Submitted:
23 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
The extracellular matrix (ECM) is a dynamic and complex three-dimensional network that provides structural support and mechanical stability to tissues. The complete repertoire of ECM and associated proteins has recently been cataloged as matrisome, which encompasses both core structural components and ECM-associated proteins. Advances in ECM biology have overturned the traditional view of the ECM as a purely passive scaffold, revealing its active involvement in a wide range of biological processes. Among these, the ECM plays a critical regulatory role in inflammation. This review examines the bidirectional interplay between the matrisome and inflammatory processes, highlighting how matrisome components shape inflammatory responses and how inflammation, in turn, drives matrisome remodeling. A deeper understanding of matrisome–inflammation in-teractions will provide important insights into immunopathology and may inform the development of novel therapeutic strategies.
Keywords:
matrisome
; matrikines
; inflammation
1. Introduction
The extracellular matrix (ECM) comprises the most abundant proteins in our body (one-third of our body mass), and its dysregulation contributes significantly to most chronic diseases [1,2]. Fibrosis exemplifies this phenomenon, accounting for nearly half of all mortality cases and representing abnormal matrix behavior and inflammation [3]. Recently, the diverse components of the ECM have been cataloged as the matrisome [4,5]. The matrisome comprises over 1,000 proteins and is organized into two main categories: the core matrisome and matrisome-associated proteins [6]. The core matrisome includes collagens, ECM glycoproteins, and proteoglycans, while the matrisome-associated proteins consist of ECM-affiliated proteins, ECM regulators, and secret factors. Fibroblasts, which are the predominant stromal cells in the tissue, play an essential role in synthesizing these matrisome components and have long been recognized for providing structural support to the tissue [7,8].
The traditional view of ECM as passive structural proteins has undergone a paradigm shift in recent years [1,2,9,10]. Immune cells attach to and inhabit the ECM within tissue environments [1,11], yet not much research investigates the connections between ECM and immune cells. Despite the ECM making up roughly one-third of tissue composition, immunologists have largely overlooked its significance. Similarly, those studying matrix biology typically ignore how immune systems regulate these sophisticated structural networks. Investigating how matrix quality and physical characteristics, including rigidity and mechanical forces, shape immune response represents a compelling research frontier.
Accumulating evidence demonstrates that matrisome are active participants in immune surveillance and inflammation, possessing the capacity to trigger danger signals, regulate inflammatory mediators, recruit immune cells, and modulate immune responses [1,10,12,13]. These immunomodulatory functions position matrisome as critical orchestrators of inflammation, influencing the initiation, progression, and resolution of inflammatory diseases.
Human skin is the most voluminous matrisome-rich tissue in the human body. The bulk of human skin is composed of dense collagen-rich matrisome, which is essential for the maintenance of skin structure, mechanical properties, and function [14,15,16]. The collagen-rich matrisome provides structural and mechanical support for skin while also modulating inflammatory responses [17,18,19]. Alterations of matrisome also creates a tissue microenvironment with many pathologic skin disorders, such as increased fragility [20], impaired vasculature support [21], poor wound healing [22,23], skin dermal aging [14,15], and cancer development [14,15,24].
This review highlights current knowledge of the immunomodulatory roles of matrisome in inflammation, with particular emphasis on its capacity to trigger and regulate immune responses. Furthermore, this review explores the therapeutic implications of these findings and discusses future research directions that could translate insights from matrisome biology into novel treatment strategies for inflammatory conditions.
2. ECM and Immune Cells Bidirectional Interactions
The ECM is not merely a static scaffold but rather a dynamic structure that profoundly influences immune cell behavior and inflammatory processes [1,2,25]. Recently, the relationship between ECM remodeling and inflammation is increasingly recognized as bidirectional and dynamic [1,11]. This reciprocal interaction creates a complex feedback loop where the physical and biochemical properties of the ECM influence inflammatory cell behavior and cytokine production, while simultaneously, inflammatory mediators drive ECM remodeling. The evolving paradigm highlights how tissue homeostasis, disease progression, and repair processes hinge on this molecular dialogue (Figure 1).
2.1. Integrin-Mediated Immune Cell-ECM Interactions
Integrins serve as the primary receptors mediating immune cell-ECM interactions [26,27]. These heterodimeric transmembrane receptors recognize specific ECM ligands and transduce bidirectional signals across the plasma membrane. The β2 integrin subfamily (CD18) is particularly important for leukocyte function [28]. It pairs with four different α subunits, αL (CD11a), αM (CD11b), αX (CD11c), and αD (CD11d), to form distinct receptors with specific ligand preferences. For instance, αMβ2 (Mac-1) expressed on neutrophils and macrophages binds to fibrinogen, iC3b, and denatured collagen, facilitating cell adhesion at inflammatory sites [29]. LFA-1 (αLβ2) primarily mediates leukocyte-endothelial and leukocyte-leukocyte interactions through ICAM-1 binding but also interacts with ECM components [30,31].
The β1 integrin family plays crucial roles in immune cell trafficking and retention [32]. α1β1 and α2β1 are collagen receptors that regulate T cell and dendritic cell positioning within tissues. α4β1 (VLA-4) binds to fibronectin and VCAM-1, enabling leukocyte rolling, firm adhesion, and transendothelial migration during recruitment to inflamed tissues [33]. α5β1 recognizes the RGD sequence in fibronectin and is important for T cell adhesion and migration [34].
This integrin-mediated crosstalk between immune cells and the ECM represents more than simple adhesion; it provides bidirectional signaling that integrates mechanical and biochemical cues from the tissue environment with intracellular signaling pathways. The specificity of integrin-ligand interactions, combined with their capacity for dynamic regulation through conformational changes and expression patterns, allows immune cells to navigate complex tissue architectures, respond appropriately to inflammatory signals, and execute localized effector functions. Understanding these integrin-ECM interactions is fundamental to comprehending immune cell biology.
2.2. Immune Cell Migration Through ECM
Immune cells must navigate through complex three-dimensional ECM networks to reach sites of infection or injury [1]. This migration involves both proteolytic and non-proteolytic mechanisms [25]. Proteolytic migration requires matrix metalloprotease (MMPs) and other proteases secreted by migrating immune cells. Neutrophils release MMP-8 and MMP-9 to degrade collagen and gelatin, creating pathways through dense ECM [35,36]. Macrophages secrete a broad spectrum of MMPs including MMP-2, MMP-9, and MMP-12, enabling them to traverse basement membranes and interstitial matrices [37]. T cells upregulate MT1-MMP (MMP-14) upon activation, facilitating their penetration into inflamed tissues [38].
Non-proteolytic migration, or amoeboid movement, allows immune cells to squeeze through pre-existing pores in the ECM using cellular deformation and contractility [39,40]. This mode is particularly important for lymphocytes and is less dependent on integrin adhesion but requires coordination of actomyosin contractility and nuclear deformability.
2.3. ECM-Sequestered Factors and Immune Regulation
The ECM serves as a reservoir for cytokines, chemokines, and growth factors that regulate immune cell function [1,9]. These factors bind to ECM components through electrostatic interactions with glycosaminoglycans or through specific protein-protein interactions. Chemokines such as CCL2, CCL5, CXCL8, and CXCL12 bind to heparan sulfate proteoglycans, creating immobilized gradients that guide immune cell migration [41]. This "hypotaxis" provides directional cues that are more stable than soluble gradients. Growth factors including TGF-β, VEGF, FGF, and PDGF are sequestered in the ECM in latent or bound forms and released upon ECM remodeling, influencing immune cell recruitment, differentiation, and resolution of inflammation [42].
2.4. Immune Cell-Mediated ECM Remodeling
Immune cells actively remodel ECM, altering tissue architecture and creating a modified microenvironment that affects subsequent immune responses [43].
Neutrophils are early responders that release granule contents including neutrophil elastase, cathepsin G, proteinase-3, and MMPs, causing rapid ECM degradation [44]. They also release neutrophil extracellular traps (NETs) composed of DNA, histones, and proteases that can degrade ECM while trapping pathogens. Simultaneously, inflammation reduces the expression of tissue inhibitors of metalloproteinases (TIMPs), further shifting the balance toward ECM degradation.
Macrophages exhibit phenotype-dependent ECM remodeling [43]. M1 macrophages secrete high levels of MMPs and produce ROS that damage ECM proteins, promoting degradation. M2 macrophages express lower MMP levels but produce TIMPs and secrete factors like TGF-β and PDGF that stimulate fibroblast ECM synthesis, supporting tissue repair and potentially fibrosis [45].
T cells influence ECM remodeling indirectly through cytokine production. Th1 cells secrete IFN-γ, which enhances macrophage MMP production, while Th2 cells produce IL-4 and IL-13, driving M2 macrophage polarization and fibrotic responses. Regulatory T cells (Tregs) can suppress excessive ECM degradation and promote resolution of inflammation [46].
Mast cells release tryptase and chymase, serine proteases that activate pro-MMPs and directly degrade ECM proteins [47]. They also secrete histamine and other mediators that increase vascular permeability, facilitating immune cell extravasation.
Inflammatory cells generate reactive oxygen species (ROS) and reactive nitrogen species (RNS) that chemically modify ECM proteins through oxidation, nitration, and glycation [48]. These modifications alter ECM mechanical properties, reduce its biological function, and create neoepitopes that can trigger further immune responses.
This multifaceted regulation of ECM remodeling by inflammation represents a double-edged sword: while acute inflammatory ECM remodeling facilitates tissue repair and pathogen clearance, chronic inflammation leads to pathological ECM changes that contribute to tissue dysfunction, fibrosis, and aging-related diseases. Thus, ECM remodeling is fundamentally intertwined with inflammation, orchestrating immune cell trafficking, activation, and function via direct receptor interactions, mechanical and biochemical signaling, and reciprocal modification by inflammatory mediators.
2.5. ECM in Immune Cell Niche Formation
The ECM creates specialized microenvironments or "niches" that support immune cell residence and function in specific tissues [49]. In lymphoid organs, the reticular network composed of collagen, fibronectin, and laminin provides structural support and creates distinct compartments for B and T cell zones [50]. Fibroblastic reticular cells produce ECM that organizes lymphocyte traffic and presents chemokines. In bone marrow, the ECM niche regulates hematopoietic stem cell maintenance and immune cell development [51,52]. Specific ECM components like osteopontin, tenascin-C, and various proteoglycans modulate stem cell quiescence versus proliferation [53,54]. In peripheral tissues, tissue-resident memory T cells and tissue-resident macrophages interact extensively with local ECM, which provides survival signals and maintains their phenotype [55].
2.6. Pathological Immune Cell-ECM Interactions
In chronic inflammation, persistent immune cell infiltration and activation lead to excessive ECM degradation or pathological ECM accumulation (fibrosis) [57,58]. The altered ECM further perpetuates inflammation by presenting damage signals and maintaining pro-inflammatory immune cell phenotypes.
In autoimmune diseases, immune cells may target ECM components directly (as in anti-collagen antibodies in rheumatoid arthritis) or indirectly damage ECM through chronic inflammation [59,60]. Modified ECM can then expose cryptic epitopes that become targets for autoimmune responses.
In cancer, tumor-associated ECM remodeling creates an immunosuppressive microenvironment [61]. Dense, cross-linked collagen networks physically exclude immune cells from tumors, while specific ECM molecules like tenascin-C and versican promote immunosuppressive cell recruitment and function [62,63,64].
In aging, accumulated ECM damage and altered composition contribute to inflammaging [65]. Degraded ECM releases pro-inflammatory fragments, while loss of ECM mechanical integrity impairs immune cell migration and function.
The intricate interplay between immune cells and the ECM represents a fundamental aspect of tissue immunity and homeostasis. These bidirectional interactions regulate immune surveillance, control inflammatory responses, and determine outcomes in tissue repair and disease. Understanding these molecular dialogues offers promising avenues for therapeutic intervention in inflammatory diseases, fibrosis, cancer, and aging-related immune dysfunction.
3. Matrisome-Associated Proteins: MMPs as Inflammatory Modulators
Dermal fibroblasts are the principal regulators of ECM homeostasis, controlling both ECM synthesis and degradation. During inflammation, ECM remodeling becomes particularly active, with fibroblasts and immune cells modulating their production of MMPs and TIMPs in response to inflammatory signals [66,67,68]. Both MMPs and TIMPs are matrisome components classified as ECM regulators within the ECM-associated proteins category.
MMPs constitute a family of zinc-dependent endopeptidases capable of degrading virtually all ECM components [69,70]. Dermal fibroblasts express numerous MMPs, including interstitial collagenases (MMP-1, MMP-8, MMP-13), gelatinases (MMP-2, MMP-9), stromelysins (MMP-3, MMP-10), membrane-type MMPs (MT-MMPs), and others [71]. Under homeostatic conditions, fibroblasts produce low levels of most MMPs, maintaining balanced ECM turnover. However, inflammatory stimulation dramatically alters MMP expression patterns.
Pro-inflammatory cytokines, particularly IL-1β and TNF-α, are potent inducers of MMP expression in fibroblasts [72,73]. These cytokines activate transcription factors including AP-1 and NF-κB that bind to MMP gene promoters, increasing transcription. IL-17, increasingly recognized as a key inflammatory mediator in skin diseases, also induces fibroblast MMP production [74]. Beyond ECM degradation, MMPs have numerous other functions relevant to inflammation. MMPs can process cytokines and chemokines, either activating them (as with pro-TNF-α cleavage by TACE/ADAM17) or inactivating them through proteolytic cleavage. MMPs can release matrix-sequestered growth factors, making them available to cells [75,76].
During acute inflammation, increased MMP production by inflammatory cytokines facilitates immune cell migration by degrading ECM physical barriers [67,77]. This enables neutrophils, monocytes, and lymphocytes to reach sites of infection or injury. The degradation of basement membranes by MMPs is particularly important for immune cell extravasation from blood vessels. However, excessive MMP activity can cause collateral tissue damage, and inflammatory conditions are associated with elevated MMP levels in both tissue and circulation.
In chronic inflammation, persistent alterations in the MMP/TIMP balance contribute to pathological tissue remodeling [78,79]. Chronic overexpression of MMPs can lead to excessive ECM degradation, as observed in chronic wounds and some inflammatory diseases [80,81]. Conversely, chronically elevated TIMP levels or suppressed MMP expression contributes to fibrosis, as seen in systemic sclerosis and hypertrophic scarring.
As such, MMPs serve as critical mediators linking ECM remodeling to inflammation in tissue. MMP regulation represents a critical determinant of whether inflammation resolves appropriately or progresses to chronic pathology, positioning MMPs as both essential inflammatory mediators and potential therapeutic targets in skin diseases (Figure 2).
4. Matrikines and Matrisome-Associated Proteins as Inflammatory Modulators
Matrikines are degradation products of the ECM such as the fragments generated from collagen, elastin, laminin, and other ECM proteins [10,82,83]. Unlike intact ECM, which primarily provides structural support and mediates cell attachment, matrikines possess distinct signaling capabilities as biologically active fragments. Matrikines interact with specific cell surface receptors, thereby modulating intracellular pathways [82]. Matrikines also act as damage-associated molecular patterns (DAMPs), thereby propagate inflammatory signaling [18]. As such, matrikines can influence diverse cellular behaviors, including proliferation, migration, differentiation, and inflammatory responses (Figure 3). Matrikines presence is especially significant in tissue remodeling, wound healing, and various pathological conditions where matrisome turnover is increased, such as arthritis, fibrosis, and cancer [2,84].
