, is the interatomic distance (nm), and is the equilibrium bond length between the atoms.
In the case of bond deformation along the longitudinal axis, the strain component is given by
and Equation (21) takes the form:
The physical, unnormalized components of the elastic strain tensor are determined according to the well-known relation [20] = , and the expression for calculating the elastic modulus is obtained after performing the normalization operation under the condition that :
The shear modulus of the representative volume of the C–C bond is determined according to the established relation:
The functional dependencies of the Young's modulus on the C–C bond diameter and the shear modulus on Poisson's ratio are shown in Figure 8 and Figure 9, respectively.
of the C–C bond.
4. Discussion
The obtained values of the transverse diameter of the C–C bond’s electron density localization region are crucial for refining the parameters of mechanical and multiscale models of carbon materials. In classical continuum models, which use the Timoshenko or Euler–Bernoulli beam approximations to describe the behavior of carbon nanostructures, the C–C bond is often treated as an element with equivalent mechanical properties, such as Young’s modulus, shear modulus, and geometric characteristics of its cross-section. However, this approach inherently assumes a simplified bond geometry and neglects the actual distribution of electron density. Studies conducted within quantum-mechanical frameworks, in particular using Density Functional Theory (DFT), show that the electron density of the σ C–C bond exhibits a spatial distribution resembling a Gaussian profile elongated along the interatomic axis. The isolines of this density display a smooth exponential decay from the bond center, as confirmed in [5]. Therefore, the actual cross-sectional area is determined not by atomic radii or tabulated parameters, but by the electron density distribution that governs the bond’s strength and stiffness. Consequently, defining an effective cross-sectional diameter corresponding to a specified fraction of the electron density is essential for the accurate interpretation of phenomenological elastic properties of the representative volume of diatomic systems.
Using the values obtained in this work,, corresponding to 90–99% of the bond’s electron density, ensures consistency between molecular-mechanical and continuum models. The relevance of accurately determining the geometric parameters of the C–C bond stems from their impact on the calculation of bending stiffness, which is highly sensitive to the moment of inertia I ∼ d4; on torsional stiffness, determined by the polar moment J ∼ d4; and on the relationship between bending and torsional modes. Accounting for these factors is crucial when modeling the behavior of carbon nanotubes and graphene fragments. In particular, studies [29], [36], [40] have shown that errors in the chosen bond diameter of even 10-20% can lead to significant deviations in predicted stiffness values, especially for small-diameter nanotubes. Table 1 presents the values of the effective C–C bond thickness, Young’s modulus, and Poisson’s ratio used in the calculations of single-walled carbon nanotubes.
Figure 10 presents a comparison of the C–C bond diameter–Poisson’s ratio correspondence obtained in this work with results reported by other authors [29,33,38,41].
The oisson’s ratio ranges from 0.149 to 0.34 (Δ = 56%), while the C–C bond diameter varies from 0.066 nm to 0.0896 nm (Δ = 26%). The relatively high discrepancies in these parameters lead to an even greater spread in the values obtained for the Young’s modulus (Δ = 61%) (Table 1). This highlights the need for further studies aimed at refining models of diatomic covalent interactions.
In conclusion, it should be noted that the values of the effective bond diameter obtained in this work correspond to the actual electron density distribution in diatomic systems and enable:
- the elimination of discrepancies between continuum and atomistic models;
- the reconciliation of the geometric and physical properties of the C–C bond with molecular mechanics parameters, ensuring compliance with the thermodynamic stability conditions of deformable solid mechanics;
- the correct and physically justified application of the effective diameter concept in the engineering analysis and calculation of the mechanical properties of carbon nanostructures.
5. Conclusions
In this study, the fundamental problem of correlating the atomic bonding parameters of the C–C bond with its mechanical properties at the continuum model level is addressed. The use of traditional hypotheses for the Euler–Bernoulli beam theory proves inapplicable when directly utilizing molecular mechanics data, since neglecting shear deformation leads to nonphysical values for the Poisson's ratio (ν.5).
To eliminate this inconsistency, an approach accounting for transverse shear based on the Timoshenko beam theory is proposed, providing an energetically consistent description of stretching, bending, and torsion of an individual C–C bond. By linking the molecular constants , , and to the structural parameters of an energetically equivalent representative-volume model, a sixth-order Equation for the effective bond diameter dependent on the Poisson’s ratio, is obtained.
The solutions of this Equation show that the diameter exhibits only a weak dependence on the Poisson’s ratio within its physically justified range of 0-0.5. Moreover, full compatibility between the atomistic and continuum descriptions of the two-atom C–C system is maintained across the entire range of these parameters. This range also corresponds to the conditions of thermodynamic stability, in which macroscopic system variables, such as temperature, pressure, volume, and entropy, undergo only small fluctuations around their equilibrium values when the system is isolated from the environment.
The proposed method therefore establishes a robust and physically consistent multiscale framework for modeling carbon nanostructures, such as carbon nanotubes and graphene, and provides a justified basis for determining effective geometric and elastic parameters of C–C bonds in continuum-based engineering analyses.
Author Contributions
Conceptualization, O.H. and S.A.; methodology, O.H. and S.A.; model validation, O.H.; formal analysis, O.H.; investigation, M.W. and S.P.; data curation, O.H.; writing—original draft preparation, O.H.; writing—review and editing, S.A.; supervision, O.H. and S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research study is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID: 554884501"Multiscale model of deformation and strength characteristics of dispersed carbon nanotube-reinforced polymers".
