
Submitted:
26 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
The c-Jun N-terminal kinase (JNK) signaling cascade is a central regulator of cellular stress responses and a validated therapeutic target for diverse pathologies, including neurodegeneration, fibrosis, and cancer. However, the development of direct JNK inhibitors has been impeded by challenges regarding isoform selectivity and on-target toxicity. Consequently, medicinal chemistry efforts have shifted upstream to the "gatekeepers" of the pathway: the dual-specificity kinases MKK4 and MKK7. This review provides a comprehensive analysis of the structural biology, physiological functions, and pharmacological inhibition of these critical signaling nodes. We highlight the non-redundant therapeutic potential of these kinases, contrasting the role of MKK4 inhibition in unlocking liver regeneration with the utility of MKK7 inhibition in suppressing fibrosis and chronic inflammation. A systematic overview of small-molecule inhibitors is presented. For MKK4, we discuss the evolution from early promiscuous chemotypes to the first-in-class clinical candidate HRX215, which has demonstrated safety and efficacy in promoting hepatocyte proliferation. For MKK7, we examine the design of covalent inhibitors exploiting the unique hinge-region cysteine (Cys218), as well as emerging modalities such as lysine-targeting binders and peptide inhibitors disrupting protein-protein interactions. Finally, we discuss remaining challenges and future opportunities, including the development of dual inhibitors and proteolysis-targeting chimeras (PROTACs), to fully exploit the therapeutic value of the MKK4/7-JNK axis.
Keywords:
MKK4 (MAP2K4)
; MKK7 (MAP2K7)
; JNK signaling
; kinase inhibitors
; liver regeneration
; covalent inhibitors
; medicinal chemistry
; fibrosis
; HRX215
1. Introduction
The mitogen-activated protein kinase (MAPK) signaling cascades serve as central integration points for transducing extracellular stimuli into specific cellular responses, governing fundamental processes such as proliferation, differentiation, apoptosis, and the inflammatory response [1,2]. Among these pathways, the c-Jun N-terminal kinase (JNK) signaling axis—often referred to as the stress-activated protein kinase pathway—plays a pivotal role in the pathogenesis of diverse human disorders, including neurodegenerative diseases, fibrotic conditions, inflammatory disorders, and cancer [3,4,5,6,7]. While the dysregulation of JNK signaling is a well-established driver of disease, the therapeutic validation of this pathway has historically proven challenging. Direct pharmacological inhibition of JNK isoforms (JNK1, JNK2, and JNK3) has been hampered by issues regarding isoform selectivity and toxicity, largely due to the high conservation of the ATP-binding cleft across the kinome and the critical physiological roles of basal JNK activity [8,9,10].
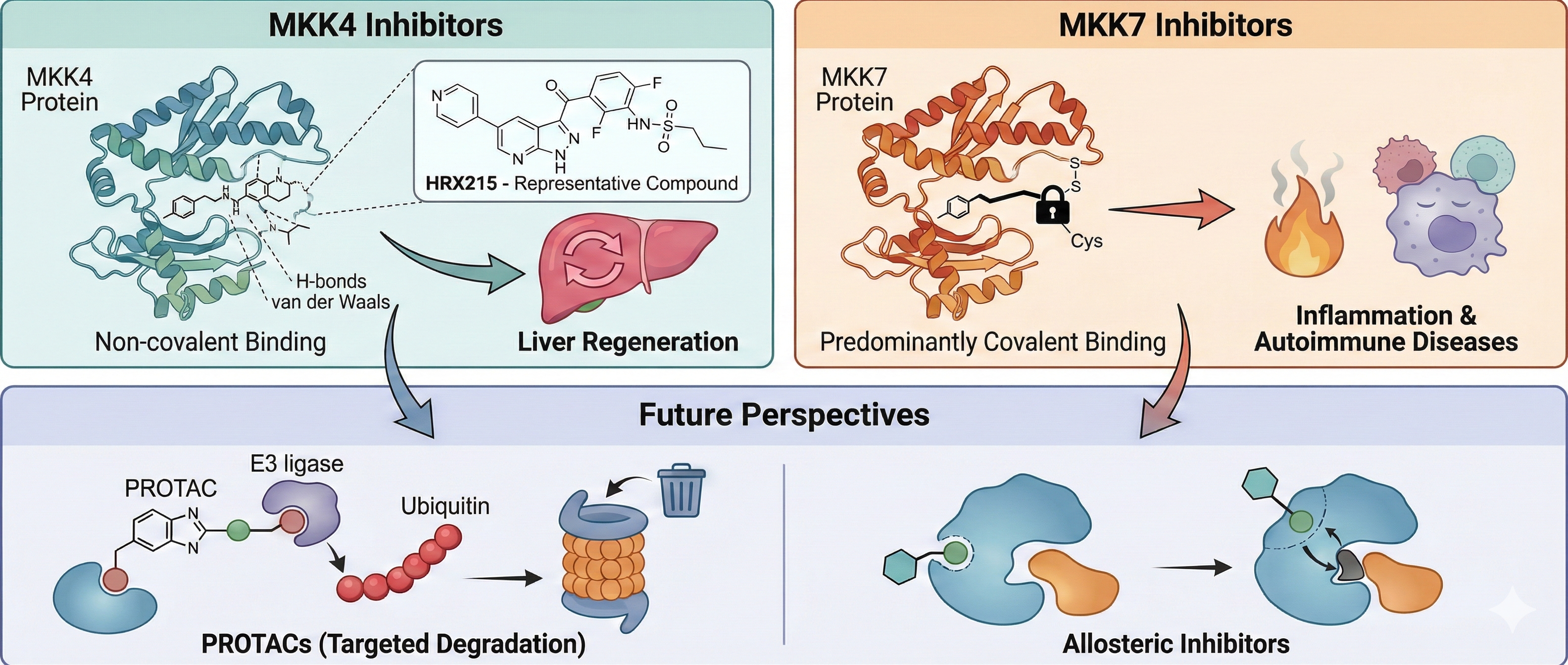
To overcome the limitations associated with direct JNK targeting, medicinal chemistry efforts have increasingly shifted upstream to the “gatekeepers” of the pathway: the mitogen-activated protein kinase kinases (MAP2Ks), specifically MKK4 (MAP2K4/SEK1) and MKK7 (MAP2K7) [11,12]. These dual-specificity kinases represent the convergence point of the three-tiered kinase cascade, linking diverse upstream MAP kinase kinase kinases (MAP3Ks) to the downstream JNK effectors. MKK4 and MKK7 are responsible for the dual phosphorylation of the Thr-Pro-Tyr motif within the activation loop of JNKs, a requisite step for full enzymatic activation [13,14,15].