4.1. Collagen-Derived Matrikines
Collagens, the most abundant matrisome proteins, have diverse effects on inflammation beyond their structural roles [16,85]. Intact fibrillar collagens (types I and III) provide anchoring points for cells through collagen-binding integrin receptors, particularly integrin α11/β1 in skin dermal fibroblasts [86]. These integrin-collagen interactions deliver survival signals to fibroblasts and other cells and influence cell phenotypes. Collagen degradation products, however, can have pro-inflammatory effects [83]. Small collagen fragments act as chemotactic agents for fibroblasts and immune cells. Collagen fragments, particularly those derived from type I and type IV collagen, can act as chemoattractant for neutrophils and monocytes and stimulate angiogenesis [87]. Type IV collagen, a major component of basement membranes, is degraded during inflammation, facilitating immune cell migration [88,89]. Basement membrane remodeling is essential for angiogenesis during wound healing and chronic inflammation. Fragments of type IV collagen possess biological activities distinct from the intact molecule, including anti-angiogenic properties (as with endostatin and tumstatin, fragments derived from collagen XVIII and IV, respectively) [90].
4.2. Fibronectin-Derived Matrikines
Glycoproteins are a potentially important source of matrikines [10,84]. Fibronectin, a matrisome glycoprotein, plays multiple roles in inflammation [91,92]. Cellular fibronectin containing alternatively spliced domains (EDA and EDB) is upregulated during inflammation and wound healing [93]. EDA-fibronectin serves as an endogenous TLR4 ligand, activating fibroblasts and immune cells [91,92,94,95]. This creates an inflammatory positive feedback loop whereby tissue damage and inflammation induce production of EDA-fibronectin, which further activates inflammatory responses. Fibronectin also contains binding sites for various growth factors, chemokines, and other bioactive molecules, serving as a reservoir that concentrates these factors at appropriate tissue locations [96]. Fibronectin fragments generated during ECM degradation can have pro-inflammatory effects [97]. These fragments can activate fibroblasts through integrin signaling and potentially through TLRs, inducing MMP and cytokine production. The accumulation of fibronectin fragments in chronic wounds and inflammatory conditions may contribute to persistent inflammation [98,99].
4.3. Hyaluronan-Derived Matrikines
Hyaluronan (hyaluronic acid, HA) belongs to the proteoglycan category of the core matrisome. Hyaluronan, a glycosaminoglycan exhibits size-dependent effects on inflammation [100]. High molecular weight HA (>1,000 kDa), predominant in healthy tissues, generally has anti-inflammatory and immunosuppressive properties. High MW HA signals through CD44 and other HA receptors to promote tissue integrity and inhibit inflammatory responses. In contrast, low molecular weight HA fragments (<200 kDa), generated during tissue injury and inflammation, have pro-inflammatory effects [101]. HA fragments activate fibroblasts, macrophages, and dendritic cells through TLR2, TLR4, and CD44, inducing cytokine and chemokine production [102]. Dermal fibroblasts both synthesize HA through hyaluronan synthases (HAS1, HAS2, HAS3) and degrade it through hyaluronidases [103,104]. During inflammation, fibroblasts alter their HA synthesis patterns, sometimes producing more HA but also generating conditions conducive to HA fragmentation through increased hyaluronidase and reactive oxygen species [105]. The balance between intact and fragmented HA significantly influences the inflammatory environment.
4.4. Elastin-Derived Matrikines
Elastin is classified as an ECM glycoprotein within the core matrisome. Elastin is secreted as tropoelastin (the soluble precursor) and then cross-linked into insoluble elastic fibers that provide elasticity and resilience to tissues, such as arteries, lungs, and skin [106]. Elastin-derived peptides (EDPs), also known as elastokines, are bioactive fragments released during the proteolytic degradation of elastin [107]. EDPs are generated primarily through the action of MMPs, particularly MMP-2, MMP-9, and MMP-12, as well as serine elastases including neutrophil elastase and cathepsins [68,108]. EDPs exert their chemotactic effects primarily through interaction with the elastin receptor complex (ERC), which consists of three subunits: the peripheral 67-kDa elastin-binding protein (EBP), a catalytically inactive splice variant of β-galactosidase; the protective protein/cathepsin A (PPCA); and neuraminidase-1 (Neu-1) [109]. This receptor complex is expressed on various immune cell types, including monocytes, macrophages, to trigger a cascade of intracellular signaling events: such as activation of phospholipase C and generation of inositol trisphosphate (IP3) and diacylglycerol (DAG; activation of small GTPases (Rho, Rac, Cdc42) leads to polymerization and formation of lamellipodia and filopodia necessary for cell movement [110,111]. The chemotactic properties of EDPs create self-perpetuating inflammatory cycles in various pathological conditions: In COPD, cigarette smoke and inflammatory mediators stimulate elastase release, degrading lung elastin [112,113]. The resulting EDPs: Recruit additional inflammatory cells (neutrophils, macrophages, monocytes) to the airways. EDPs represent critical pathogenic mediators linking tissue degradation, inflammation, vascular disease, and aging [108]. Their ability to recruit immune cells creates self-amplifying cycles of matrix destruction and aberrant remodeling that characterize numerous chronic diseases. Recent evidence indicates that age-related elastin-derived fragments can evoke systemic inflammaging (inflammaging) that drives aging processes [114,115]. Their ability to recruit immune cells creates self-amplifying cycles of matrix destruction and aberrant remodeling that characterize numerous chronic diseases. Targeting these molecules and their signaling pathways offers therapeutic strategies for conditions ranging from atherosclerosis to emphysema. Future research must focus on developing specific EDP antagonists, preventing elastin degradation, and promoting regenerative repair of elastic tissues to break the pathological cycles driven by these bioactive peptides.
4.5. Proteoglycans
Proteoglycans are one of the three core matrisome categories. Proteoglycans, consisting of core proteins with attached glycosaminoglycan chains, serve multiple functions in inflammation [116]. Proteoglycans bind chemokines to create haptotactic gradients that direct immune cell migration toward inflamed tissue niches [117]. For instance, heparan sulfate and syndecan-1 can sequester chemokines such as CXCL8 (IL-8), establishing gradients that enhance neutrophil infiltration [118]. Small leucine-rich proteoglycans (SLRPs) such as decorin, biglycan, and lumican are abundant in tissue stroma and can act as a DAMP, signaling through TLR2 and TLR4 to induce inflammatory responses [119,120]. Soluble biglycan, released during ECM degradation or actively secreted, activates immune cells and fibroblasts [121]. The pro-inflammatory effects of biglycan contribute to sterile inflammation. Lumican also has immunomodulatory properties, influencing immune cell migration and inflammatory responses [122].
Versican, a large chondroitin sulfate proteoglycan, is upregulated in inflamed and remodeling tissues [123]. Versican influences cell adhesion, migration, and proliferation. Versican fragments, particularly the G1 domain fragment versikine, have pro-inflammatory properties and can amplify inflammation [124]. Fibroblast production of versican is induced by inflammatory cytokines and growth factors, contributing to ECM changes during inflammation.
4.6. Matricellular Proteins
Matricellular proteins are a specific subset of matrisome-associated proteins that include: CCN family (CCN1/Cyr61, CCN2/CTGF, CCN3/NOV, CCN4, CCN5, CCN6), Thrombospondins (TSP-1, TSP-2), SPARC (secreted protein acidic and rich in cysteine), Tenascins (tenascin-C, tenascin-X), Osteopontin, and Periostin [125]. These proteins are secreted into the ECM but do not provide structural support. Instead, they modulate cell-matrix interactions and cell signaling, and regulate ECM assembly, remodeling, and cell behavior. Matricellular proteins are typically expressed at low levels in healthy adult tissues but are dramatically upregulated during development, wound healing, and inflammation.
CCN family proteins are a group of six secreted, matricellular proteins that play crucial roles in cellular communication and tissue regulation [126,127]. CCN proteins have emerged as important regulators of inflammatory responses, though their effects can be context dependent [125,128,129]. Several CCN proteins, particularly CCN1 and CCN2, can promote inflammation by stimulating the production of pro-inflammatory cytokines like IL-6, TNF-α, and IL-1β in immune cells and tissue-resident cells [130,131,132]. CCN1 can activate inflammatory signaling pathways including NF-κB, which is central to inflammatory gene expression. These proteins also promote leukocyte recruitment and adhesion to inflamed tissues [133]. They help coordinate the transition from acute inflammation to healing by promoting angiogenesis, fibroblast activation, and extracellular matrix remodeling. Dysregulated CCN protein expression is implicated in chronic inflammatory diseases including rheumatoid arthritis, atherosclerosis, inflammatory bowel disease, and fibrotic conditions. Their dual nature makes them interesting therapeutic targets, though careful consideration of context is essential.
Tenascin-C is particularly interesting in the context of inflammation [134]. It is rapidly upregulated in response to tissue injury and inflammation and can activate TLR4, inducing inflammatory responses [135,136]. Tenascin-C influences cell adhesion and migration and can modulate growth factor signaling. Fibroblasts are major producers of tenascin-C during inflammation, and its expression correlates with inflammatory activity in various diseases. Conversely, certain matricellular proteins such as thrombospondin-1 possess anti-inflammatory properties and promote resolution [137,138].
5. ECM Mechanical Properties as Inflammatory Modulators
Cells respond to both biochemical signals and mechanical cues within their environment [139,140]. The mechanical properties of the ECM, including its stiffness, elasticity, and topography, actively modulates immune cells through mechanical signals [141,142]. ECM mechanical properties can activate inflammatory signaling pathways, contributing to both the initiation and persistence of inflammation, particularly in conditions characterized by tissue fibrosis or abnormal mechanical loading [143,144] (Figure 4).
Macrophages are especially sensitive to substrate rigidity, altering their gene expression profiles and polarization states: stiffer, fibrotic matrices promote the pro-inflammatory M1 phenotype, characterized by enhanced production of cytokines such as TNF-α and IL-6, whereas softer matrices favor the anti-inflammatory M2 phenotype, which secretes IL-10 and participates in tissue repair [145,146]. This mechanical sensitivity is also noted in other cell types such as dendritic cells, which modulate their activation in response to changes in ECM compliance [147,148]. Furthermore, mechanical cues can influence the threshold and duration of immune cell activation, serving as a “danger signal” in the context of tissue injury and fibrosis [149].
Mechanosensitive pathways, encompassing integrins, focal adhesion complexes, and transcriptional regulators, enable cells to detect changes in tissue stiffness, stretch, and deformation [150]. The Hippo pathway effectors Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) have emerged as critical mechanosensitive transcriptional regulators in cells [151]. Unlike traditional transcription factors, YAP/TAZ lack DNA-binding domains and instead function as transcriptional coactivators by partnering with TEAD family transcription factors to regulate genes involved in cell proliferation, survival, and ECM remodeling [152]. The subcellular localization of YAP/TAZ is exquisitely sensitive to mechanical cues: high mechanical tension promotes nuclear accumulation and transcriptional activity, while low tension or soft substrates result in cytoplasmic sequestration and degradation. Nuclear YAP/TAZ enhances expression of pro-inflammatory cytokines including IL-6, IL-8, and CCL2, creating a mechanically driven inflammatory milieu [153]. Additionally, YAP/TAZ drive the expression of connective tissue growth factor (CTGF), transforming growth factor-β (TGF-β), and matrix metalloproteinases (MMPs), establishing a pro-fibrotic program that sustains fibroblast activation and amplifies inflammatory signaling [154,155,156].
YAP/TAZ signaling in dermal fibroblasts directly upregulates numerous inflammatory mediators [157]. The age-related decline in YAP/TAZ activity, driven by mechanical deterioration of the dermal ECM, represents a key mechanism underlying inflammaging in skin aging [65]. Single-cell RNA-seq analysis revealed that decreased expression of the YAP/TAZ signature gene set represents a primary hallmark of aged mouse dermal fibroblasts, strongly supporting the role of ECM mechanical defects in regulating YAP/TAZ activity and inflammaging. Consistent with this, YAP/TAZ signaling is impaired in aged human skin, which exhibits characteristic inflammaging features [15,156]. This impairment results from age-related increases in collagen fragmentation, a prominent feature of dermal aging. This mechanical-inflammatory coupling creates a pathological feedforward loop in which initial mechanical stress triggers inflammatory responses that further alter tissue mechanics, thereby sustaining chronic inflammation and progressive tissue dysfunction.
Mechanical stress activates multiple inflammatory signaling pathways in dermal fibroblasts beyond YAP/TAZ. Stretch-activated ion channels, particularly transient receptor potential (TRP) channels and Piezo channels, respond to membrane deformation by permitting calcium influx, which triggers downstream inflammatory cascades including NF-κB and MAPK pathways [158,159]. Mechanical activation of integrin-FAK complexes stimulates phosphoinositide 3-kinase (PI3K)/Akt signaling, which cross-talks with inflammatory pathways to enhance production of prostaglandins, leukotrienes, and reactive oxygen species [160,161].
The inflammasome, particularly the NLRP3 complex, can be activated by mechanical stress in fibroblasts through mechanisms involving mitochondrial dysfunction, ATP release, and potassium efflux [162]. Mechanically stressed fibroblasts exhibit increased caspase-1 activation and secretion of mature IL-1β and IL-18, pro-inflammatory cytokine that amplify local inflammatory responses and recruit immune cells [163,164]. Furthermore, mechanical stress enhances expression and activity of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), generating inflammatory lipid mediators and reactive nitrogen species that contribute to tissue damage and sustained inflammation [165,166].
6. Matrisome-Related Senescence-Associated Secretory Phenotype (SASP) Factors as Inflammatory Modulators
The Senescence-Associated Secretory Phenotype (SASP) refers to the complex mixture of factors that senescent cells secrete into their surrounding environment [167]. When cells become senescent (they stop dividing but remain metabolically active), they become highly secretory, releasing SASP factors. Senescent fibroblasts accumulate in chronically inflamed and aged skin and exhibit a SASP characterized by persistent secretion of inflammatory cytokines, chemokines, and MMPs [168]. These senescent fibroblasts contribute to chronic low-grade inflammation, a phenomenon known as inflammaging, which represents a hallmark of aging [169]. The SASP comprises several matrisome components, including ECM regulators such as MMPs, particularly MMP-1, MMP-3, and MMP-9, which establish a pro-inflammatory microenvironment that influences ECM remodeling and sustains chronic inflammation. Furthermore, senescent fibroblasts exhibit impaired core matrisome synthesis with reduced production of type I and type III collagen, elastin, and proteoglycans, while simultaneously increasing ECM degradation, thereby driving progressive matrix deterioration including altered matrix mechanical properties [170].
Collectively, these alterations in ECM composition and mechanical properties create a tissue microenvironment that modulates immune cell behavior, including recruitment, activation state, and functional responses, potentially perpetuating inflammatory signaling and impairing tissue repair. One convincing example is a recent report showing that senescence-associated mechano-defective dermal ECM impairs YAP/TAZ activity, which in turn unleashes cGAS–STING signaling and leads to skin dermal aging in mice [65]. YAP/TAZ activity is indeed impaired in aged human skin due to age-related decline in dermal ECM mechanical properties, contributing to skin aging through diminished collagenous ECM production [14,15,156,171,172]. This mechanosensitive signaling pathway, which responds to ECM stiffness and mechanical cues, becomes progressively dysregulated with advancing age as the dermis loses structural integrity and biomechanical properties. Specifically, when YAP/TAZ transcriptional activity is diminished due to mechano-defective dermal ECM, cytoplasmic DNA accumulates and activates the cyclic GMP-AMP synthase (cGAS), which then produces the second messenger cGAMP. This molecule binds to and activates STING (stimulator of interferon genes), triggering a cascade of inflammatory responses including the secretion of pro-inflammatory cytokines and type I interferons. This mechanically-regulated cGAS–STING axis represents a critical link between mechano-defective ECM and the SASP that characterizes aged skin. The loss of YAP/TAZ-mediated mechanotransduction not only impairs fibroblast synthetic capacity for ECM components like collagen and elastin, but also creates a feed-forward loop wherein reduced ECM production further diminishes mechanical signaling, perpetuating inflammation and accelerating tissue aging. Understanding this mechanobiological mechanism has important implications for developing interventions that target ECM restoration, YAP/TAZ reactivation, or modulation of the cGAS–STING pathway to ameliorate age-related skin degeneration (Figure 5).