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| C-C | Covalent bond between two carbon atoms |
| Gaussian distribution of the electronic-density function in the transverse cross-section of the C–C bond | |
| Generalized coordinate in the radial direction of the C–C bond’s transverse cross-section, nm | |
| Effective bond diameter, nm | |
| Average effective bond diameter, nm | |
| Effective Poisson’s ratio of the bond | |
| Force constant of molecular mechanics (tension), | |
| Force constant of molecular mechanics (bending), | |
| Force constant of molecular mechanics (torsion), | |
| I | Moment of inertia, nm4 |
| J | Polar moment of inertia, nm4 |
| ELF | Electron Localization Function |
| F(v,d) | Residual function |
| Shear coefficient | |
| Potential energy of the beam under tension, bending, and torsion, J | |
| Young’s modulus, GPa | |
| Shear modulus, GPa | |
| Cross-sectional area, nm2 | |
| Bond length, nm | |
| ∆L | Bond elongation, nm |
| α | Bond bending angle increment, rad |
| ∆β | Bond torsion angle increment, rad |
References
- Li, Ch.; Chou, T.W. A structural mechanics approach for the analysis of carbon nanotubes. International Journal of Solids and Structures 2003, Volume 40(Issue 10), 2487–2499. [Google Scholar] [CrossRef]
- Tserpes, K.I.; Papanikos, P. Finite element modeling of single-walled carbon nanotubes. Composites Part B: Engineering 2005, Volume 36(Issue 5), 468–477. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Qian, Ch.; McLean, B.; Hedman, D.; Ding, F. A comprehensive assessment of empirical potentials for carbon materials. APL Mater 2021, 061102. [Google Scholar] [CrossRef]
- Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 1988, 37, 6991–7000. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys Rev B Condens Matter 1990, 42(15), 9458–9471. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.W.; Shenderova, O.A; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14 783. [Google Scholar] [CrossRef]
- Dilrukshi, K.G.S.; Dewapriya, M.A.N.; Puswewala, U.G.A. Size dependency and potential field influence on deriving mechanical properties of carbon nanotubes using molecular dynamics. Theoretical and Applied Mechanics Letters 2015, Volume 5(Issue 4), 167–172. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. The Journal of chemical physics 2000, 112(14), 6472–6486. [Google Scholar] [CrossRef]
- Willman, J.T.; Gonzalez, J.M.; Nguyen-Cong, K.; Hamel, S.; Lordi, V.; Oleynik, I.I. Accuracy, transferability, and computational efficiency of interatomic potentials for simulations of carbon under extreme conditions. J. Chem. Phys. 2024, 161(8), 084709. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casemit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic-table force-field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, R. Nanomechanics of graphene. Natl Sci Rev. 2018, 6(2), 324–348. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Ajith, K. M.; Lee; Uck, Sang; Valsakumar, M. C. Assessment of the mechanical properties of monolayer graphene using the energy and strain-fluctuation methods. RSC Adv. 2018, 8(27283), 27283–27292. [Google Scholar] [CrossRef]
- Gondlyakh, A.V.; Sokolskiy, A. L.; Kolosov, A. E.; Chemeris, A. O.; Shcherbina, V. Y.; Antonyuk, S. I. Modeling the mechanisms of fracture formation in nanomodified polymers. Int. Conf. Nanomaterials: Applications & Properties (NAP) 2020, 02TM06-1-02TM06-7. [Google Scholar] [CrossRef]
- Torkaman-Asadi, M.A.; Kouchakzadeh, M.A. Atomistic simulations of mechanical properties and fracture of graphene: A review. J. Computational Materials Science 2022, Vol. 210, 111457. [Google Scholar] [CrossRef]
- Lu, X.; Hu, Z. Mechanical property evaluation of single-walled carbon nanotubes by finite element modeling. J Composites 2012, Part B Engineering 43(4), 1902–1913. [CrossRef]
- Lee, Ch.; Wei, X.; Kysar, J. W.; Hone, J. Measurement of the elastic properties and intrinsic graphene strength of monolayer. Science 2008, Vol. 321(Issue 5887), 385–388. [Google Scholar] [CrossRef]
- Cao, K.; Feng, S.; Han, Y.; et al. Elastic straining of free-standing monolayer graphene. Nat Commun 2020, 11, 284. [Google Scholar] [CrossRef]
- Jeon, S.K.; et al. Mechanical test method and properties of a carbon nanomaterial with a high aspect ratio. J. Nano Convergence 2016, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Mauri, F.; Louie, S.G. Magnetic susceptibility of insulators from first principles. Phys. Rev. Lett. 1996, 76, 4246. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Stalke, D. Charge Density and Chemical Bonding. In The chemical bond I. Structure and bonding; Mingos, D., Ed.; Springer, 2016; Vol 169. [Google Scholar] [CrossRef]
- Ma, Zh.; Tan, Y.; Cai, X.; Chen, X.; Shi, T.; Jin, J.; Ouyang, Y; Peng, Q. Assessment of classical force-fields for graphene mechanics. Crystals 2024, 14, 960. [Google Scholar] [CrossRef]
- Gondlyakh, A.; Sokolskiy, A.; Shylovych, T.; Shylovych, Y.; Chemeris, A.; Antonyuk, S. Numerical determination of the strength of nanomodified ceramics. Int. Conf. Nanomaterials: Applications & Properties (NAP), 2021; 24, pp. 1–6. [Google Scholar] [CrossRef]
- Hirsch, A. Functionalization of Single-Walled Carbon Nanotubes. Angewandte. Chem, Int. 2002, 41(11), 1853–1859. [Google Scholar] [CrossRef]
- Gondlyakh, A.; Kolosov, A.; Scherbina, V.; Mamchur, O.; Shilovich, Y. Crack Resistance Parameters of Nano-reinforced Rubber Products in Mechanical Engineering. In Lecture Notes in Mechanical Engineering; Springer, 2022; pp. 272–281. [Google Scholar] [CrossRef]
- Tersoff, J. Modeling solid-state chemistry: Interatomic potentials for multicomponent systems. Phys. Rev. B 1990, 41, 3248. [Google Scholar] [CrossRef] [PubMed]
- Fthenakis; Evangelakis, P. S.; Fthenakis, A. I. Evaluating the performance of ReaxFF potentials for sp2 carbon. (open-access report) . 2022. Available online: https://helios.eie.gr/helios/bitstream/10442/18419/5/.
- Yakobson, B.I.; Brabec, C.J.; Bernholc, J. Nanomechanics of carbon tubes: Instabilities beyond linear response. Phys. Rev. Lett. 1996, 76, 2511. [Google Scholar] [CrossRef]
- Kudin, K. N.; Scuseria, G. E.; Yakobson, B. I. C2F, BN, and C nanoshell elasticity from ab initio computations. Phys. Rev. B 2001, Vol. 64(Issue 23), 235406. [Google Scholar] [CrossRef]
- Chang, T.; Gao, H. Size-dependent elastic properties of a single-walled carbon nanotube via a molecular mechanics model. J. Mech. Phys. Solids 2003, Vol. 51, 1059–1074. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, J; Hwang, K. C. Thickness of graphene and single-wall carbon nanotubes. Phys. Rev. B 2006, 74, 245413. [Google Scholar] [CrossRef]
- Scarpa, F.; Adhikari, S. A mechanical equivalence for Poisson's ratio and thickness of C–C bonds in single wall carbon nanotubes. J. Phys. D: Appl. Phys. 2008, 41 085306. [Google Scholar] [CrossRef]
- Cowper, G. R. The shear coefficient in Timoshenko’s beam theory. J. Appl. Mech 1966, Vol. 33(No.2), 335–340. [Google Scholar] [CrossRef]
- Love, A. E. H. A treatise on the mathematical theory of elasticity Chapter 16, especially p. 358; Cambridge University Press: Cambridge, England, 1952. [Google Scholar]
- Sears, A.; Batra, R.C. Macroscopic properties of carbon nanotubes from molecular-mechanics simulations. Phys. Rev B 2004, 69, 235406. [Google Scholar] [CrossRef]
- Przemieniecki, J. S. Theory of matrix structural analysis. 1968 by McGraw-Hill. In Library of Congress Catalog Card; Volume Number 67-19151, p. 50904, 1234567890MAMM7432106987.