Crucially, MKK4 and MKK7 are not functionally redundant, offering distinct therapeutic opportunities. MKK7 functions as a specific activator of JNKs, mediating inflammatory and stress responses, making it an attractive target for inflammatory and autoimmune indications [16,17,18]. In contrast, MKK4 exhibits a broader substrate profile, capable of activating both the JNK and p38 MAPK pathways [19,20]. Recent pharmacological and genetic studies have highlighted a unique role for MKK4 inhibition in promoting hepatocyte proliferation and liver regeneration, sparking renewed interest in this kinase as a regenerative medicine target [21,22]. This divergence in biological function underscores the necessity for highly selective chemical probes that can discriminate not only between MKK4 and MKK7 but also against other closely related MAP2Ks.
Despite the therapeutic promise, the development of potent and selective small-molecule inhibitors for MKK4 and MKK7 has lagged behind that of downstream MAPKs. However, recent advances in structural biology and fragment-based drug design have led to the identification of novel chemotypes and binding modes, ranging from classical ATP-competitive inhibitors to covalent modifiers targeting non-catalytic cysteines [11].
This review aims to provide a comprehensive analysis of the current landscape of MKK4 and MKK7 inhibitors. We will discuss the structural characteristics of these kinases that facilitate ligand binding, examine the medicinal chemistry strategies employed to improve potency and selectivity—including the evolution of key chemical scaffolds—and critically evaluate the therapeutic potential of targeting these upstream kinases in the context of liver disease, oncology, and inflammation. By synthesizing data from structural studies and pharmacological evaluations, we highlight the remaining challenges and future directions for drugging these critical nodes of the JNK signaling architecture.
2. The Biological Functions and Therapeutic Potential of MKK4/7
2.1. The JNK Signaling Architecture
The JNK pathway represents an evolutionarily conserved signaling cascade that orchestrates cellular responses to environmental stress, pro-inflammatory cytokines, and growth factors. Functioning within the canonical three-tiered MAPK module, the signal transmission is initiated by a diverse group of MAP3Ks (Table 1), such as ASK1, TAK1, and MEKK1, which sense upstream stimuli and phosphorylate the activation loops of the dual-specificity kinases, MKK4 and MKK7 (Figure 1) [23,24,25]. MKK4 and MKK7 share a kinase motif known as Ser–Xaa–Ala–Lys–Thr (S–X–A–K–T), which is located in the T-loop of the kinase domain [26]. Phosphorylation of both hydroxy residues (MKK4: Ser257 and Thr261; MKK7: Ser271 and Thr275) is required for the full activation of both proteins [27,28]. Studies have also demonstrated that Ser257 phosphorylation is essential, and Thr261 phosphorylation is required for full MKK4 activity [29].
As the central nodes of this cascade, MKK4 and MKK7 serve as the exclusive activators of JNK proteins (JNK1, JNK2, and JNK3). Activation occurs via dual phosphorylation of the conserved Threonine-Proline-Tyrosine (TxY) motif located within the JNK activation loop [13,14,15]. Importantly, while both MKK4 and MKK7 can mediate dual phosphorylation of JNKs in vitro, this process is inefficient individually, as MKK4 exhibits a striking preference for the tyrosine residue and MKK7 for the threonine residue, suggesting that both kinases are required for efficient dual phosphorylation in cells [30,31]. The dual phosphorylation event is estimated to induce a conformational change in the JNK activation loop, which creates a functional active site by realigning the N- and C-terminal domains, thus stabilizing the kinase in a conformation suitable for substrate binding [32,33]. Once activated, JNKs translocate to the nucleus or target mitochondrial proteins to phosphorylate specific substrates, most notably c-Jun, a component of the AP-1 transcription factor complex. This phosphorylation event (e.g., at Ser63 and Ser73 of c-Jun) drives the transcriptional expression of genes regulating apoptosis, survival, and cellular differentiation [33,34].
Crucially, the architecture of this pathway exhibits an “hourglass” topology: while there are over a dozen upstream MAP3Ks and multiple downstream JNK isoforms and substrates, the signal must funnel through the bottleneck of just two MAP2Ks (MKK4 and MKK7). This structural feature highlights MKK4 and MKK7 as strategic intervention points for therapeutically modulating JNK signaling output without engaging the complex redundancy found at the MAP3K level.
2.2. Biological Functions and Therapeutic Potential
The validation of MKK4 and MKK7 as therapeutic targets is grounded in their non-redundant physiological roles and their involvement in distinct pathological signaling axes. While both kinases serve as the proximal activators of JNK, their contributions to embryonic development, tissue homeostasis, and disease progression are markedly different.
2.2.1. MKK4 in Liver Regeneration and Hepatoprotection
Liver regeneration is a complex, orchestrated process where hepatocytes re-enter the cell cycle to restore functional liver mass following injury or resection. While the liver possesses a unique innate regenerative capacity, this process is often blunted in severe acute liver failure (ALF) or chronic liver diseases (CLD) [35,36]. Recent functional genetic screens utilizing in vivo RNA interference (RNAi) have identified MKK4 as a master negative regulator [37]. Under both physiological and stress conditions, MKK4 limits the extent of hepatocyte proliferation, while silencing or pharmacological inhibition of MKK4 releases this brake, resulting in a robust increase in hepatocyte DNA synthesis, faster cell cycle entry, and enhanced overall regenerative capacity (Figure 2). The mechanism by which MKK4 inhibition promotes regeneration is counterintuitive, as JNK signaling is traditionally associated with apoptosis. The current prevailing model involves a “rerouting” of stress signaling. When MKK4 activity is inhibited, upstream stress signals (from cytokines like TNF-α or growth factors) can no longer flow through MKK4. Consequently, the signaling flux is rerouted predominantly through MKK7. This MKK7-driven axis preferentially activates JNK1. Unlike the apoptotic AP-1 complex often driven by MKK4, the specific MKK7-JNK1 axis leads to the phosphorylation and activation of distinct transcription factors, most notably activating transcription factor 2 (ATF2) and ELK1. These transcription factors drive the expression of pro-proliferative genes (e.g., Ccnd1 encoding Cyclin D1) and survival factors, thereby accelerating the G1/S phase transition in hepatocytes [20,38]. Moreover, suppression of MKK4 expression under steady-state conditions confers resistance to Fas-mediated apoptosis in hepatocytes [39].