7. Matrisome-Targeted Therapeutic Implications
The recognition of ECM components as active orchestrators rather than passive bystanders in inflammation has catalyzed the development of multiple therapeutic avenues targeting the immune-matrisome/ECM interface [1,83,173]. These strategies range from direct blockade of immune cell-matrisome/ECM interactions to sophisticated modulation of matrisome/ECM composition, mechanical properties, and matrikines (Figure 6).
7.1. Integrin-Based Therapeutics: Disrupting Immune Cell-ECM Adhesion
Integrin-blocking antibodies represent one of the most clinically advanced approaches for targeting immune cell-ECM interactions [174,175]. By preventing integrin-mediated adhesion and migration, these agents may effectively reduce immune cell trafficking into inflamed tissues. The clinical success of natalizumab in multiple sclerosis and Crohn's disease validates this mechanism, demonstrating that selectively inhibiting specific integrin pathways (α4β1/α4β7) can effectively reduce pathological immune trafficking while maintaining acceptable safety profiles [176,177,178]. In dermatology, efalizumab previously targeted αLβ2 integrin (LFA-1) to limit T-cell activation and migration in psoriasis, though it was subsequently withdrawn due to safety concerns regarding progressive multifocal leukoencephalopathy risk [179]. Small-molecule integrin antagonists and more selective antibodies targeting β1, β2, and αv integrins are currently in preclinical and clinical development for various inflammatory skin conditions [174,180].
Future therapeutic development should focus on designing highly selective integrin antagonists that block pathological immune cell migration in specific tissue contexts while minimizing systemic immunosuppression, potentially through tissue-specific delivery systems, temporary inhibition strategies, or targeting integrins preferentially expressed on pathogenic immune cell subsets rather than those essential for protective immunity.
7.2. Targeting ECM Composition to Modulate Immune Cell Behavior
Beyond direct pharmacological intervention, modulating ECM composition offers an indirect yet powerful strategy for controlling immune cell behavior [181,182]. In tissue engineering and regenerative medicine applications, biomaterials and scaffolds engineered to recapitulate native ECM characteristics can profoundly influence immune responses [183]. Matrix stiffness, topography, ligand density, and degradation kinetics collectively determine whether an implanted material promotes inflammatory or pro-regenerative immune polarization [184,185]. Soft, compliant hydrogels mimicking the mechanical properties of healthy dermis tend to favor M2 macrophage polarization and regulatory T-cell recruitment, whereas stiff matrices promote M1 activation and pro-inflammatory cytokine production [186,187,188]. These principles are being translated into therapeutic applications for chronic wounds, where bioengineered ECM scaffolds incorporating anti-inflammatory signals can redirect dysfunctional immune responses and promote healing [189,190].
Future therapeutic strategies targeting ECM composition should focus on developing "smart" biomaterials that dynamically respond to the inflammatory microenvironment to actively guide immune cell behavior toward resolution rather than chronicity. Incorporating bioactive ECM fragments, growth factors, or immunomodulatory peptides with controlled release kinetics could provide temporal control over immune responses, delivering pro-regenerative signals precisely when needed during different healing phases.
7.3. Targeting Pathological ECM Remodeling and Mechanical Properties
Therapeutic strategies aimed at reversing or preventing pathological ECM remodeling are advancing on multiple fronts, with particular relevance for fibrotic skin diseases and tumor-associated inflammation [173,191]. Lysyl oxidase (LOX) and LOX-like enzymes catalyze collagen and elastin cross-linking, contributing to tissue fibrosis and creating a pro-tumorigenic microenvironment. Development of highly selective LOX inhibitors designed to disrupt pathological collagen cross-linking and fibrotic processes while preserving normal extracellular matrix remodeling and homeostatic tissue maintenance [192,193]. Delivery strategies ensuring tissue-specific or cell-type-specific targeting will be critical for clinical success. ECM-degrading enzymes, including recombinant collagenase and hyaluronidase, are being investigated for their ability to normalize fibrotic tissue mechanical properties and architecture as well as enhance immune cell infiltration in solid tumors [173,194,195]. By breaking down excessive or aberrantly cross-linked matrix, these enzymes can restore tissue compliance and improve accessibility for therapeutic immune cells in immunotherapy contexts. Small-molecule LOX inhibitors and neutralizing antibodies have shown promise in preclinical models of fibrotic diseases and cancer by reducing matrix stiffness, normalizing tissue mechanics, and improving immune cell infiltration [196,197,198].
MMP modulation presents a more nuanced therapeutic challenge, as these enzymes can both promote and resolve inflammation depending on context. While broad-spectrum MMP inhibitors have largely failed in clinical trials due to lack of specificity and disruption of homeostatic ECM turnover, selective targeting of specific MMPs implicated in pathological remodeling (such as MMP-9 in chronic wounds or MMP-2 in keloids) remains under investigation [199,200,201]. Tissue inhibitors of metalloproteinases (TIMPs) have also been explored as therapeutic agents, though their pleiotropic effects on multiple MMPs complicate clinical translation [202].
Future therapeutic strategies for pathological ECM remodeling should focus on developing highly selective LOX inhibitors with tissue-specific delivery systems to disrupt fibrotic cross-linking while preserving normal matrix homeostasis, particularly for fibrotic skin diseases and tumor microenvironments. Combining ECM-degrading enzymes (recombinant collagenase, hyaluronidase) with immunotherapy represents a promising approach to normalize tissue mechanics, reduce matrix stiffness, and enhance immune cell infiltration in solid tumors. Rather than broad-spectrum MMP inhibition, future efforts should target specific MMPs involved in pathological remodeling (MMP-9 in chronic wounds, MMP-2 in keloids) using context-dependent strategies that account for their dual pro-inflammatory and resolving roles. Success will require precision medicine approaches that consider disease stage, tissue context, and the balance between pathological remodeling and essential physiological ECM turnover, potentially utilizing combination therapies that coordinate matrix normalization with immune modulation or regenerative interventions.
7.4. Matricellular Proteins as Therapeutic Targets
Matricellular proteins, including thrombospondins, tenascins, osteopontin, and CCN family members, present promising therapeutic opportunities for inflammatory and fibrotic skin diseases due to their regulatory roles in immune cell recruitment, activation, and ECM deposition without providing structural support [125,203]. Thrombospondins, tenascins, osteopontin, and members of the CCN family are upregulated during skin inflammation and fibrosis, where they promote immune cell recruitment, activation, and ECM deposition [204]. Neutralizing antibodies or small molecules targeting these proteins have demonstrated efficacy in preclinical models of inflammatory and fibrotic skin diseases [205]. For instance, antibodies against tenascin-C reduce inflammation and fibrosis in experimental models of scleroderma, while osteopontin blockade attenuates psoriasiform inflammation [206,207].
Future therapeutic strategies could focus on developing humanized monoclonal antibodies or small molecule inhibitors targeting these proteins, building upon preclinical successes such as tenascin-C blockade in scleroderma models and osteopontin inhibition in psoriasiform inflammation. Combination approaches that simultaneously target multiple matricellular proteins may offer synergistic benefits, while biomarker-driven patient stratification could identify individuals most likely to respond to specific matricellular protein-targeted therapies. Additionally, developing reversible inhibitors with favorable pharmacokinetic profiles and exploring topical delivery systems could enhance therapeutic efficacy while minimizing systemic side effects, ultimately translating these promising preclinical findings into effective clinical interventions for conditions like scleroderma, psoriasis, and other inflammatory dermatoses.
7.5. Matrikine-Based Therapeutics
Synthetic matrikines or matrikine antagonists could modulate inflammation with potentially fewer side effects than systemic immunosuppression. Both pro-regenerative matrikines (promoting M2 polarization and tissue repair) and anti-inflammatory matrikines (neutralizing DAMP signaling) deserve clinical investigation [208]. Engineered hybrid molecules combining matrikine sequences with targeting moieties could achieve localized immunomodulation.
Hyaluronan oligosaccharides of specific molecular weights have demonstrated ability to modulate macrophage polarization, reduce neutrophil recruitment, and promote tissue repair in experimental models of skin inflammation and wound healing [209]. The small leucine-rich proteoglycan decorin exerts anti-inflammatory and anti-fibrotic effects by sequestering TGF-β, modulating collagen fibrillogenesis, and regulating immune cell behavior [210]. Recombinant decorin and decorin-derived peptides are being explored as therapeutics for fibrotic skin diseases and tumor immunotherapy enhancement [210].
DAMPs derived from matrikines during tissue injury or inflammation constitute critical initiators and amplifiers of immune responses. Low-molecular-weight hyaluronan fragments, fibronectin fragments containing extra domain A (EDA-FN), and versican fragments all signal through pattern recognition receptors such as TLR2, TLR4, and RAGE to activate innate immunity [12,18,211]. Therapeutic strategies to neutralize these ECM-derived DAMPs include antibodies targeting specific fragments, decoy receptors, or inhibitors of the proteases that generate immunostimulatory fragments [84]. Such approaches have shown promise in attenuating chronic inflammation in preclinical models of wound healing disorders and autoimmune skin diseases.
Other ECM-derived peptides with immunomodulatory activity include matrikines generated from collagen (such as Pro-Gly-Pro tripeptides), elastin (elastin-derived peptides), and laminin (laminin-derived sequences like YIGSR and IKVAV) [208,212,213]. While most therapeutic development has focused on wound healing and tissue regeneration, the immunomodulatory properties of these bioactive fragments suggest potential applications in inflammatory dermatoses.
Future matrikine-based therapeutics hold promise for treating inflammatory disorders through targeted immunomodulation with reduced systemic side effects. Clinical development should prioritize synthetic matrikines or antagonists that either promote tissue repair via M2 macrophage polarization or block DAMP-mediated inflammation through pattern recognition receptor inhibition. Engineered hybrid molecules combining matrikine sequences with tissue-targeting moieties could enable localized delivery, while specific molecular weight hyaluronan oligosaccharides and decorin-derived peptides show potential for modulating fibrotic diseases and chronic inflammation. Therapeutic strategies neutralizing ECM-derived DAMPs, such as antibodies against hyaluronan fragments, fibronectin EDA domains, or versican fragments, along with protease inhibitors preventing immunostimulatory fragment generation, warrant investigation in inflammatory dermatoses and wound healing disorders. Additionally, bioactive peptides from collagen, elastin, and laminin merit translation from regenerative medicine to immunomodulatory applications in skin disease, particularly given their demonstrated ability to regulate innate immunity while promoting physiologic tissue repair.
7.6. Targeting YAP/TAZ Mechanosensitive Signaling Pathway
The mechanical properties of the ECM have emerged as powerful regulators of inflammation through mechanotransduction pathways, particularly involving YAP/TAZ signaling. Therapeutic strategies such as restoring dermal mechanical properties through ECM scaffolds or crosslinking agents, directly enhancing YAP/TAZ activity through mechanical or biochemical means, or pharmacologically inhibiting STING signaling represent promising approaches for combating age-related skin dysfunction and reversing the inflammatory milieu that perpetuates tissue deterioration.
Future interventions could focus on biomaterial scaffolds engineered to restore optimal tissue stiffness and ECM architecture, thereby reactivating mechanotransduction signaling in aged skin dermis. Small molecule activators of YAP/TAZ or inhibitors of their negative regulators (such as LATS1/2 kinases) may enhance fibroblast function and tissue repair capacity. Additionally, combination approaches integrating mechanical restoration through crosslinking agents or topical formulations that modulate substrate stiffness alongside pharmacological STING pathway inhibition could synergistically reduce chronic inflammation while promoting tissue regeneration.
7.7. Elimination of Senescent Fibroblasts
Elimination of senescent fibroblasts through senolytic agents (compounds that selectively induce apoptosis in senescent cells) or suppression of the SASP through senomorphic compounds (which inhibit SASP secretion without killing senescent cells) represents a promising therapeutic strategy for mitigating chronic inflammation and age-related skin disorders [214,215]. Preclinical studies have demonstrated that senolytic combinations such as dasatinib plus quercetin, or novel agents like fisetin and navitoclax, can effectively clear senescent cells and improve tissue function in aged skin models [216]. Similarly, senomorphic approaches targeting NF-κB signaling, mTOR, or JAK/STAT pathways have shown efficacy in suppressing SASP production [217]. Topical and systemic delivery of these agents is under investigation for treating photoaging, chronic wounds, and inflammatory dermatoses, with early clinical trials showing encouraging results in improving skin appearance, reducing inflammatory markers, and enhancing wound healing capacity in aged individuals [217,218].
Based on these findings, future therapeutic strategies should focus on developing targeted senolytic and senomorphic interventions for age-related skin conditions. Priority areas include optimizing topical and systemic delivery systems for compounds like dasatinib-quercetin combinations, fisetin, and navitoclax to enhance skin penetration and bioavailability while minimizing systemic side effects. Additionally, developing biomarkers to monitor senescent cell burden and SASP activity in skin tissue would enable personalized treatment approaches and real-time assessment of therapeutic efficacy. Combination strategies that integrate senolytics with senomorphics targeting specific pathways (NF-κB, mTOR, JAK/STAT) may offer synergistic benefits, warranting investigation in both preclinical models and clinical settings to maximize anti-inflammatory effects while preserving tissue regenerative capacity.
8. Future Directions
The matrisome represents far more than the sum of its molecular components ECM and ECM-associated proteins. As an integrated system, it serves as a critical interface between tissue structure and immune function, translating physical and biochemical changes in the tissue environment into inflammatory responses and, conversely, allowing inflammatory processes to reshape tissue architecture. Advancing our understanding of these intricate relationships holds transformative potential for treating inflammatory diseases, fibrosis, cancer, aging-related disorders, and regenerative medicine applications. The coming years promise exciting discoveries as technological advances enable unprecedented resolution of matrisome-immune interactions, mechanistic insights drive therapeutic innovation, and clinical translation brings hope to patients suffering from conditions rooted in dysregulated matrisome-inflammation crosstalk. The future of immunology and tissue biology lies in appreciating the matrisome not as a passive backdrop, but as an active protagonist in the drama of inflammation and disease.
8.1. High-Resolution Mapping of Matrisome-Immune Interactions
Future research must leverage advanced technologies to achieve spatiotemporal resolution of matrisome-immune cell interactions at the molecular level. Single-cell RNA sequencing combined with spatial transcriptomics can reveal how individual immune cell subsets respond to specific matrisome components within distinct tissue microenvironments. Multiplexed imaging technologies enabling simultaneous visualization of dozens of matrisome proteins, immune markers, and signaling molecules will provide unprecedented insight into the architectural organization of inflammatory niches. Mass spectrometry-based proteomics can comprehensively catalog matrikine species generated during different phases of inflammation, identifying novel bioactive fragments and their receptor targets. Such high-resolution mapping will enable identification of tissue-specific and disease-specific matrisome signatures that could serve as diagnostic biomarkers and therapeutic targets.