- Yakobson, B.I.; Avouris, P. Mechanical properties of carbon nanotubes. In Carbon Nanotubes: Synthesis, Structure, Properties, and Applications; Dresselhaus, M. S., Dresselhaus, G., Avouris, P., Eds.; Springer-Verlag, 2001; pp. 287–327. [Google Scholar]
- Zhou, X.; Zhou, J; Ou-Yang, Z.C. Strain energy and Young’s modulus of single-wall carbon nanotubes calculated from electronic energy-band theory. Phys. Rev. B 2000, 62, 13692. [Google Scholar] [CrossRef]
- Lu, J.P. Elastic properties of carbon nanotubes and nanoropes. Phys. Rev. letters 1997, 79(7), 1297. [Google Scholar] [CrossRef]
- Tu, Zhan-chun; Ou-Yang, Zhong-can. Single-walled and multiwalled carbon nanotubes viewed as elastic tubes with the effective Young’s moduli dependent on layer number. Phys. Rev. B 2002, 65, 233407. [Google Scholar] [CrossRef]
- Pantano, A.; Parks, D.M.; Boyce, M.C. Mechanics of deformation of single- and multi-wall carbon nanotubes. Journal of the Mechanics and Physics of Solids 2004, Volume 52(Issue 4), 789–821. [Google Scholar] [CrossRef]
Figure 1.
Interaction schemes: (a) Interatomic; (b) Beam analogue.
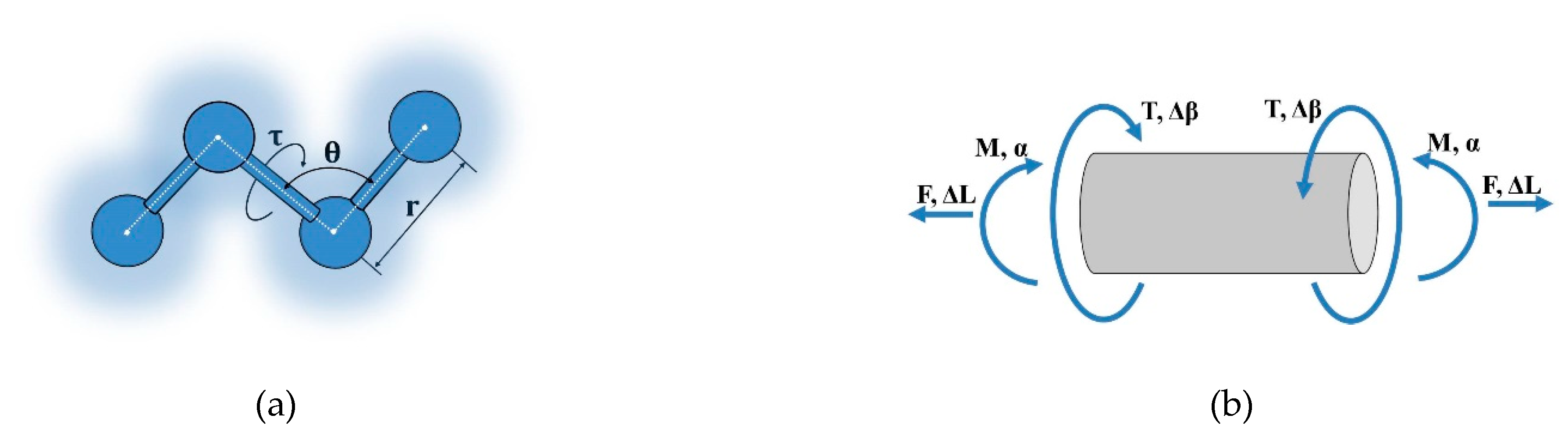
Figure 2.
Dependence of the C–C bond diameter
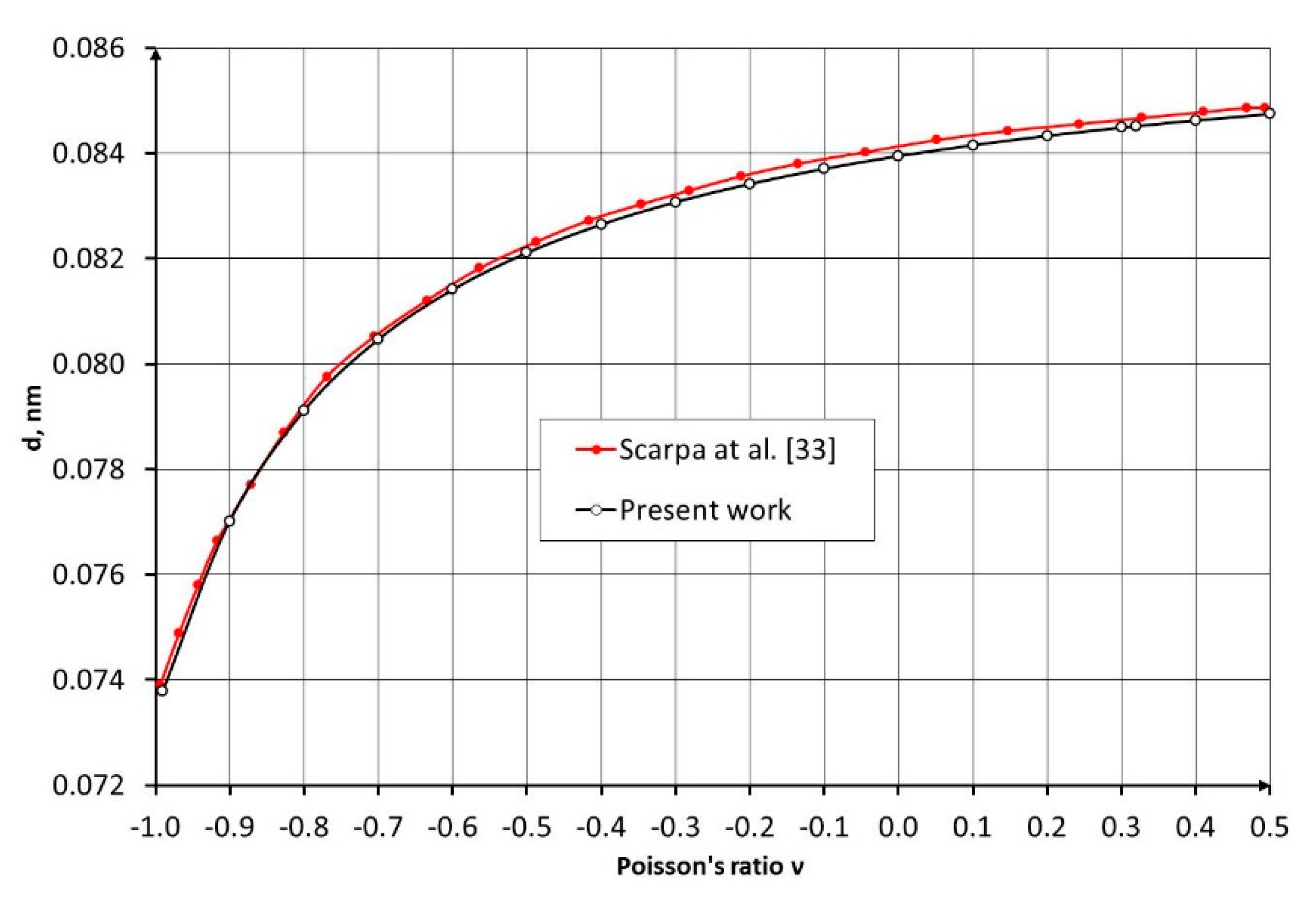
Figure 3.