A critical concern in promoting regeneration is the risk of tumorigenesis. However, MKK4 inhibition appears to induce cell cycle arrest in hepatocellular carcinoma (HCC) cells. In tumor cells, MKK4 inhibition may lead to the hyperactivation of the p38 MAPK pathway (due to loss of negative feedback or crosstalk), which suppresses the cell cycle regulator CDC25C, thereby inhibiting tumor growth. This “context-dependent” signaling makes MKK4 an exceptionally attractive target [40].
2.2.2. The Role of MKK7 in Cancer
The role of MKK7 in tumorigenesis is highly context-dependent, functioning as a tumor suppressor in early-stage development and genomic stability maintenance, while acting as a pro-tumorigenic or pro-metastatic factor in established malignancies and specific genetic contexts (Figure 3A). As a tumor suppressor, MKK7 is essential for maintaining genomic stability and facilitating stress-induced senescence. In KRasG12D-driven lung carcinomas and NeuT-driven mammary tumors, MKK7 senses oncogenic stress and phosphorylates p53, which prevents p53 degradation. Loss of MKK7 leads to p53 instability, thereby accelerating tumor onset and reducing overall survival [41]. Furthermore, MKK7 functions cooperatively with p53 to regulate Polo-like kinase 4 (PLK4) activity, which prevents centrosome overduplication during mitosis and protects cells from aneuploidy, a hallmark of cancer genomic instability [42]. In prostate cancer models, MKK7 deficiency has also been shown to synergize with PTEN loss, driving the rapid development of invasive adenocarcinoma [43,44].
However, the role of MKK7 is double-edged. In established malignancies and specific genetic contexts, MKK7 acts as a pro-tumorigenic or pro-metastatic factor. In T-cell acute lymphoblastic leukemia (T-ALL), the transcription factor Kruppel-like factor 4 (KLF4) typically represses the MAP2K7 gene. However, the loss of KLF4 (often via DNA methylation) results in the overexpression of MKK7, which drives excessive JNK pathway activation and leukemic cell proliferation [45]. In the context of metastasis, a specific rare polymorphism in MKK7 (p.Glu116Lys) found in lung cancer patients is significantly associated with increased metastatic potential. Mechanistically, this mutation alters the expression of cancer-associated genes, specifically upregulating STC2, SLC1A3, and BCL10, while downregulating SAA1 and CDH5 [46]. Additionally, in colorectal cancer, MKK7 is a direct target of the metastasis-suppressor miR-493, which binds to the 3′-UTR of MKK7 mRNA. Loss of this regulatory interaction leads to MKK7 upregulation, thereby promoting liver metastasis [47].
The activity of MKK7 is further modulated by specific protein-protein interactions and post-translational modifications that cancer cells exploit to evade apoptosis. In multiple myeloma, the NF-β pathway upregulates GADD45β, which physically binds to MKK7 and blocks ATP access to its catalytic pocket, thereby inhibiting the MKK7-JNK apoptotic axis and promoting cell survival [48,49]. Similarly, in HCC, the TOR signaling regulator TIPRL binds to MKK7 and recruits the phosphatase PP2Ac to form a complex that facilitates MKK7 dephosphorylation. This inactivation of MKK7 contributes to resistance against TRAIL-induced apoptosis [50]. MKK7 activity in breast cancer is also suppressed by neddylation mediated by the E3 ligase RanBP2, where RanBP2 knockdown has been shown to restore MKK7 kinase activity and inhibit cell proliferation [51]. Other regulatory interactions include RACK1, which acts as a scaffold to enhance MKK7 activity in HCC, and RASSF7, which binds specifically to phosphorylated MKK7 to potentially hinder its interaction with downstream substrates, thereby negatively regulating pro-apoptotic signaling [52,53].
2.2.3. The Role of MKK7 in Inflammation
MKK7 plays a pivotal role in the innate immune system, acting as a central regulator of macrophage biology and the inflammatory response (Figure 3B). Upon stimulation by pathogens or pro-inflammatory cytokines, MKK7 is indispensable for the production of key inflammatory cytokines, including TNF-α, IL-1α, IL-1β, and IL-6, particularly in response to bacterial lipopolysaccharides (LPS). While the related kinase MKK4 also contributes to this signaling network, its influence on cytokine production is modest compared to the dominant role of MKK7. Beyond cytokine secretion, MKK7 governs the physical mobilization of immune cells. Deficiency of MKK7 leads to a significant blockade in macrophage migration and invasion capabilities. Furthermore, MKK7 is essential for macrophage polarization towards the M1 pro-inflammatory phenotype, thereby orchestrating the initiation of an effective immune defense [17,54].
Mechanistically, MKK7 functions through both canonical and non-canonical signaling pathways to execute these inflammatory responses. Classically, MKK7 is known as the specific upstream activator of the JNK pathway, which drives the transcriptional activation of cytokine genes. However, recent evidence indicates a critical cross-talk mechanism where MKK7 is also required for the full activation of p38 MAPK in macrophages, challenging the conventional understanding that MKK3/6 are the sole activators of p38 [54]. This dual regulation of both JNK and p38 pathways suggests that MKK7 acts as a master node in the inflammatory signaling network. Additionally, the activation of this pathway often relies on the coordination of scaffold proteins, such as JNK-interacting protein 1 (JIP1), which facilitate the interaction between MKK7 and its upstream or downstream partners to ensure robust signaling [55].