8.2. Mechanobiology of Inflammation
The mechanical regulation of inflammation through matrisome properties requires deeper investigation. Key questions include: How do specific changes in matrix stiffness, viscoelasticity, and topography influence different immune cell subsets? What are the precise molecular mechanisms linking mechanical sensing through integrins, YAP/TAZ, Piezo channels, and other mechanosensors to inflammatory gene expression programs? How do aging-related changes in dermal mechanical properties contribute to immunosenescence and inflammaging? Can therapeutic restoration of tissue mechanical properties reverse pathological inflammation? Advanced biomaterials with tunable mechanical properties, combined with real-time imaging of mechanotransduction events and genetic manipulation of mechanosensitive pathways, will be essential for addressing these questions. Understanding mechanobiology could reveal entirely new classes of anti-inflammatory interventions based on manipulating tissue physical properties rather than blocking specific molecular targets.
8.3. Matrikine Signaling Networks
The signaling mechanisms and receptor repertoires for matrikines remain incompletely characterized. Many matrikines likely act through multiple receptors and exhibit context-dependent activities that vary with concentration, co-presentation with other signals, and cellular phenotype. Future research should systematically identify receptors for known matrikines, elucidate downstream signaling cascades, and determine how matrikine signals integrate with cytokine and growth factor pathways. Structure-function studies can identify minimal bioactive sequences within matrikines, enabling development of synthetic peptides or peptidomimetics as therapeutic agents. Understanding whether specific matrikines exhibit tissue-specific or cell-type-specific activities could enable targeted immunomodulation. Additionally, investigating how post-translational modifications of matrikines (glycosylation, sulfation, phosphorylation) alter their biological activities represents an unexplored frontier.
9. Conclusions
The matrisome has emerged as a dynamic and multifaceted regulator of inflammation, fundamentally reshaping our understanding of tissue immunity and inflammatory disease pathogenesis. Far from serving merely as a passive structural scaffold, the matrisome actively orchestrates immune responses through diverse mechanisms: integrin-mediated cellular interactions that guide immune cell trafficking and activation, mechanical signaling that translates tissue stiffness into inflammatory programs, bioactive matrikines that function as damage signals and immunomodulators, and sequestration of cytokines and growth factors that create localized inflammatory microenvironments.
The bidirectional relationship between matrisome and inflammation represents a critical paradigm in modern immunology. Inflammatory mediators drive extensive matrisome remodeling through altered synthesis, increased proteolytic degradation, and chemical modification of ECM components. Reciprocally, this remodeled matrisome perpetuates and amplifies inflammatory responses by releasing pro-inflammatory fragments, exposing cryptic epitopes, altering mechanical properties that activate mechanosensitive pathways, and creating tissue microenvironments that sustain pathological immune cell phenotypes. This creates self-reinforcing cycles that underline chronic inflammatory diseases, fibrosis, impaired wound healing, and aging-related tissue dysfunction.
Understanding the immunomodulatory roles of matrisome provides important insights into numerous therapeutic opportunities. Targeting matrisome, modulating matrisome-immune interactions, inhibiting pathological ECM remodeling, and manipulating ECM properties represent promising strategies for treating inflammatory diseases. As technologies advance and our understanding deepens, matrisome-directed therapies may achieve more precise control of tissue inflammation with improved efficacy and safety profiles. The integration of advanced technologies including matrisome proteomics combine with single-cell omics, and spatial transcriptomics will accelerate progress toward these goals. Ultimately, recognizing matrisome as dynamic immunomodulators rather than passive structural protein fundamentally reshapes our understanding of inflammation and offers hope for improved therapeutic strategies.
Funding
This work was supported by the National Institute of Health (RO1ES014697, ES01469730S1, RO1AG081805, RO1AG083378), and the Dermatology Foundation Research grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. .
Acknowledgments
The author thanks Gary J. Fisher and the Department of Dermatology, University of Michigan, for their support. The author thanks Hehui Quan for editorial support.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Sutherland, T.E.; Dyer, D.P.; Allen, J.E. The extracellular matrix and the immune system: A mutually dependent relationship. Science 2023, 379, eabp8964. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 2012, 4, a004903. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the "omics" era. Matrix Biol 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Naba, A.; Hoersch, S.; Hynes, R.O. Towards definition of an ecm parts list: An advance on go categories. Matrix Biol 2012, 31, 371–372. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular matrix and dermal fibroblast function in the healing wound. Adv Wound Care (New Rochelle) 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Maquart, F.X.; Pasco, S.; Ramont, L.; Hornebeck, W.; Monboisse, J.C. An introduction to matrikines: Extracellular matrix-derived peptides which regulate cell activity - implication in tumor invasion. Crit Rev Oncol Hemat 2004, 49, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nature Reviews Immunology 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J Histochem Cytochem 2018, 66, 213–227. [Google Scholar] [CrossRef]
- Boyd, D.F.; Thomas, P.G. Towards integrating extracellular matrix and immunological pathways. Cytokine 2017, 98, 79–86. [Google Scholar] [CrossRef]
- Quan, T.; Xia, W.; He, T.; Calderone, K.; Bou-Gharios, G.; Voorhees, J.J.; Dlugosz, A.A.; Fisher, G.J. Matrix metalloproteinase-1 expression in fibroblasts accelerates dermal aging and promotes papilloma development in mouse skin. J Invest Dermatol 2023, 143, 1700–1707 e1701. [Google Scholar] [CrossRef]
- Quan, T. Molecular insights of human skin epidermal and dermal aging. J Dermatol Sci 2023, 112, 48–53. [Google Scholar] [CrossRef]
- McCabe MC, H.R.; Calderone, K; Cui, Y; Yan, Y; Quan, T; Fisher, GJ; Hansen, KC. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biology Plus, in press 2020. [Google Scholar]
- Pfisterer, K.; Shaw, L.E.; Symmank, D.; Weninger, W. The extracellular matrix in skin inflammation and infection. Front Cell Dev Biol 2021, 9, 682414. [Google Scholar] [CrossRef]
- Bale, S.; Verma, P.; Varga, J.; Bhattacharyya, S. Extracellular matrix-derived damage-associated molecular patterns (damp): Implications in systemic sclerosis and fibrosis. Journal of Investigative Dermatology 2023, 143, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, O.; Ayyangar, U.; Kurbet, A.S.; Ashok, D.; Raghavan, S. Unraveling the ecm-immune cell crosstalk in skin diseases. Frontiers in Cell and Developmental Biology 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Sachs, D.L.; Voorhees, J.J. Ageing: Collagenase-mediated collagen fragmentation as a rejuvenation target. Br J Dermatol 2014, 171, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.P. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 2003, 57, 195–202. [Google Scholar] [CrossRef]
- Gould, L.; Abadir, P.; Brem, H.; Carter, M.; Conner-Kerr, T.; Davidson, J.; DiPietro, L.; Falanga, V.; Fife, C.; Gardner, S.; et al. Chronic wound repair and healing in older adults: Current status and future research. J Am Geriatr Soc 2015, 63, 427–438. [Google Scholar] [CrossRef]
- Khalid, K.A.; Nawi, A.F.M.; Zulkifli, N.; Barkat, M.A.; Hadi, H. Aging and wound healing of the skin: A review of clinical and pathophysiological hallmarks. Life (Basel) 2022, 12. [Google Scholar] [CrossRef]
- Dotto, G.P. Multifocal epithelial tumors and field cancerization: Stroma as a primary determinant. J Clin Invest 2014, 124, 1446–1453. [Google Scholar] [CrossRef]
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol 2011, 21, 736–744. [Google Scholar] [CrossRef]
- Moreno-Layseca, P.; Icha, J.; Hamidi, H.; Ivaska, J. Integrin trafficking in cells and tissues. Nature Cell Biology 2019, 21, 122–132. [Google Scholar] [CrossRef]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider's guide to leukocyte integrin signalling and function. Nature Reviews Immunology 2011, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Blythe, E.N.; Weaver, L.C.; Brown, A.; Dekaban, G.A. Β2 integrin cd11d/cd18: From expression to an emerging role in staged leukocyte migration. Front Immunol 2021, 12, 775447. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. Β2 integrins-multi-functional leukocyte receptors in health and disease. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Nordenfelt, P.; Moore, T.I.; Mehta, S.B.; Kalappurakkal, J.M.; Swaminathan, V.; Koga, N.; Lambert, T.J.; Baker, D.; Waters, J.C.; Oldenbourg, R.; et al. Direction of actin flow dictates integrin lfa-1 orientation during leukocyte migration. Nature Communications 2017, 8, 2047. [Google Scholar] [CrossRef]
- Shi, H.; Shao, B. Lfa-1 activation in t-cell migration and immunological synapse formation. Cells 2023, 12, 1136. [Google Scholar] [CrossRef]
- Evans, R.; Patzak, I.; Svensson, L.; De Filippo, K.; Jones, K.; McDowall, A.; Hogg, N. Integrins in immunity. Journal of Cell Science 2009, 122, 215–225. [Google Scholar] [CrossRef]
- Chan, P.Y.; Aruffo, A. Vla-4 integrin mediates lymphocyte migration on the inducible endothelial cell ligand vcam-1 and the extracellular matrix ligand fibronectin. J Biol Chem 1993, 268, 24655–24664. [Google Scholar] [CrossRef] [PubMed]
- Hauzenberger, D.; Klominek, J.; Sundqvist, K.G. Functional specialization of fibronectin-binding beta 1-integrins in t lymphocyte migration. The Journal of Immunology 1994, 153, 960–971. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, Y.; Ji, Q.; Fu, S.; Gu, J.; Tai, N.; Wang, X. Interplay between extracellular matrix and neutrophils in diseases. J Immunol Res 2021, 2021, 8243378. [Google Scholar] [CrossRef]
- Ong, C.W.; Elkington, P.T.; Brilha, S.; Ugarte-Gil, C.; Tome-Esteban, M.T.; Tezera, L.B.; Pabisiak, P.J.; Moores, R.C.; Sathyamoorthy, T.; Patel, V.; et al. Neutrophil-derived mmp-8 drives ampk-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog 2015, 11, e1004917. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overall, C.M. Macrophage-specific metalloelastase (mmp-12) truncates and inactivates elr+ cxc chemokines and generates ccl2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef]
- Esparza, J.; Vilardell, C.; Calvo, J.; Juan, M.; Vives, J.; Urbano-Márquez, A.; Yagüe, J.; Cid, M.C. Fibronectin upregulates gelatinase b (mmp-9) and induces coordinated expression of gelatinase a (mmp-2) and its activator mt1-mmp (mmp-14) by human t lymphocyte cell lines. A process repressed through ras/map kinase signaling pathways. Blood 1999, 94, 2754–2766. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Müller, R.; Borgmann, S.; Bröcker, E.B.; Friedl, P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by mmps and other proteases. Blood 2003, 102, 3262–3269. [Google Scholar] [CrossRef]
- Ullo, M.F.; D’Amico, A.E.; Lavenus, S.B.; Logue, J.S. The amoeboid migration of monocytes in confining channels requires the local remodeling of the cortical actin cytoskeleton by cofilin-1. Sci Rep-Uk 2024, 14, 10241. [Google Scholar] [CrossRef]
- Crijns, H.; Vanheule, V.; Proost, P. Targeting chemokine-glycosaminoglycan interactions to inhibit inflammation. Front Immunol 2020, 11, 483. [Google Scholar] [CrossRef]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory role of the extracellular matrix within the liver disease microenvironment. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines (vol 41, pg 14, 2014). Immunity 2014, 41, 339–340. [Google Scholar] [CrossRef]
- Wynn, T.A. Type 2 cytokines: Mechanisms and therapeutic strategies. Nature Reviews Immunology 2015, 15, 271–282. [Google Scholar] [CrossRef]
- Tchougounova, E.; Lundequist, A.; Fajardo, I.; Winberg, J.O.; Åbrink, M.; Pejler, G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. Journal of Biological Chemistry 2005, 280, 9291–9296. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative stress and inflammation: What polyphenols can do for us? Oxidative Medicine and Cellular Longevity 2016, 2016, 7432797. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Link, A.; Vogt, T.K.; Favre, S.; Britschgi, M.R.; Acha-Orbea, H.; Hinz, B.; Cyster, J.G.; Luther, S.A. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive t cells. Nature Immunology 2007, 8, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.S.; et al. Recruitment of stem and progenitor cells from the bone marrow niche requires mmp-9 mediated release of kit-ligand. Cell 2002, 109, 625–637. [Google Scholar] [CrossRef]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grünewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. Journal of Experimental Medicine 2005, 201, 1781–1791. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Orend, G.; Chiquet, M.; Tucker, R.P.; Midwood, K.S. Tenascins in stem cell niches. Matrix Biology 2014, 37, 112–123. [Google Scholar] [CrossRef]
- Di Vito, A.; Donato, A.; Bria, J.; Conforti, F.; La Torre, D.; Malara, N.; Donato, G. Extracellular matrix structure and interaction with immune cells in adult astrocytic tumors. Cell Mol Neurobiol 2024, 44. [Google Scholar] [CrossRef]
- Huse, M. Mechanical forces in the immune system. Nature Reviews Immunology 2017, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biology 2018, 68-69, 81-93. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Mechanisms of disease the pathogenesis of rheumatoid arthritis. New Engl J Med 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Trouw, L.A.; Rispens, T.; Toes, R.E.M. Beyond citrullination: Other post-translational protein modifications in rheumatoid arthritis. Nature Reviews Rheumatology 2017, 13, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Research 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of t cells into the stroma of human lung tumors. Journal of Clinical Investigation 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Chitty, J.L.; Yam, M.; Perryman, L.; Parker, A.L.; Skhinas, J.N.; Setargew, Y.F.I.; Mok, E.T.Y.; Tran, E.; Grant, R.D.; Latham, S.L.; et al. A first-in-class pan-lysyl oxidase inhibitor impairs stromal remodeling and enhances gemcitabine response and survival in pancreatic cancer. Nat Cancer 2023, 4, 1326–+. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.M.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Sladitschek-Martens, H.L.; Guarnieri, A.; Brumana, G.; Zanconato, F.; Battilana, G.; Xiccato, R.L.; Panciera, T.; Forcato, M.; Bicciato, S.; Guzzardo, V.; et al. Yap/taz activity in stromal cells prevents ageing by controlling cgas-sting. Nature 2022, 607, 790–798. [Google Scholar] [CrossRef]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of matrix metalloproteinases in angiogenesis and cancer. Front Oncol 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, L.; Kähäri, V.M. Matrix metalloproteinases in inflammation. Bba-Gen Subjects 2014, 1840, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. Mmp generated matrikines. Matrix Biol 2015, 44-46, 122–129. [Google Scholar] [CrossRef]
- Köhrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (mmps) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. Bmc Cancer 2009, 9. [Google Scholar] [CrossRef]
- de Almeida, L.G.N.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix metalloproteinases: From molecular mechanisms to physiology, pathophysiology, and pharmacology. Pharmacological Reviews 2022, 74, 714–770. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J Investig Dermatol Symp Proc 2009, 14, 20–24. [Google Scholar] [CrossRef]
- Siwik, D.A.; Chang, D.L.F.; Colucci, W.S. Interleukin-1β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circulation Research 2000, 86, 1259–1265. [Google Scholar] [CrossRef]
- Dasu, M.R.; Barrow, R.E.; Spies, M.; Herndon, D.N. Matrix metalloproteinase expression in cytokine stimulated human dermal fibroblasts. Burns 2003, 29, 527–531. [Google Scholar] [CrossRef]
- Cortez, D.M.; Feldman, M.D.; Mummidi, S.; Valente, A.J.; Steffensen, B.; Vincenti, M.; Barnes, J.L.; Chandrasekar, B. Il-17 stimulates mmp-1 expression in primary human cardiac fibroblasts via p38 mapk- and erk1/2-dependent c/ebp-beta, nf-kappab, and ap-1 activation. Am J Physiol Heart Circ Physiol 2007, 293, H3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Becker-Pauly, C.; Rose-John, S. Tnfα cleavage beyond tace/adam17: Matrix metalloproteinase 13 is a potential therapeutic target in sepsis and colitis. EMBO Molecular Medicine 2013, 5, EMMM201302899. [Google Scholar] [CrossRef] [PubMed]
- Elkington, P.T.; O'Kane, C.M.; Friedland, J.S. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol 2005, 142, 12–20. [Google Scholar] [CrossRef]
- Liu, J.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigational and therapeutic tools in unrestrained tissue remodeling and pathological disorders. Prog Mol Biol Transl Sci 2017, 148, 355–420. [Google Scholar]
- Kazemi, A.; Fathy, M.; Jahanian, A.; Khanali, J.; Ostadi, Y.; Babajani, A.; Tayebi, T.; Niknejad, H. The role of mmps and timps in regenerative medicine: From pathological ecm remodeling to therapeutic applications. Biomed Pharmacother 2025, 191, 118457. [Google Scholar] [CrossRef] [PubMed]
- Caley, M.P.; Martins, V.L.; O'Toole, E.A. Metalloproteinases and wound healing. Adv Wound Care (New Rochelle) 2015, 4, 225–234. [Google Scholar] [CrossRef]
- Leong, E.; Bezuhly, M.; Marshall, J.S. Distinct metalloproteinase expression and functions in systemic sclerosis and fibrosis: What we know and the potential for intervention. Front Physiol 2021, 12, 727451. [Google Scholar] [CrossRef]
- Jariwala, N.; Ozols, M.; Bell, M.; Bradley, E.; Bradley, E.; Gilmore, A.; Debelle, L.; Sherratt, M.J. Matrikines as mediators of tissue remodelling. Adv Drug Deliver Rev 2022, 185. [Google Scholar] [CrossRef] [PubMed]
- Adair-Kirk, T.L.; Senior, R.M. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell B 2008, 40, 1101–1110. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biology 2019, 75-76, 170-189. [Google Scholar] [CrossRef]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. Journal of Cell Science 2007, 120, 1955–1958. [Google Scholar] [CrossRef]
- Quan, T.; Qin, Z.; He, T.; Fisher, G.J. Integrin alpha11beta1 as a key collagen receptor in human skin dermis: Insight into fibroblast function and skin dermal aging. J Invest Dermatol 2025, 145, 2449–2463. [Google Scholar] [CrossRef] [PubMed]
- Senior, R.M.; Hinek, A.; Griffin, G.L.; Pipoly, D.J.; Crouch, E.C.; Mecham, R.P. Neutrophils show chemotaxis to type-iv collagen and its 7s domain and contain a 67-kd type-iv collagen binding-protein with lectin properties. Am J Resp Cell Mol 1989, 1, 479–487. [Google Scholar] [CrossRef]
- Jensen, C.; Sinkeviciute, D.; Madsen, D.H.; Önnerfjord, P.; Hansen, M.; Schmidt, H.; Karsdal, M.A.; Svane, I.M.; Willumsen, N. Granzyme b degraded type iv collagen products in serum identify melanoma patients responding to immune checkpoint blockade. Cancers 2020, 12, 2786. [Google Scholar] [CrossRef] [PubMed]
- Alexdottir, M.S.; Pehrsson, M.; Domislovic, V.; Godskesen, L.E.; Krag, A.; Kjeldsen, J.; Brinar, M.; Barisic, A.; Bay-Jensen, A.-C.; Karsdal, M.A.; et al. Neutrophil-mediated type iv collagen degradation is elevated in patients with mild endoscopic ulcerative colitis reflecting early mucosal destruction. Sci Rep-Uk 2024, 14, 1641. [Google Scholar] [CrossRef]
- Walia, A.; Yang, J.F.; Huang, Y.H.; Rosenblatt, M.I.; Chang, J.H.; Azar, D.T. Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim Biophys Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.I.I.I. The extra domain a of fibronectin activates toll-like receptor 4. Journal of Biological Chemistry 2001, 276, 10229–10233. [Google Scholar] [CrossRef]
- Lasarte, J.J.; Casares, N.; Gorraiz, M.; Hervás-Stubbs, S.; Arribillaga, L.; Mansilla, C.; Durantez, M.; Llopiz, D.; Sarobe, P.; Borrás-Cuesta, F.; et al. The extra domain a from fibronectin targets antigens to tlr4-expressing cells and induces cytotoxic t cell responses in vivo. Journal of Immunology 2007, 178, 748–756. [Google Scholar] [CrossRef]
- White, E.S.; Muro, A.F. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011, 63, 538–546. [Google Scholar] [CrossRef]
- Gondokaryono, S.P.; Ushio, H.; Niyonsaba, F.; Hara, M.; Takenaka, H.; Jayawardana, S.T.M.; Ikeda, S.; Okumura, K.; Ogawa, H. The extra domain a of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukocyte Biol 2007, 82, 657–665. [Google Scholar] [CrossRef]
- Lefebvre, J.S.; Lévesque, T.; Picard, S.; Paré, G.; Gravel, A.; Flamand, L.; Borgeat, P. Extra domain a of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of toll-like receptor 4. Arthritis Rheum 2011, 63, 1527–1533. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. Journal of Cell Science 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed]
- Barilla, M.L.; Carsons, S.E. Fibronectin fragments and their role in inflammatory arthritis. Semin Arthritis Rheu 2000, 29, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Grinnell, F.; Zhu, M. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, alpha1-proteinase inhibitor, and alpha2-macroglobulin. J Invest Dermatol 1996, 106, 335–341. [Google Scholar] [CrossRef]
- Grinnell, F.; Ho, C.H.; Wysocki, A. Degradation of fibronectin and vitronectin in chronic wound fluid: Analysis by cell blotting, immunoblotting, and cell adhesion assays. J Invest Dermatol 1992, 98, 410–416. [Google Scholar] [CrossRef]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular size-dependent signaling and biological functions in inflammation and cancer. 2019, 286, 2883–2908. [Google Scholar] [CrossRef]
- Petrey, A.C.; de la Motte, C.A. Hyaluronan, a crucial regulator of inflammation. Front Immunol 2014, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging tlr2. J Immunol 2006, 177, 1272–1281. [Google Scholar] [CrossRef]
- Žádníková, P.; Šínová, R.; Pavlík, V.; Šimek, M.; Šafránková, B.; Hermannová, M.; Nešporová, K.; Velebný, V. The degradation of hyaluronan in the skin. Biomolecules 2022, 12, 251. [Google Scholar] [CrossRef]
- Terazawa, S.; Nakajima, H.; Tobita, K.; Imokawa, G. The decreased secretion of hyaluronan by older human fibroblasts under physiological conditions is mainly associated with the down-regulated expression of hyaluronan synthases but not with the expression levels of hyaluronidases. Cytotechnology 2015, 67, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiological Reviews 2011, 91, 221–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Meng, X.G.; Guo, Z.K. Elastin structure, synthesis, regulatory mechanism and relationship with cardiovascular diseases. Frontiers in Cell and Developmental Biology 2021, 9. [Google Scholar] [CrossRef]
- Duca, L.; Floquet, N.; Alix, A.J.P.; Haye, B.; Debelle, L. Elastin as a matrikine. Crit Rev Oncol Hemat 2004, 49, 235–244. [Google Scholar] [CrossRef]
- Le Page, A.; Khalil, A.; Vermette, P.; Frost, E.H.; Larbi, A.; Witkowski, J.M.; Fulop, T. The role of elastin-derived peptides in human physiology and diseases. Matrix Biology 2019, 84, 81–96. [Google Scholar] [CrossRef]
- Tembely, D.; Henry, A.; Vanalderwiert, L.; Toussaint, K.; Bennasroune, A.; Blaise, S.; Sartelet, H.; Jaisson, S.; Gales, C.; Martiny, L.; et al. The elastin receptor complex: An emerging therapeutic target against age-related vascular diseases. Front Endocrinol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Brassart, B.; Hinek, A. Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. Journal of Biological Chemistry 2002, 277, 44854–44863. [Google Scholar] [CrossRef]
- Scandolera, A.; Odoul, L.; Salesse, S.; Guillot, A.; Blaise, S.; Kawecki, C.; Maurice, P.; El Btaouri, H.; Romier-Crouzet, B.; Martiny, L.; et al. The elastin receptor complex: A unique matricellular receptor with high anti-tumoral potential. Frontiers in Pharmacology 2016, 7. [Google Scholar] [CrossRef]
- Cantor, J. Elastin peptides as a potential disease vector in the pathogenesis of pulmonary emphysema: An investigation of this hypothesis. Life 2025, 15, 356. [Google Scholar] [CrossRef]
- Mecham, R.P. Elastin in lung development and disease pathogenesis. Matrix Biol 2018, 73, 6–20. [Google Scholar] [CrossRef]
- Yi, J.Z.; Wang, Y.X.; Sui, H.R.; Chen, Z.C.; Ye, T.N.; Zhong, Y.L.; Qian, J.Y.; Wu, B.B.; Huang, J.Y.; Tian, T.; et al. Elastin-derived extracellular matrix fragments drive aging through innate immune activation. Nature Aging 2025. [Google Scholar] [CrossRef]
- Jin, S.M.; Elisseeff, J.H. Aging as a glitch in the matrix. In Nature Aging; 2025. [Google Scholar]
- Gill, S.; Wight, T.N.; Frevert, C.W. Proteoglycans: Key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat Rec (Hoboken) 2010, 293, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Rajarathnam, K.; Desai, U.R. Structural insights into how proteoglycans determine chemokine-cxcr1/cxcr2 interactions: Progress and challenges. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Parks, W.C.; Park, P.W. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered cxc chemokines. Blood 2009, 114, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Nastase, M.V.; Janicova, A.; Roedig, H.; Hsieh, L.T.; Wygrecka, M.; Schaefer, L. Small leucine-rich proteoglycans in renal inflammation: Two sides of the coin. J Histochem Cytochem 2018, 66, 261–272. [Google Scholar] [CrossRef]
- Merline, R.; Moreth, K.; Beckmann, J.; Nastase, M.V.; Zeng-Brouwers, J.; Tralhão, J.G.; Lemarchand, P.; Pfeilschifter, J.; Schaefer, R.M.; Iozzo, R.V.; et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through pdcd4 and microrna-21. Sci Signal 2011, 4, ra75. [Google Scholar] [CrossRef]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J Histochem Cytochem 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Lohr, K.; Sardana, H.; Lee, S.; Wu, F.; Huso, D.L.; Hamad, A.R.; Chakravarti, S. Extracellular matrix protein lumican regulates inflammation in a mouse model of colitis. Inflamm Bowel Dis 2012, 18, 143–151. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Merrilees, M.J. Versican and the control of inflammation. Matrix Biol 2014, 35, 152–161. [Google Scholar]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-a critical extracellular matrix regulator of immunity and inflammation. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Eun, K.; Kim, A.Y.; Ryu, S. Matricellular proteins in immunometabolism and tissue homeostasis. Bmb Rep 2024, 57, 400–416. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The ccn family of proteins: Structure–function relationships. Trends in Biochemical Sciences 2008, 33, 461–473. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the ccn family: Essential matricellular signaling modulators emerge from the bunker. Journal of Cell Science 2006, 119, 4803–4810. [Google Scholar] [CrossRef]
- Kular, L.; Pakradouni, J.; Kitabgi, P.; Laurent, M.; Martinerie, C. The ccn family: A new class of inflammation modulators? Biochimie 2011, 93, 377–388. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: Ccn proteins as emerging therapeutic targets. Nat Rev Drug Discov 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.Q.; Tang, J.F.; Zhang, X.H.; He, Y.J.; Yu, Z.Q.; Chen, S.H.; Xia, J.F.; Lin, J.P.; Ou, Q.S. Ccn1 promotes inflammation by inducing il-6 production α6β1/pi3k/akt/nf-κb pathway in autoimmune hepatitis. Frontiers in Immunology 2022, 13. [Google Scholar] [CrossRef]
- Bai, T.; Chen, C.C.; Lau, L.F. Matricellular protein ccn1 activates a proinflammatory genetic program in murine macrophages. J Immunol 2010, 184, 3223–3232. [Google Scholar] [CrossRef]
- Leask, A. Conjunction junction, what’s the function? Ccn proteins as targets in fibrosis and cancers. Am J Physiol-Cell Ph 2020, 318, C1046–C1054. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel hippo pathway component that inhibits yap oncoprotein. Genes Dev 2011, 25, 51–63. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K. Tenascin-c in heart diseases-the role of inflammation. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-c is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Midwood, K.S.; Orend, G. The role of tenascin-c in tissue injury and tumorigenesis. J Cell Commun Signal 2009, 3, 287–310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Olonisakin, T.F.; Xiong, Z.; Hulver, M.; Sayeed, S.; Yu, M.T.; Gregory, A.D.; Kochman, E.J.; Chen, B.B.; Mallampalli, R.K.; et al. Thrombospondin-1 restrains neutrophil granule serine protease function and regulates the innate immune response during klebsiella pneumoniae infection. Mucosal Immunology 2015, 8, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.W.; Liu, C.; Li, Y.J.; Wang, W.F.; Dou, Q.L. The dual role of thrombospondin-1 in inflammatory regulation during acute respiratory distress syndrome: A mini-review. Frontiers in Immunology 2025, 16. [Google Scholar] [CrossRef]
- Di, X.; Gao, X.; Peng, L.; Ai, J.; Jin, X.; Qi, S.; Li, H.; Wang, K.; Luo, D. Cellular mechanotransduction in health and diseases: From molecular mechanism to therapeutic targets. Signal Transduction and Targeted Therapy 2023, 8, 282. [Google Scholar] [CrossRef]
- Yusko, E.C.; Asbury, C.L. Force is a signal that cells cannot ignore. Mol Biol Cell 2014, 25, 3717–3725. [Google Scholar] [CrossRef] [PubMed]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating extracellular matrix stiffness: A strategic approach to boost cancer immunotherapy. Cell Death Dis 2024, 15, 307. [Google Scholar] [CrossRef]
- Jiang, T.; Zheng, M.T.; Li, R.M.; Ouyang, N.J. The effects of matrix stiffness on immune cells in bone biology. Mechanobiol Med 2024, 2. [Google Scholar] [CrossRef]
- Tiskratok, W.; Chuinsiri, N.; Limraksasin, P.; Kyawsoewin, M.; Jitprasertwong, P. Extracellular matrix stiffness: Mechanotransduction and mechanobiological response-driven strategies for biomedical applications targeting fibroblast inflammation. Polymers 2025, 17, 822. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Müller, D.; Clark, A.G. Mechanosensory feedback loops during chronic inflammation. Front Cell Dev Biol 2023, 11, 1225677. [Google Scholar] [CrossRef]