F(v,d) plotted as a function of
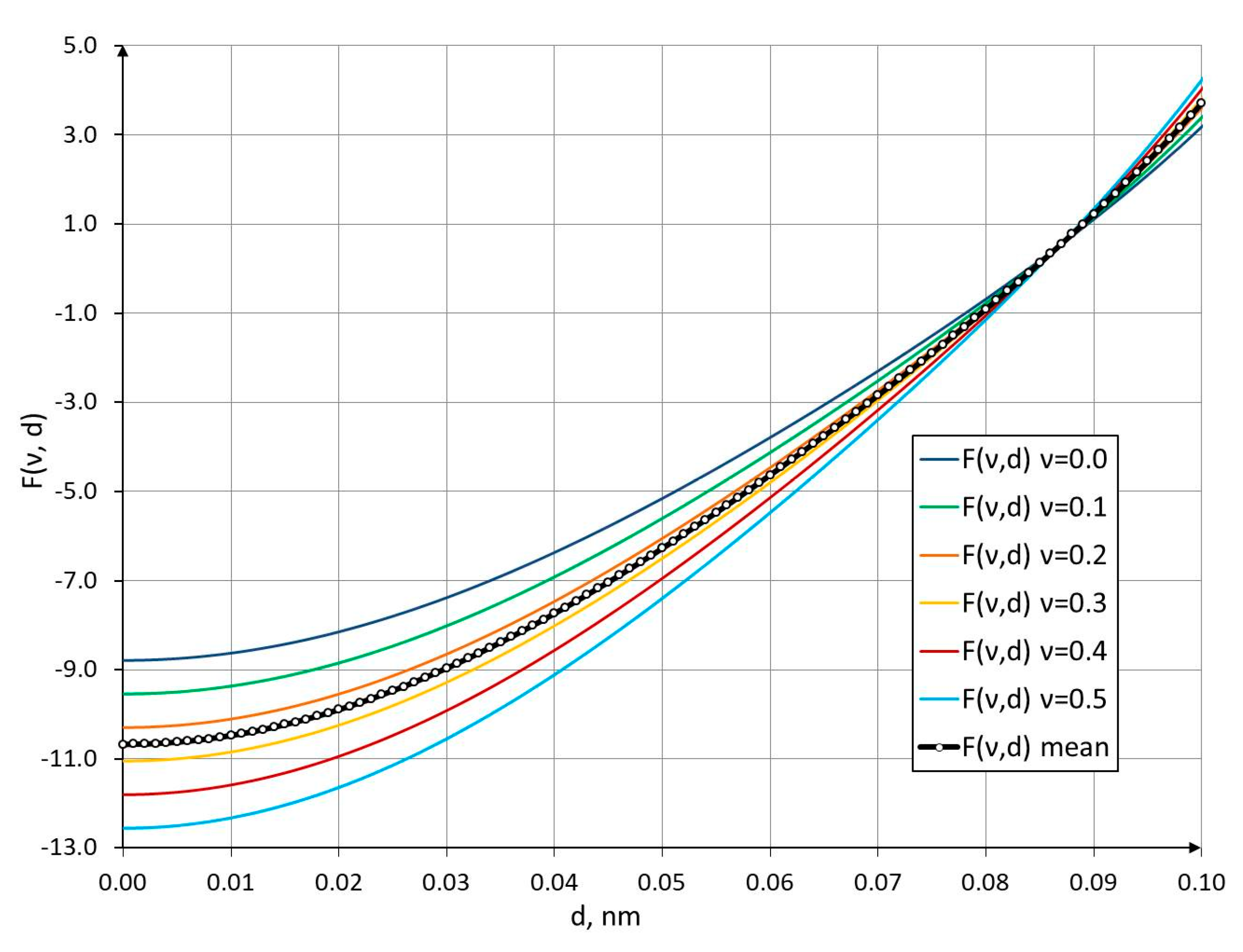
Figure 4.