The regulatory power of MKK7 makes it a significant contributor to the pathology of chronic inflammatory diseases such as rheumatoid arthritis (RA) and sepsis. In the context of RA, MKK7 drives joint destruction not only by promoting cytokine release but also by regulating matrix metalloproteinases (MMPs), the enzymes responsible for degrading the extracellular matrix. Consequently, therapeutic strategies targeting MKK7 have shown promise. For instance, the inhibition of MKK7 using antisense oligonucleotides (ASO) has been demonstrated to decrease disease severity in mouse models of serum transfer arthritis [56]. These findings highlight MKK7 as a promising therapeutic target for managing uncontrolled inflammatory responses, preventing the transition from acute to chronic inflammation, and mitigating tissue damage in autoimmune conditions.
3. Structures of MKK4 and MKK7
MKK4 and MKK7 proteins are encoded in humans by the MAP2K4 (on chromosome 17p12 with 11 exons) and MAP2K7 (on chromosome 19p13 with 12 exons) genes. In humans, alternative splicing of MAP2K4 gives rise to two isoforms: isoform 2, the longer variant, includes an additional in-frame exon in the 5′ coding region compared to isoform 1. In mice, alternative splicing of the MAP2K7 gene produces a set of protein isoforms with distinct N-terminal (α, β, γ) and C-terminal (1, 2) variants. These murine isoforms have predicted molecular weights ranging from 38 to 52 kDa and consist of approximately 345 to 467 amino acid residues [17,57,58]. Both MKK4 and MKK7 exhibit the canonical structural architecture of the MAP2K subfamily and share 54% sequence identity. Their three-dimensional structures fold into two distinct lobes: a smaller N-lobe, comprising five β-sheets and one α-helix, and a larger C-lobe primarily composed of α-helices. A flexible segment connecting the two lobes houses the ATP-binding site. Functionally, MKK proteins are organized into three primary parts (Figure 4A, B): the N-terminal docking domain (D domain), the central kinase domain, and the C-terminal domain for versatile docking (DVD) [17,20,59].
The D domain mediates interactions with downstream substrates, particularly JNK, and also serves as a binding site for scaffold proteins such as Arrestin-1 and -2 (in the case of MKK4) [60]. Structurally, the D domains of MKK4 and MKK7 consist of a cluster of two to three basic residues, followed by a short spacer (1–2 residues) and a hydrophobic–X–hydrophobic motif [58,61]. Both MKK4 and MKK7 interact with JNK through their D domains, but they differ in valency. The three MKK7 splice variants exhibit differential activity toward JNK: MKK7α displays lower activity compared to β and γ isoforms. This difference likely arises because the β and γ isoforms contain three D domains that bind to the JNK DRS (Figure 4A), whereas the α isoform possesses only a single D domain [57]. Similarly, MKK4 engages JNK through a single N-terminal D domain.
The central ATP binding pocket is highly conserved across the MAP2K family, yet MKK4 and MKK7 possess distinct characteristics that confer functional specificity (Figure 4D). For example, the MKK4 hinge region specifically contains neutral residues Leu180 and Ser182, unlike homologous positions in MKK2 which contain charged residues [62]. MKK7 has the smallest volume and a significantly shallower depth among all MEK isoforms with available crystal structures (Figure 4C). Its active site is characterized by a high proportion of hydrophobic residues and fewer charged residues compared to MKK1/2/4/6. While MKK1, MKK2, MKK4, and MKK6 binding cavities are predominantly negatively charged, the MKK7 cavity exhibits patches of partial positive and negative charges [63]. Both MKK4 and MKK7 possess a highly conserved cysteine located immediately before the DFG motif (Cys246 in MKK4 and Cys261 in MKK7). However, unlike other MEK family members, MKK7 is unique in containing a Cys218 in the hinge region, a position where only 11 human kinases share an equivalent cysteine [64].
MKK4 and MKK7 activity is increased following phosphorylation at Ser and Thr residues within a Ser–Xaa–Ala–Lys–Thr (S–X–A–K–T) motif in the T-loop by multiple MAP3Ks. Similar to MKK3 and MKK6, the amino acid following the Ser residue in MKK4 is a hydrophobic residue (Ile), whereas in MKK7, it is a basic residue (Lys). This distinction may, at least in part, account for the differential substrate specificities exhibited by certain MAP3Ks toward the MKK4 and MKK7 [26]. For instance, MEKK1 phosphorylates both MKK4 and MKK7, whereas MEKK4 selectively activates MKK4. This specificity is primarily mediated by a C-terminal DVD domain of approximately 24 amino acids in MKK4 and MKK7. Association with upstream kinases via DVD contributes to the conformational changes in the activation loop of MKK proteins, so that the Ser and Thr residues within the activation motif become accessible for efficient phosphorylation [20]. Beyond kinase interactions, the MKK4 DVD also binds scaffold proteins such as JIP-3 and plenty of SH3 (POSH), forming stress-responsive signaling complexes [65,66].
Structurally, MKK4 adopts an inactive, autoinhibited conformation and may exist as a dimer. This conformation restricts access to the ATP-binding pocket and is destabilized upon phosphorylation [67]. In contrast, MKK7 exhibits significant plasticity in its catalytic domain. Crystal structures have captured MKK7 in multiple inactive states, including one where the hinge region Leu214 is in an unprecedented DFG-in/Leu-in conformation, and one where it assumes an inactive “out” DFG conformation [68,69]. Furthermore, the transition to the active state in MKK7 involves an N-terminal regulatory helix that acts allosterically to stabilize the active conformation, a mechanism likely shared across the MAP2K subfamily [70].
4. MKK4 and MKK7 Inhibitors
4.1. MKK4 Inhibitors
Given the structural features discussed above, particularly the conservation of the ATP pocket, achieving selectivity has been a primary challenge in MKK4 inhibitor design. Furthermore, structure-based design is challenging because the available MKK4 crystal structures (PDB: 3ALN, 3ALO, 3OUT) do not allow for the establishment of a reliable docking method. This may be due to inconsistencies in the ATP-binding pocket or the absence of co-crystallized ligands. Therefore, the development of selective MKK4 inhibitors often requires detailed SAR research. To develop MKK4 inhibitors with BBB penetration capability, Ogura et al. designed and synthesized a series of prenylated quinolinecarboxylic acid (PQA) derivatives based on 1 (Figure 5A), a secondary metabolite derived from Polysphondylium pseudocandidum [72]. Screening using a hippocampal neuronal glutamate toxicity model revealed that 2 (Figure 5A) exhibited the strongest neuroprotective activity (IC50 = 127 nM). In cellular models of Parkinson’s diseases (PD), Alzheimer’s disease (AD) and glutamate-induced cell death, 2 significantly inhibited caspase-3 activation induced by glutamate, MPP+, and amyloid-beta (Aβ), while also reducing the expression levels of p-JNK and p-MKK4. Furthermore, 2 selectively bound to the resting MKK4 (Kd = 11.5 nM) rather than activated MKK4. Further in vitro kinase assays confirmed that it inhibited the autophosphorylation of MKK4 at Thr2661 by competitively blocking the binding of sphingosine to MKK4. In a MPTP-induced neurodegenerative mouse model, treatment with 2 restored the number of tyrosine hydroxylase (TH)-positive neurons in the substantia nigra. Notably, compound 2 was rapidly detected in brain tissue within four hours of intraperitoneal administration, indicating its effective blood-brain barrier (BBB) penetration.