- Mei, F.; Guo, Y.; Wang, Y.; Zhou, Y.; Heng, B.C.; Xie, M.; Huang, X.; Zhang, S.; Ding, S.; Liu, F.; et al. Matrix stiffness regulates macrophage polarisation via the piezo1-yap signalling axis. Cell Prolif 2024, 57, e13640. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.; Heyward, C.; Cameron, J.; Leifer, C. Toll-like receptor signaling in macrophages is regulated by extracellular substrate stiffness and rho-associated coiled-coil kinase (rock1/2). Int Immunol 2018, 30, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Dolgova, N.V.; Chancellor, T.J.; Acharya, A.P.; Karpiak, J.V.; Lele, T.P.; Keselowsky, B.G. The effect of cyclic mechanical strain on activation of dendritic cells cultured on adhesive substrates. Biomaterials 2013, 34, 9063–9070. [Google Scholar] [CrossRef]
- Lee, M.G.; Du, H.X.; Winer, D.A.; Clemente-Casares, X.; Tsai, S. Mechanosensing in macrophages and dendritic cells in steady-state and disease. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef]
- Du, H.; Bartleson, J.M.; Butenko, S.; Alonso, V.; Liu, W.F.; Winer, D.A.; Butte, M.J. Tuning immunity through tissue mechanotransduction. Nature Reviews Immunology 2023, 23, 174–188. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T.; Ingber, D.E. Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci 2012, 125, 3061–3073. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. Yap and taz: A nexus for hippo signaling and beyond. Trends Cell Biol 2015, 25, 499–513. [Google Scholar] [CrossRef]
- Meli, V.S.; Veerasubramanian, P.K.; Downing, T.L.; Wang, W.; Liu, W.F. Mechanosensation to inflammation: Roles for yap/taz in innate immune cells. Sci Signal 2023, 16, eadc9656. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through yap and taz drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015, 308, L344–357. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in fibrosis: Tgf-β, wnt, and yap/taz converge. Front Med (Lausanne) 2015, 2, 59. [Google Scholar] [CrossRef]
- Qin, Z.; He, T.; Guo, C.; Quan, T. Age-related downregulation of ccn2 is regulated by cell size in a yap/taz-dependent manner in human dermal fibroblasts: Impact on dermal aging. JID Innov 2022, 2, 100111. [Google Scholar] [CrossRef]
- Chu, C.-Q.; Quan, T. Fibroblast yap/taz signaling in extracellular matrix homeostasis and tissue fibrosis. J Clin Med 2024, 13, 3358. [Google Scholar]
- Pirri, C. Piezo channels in mechano-inflammation: Gatekeepers of neuroimmune crosstalk. Diseases 2025, 13. [Google Scholar] [CrossRef]
- Gao, W.N.; Hasan, H.; Anderson, D.E.; Lee, W.S. The role of mechanically-activated ion channels piezo1, piezo2, and trpv4 in chondrocyte mechanotransduction and mechano-therapeutics for osteoarthritis. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Changotra, H.; Wergedal, J.E.; Kumar, A. Regulation of phosphatidylinositol 3-kinase (pi3k)/akt and nuclear factor-kappa b signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol 2006, 208, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.C.; Pan, Y.L.; Liang, Z.Q.; Yang, G.L.; Wu, C.J.; Zeng, L.; Wang, L.N.; Sun, J.; Liu, M.M.; Yuan, Y.F.; et al. Mechanical stretch promotes macrophage polarization and inflammation via the rhoa-rock/nf-κb pathway. Biomed Res Int 2022, 2022, 6871269. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.K.; Park, H.; Lee, Y.J.; Park, S.H.; Lee, K.J.; Lee, D.G.; Kang, H.; Kim, J.E. Contribution of autophagy-notch1-mediated nlrp3 inflammasome activation to chronic inflammation and fibrosis in keloid fibroblasts. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. Nlrp3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun 2019, 10, 4375. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P.D. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum 2011, 63, 3563–3574. [Google Scholar] [CrossRef]
- Weinberg, J.B. Nitric oxide synthase 2 and cyclooxygenase 2 interactions in inflammation. Immunol Res 2000, 22, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Seed, M.P.; Willoughby, D.A. Cox-2, ho no! Cyclooxygenase-2, heme oxygenase and nitric oxide synthase: Their role and interactions in inflammation. Inflamm Res 1996, 46, 279–281. [Google Scholar] [CrossRef]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Bio 2024, 25, 958–978. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Nguyen, T.Q.T.; Cho, K.A. Targeting immunosenescence and inflammaging: Advancing longevity research. Experimental & Molecular Medicine 2025, 57, 1881–1892. [Google Scholar] [CrossRef]
- Mavrogonatou, E.; Papadopoulou, A.; Pratsinis, H.; Kletsas, D. Senescence-associated alterations in the extracellular matrix: Deciphering their role in the regulation of cellular function. Am J Physiol-Cell Ph 2023, 325, C633–C647. [Google Scholar] [CrossRef]
- Xiang, Y.; Qin, Z.; Yang, Y.; Fisher, G.J.; Quan, T. Age-related elevation of hgf is driven by the reduction of fibroblast size in a yap/taz/ccn2 axis-dependent manner. J Dermatol Sci 2021, 102, 36–46. [Google Scholar] [CrossRef]
- Chu, C.Q.; Quan, T.H. Fibroblast yap/taz signaling in extracellular matrix homeostasis and tissue fibrosis. J Clin Med 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.; Cox, T.R.; Parker, A.L.; Willumsen, N.; Sand, J.M.B.; Jenkins, G.; Hansen, H.H.; Oldenburger, A.; Geillinger-Kaestle, K.E.; Larsen, A.T.; et al. Advances in extracellular matrix-associated diagnostics and therapeutics. J Clin Med 2025, 14. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct Target Ther 2023, 8, 1. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat Rev Drug Discov 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Selewski, D.T.; Shah, G.V.; Segal, B.M.; Rajdev, P.A.; Mukherji, S.K. Natalizumab (tysabri). AJNR Am J Neuroradiol 2010, 31, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Khoy, K.; Mariotte, D.; Defer, G.; Petit, G.; Toutirais, O.; Le Mauff, B. Natalizumab in multiple sclerosis treatment: From biological effects to immune monitoring. Front Immunol 2020, 11, 549842. [Google Scholar] [CrossRef]
- Klotz, L.; Berger, T.; Brownlee, W.J.; Chan, A.; Lycke, J.; Oreja-Guevara, C.; Palavra, F.; Sacca, F.; Sejbaek, T.; Weber, M.S.; et al. Twenty years of natalizumab in multiple sclerosis: Lessons learned and future outlook. Ther Adv Neurol Diso 2025, 18. [Google Scholar] [CrossRef]
- Lebwohl, M.; Tyring, S.K.; Hamilton, T.K.; Toth, D.; Glazer, S.; Tawfik, N.H.; Walicke, P.; Dummer, W.; Wang, X.; Garovoy, M.R.; et al. A novel targeted t-cell modulator, efalizumab, for plaque psoriasis. New Engl J Med 2003, 349, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discov 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- O'Reilly, P.J.; Gaggar, A.; Blalock, J.E. Interfering with extracellular matrix degradation to blunt inflammation. Curr Opin Pharmacol 2008, 8, 242–248. [Google Scholar] [CrossRef]
- Closset, L.; Gultekin, O.; Salehi, S.; Sarhan, D.; Lehti, K.; Gonzalez-Molina, J. The extracellular matrix-immune microenvironment crosstalk in cancer therapy: Challenges and opportunities. Matrix Biology 2023, 121, 217–228. [Google Scholar] [CrossRef]
- Assunção, M.; Dehghan-Baniani, D.; Yiu, C.H.K.; Später, T.; Beyer, S.; Blocki, A. Cell-derived extracellular matrix for tissue engineering and regenerative medicine. Front Bioeng Biotechnol 2020, 8, 602009. [Google Scholar] [CrossRef]
- Qu, F.; Guilak, F.; Mauck, R.L. Cell migration: Implications for repair and regeneration in joint disease. Nature Reviews Rheumatology 2019, 15, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.N.; Zeng, X.Y.; Deng, X.K.; Yang, F.L.; Ma, X.Y.; Gao, W. Smart biomaterials: As active immune modulators to shape pro-regenerative microenvironments. Frontiers in Cell and Developmental Biology 2025, 13. [Google Scholar] [CrossRef]
- Coser, C.; Ghaemmaghami, A.M.; Yang, J. Soft tissue-mimicking hydrogel stiffness modulates polarisation of human monocyte-derived macrophages. Biomater Sci-Uk 2025, 13, 6637–6651. [Google Scholar] [CrossRef] [PubMed]
- Butenko, S.; Nagalla, R.R.; Guerrero-Juarez, C.F.; Palomba, F.; David, L.-M.; Nguyen, R.Q.; Gay, D.; Almet, A.A.; Digman, M.A.; Nie, Q.; et al. Hydrogel crosslinking modulates macrophages, fibroblasts, and their communication, during wound healing. Nature Communications 2024, 15, 6820. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Tan, B.; Zhu, L.; Wang, Y.; Liao, J. A review on the design of hydrogels with different stiffness and their effects on tissue repair. Front Bioeng Biotechnol 2022, 10, 817391. [Google Scholar] [CrossRef]
- Da, L.C.; Huang, Y.Z.; Xie, H.Q.; Zheng, B.H.; Huang, Y.C.; Du, S.R. Membranous extracellular matrix-based scaffolds for skin wound healing. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef]
- Romero, D.J.; Hussey, G.; Capella-Monsonís, H. Immune response to extracellular matrix bioscaffolds: A comprehensive review. Biologics 2025, 5, 28. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Chen, J.; Lee, K.-J.; Donahue, L.R.; Kacaj, D.; McDonough, S.P.; White, A.C. Therapy-induced ecm remodeling creates a transient immune barrier in residual melanoma. Advanced Science 2025, 12, e08451. [Google Scholar] [CrossRef]
- Perryman, L.; Findlay, A.; Baskar, J.; Charlton, B.; Foot, J.; Hamilton, R.; Hamprecht, D.; Joshi, A.; Stolp, J.; Turner, C.; et al. The small molecule loxl2 inhibitor snt-5382 reduces cardiac fibrosis and achieves strong clinical target engagement. Sci Rep-Uk 2025, 15, 22653. [Google Scholar] [CrossRef]
- Mascharak, S.; Guo, J.L.; Griffin, M.; Berry, C.E.; Wan, D.C.; Longaker, M.T. Modelling and targeting mechanical forces in organ fibrosis. Nat Rev Bioeng 2024, 2, 305–323. [Google Scholar]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ecm) stiffness and degradation as cancer drivers. J Cell Biochem 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule loxl2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef]
- Burchard, P.R.; Ruffolo, L.I.; Ullman, N.A.; Dale, B.S.; Dave, Y.A.; Hilty, B.K.; Ye, J.; Georger, M.; Jewell, R.; Miller, C.; et al. Pan-lysyl oxidase inhibition disrupts fibroinflammatory tumor stroma, rendering cholangiocarcinoma susceptible to chemotherapy. Hepatology Communications 2024, 8, e0502. [Google Scholar] [CrossRef]
- Badarau, E.; Wang, Z.; Rathbone, D.L.; Costanzi, A.; Thibault, T.; Murdoch, C.E.; El Alaoui, S.; Bartkeviciute, M.; Griffin, M. Development of potent and selective tissue transglutaminase inhibitors: Their effect on tg2 function and application in pathological conditions. Chem Biol 2015, 22, 1347–1361. [Google Scholar] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nature Reviews Drug Discovery 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Mmps as therapeutic targets--still a viable option? Semin Cell Dev Biol 2008, 19, 61–68. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Molecular Cancer Therapeutics 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Coates-Park, S.; Rich, J.A.; Stetler-Stevenson, W.G.; Peeney, D. The timp protein family: Diverse roles in pathophysiology. Am J Physiol-Cell Ph 2024, 326, C917–C934. [Google Scholar]
- Leask, A. Ccn1: A saspy protein that plays multifaceted roles in fibrogenesis. Matrix Biology 2026, 143, 14–17. [Google Scholar] [CrossRef]
- Feng, D.; Gerarduzzi, C. Emerging roles of matricellular proteins in systemic sclerosis. International Journal of Molecular Sciences 2020, 21, 4776. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Johnston, A.; Gudjonsson, J.E.; Fisher, G.J. Cyr61/ccn1: A novel mediator of epidermal hyperplasia and inflammation in psoriasis? J Invest Dermatol 2015, 135, 2562–2564. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Hammarström, C.; Haraldsen, G. Tenascins in fibrotic disorders-from bench to bedside. Cell Adhes Migr 2015, 9, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, D.F.; Borkner, L.; Scheurmann, J.; Singh, K.; Scharffetter-Kochanek, K.; Weiss, J.M. Osteopontin deficiency affects imiquimod-induced psoriasis-like murine skin inflammation and lymphocyte distribution in skin, draining lymph nodes and spleen. Exp Dermatol 2015, 24, 305–307. [Google Scholar] [CrossRef]
- Sirois, J.P.; Heinz, A. Matrikines in the skin: Origin, effects, and therapeutic potential. Pharmacol Therapeut 2024, 260. [Google Scholar] [CrossRef]
- Tolg, C.; Telmer, P.; Turley, E. Specific sizes of hyaluronan oligosaccharides stimulate fibroblast migration and excisional wound repair. PLoS One 2014, 9, e88479. [Google Scholar] [CrossRef]
- Baghy, K.; Szakadáti, H.; Kovalszky, I. Decorin the antifibrotic proteoglycan and its progression in therapy. Am J Physiol Cell Physiol 2025, 328, C1853–c1865. [Google Scholar] [CrossRef]
- Avenoso, A.; Bruschetta, G.; D'Ascola, A.; Scuruchi, M.; Mandraffino, G.; Gullace, R.; Saitta, A.; Campo, S.; Campo, G.M. Hyaluronan fragments produced during tissue injury: A signal amplifying the inflammatory response. Arch Biochem Biophys 2019, 663, 228–238. [Google Scholar] [CrossRef]
- Patel, D.F.; Snelgrove, R.J. The multifaceted roles of the matrikine pro-gly-pro in pulmonary health and disease. Eur Respir Rev 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Moore, E. Yigsr, a laminin-derived peptide, dictates a concentration-dependent impact on macrophage phenotype response. Cell Mol Bioeng 2024, 17, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Thau, H.; Gerjol, B.P.; Hahn, K.; von Gudenberg, R.W.; Knoedler, L.; Stallcup, K.; Emmert, M.Y.; Buhl, T.; Wyles, S.P.; Tchkonia, T.; et al. Senescence as a molecular target in skin aging and disease. Ageing Research Reviews 2025, 105. [Google Scholar] [CrossRef] [PubMed]
- Saliev, T.; Singh, P.B. Targeting senescence: A review of senolytics and senomorphics in anti-aging interventions. Biomolecules 2025, 15. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. Journal of Internal Medicine 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. The FEBS Journal 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Gerdes, E.O.W.; Misra, A.; Netto, J.M.E.; Tchkonia, T.; Kirkland, J.L. Strategies for late phase preclinical and early clinical trials of senolytics. Mechanisms of Ageing and Development 2021, 200. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Bidirectional crosstalk between the ECM and immune cells. The ECM and immune cells engage in dynamic, reciprocal signaling that shapes tissue function in health and disease. ECM components, including structural proteins, proteoglycans, and matricellular proteins, regulate immune cell recruitment, activation, and effector functions through biochemical and mechanical cues. Conversely, immune cells secrete cytokines, chemokines, and matrix-remodeling enzymes that alter ECM composition, organization, and stiffness. These bidirectional interactions govern immune surveillance, modulate inflammatory responses, and influence outcomes in tissue repair and pathological conditions.
Figure 1.
Bidirectional crosstalk between the ECM and immune cells. The ECM and immune cells engage in dynamic, reciprocal signaling that shapes tissue function in health and disease. ECM components, including structural proteins, proteoglycans, and matricellular proteins, regulate immune cell recruitment, activation, and effector functions through biochemical and mechanical cues. Conversely, immune cells secrete cytokines, chemokines, and matrix-remodeling enzymes that alter ECM composition, organization, and stiffness. These bidirectional interactions govern immune surveillance, modulate inflammatory responses, and influence outcomes in tissue repair and pathological conditions.