The C-C bond diameter
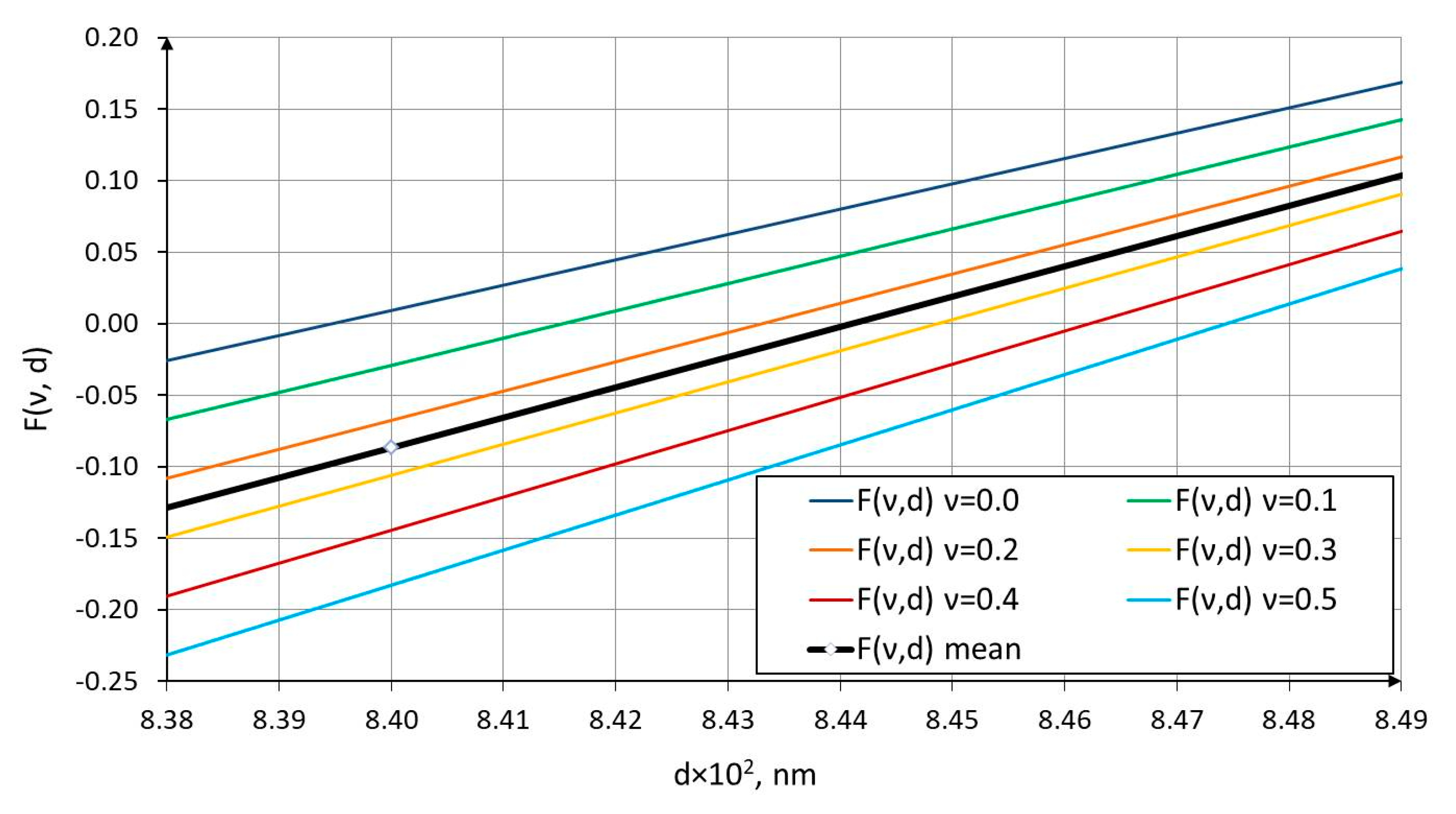
Figure 5.
Two-dimensional moiré cross sections of the electron density distribution for the
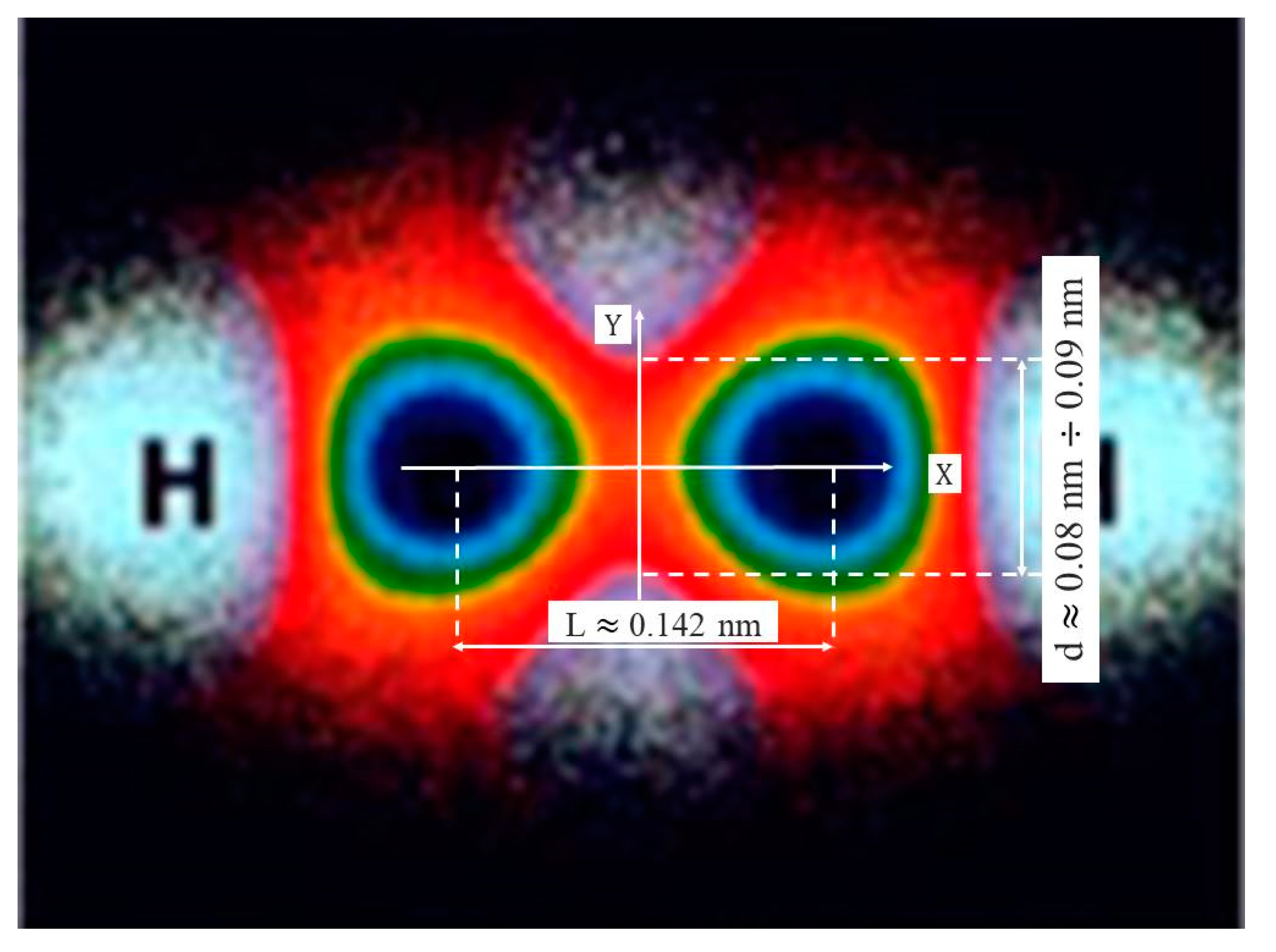
Figure 6.
Normalized distribution of the electron density function (ELF) in the C–C bond cross-section.
Figure 6.
Normalized distribution of the electron density function (ELF) in the C–C bond cross-section.