Leveraging the previously established MEK kinase screening platform, Scheidt’s group identified lead compound 3 (Figure 5B) featuring a 3-arylindazole scaffold [73]. Compound 3 competitively bound to the ATP-binding site of MKK4 and, notably, stabilized both its active and inactive forms to a similar extent. Subsequent structure-activity relationship (SAR) studies showed that modifications at the 5- and 6-positions of the indazole ring significantly enhanced inhibitory activity, with the 6-fluoro-substituted derivative 4 (Figure 5B) displaying the optimal activity. Notably, the N–H group in the indazole ring and carboxylic acid moiety were crucial for maintaining activity. Selectivity evaluation indicated that 4 exhibited more than 150-fold selectivity over other MEK family members, but displayed a broad inhibition profile in kinome screening (S(35) = 0.21). Cellular studies revealed compound 4 failed to suppress cancer cell migration in a wound healing assay, a phenotype likely attributable to reduced membrane permeability due to the highly polar carboxyl group. To resolve this, Scheidt’s group employed a bioisostere strategy to structurally modify the carboxylic acid group and successfully obtained the para-substituted sulfonamide derivative 5 and 6 (Figure 5B) [74]. In CD18 and MiaPaCa2 pancreatic cancer cell lines, both 5 and 6 demonstrated significant antiproliferative activity and upregulated phosphorylation level of ERK1/2. In addition, co-administration of 5 with the MEK1/2 inhibitor significantly enhanced antiproliferative effects, achieving a coefficient of drug interaction (CDI) of 0.6. However, compound 6 exhibited significantly broader kinase inhibition (28/97 kinases at 10 μM) and reduced selectivity within the MEK family.
Considering the off-target activity of Vemurafenib (7, the FDA-approved BRAFV600E inhibitor, Figure 6A) towards MKK4, Laufer’s group employed an iterative multi-parameter optimization approach to systematically investigate the impact of key structural domains within 7 on the affinity and selectivity toward MKK4 [75]. The optimization process was based on binding affinity assessments using KINOMEscan technology, with data expressed as percentage of control (POC) values (where POC = 0 indicates complete binding, and POC = 100 indicates no binding). The final compound 8 (Figure 6A) exhibited improved MKK4 affinity and kinase selectivity, inhibiting only 5 out of 97 kinases with ≥65% inhibition at 1 μM, and none with ≥90% inhibition. In 2021, Laufer’s group replaced the 7-azaindole moiety in 7 with 1H-pyrazolo [3,4-b]pyridine and conducted a systematic SAR analysis (Figure 6B) [76]. Compound 9 (Figure 6A), featuring a para-sulfonamide group, was selected as a new lead compound due to its high affinity for MKK4. Attempts to further enhance selectivity for BRAF by introducing nonpolar substituents at the ortho position were unsuccessful. The authors then shifted their focus to modifying the sulfonamide side chain on the opposite side. The introduction of a benzyl group resulted in compound 10 with preserved activity while only inhibiting two kinases (AURKB and SNARK, POC < 35%) in a 97-kinase panel under 1 μM concentration.
In 2024, Laufer, Nyberg and Zender’s group reported the preclinical the phase I clinical studies of first-in-class MKK4 inhibitor HRX215 (11, Figure 6A), which incorporates 2,6-difluorophenyl and pyridine moieties and exhibits improved water solubility and >100 selectivity against JNK1, BRAF, and MKK7 [21]. In preclinical studies, 11 demonstrated a significant pro-regenerative effect on the liver. Administering 11 (10 mg) to mice 12 hours before and immediately prior to partial hepatectomy increased hepatocyte proliferation by 5.3-fold (26.3% vs 4.93% in the control group), an effect that remained effective even in fibrotic livers. Notably, no induction of hepatocyte proliferation or increase in liver-to-body weight ratio was observed when 11 was administered to wild-type mice, indicating that activation of stress signaling pathways in hepatocytes is required to unlock regenerative potential. Similarly, in a porcine model subjected to 80% hepatectomy, 43 hours post-surgery the regeneration index (RI) in the 11 pre-treatment group was 21% higher than in the control group (2.57 vs 2.12), and the regenerated liver volume increased by 57% (251.8 mL vs 160.0 mL). In cases of fatal 85% hepatectomy, the survival rates in the 11 pre-treatment and post-treatment groups (66.7% [4/6], 83% [5/6]) were significantly higher than in the control group (17% [1/6]). Surprisingly, 11-treated pigs did not exhibit increased intracranial pressure post-surgery, and the treatment prevented elevations in plasma ammonium, bilirubin, and hepatic aspartate aminotransferase. Additionally, 11 exhibited notable hepatoprotective effects. In a CCl4-induced liver injury model, 11 (0.4 mg/kg) significantly reduced apoptotic cells. Notably, it also demonstrated anti-steatotic and anti-fibrotic efficacy in models of alcoholic steatohepatitis (ASH) and chronic CCl4-induced liver fibrosis, respectively. The authors also investigated its inhibitory effects on HCC. In a mouse NASH-HCC model, 11 neither promoted tumorigenesis nor accelerated the progression of established liver tumors compared to the control group. Furthermore, it showed a significant anti-steatotic effect and a trend toward anti-fibrotic activity. Unlike the pro-regenerative signaling induced by MKK4 inhibition in hepatocytes, MKK4 inhibition in HCC cells led to significant downregulation of MKK7 and JNK1 along with strong activation of p38, followed by downregulation of Cyclin B1/D1/A2 and subsequent cell cycle arrest. In a Phase I clinical trial (EudraCT 2021-000193-28), a single or multiple dose administration (5–500 mg) of 11 to 48 healthy male volunteers resulted in an overall adverse event rate comparable to that of the placebo group (36.1% vs 41.7%), with no severe adverse reactions observed. Pharmacokinetic (PK) analysis revealed dose-dependent absorption of compound 11, and bioavailability was increased by 2.1-fold when administered after a meal.