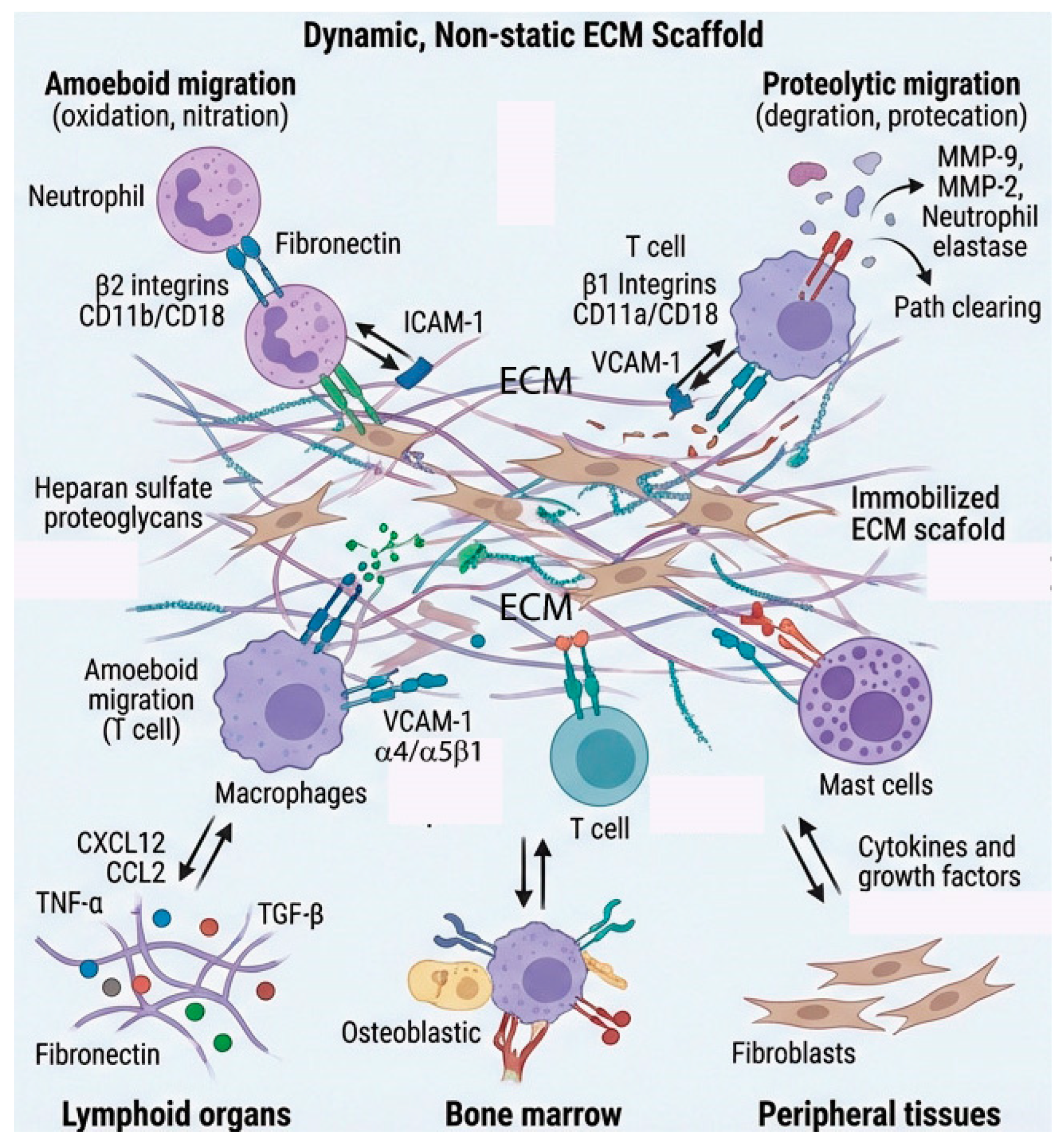
Figure 2.
MMPs as mediators of inflammation. Pro-inflammatory cytokines (IL-1β, TNF-α, IL-17) activate transcription factors NFκ-B and AP-1 to upregulate MMP expression. Secreted MMPs execute three primary functions that determine tissue fate: (1) ECM degradation, breaking down collagen and basement membrane components to facilitate immune cell migration and extravasation during acute inflammation; (2) Cytokine processing, proteolytically activating cytokines and chemokines while modulating inflammatory signaling cascades for inflammatory regulation; and (3) Growth factor release, liberating sequestered growth factors from the ECM to enable cell-ECM-growth factor interactions critical for repair and remodeling. The balance between MMPs and TIMPs dictates three distinct pathological outcomes: balanced MMP/TIMP activity promotes normal wound healing and tissue homeostasis; excessive MMP activity with insufficient TIMP inhibition leads to pathologic ECM degradation, resulting in chronic non-healing wounds and tissue damage; conversely, reduced MMP activity with elevated TIMPs drives excessive matrix deposition, hypertrophic scarring, fibrosis, and systemic sclerosis.
Figure 2.
MMPs as mediators of inflammation. Pro-inflammatory cytokines (IL-1β, TNF-α, IL-17) activate transcription factors NFκ-B and AP-1 to upregulate MMP expression. Secreted MMPs execute three primary functions that determine tissue fate: (1) ECM degradation, breaking down collagen and basement membrane components to facilitate immune cell migration and extravasation during acute inflammation; (2) Cytokine processing, proteolytically activating cytokines and chemokines while modulating inflammatory signaling cascades for inflammatory regulation; and (3) Growth factor release, liberating sequestered growth factors from the ECM to enable cell-ECM-growth factor interactions critical for repair and remodeling. The balance between MMPs and TIMPs dictates three distinct pathological outcomes: balanced MMP/TIMP activity promotes normal wound healing and tissue homeostasis; excessive MMP activity with insufficient TIMP inhibition leads to pathologic ECM degradation, resulting in chronic non-healing wounds and tissue damage; conversely, reduced MMP activity with elevated TIMPs drives excessive matrix deposition, hypertrophic scarring, fibrosis, and systemic sclerosis.
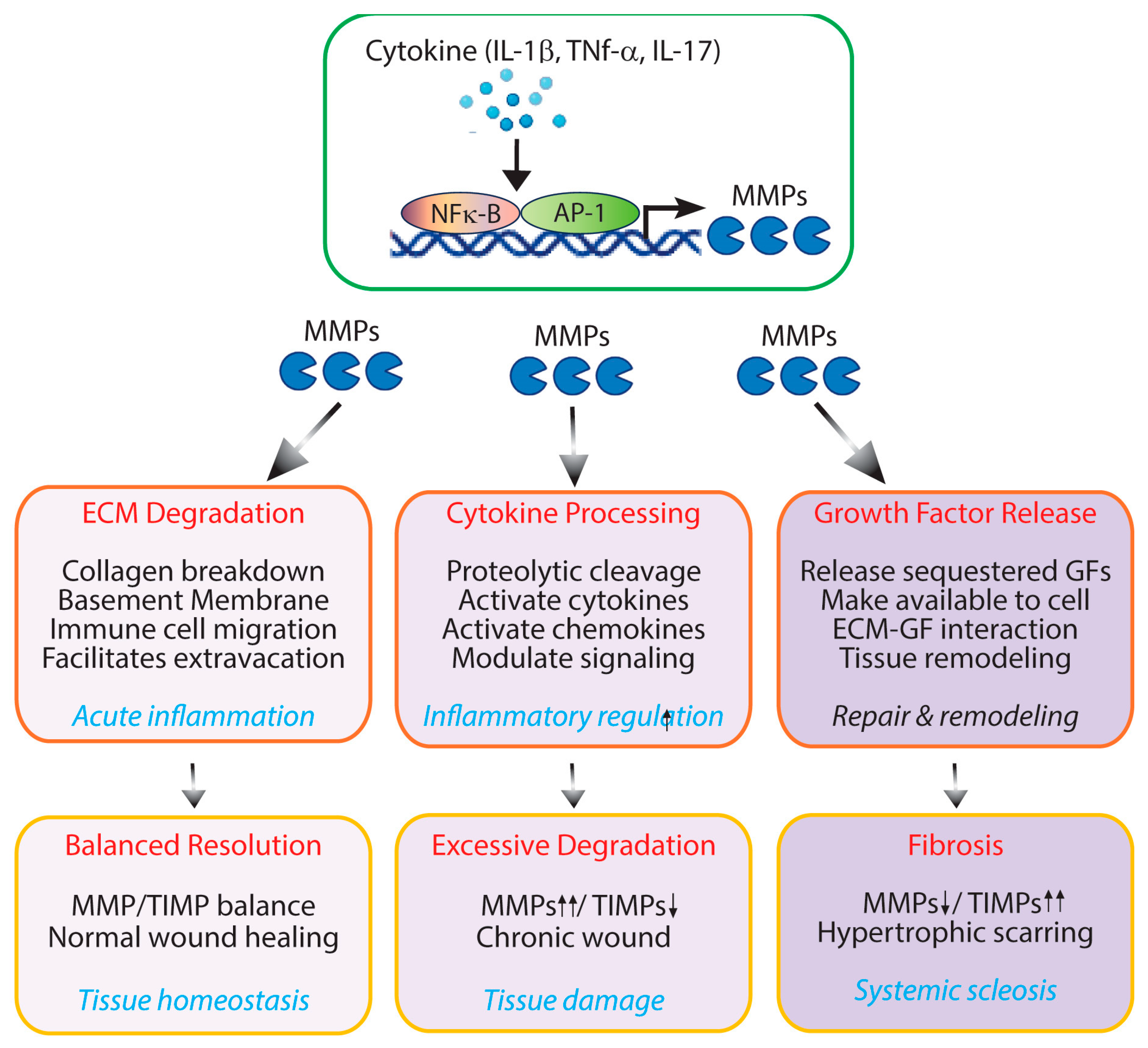
Figure 3.
Matrikines regulate immune cell functions. Matrikines are bioactive ECM fragments generated by proteolytic cleavage of structural proteins including collagen, elastin, fibronectin, laminin, and hyaluronan by MMPs and other enzymes. These fragments bind specific receptors (integrins, TLRs, CD44, RAGE) on immune cells such as neutrophils, macrophages, dendritic cells, and lymphocytes, activating signaling pathways (MAPK, NF-κB, PI3K/Akt) that regulate chemotaxis, cytokine production, and cellular activation. Matrikine-immune cell interactions modulate inflammatory responses and tissue remodeling outcomes, influencing the balance between acute inflammation, tissue repair, fibrosis, and restoration of homeostasis.
Figure 3.
Matrikines regulate immune cell functions. Matrikines are bioactive ECM fragments generated by proteolytic cleavage of structural proteins including collagen, elastin, fibronectin, laminin, and hyaluronan by MMPs and other enzymes. These fragments bind specific receptors (integrins, TLRs, CD44, RAGE) on immune cells such as neutrophils, macrophages, dendritic cells, and lymphocytes, activating signaling pathways (MAPK, NF-κB, PI3K/Akt) that regulate chemotaxis, cytokine production, and cellular activation. Matrikine-immune cell interactions modulate inflammatory responses and tissue remodeling outcomes, influencing the balance between acute inflammation, tissue repair, fibrosis, and restoration of homeostasis.
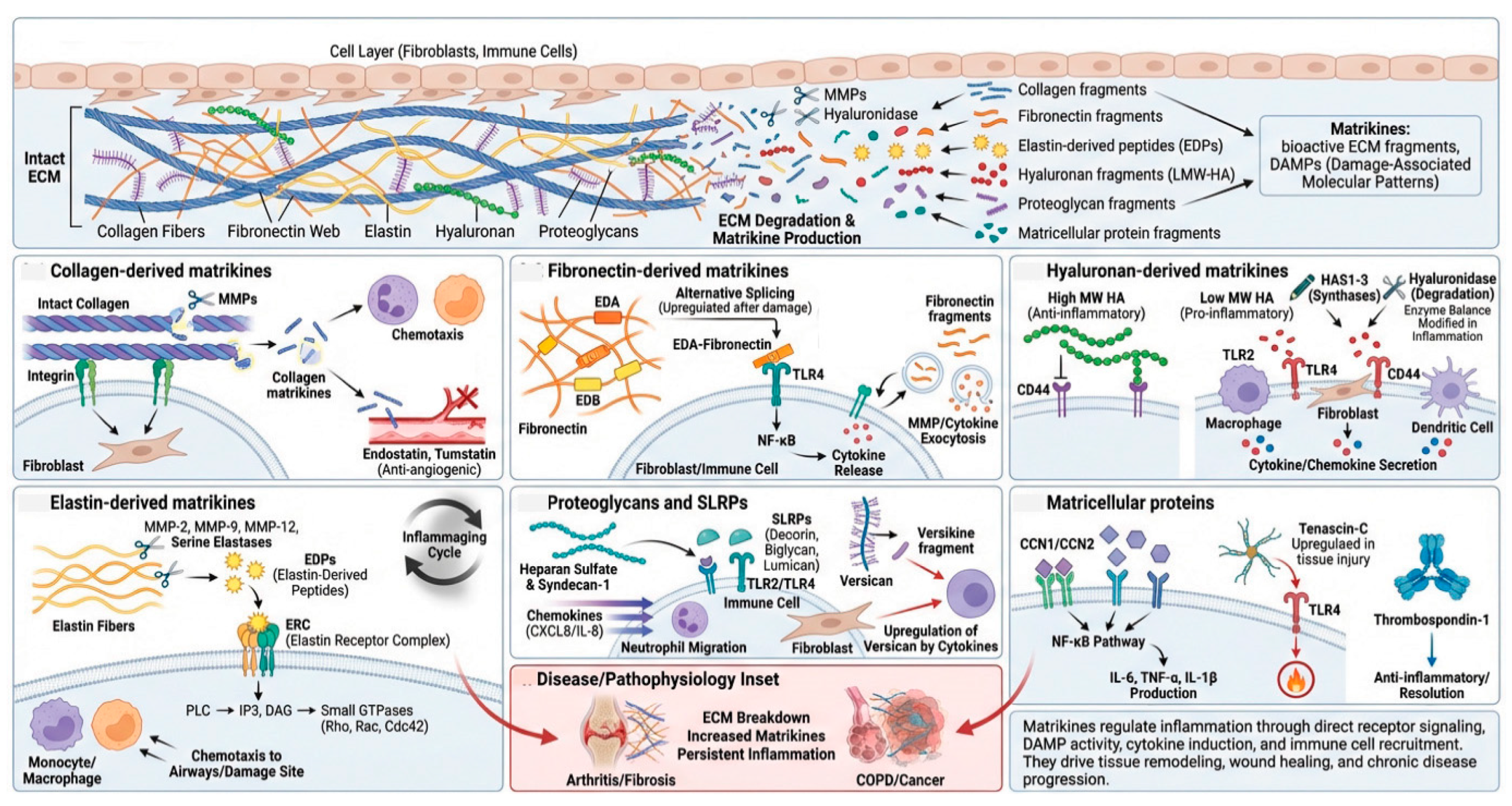
Figure 4.
ECM mechanical properties as inflammatory modulators. ECM stiffness regulates inflammatory signaling through mechanotransduction. Stiff, fibrotic ECM (left) activates YAP/TAZ, Integrin-FAX-PI3K/Akt, NF-κB/MAPK, and NLRP3 inflammasome pathways, driving pro-inflammatory cytokine production (TNF-α, IL-6, IL-8), M1 macrophage polarization, and pro-fibrotic factors (CTGF, TGF-β) that perpetuate inflammation and fibrosis. Soft, healthy ECM (right) sequesters YAP/TAZ and reduces pathway activation, promoting anti-inflammatory IL-10 production, M2 macrophage polarization, tissue repair, and ECM homeostasis. ECM mechanical properties thus serve as critical modulators of tissue inflammation in fibrotic diseases and conditions with abnormal mechanical loading.