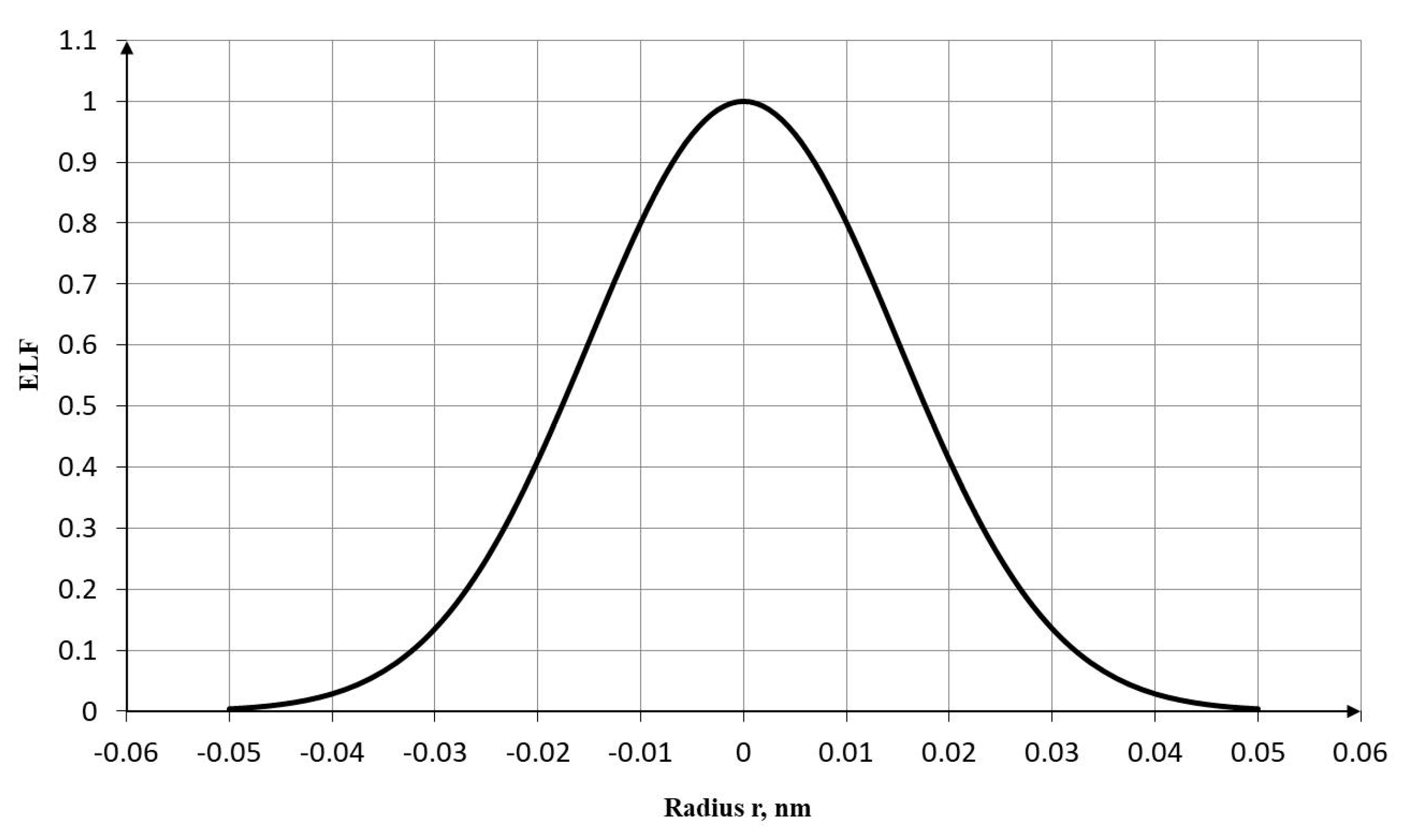
Figure 7.
Moiré pattern of the electron density distribution in the transverse section of the C–C bond. White dashed lines indicate normalized density isolines at 0.9, 0.5, and 0.2 of the maximum value.
Figure 7.
Moiré pattern of the electron density distribution in the transverse section of the C–C bond. White dashed lines indicate normalized density isolines at 0.9, 0.5, and 0.2 of the maximum value.
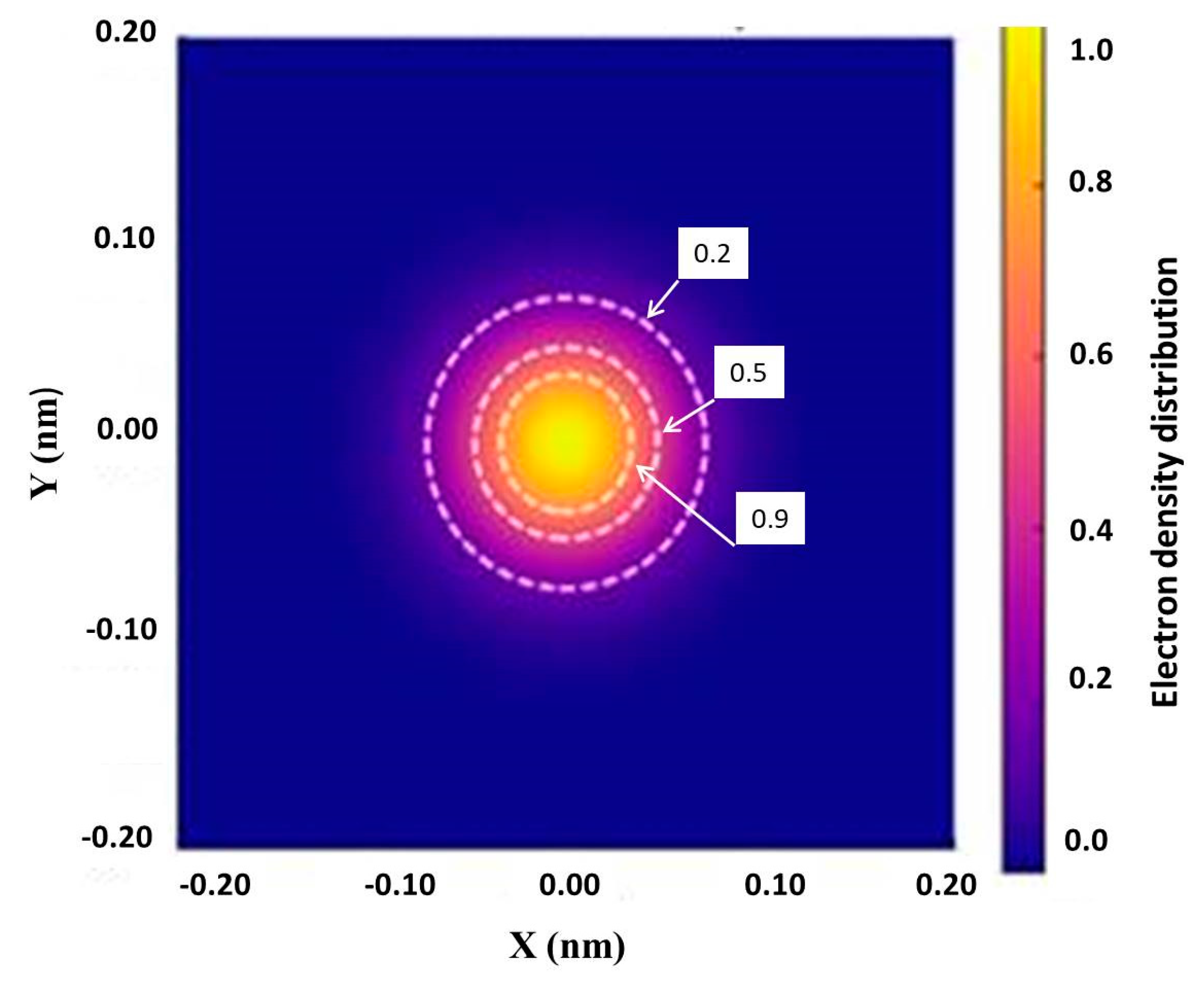
Figure 8.