In 2022, Laufer’s group also demonstrated that α-carboline can replace the 7-azaindole in 7 as a novel scaffold to drive selectivity [77]. The increased steric demand and its rigid structure proved to be a highly robust selectivity-inducing structural element without compromising potency. Representative compounds exhibited favorable profiles: compound 12 (Figure 7A) was non-cytotoxic in HepG2 cells (10 μM, 72 h), and compound 13 demonstrated high kinase selectivity across 320 kinases (S(20) = 0.103 at 100 nM). The authors propose that the rigid structure of α-carboline may occupy the hydrophobic pocket of MKK4 while creating steric hindrance with off-target kinases such as BRAF and ZAK. In summary, α-carboline supports the incorporation of various activity-enhancing groups without sacrificing selectivity, offering broad flexibility for PK optimization. Additionally, Laufer’s group designed novel fluorescent labeling compounds based on the α-carboline scaffold for high-throughput fluorescence polarization (FP) screening of MKK4 [78]. In the crystal structures of 7 binding to BRAF, the para-chlorophenyl group extends toward the solvent-accessible region, providing a proper site to link a fluorophore for avoiding steric clashes. Thus, a 5-TAMRA fluorophore was connected to the para-chlorophenyl group via carbon chains of varying lengths. The resulting probe 14 (Figure 7A) exhibited significantly enhanced binding to MKK4 and almost no binding to BRAF. In FP competition assays, probe 14 (20 nM) was able to distinguish MKK4 inhibitors with different affinities, suggesting its potential as a high-throughput screening (HTS) probe.
Recently, Laufer’s group reported the first highly selective 1,4-dihydropyrido[3,4-b]pyrazin-3(2H)-one MKK4 inhibitor [79]. This study was based on BI-D1870 (15, Figure 7B), a reversible ribosomal S6 kinase (RSK) inhibitor with broad off-target activity. After a systematic SAR exploration, the resulting compound 16 only inhibited three kinases at a concentration of 1 μM, significantly outperforming BI-D1870. It also showed excellent selectivity for RSK4 (MKK4 IC50 = 78 nM, RSK4 IC50 = 2920 nM, SI = 37.4). The thermal shift assay (TSA) and cellular thermal shift assay (CETSA) results confirmed the strong binding affinity of compound 16 to MKK4. In mouse HCC and human non-small cell lung cancer (NSCLC) cells, compound 16 displayed no significant cytotoxicity within the tested concentration range (IC50 > 30 μM), a feature attributable to its markedly reduced off-target activity. Furthermore, after incubation in mouse liver microsomes (MLM) for 2 hours, compound 16 did not undergo metabolic degradation, indicating favorable drug-like properties.
4.2. MKK7 Inhibitors
4.2.1. Covalent Inhibitors
Unlike MKK4, the specific ligands for MKK7 are predominantly targeted covalent kinase inhibitors (TCKIs) since it is the only MEK isoform that possesses a cysteine residue in the hinge region (Cys218) [80]. The fungal natural product 5(Z)-7-oxozeaenol (17, Figure 8A) is the first reported covalent inhibitor of MKK7 [64,81,82]. Compound 17 moderately inhibits MKK7 (IC50 = 1.3 μM) and its α,β-unsaturated ketone moiety covalently binds to the Sγ of Cys218, which is located at the end of the hinge region. Even in the MKK7-C218S mutant, 17 does not form a covalent bond with Cys276, the gatekeeper cysteine adjacent to the DFG motif, likely due to steric hindrance by Met212. Additionally, the resorcinol moiety forms two hydrogen bonds with the backbone of hinge residue Met215. The two hydroxyl groups on the 14-membered ring also form hydrogen bonds with the backbone carbonyl groups of Thr146 and Ser263, as well as with the nitrogen atom of Lys165. Notably, there is a unique large hydrophobic pocket located behind the allyl group attached to the phenyl moiety.
London’s group conducted a virtual screening (VS) of a 117,667-compound acrylamide library with the aim of targeting the non-conserved Cys218 in MKK7 [83]. Candidates were required to form at least two hydrogen bonds with the kinase hinge region. The best hit, 18 (Figure 8B), exhibited potent inhibitory activity against MKK7 (IC50 = 11 nM), albeit with a micromolar range cellular activity in HEK293 and 3T3 cells. Based on the docking model, the authors optimized the lead compound using an indazole scaffold and found that substitutions were well tolerated at the 6-position but not at the 7-position. Although the resulting inhibitor 19 (Figure 8B) showed enhanced selectivity over MKK4, it was found to inhibit 4 out of 76 kinases in the panel (Aurora B, LRRK2, MKK4, and FLT3) by more than 75% at 1 μM, and exhibited poor liver microsomal stability (t1/2 < 5 min). In addition to the covalent binding to Cys218, the indazole nitrogen mediated two hydrogen bonds in the hinge region in the crystal structure. Efforts to reduce lipophilicity and improve metabolic stability led to the discovery of compound 20 (Figure 8B), which showed good selectivity in proteomic profiling of bacterial and human cell lysates (MDA-MB-231 cells) but also displayed some cytotoxicity. In an assay blocking activation of primary mouse B Cells, 19 (10 μM) suppressed LPS-induced CD86 expression (a marker of B-cell activation) by 60%, whereas 20 achieved 90% inhibition.