Figure 4.
ECM mechanical properties as inflammatory modulators. ECM stiffness regulates inflammatory signaling through mechanotransduction. Stiff, fibrotic ECM (left) activates YAP/TAZ, Integrin-FAX-PI3K/Akt, NF-κB/MAPK, and NLRP3 inflammasome pathways, driving pro-inflammatory cytokine production (TNF-α, IL-6, IL-8), M1 macrophage polarization, and pro-fibrotic factors (CTGF, TGF-β) that perpetuate inflammation and fibrosis. Soft, healthy ECM (right) sequesters YAP/TAZ and reduces pathway activation, promoting anti-inflammatory IL-10 production, M2 macrophage polarization, tissue repair, and ECM homeostasis. ECM mechanical properties thus serve as critical modulators of tissue inflammation in fibrotic diseases and conditions with abnormal mechanical loading.
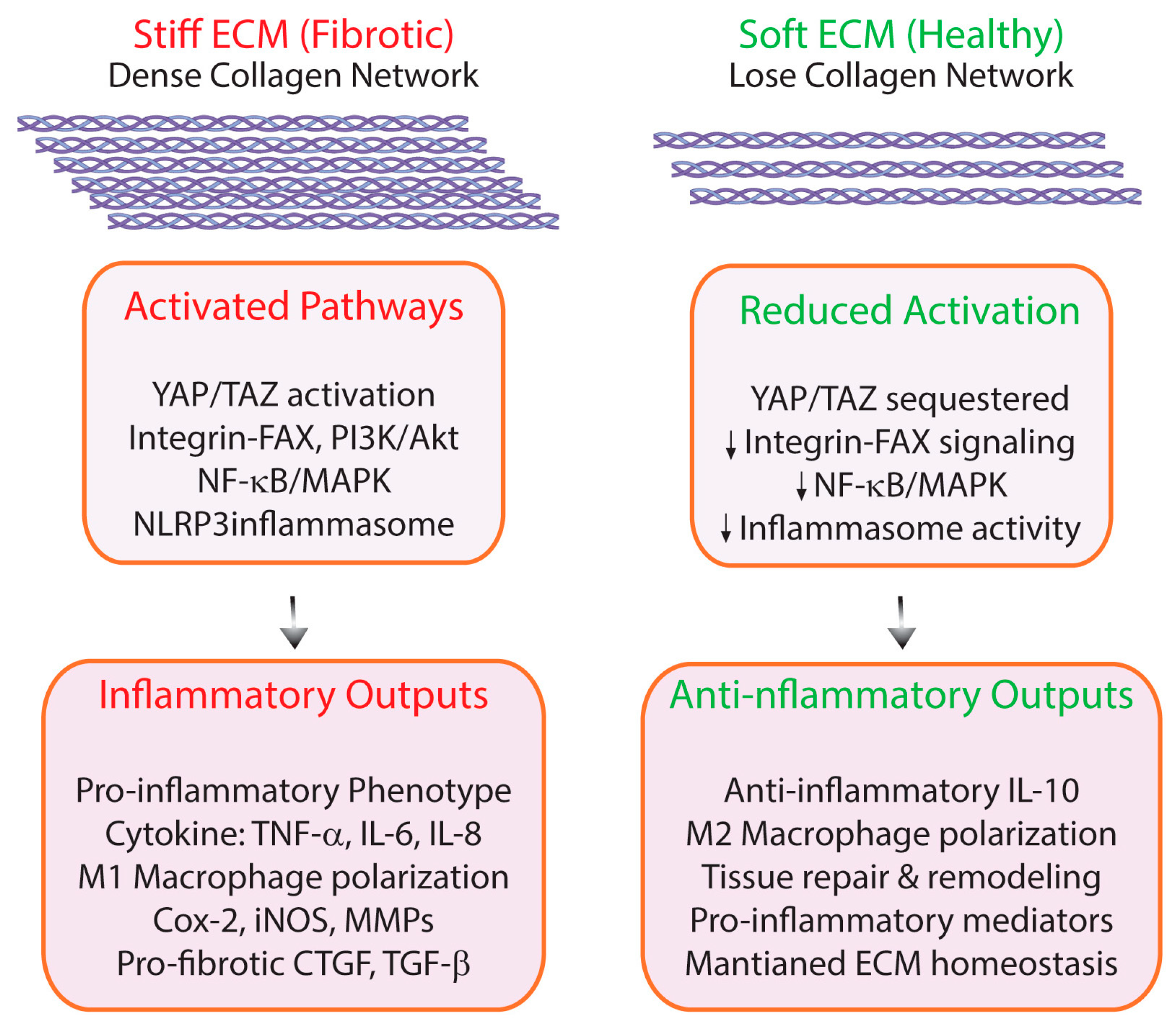
Figure 5.
Matrisome-related SASP factors as inflammatory modulators. Senescent fibroblasts accumulate with age and secretes SASP factors, which drives chronic low-grade inflammation (inflammaging), a hallmark of aging. Key matrisome-related SASP components include ECM-degrading enzymes (MMP-1, MMP-3, MMP-9), ECM regulatory proteins (TIMPs, matricellular proteins), and pro-inflammatory mediators (IL-6, IL-8, IL-1, CCL2, TGF-β, VEGF). The diagram illustrates how senescent fibroblasts create a pro-inflammatory microenvironment that perpetuates ECM degradation, sustains chronic inflammation, and contributes to age-related tissue dysfunction, representing a potential therapeutic target for healthy aging interventions.
Figure 5.
Matrisome-related SASP factors as inflammatory modulators. Senescent fibroblasts accumulate with age and secretes SASP factors, which drives chronic low-grade inflammation (inflammaging), a hallmark of aging. Key matrisome-related SASP components include ECM-degrading enzymes (MMP-1, MMP-3, MMP-9), ECM regulatory proteins (TIMPs, matricellular proteins), and pro-inflammatory mediators (IL-6, IL-8, IL-1, CCL2, TGF-β, VEGF). The diagram illustrates how senescent fibroblasts create a pro-inflammatory microenvironment that perpetuates ECM degradation, sustains chronic inflammation, and contributes to age-related tissue dysfunction, representing a potential therapeutic target for healthy aging interventions.
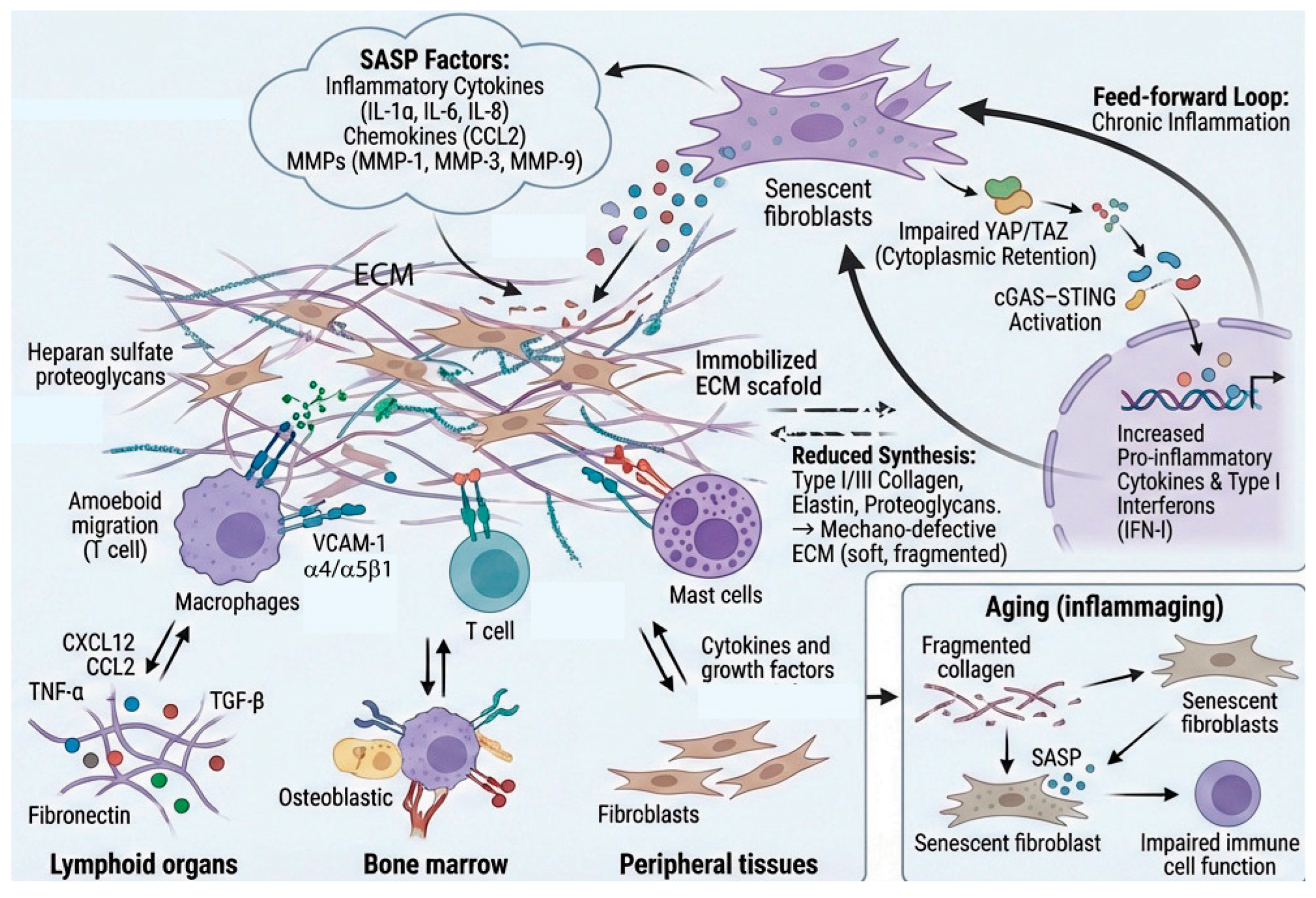
Figure 6.
Therapeutic strategies targeting the immune-matrisome interface. The matrisome has emerged as a critical regulator of inflammation, shifting from a view of the ECM as a passive structural scaffold to an active participant in immune responses. This paradigm shift has enabled development of diverse therapeutic approaches that target immune cell-matrisome interactions: (1) direct inhibition of immune cell adhesion and migration through blockade of integrin-ECM or CD44-hyaluronan interactions; (2) modulation of ECM composition via MMP inhibitors or crosslinking enzyme antagonists; (3) manipulation of ECM mechanical properties to alter immune cell behavior; (4) targeting bioactive matrikines that propagate inflammatory signals; and (5) delivery of engineered biomaterials or decellularized matrices to redirect tissue remodeling. These matrisome-targeted interventions hold promise across autoimmune disorders, fibrotic diseases, cancer immunotherapy, and regenerative medicine applications where dysregulated immune-ECM crosstalk drives pathology.
Figure 6.
Therapeutic strategies targeting the immune-matrisome interface. The matrisome has emerged as a critical regulator of inflammation, shifting from a view of the ECM as a passive structural scaffold to an active participant in immune responses. This paradigm shift has enabled development of diverse therapeutic approaches that target immune cell-matrisome interactions: (1) direct inhibition of immune cell adhesion and migration through blockade of integrin-ECM or CD44-hyaluronan interactions; (2) modulation of ECM composition via MMP inhibitors or crosslinking enzyme antagonists; (3) manipulation of ECM mechanical properties to alter immune cell behavior; (4) targeting bioactive matrikines that propagate inflammatory signals; and (5) delivery of engineered biomaterials or decellularized matrices to redirect tissue remodeling. These matrisome-targeted interventions hold promise across autoimmune disorders, fibrotic diseases, cancer immunotherapy, and regenerative medicine applications where dysregulated immune-ECM crosstalk drives pathology.
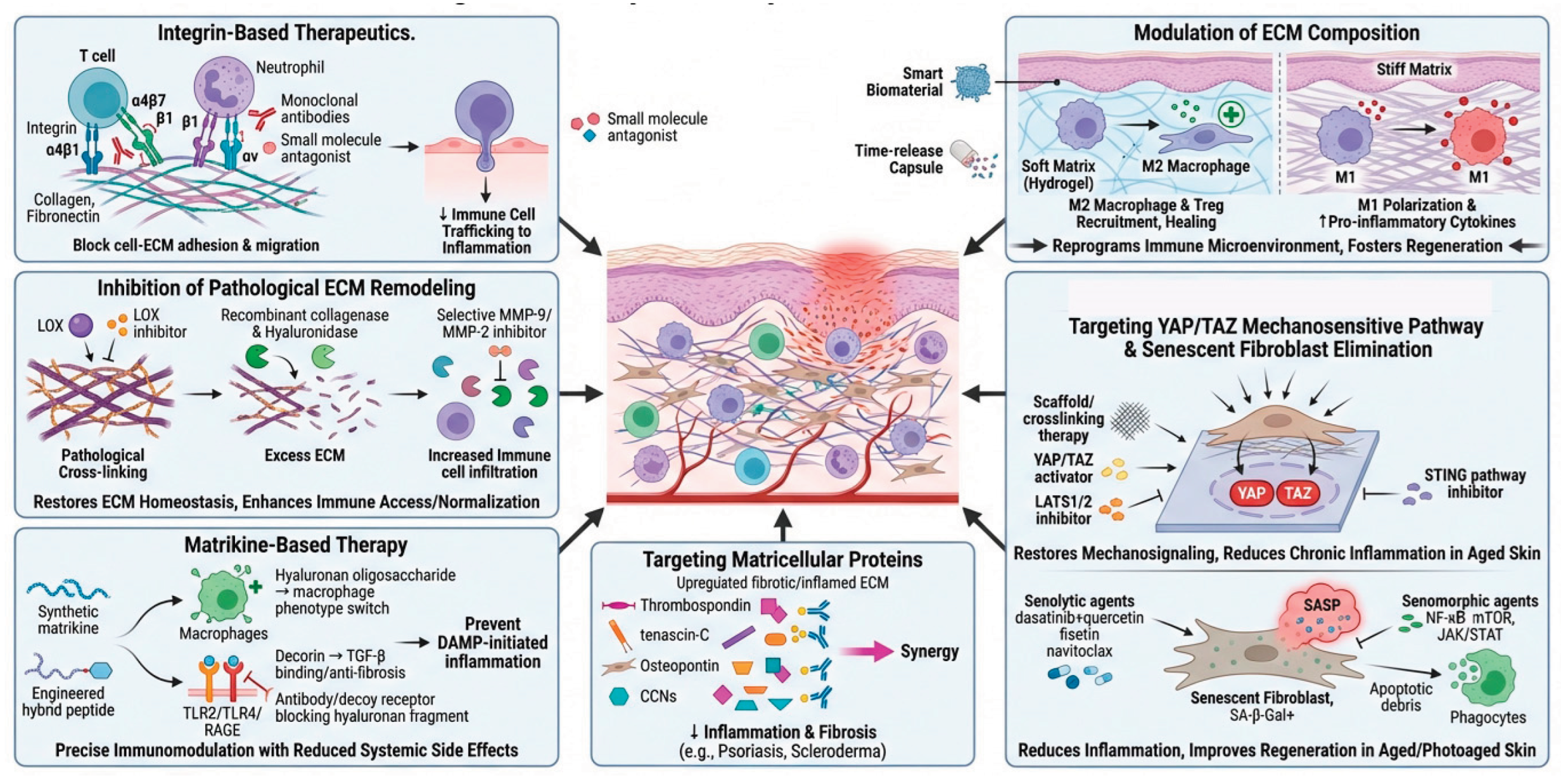
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