Dependence of the Young’s modulus on the C–C bond diameter.
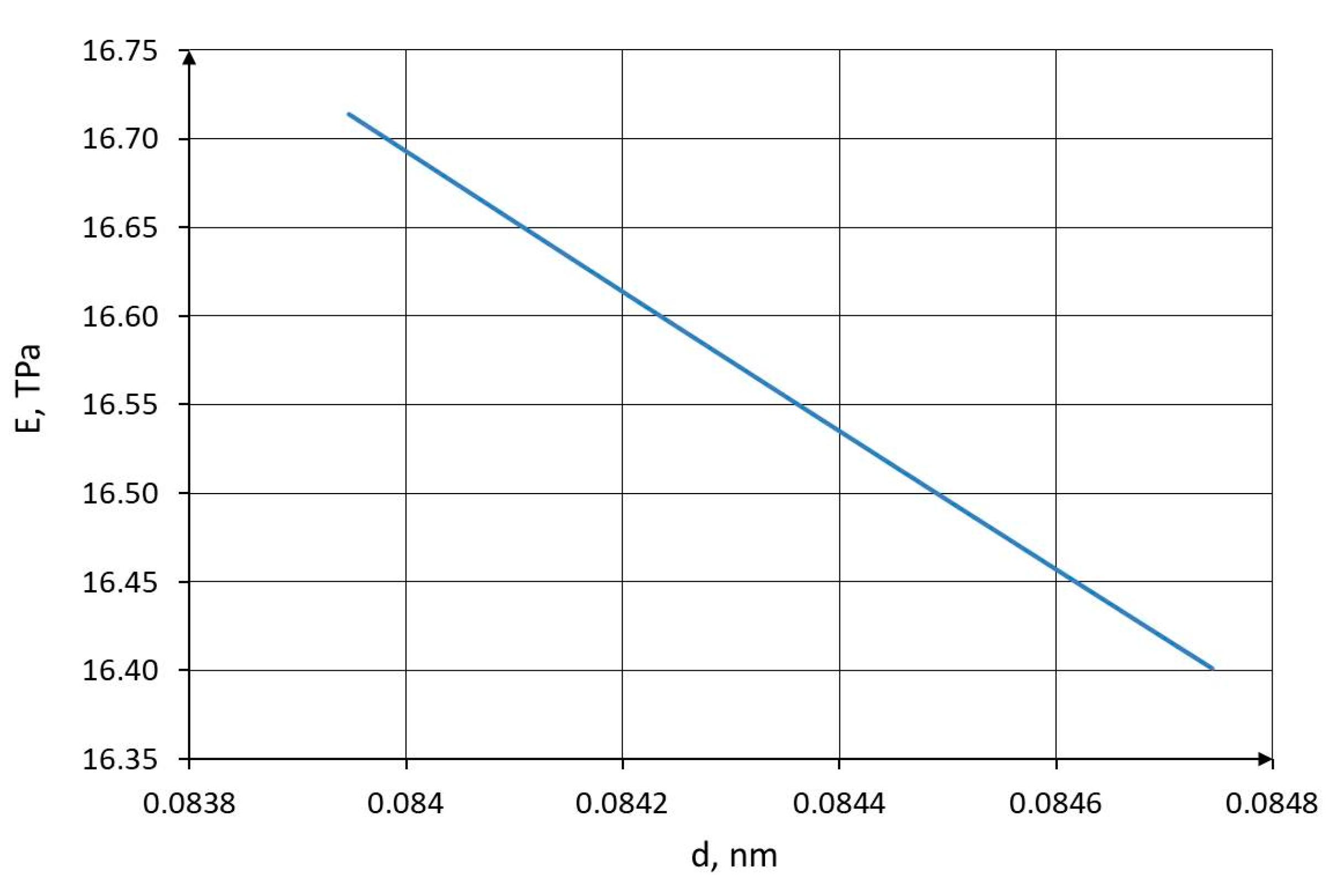
Figure 9.
Dependence of the shear modulus G on the Poisson’s ratio
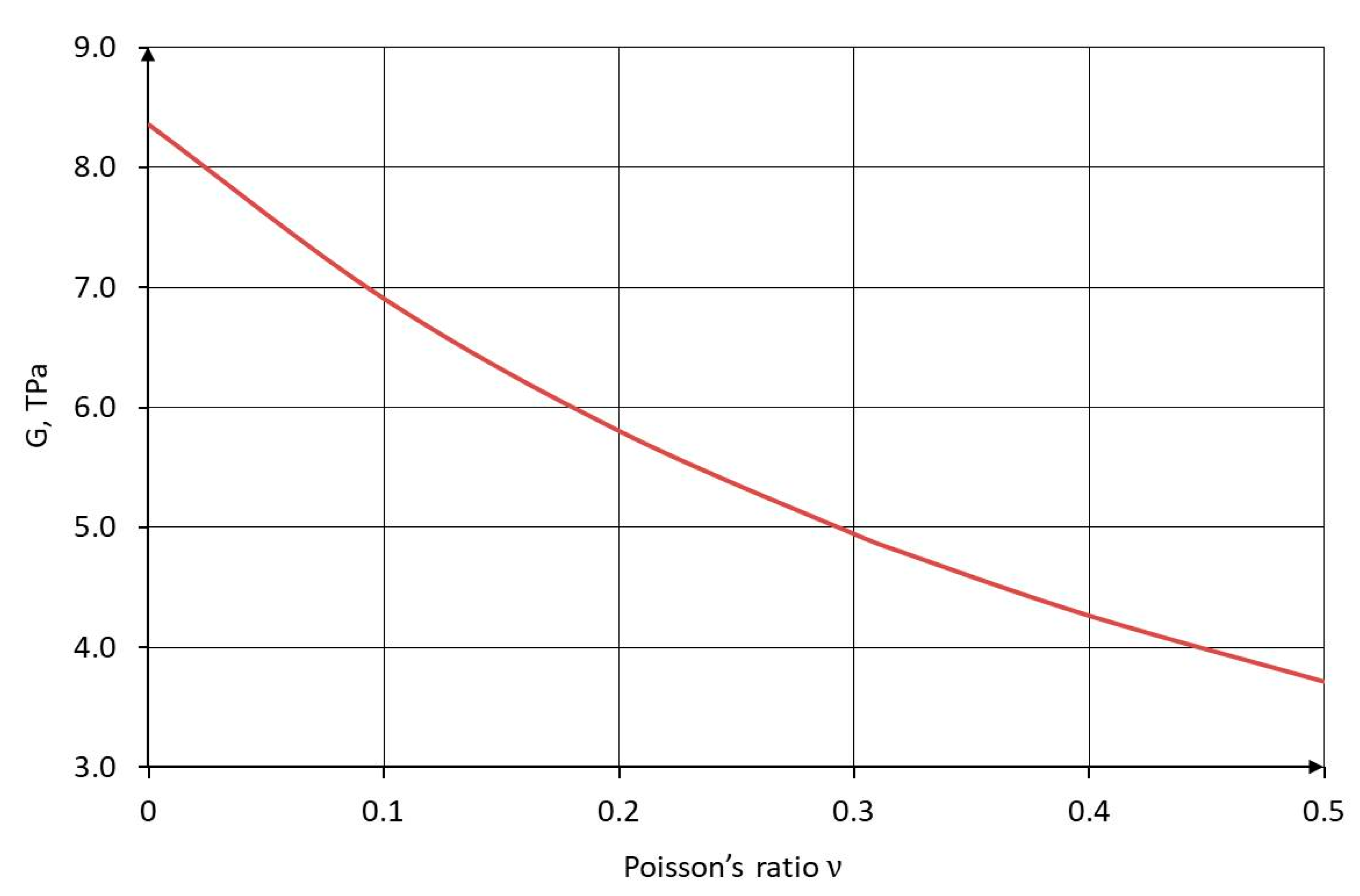
Figure 10.