Due to the structural similarity between EGFR and MKK7, Rauh’s group designed a series of MKK7 covalent inhibitors based on the pyrazolopyrimidine scaffold of EGFR inhibitor 21 (MKK7 IC50 = 302 nM) [68]. Replacing the indole group in 21 with an alkyne significantly enhanced both activity and selectivity (22, IC50 = 40 nM). The co-crystal structure of 22 with MKK7 revealed additional space between the Met212 gatekeeper in the MKK7 back pocket and the catalytic Lys165 (Figure 9). Utilizing the alkyne for copper-catalyzed click chemistry to introduce a para-ketophenyl derivative (23) markedly improved activity and selectivity against EGFR. Additionally, the authors employed Robotics-Assisted Screening Platform for Efficient Ligand Discovery (RASPELD) to conduct a focused screening of 101 EGFR-targeted compounds [84]. Hit 24 (with a para-phenolic group) and 25 (with an pyrrolo[2,3-b]pyridine substitution) exhibited excellent MKK7 inhibitory activity (Figure 9). SAR studies highlighted the importance of covalent inhibition, as non-covalent or reversibly covalent inhibitors lacking the acrylamide warhead showed no activity. The covalent inhibitors 23, 24, and 25 exhibited time-dependent engagement with MKK7, a process significantly delayed by ATP competition. Their crystal structures elucidated the binding mode, revealing two key interactions: the pyrazolopyrimidine core forms conserved hydrogen bonds with the Glu213 and Met215, while the acrylamide warhead covalently binds Cys218 and its carbonyl group hydrogen-bonds with Lys221. Differently, the phenolic hydroxyl group of 24 forms an additional hydrogen bond with Asp182, and the nitrogen atoms of the pyrrolopyridine in 25 interacts with the hinge region, explaining their enhanced activity. Notably, although the binding modes of these compounds were similar to those observed for EGFR inhibitors, the orientation of the flexible methionine gatekeeper side chain (Met212 in MKK7) was slightly different. A kinase selectivity profile across 320 kinases showed that 23 at 1 μM inhibited only seven off-target kinases (BLK, BMX, BTK, ITK, JAK3, mTOR, S6K), all of which contain a conserved Cys residue. In cellular assays, 23 completely suppressed the phosphorylation of JNK and c-Jun. Compound 23 exhibited low lipophilicity (cLogP = 1.78), poor aqueous solubility (8 μM), but favorable ligand lipophilic efficiency (LLE = 6.22). It demonstrated good drug absorption in Caco-2 permeability assays and exhibited stability in human and mouse plasma, albeit with moderate microsomal stability.
In 2022, Rauh and London’s group employed the robust copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) reaction for late-stage functionalization (LSF) at the nanomolar scale in a plate-based format [85]. This workflow is compatible with efficient large-scale cellular and biochemical assays for high-throughput testing of covalent inhibitors. The authors selected compound 20 and 22 as starting points for the workflow, reacting them with a library of 448 commercially available azides via CuAAC. The In-Cell Western assay (ICW) was subsequently used to perform high-throughput detection of the phosphorylation of JNK by the products. The most potent derivative 26 (Figure 10A) demonstrated superior target inhibition and intracellular activity (IC50 = 4 nM, EC50 = 265 nM) compared to 22, likely driven by additional lipophilic interactions within the MKK7 back pocket. In contrast, compound 27 (derivative of 20, Figure 10B) showed significantly reduced activity at both the protein and cellular levels. However, targeting the solvent-exposed region also offer hope for the future design of potential exit vectors, serving as a blueprint for the design of PROTACs or other bivalent compounds. Co-crystal structures revealed that the triazole moiety did not form additional interactions either within the back pocket (for 22 derivatives) or in the solvent-exposed periphery of the binding site (for 20 derivatives). Interestingly, Asp182 is pushed away in the 26/MKK7 complex to accommodate the extended compounds. Compared to 22, 26 exhibited better kinetic solubility (174.1 μM), reduced efflux, similar PAMPA and plasma stability values (Plasma stability human/mouse = 116/94%remaining), but poorer intestinal permeability (Caco-2 Papp(A−B) = 11.81×10−6 cm/s) and HLM stability (CLint = 308.1 nL/min/mg).
While covalent drugs offer advantages like high potency, most have historically targeted the cysteine residue. Lysine is an attractive alternative target due to its abundance, but its high flexibility and different chemical properties (like pKa) make it difficult to design binders. Recently, Rauh and London’s group presented two new computational methods, developed within the Rosetta software framework, for designing lysine-targeting covalent inhibitors [86]. In a “ligand-side” approach, a virtual library of “electrophilic analogs” is created by adding different warheads to different positions on the ligand and is subsequently docked into the target protein. In a “protein-side” approach, an electrophile is installed on the target lysine and model its conformational space to find suitable installation vectors on the ligand. Both of these protocols were applied retrospectively to a data set of electrophilic ligands and to a data set of vitamin B6 covalently bound to a receptor lysine residue. The ligand-side protocol successfully identified the known covalent binder in 80% and 86% of cases, while the protein-side protocol achieved identification rates of 56% and 82%, respectively. In the context of the MKK7 kinase, both protocols predicted that compound 28 (a derivative of 18) and 29 would be capable of targeting Lys221 (Figure 10C). Compounds 28 and 29 were synthesized and successfully inhibited MKK7, with IC50 values of 738 nM and 695 nM, respectively. Mass spectrometry confirmed that compound 28 formed a reversible covalent bond with MKK7. The cocrystal structure of compound 28 with MKK7 (PDB: 9hz0) confirmed Lys221 as the binding site. Although the electron density for the covalent linkage was poor, the structure revealed a large movement of the lysine side chain.
Jiang et al. employed a structural modification strategy to optimize the pan-kinase inhibitor 30 [87] into a covalent inhibitor targeting the conserved cysteine residues (Cys247 in MKK4 and Cys261 in MKK7) [62]. This design retained the acrylamide warhead while replacing the pyrimidine core with a pyrrolo[2,3-d]pyrimidine scaffold. This modification maintained dual hydrogen-bonding interactions with hinge region (Met181/Glu179) in MKK4 and simultaneously optimized the spatial proximity between the acrylamide and the target cysteine. The reversible binding affinity in 30 was eliminated by removing the phenylpiperazine moiety. Further introduction of a chlorine atom at the 5-position of the pyrrolopyrimidine core ultimately yielded the potent covalent inhibitor 31 (Figure 11A, MKK4 IC50 = 4 nM; MKK7 IC50 = 181 nM). Competitive pull-down assays demonstrated that 5 μM 31 effectively blocked the binding of the biotinylated probe to MKK4. The pan-kinome profiling revealed that in MDA-MB-231 cells, this compound selectively occupied MKK4 (90.2%) and MKK7 (87.0%) with no impact on other kinases. Treatment with 10 μM 31 completely inhibited anisomycin-induced JNK phosphorylation without affecting the phosphorylation levels of p38, ERK1/2, or ERK5, confirming its favorable pathway selectivity. Notably, even in MKK4- or MKK7-knockdown cells, 31 still fully blocked JNK phosphorylation, an effect dependent on its covalent binding properties. Although the compound alone did not significantly inhibit the proliferation of MDA-MB-231 cells, it exhibited strong synergistic effects when combined with a JNK inhibitor (CI = 0.7–0.9). Recently, Okada’s group reported that at concentrations that did not affect the viability of normal fibroblasts, 31 effectively inhibited JNK activity and reduced the expression of stem cell markers in glioma stem cells (GSCs) [88]. Furthermore, the capacity of GSCs that survived the 31 treatment to form spheres and initiate tumors was impaired.