Comparison of the dependence of the C–C bond diameter on the Poisson’s ratio with results from other authors.
Figure 10.
Comparison of the dependence of the C–C bond diameter on the Poisson’s ratio with results from other authors.
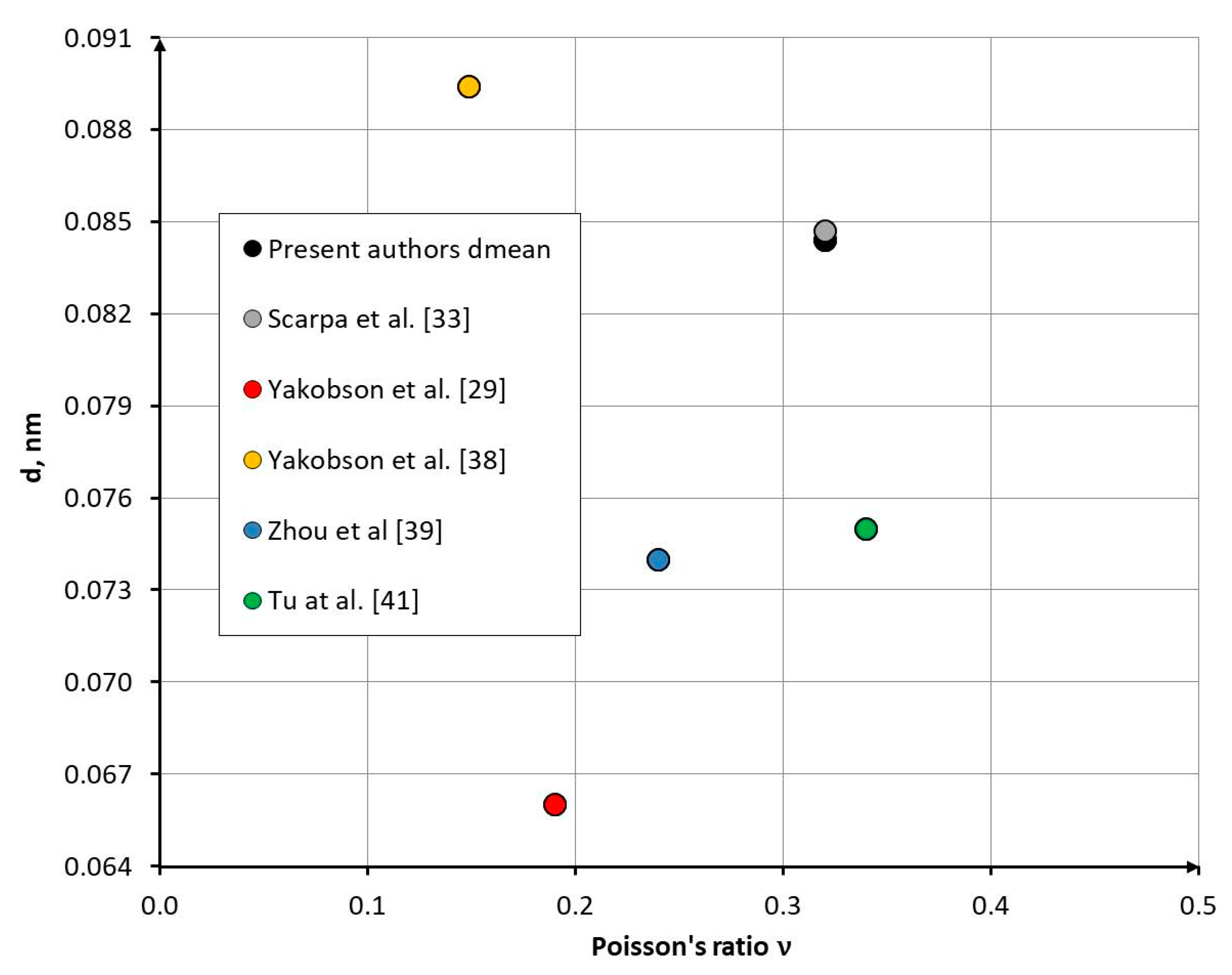

Table 1.
Typical values of Young’s modulus, Poisson’s ratio ν, and effective wall thickness h of single-walled carbon nanotubes reported by different authors.
Table 1.
Typical values of Young’s modulus, Poisson’s ratio ν, and effective wall thickness h of single-walled carbon nanotubes reported by different authors.
| Authors | Young’s modulus, TPa | Poisson’s ratio ν | h, nm |
|---|---|---|---|
| Yakobson et al. [29] | 5.5 | 0.19 | 0.066 |
| Yakobson et al. [38] | 3.859 | 0.149 | 0.0894 |
| Zhou et al. [39] | 5.1 | 0.24 | 0.074 |
| Lu [40] | 1.0 | 0.28 | 0.34 |
| Tu et al. [41] | 4.7 | 0.34 | 0.075 |
| Pantano et al. [42] | 4.84 | 0.075 | |
| Kudin et al. [30] | 3.859 | 0.0894 | |
| Present authors | 16.4 | 0.32 | 0.0844 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