Scheidt’s group employed sub-micromolar off-target inhibition of MKK7 by the FMS-like tyrosine kinase 3 (FLT3) inhibitor AST-487 (32, Figure 11B) and designed novel covalent MKK7 inhibitors through covalent modifications [89]. After truncating the structure of 32 to retain the phenoxypyrimidine core and introducing an α-chloroacetamide as a covalent warhead targeting Cys218, they explored the chemical space around the aminopyrimidine moiety. Among the derivatives, benzylamine derivative 33 (Figure 11B) exhibited the highest potency (IC50 = 10 nM), while N-methylation or ortho-substitution significantly reduced activity. Although compound 33 showed no activity against MKK3/4/5/6, it displayed notable binding to EGFR and EGFR-L858R in a 97-kinase screening panel. In T cell acute lymphoblastic leukemia (T-ALL) cell lines, compound 33 demonstrated potent cytotoxicity within 48 hours (EC50 = 1.1–2.9 μM), and the cytotoxic effect persisted after compound removal, supporting an irreversible inhibition mechanism.
Schröder et al. used melting thermal shift (ΔTm) assay to screen an in-house library of 360 compounds against MKK7 [70]. These efforts identified nine Type I and Type II covalent inhibitors, among which the Type I inhibitor 34 (Figure 11C) exhibited an IC50 value of 0.08 μM against MKK7. The Type II compound 35 (Figure 11C) showed the highest thermal shift of 24.7 °C and displayed similar activity (IC50 = 0.12 μM). Compounds 34 and 35 significantly suppressed the levels of phosphorylated JNK and c-Jun in sorbitol-treated THP-1 cells at nanomolar concentrations. Co-crystal structures revealed that 34 forms a covalent bond with Cys218 and establishes three hydrogen bonds through its 2-aminopyrolopyrimidine and aminopyrazole scaffolds, thereby stabilizing the DFG-in conformation. In contrast, 35 induces an outward movement (~23°) of the αC-helix and promotes a DFG-out conformation by covalently binding to Cys218, creating a hydrophobic pocket required for its trifluoromethylphenyl moiety. Unexpectedly, ibrutinib (36, Figure 11C) was found to bind not only in the ATP pocket but also at an allosteric site located at the top of the kinase N-lobe. Significant structural alterations for the pocket formation included an ‘‘out’’ swing of Phe202, an outward movement of the helical turn preceding the β1 that harbors Ile133 as well as a small alteration of the Trp151 side chain. Isothermal titration calorimetry (ITC) confirmed that this binding is independent of the ATP site, providing a new strategy for the development of allosteric Type III inhibitors.
4.2.2. Other Inhibitors
Given that naphthoquinone 37 is a reversible weak inhibitor of MKK7 (Kd = 1.1 μM) [90], Quinn’s group conducted a systematic SAR study of natural and synthetic naphthoquinone derivatives (Figure 12) [91]. Among the 33 naphthoquinone compounds evaluated, compounds 38 and 39 exhibited the highest affinities (38: Kd = 230 nM; 39: Kd = 1.9 μM) and showed no activity against MKK4. The SAR analysis revealed that the length of the sulfur-containing side chain is critically important for activity—compound 38 (with a propionic acid chain) displayed greater potency than those with an acetic acid or isobutyric acid chain. Additionally, methoxy substitution or methoxylation at the 5,8-positions of the naphthoquinone ring significantly reduced activity. Compounds 38 and 39 demonstrated antiproliferative activity against various tumor cell lines. Docking studies showed that the thio-propionic acid chain of 38 formed hydrogen bonds with Lys165 and Asp277, while the quinone carbonyl group interacts with Cys276 and Met215.
DTP3 (Ac-D-Tyr-D-Arg-D-PheNH2) is a D-configured tripeptide identified through the screening of a combinatorial library of synthetic peptides targeting the GADD45β/MKK7 complex [48]. In multiple myeloma (MM), NF-κB upregulates GADD45β, which binds to MKK7 and blocks its enzymatic activity, thereby suppressing JNK-mediated apoptosis. DTP3 specifically binds to MKK7 with high affinity, inducing a conformational rearrangement that effectively displaces GADD45β. This “relieves” the inhibition of MKK7, allowing it to activate the JNK signaling cascade and trigger cancer-cell-selective apoptosis. Computational and NMR studies indicate that DTP3 interacts with a shallow pocket on the N-terminal region of MKK7, located proximally to the ATP pocket. The aromatic side chains (D-Tyr and D-Phe) are the primary drivers of this interaction, while the central D-Arg residue contributes minimally [49,92]. DTP3 is highly selective for cancer cells because the GADD45β/MKK7 survival module is non-redundant in these cells, whereas most normal cells do not require GADD45β for survival. In preclinical mouse models, systemic administration of DTP3 induced complete regression of established MM xenografts. In addition, DTP3 has shown the capacity to synergize with bortezomib and can bypass drug resistance to conventional treatments like steroids and immunomodulatory agents [93]. DTP3 exhibits linear systemic exposure, a long terminal plasma half-life (approximately 11–19 hours in preclinical species), and good bioavailability. It distributes extensively to most tissues but notably does not cross the blood-brain barrier. Currently, DTP3 has advanced into Phase I/IIa clinical trials for patients with relapsed or refractory MM, where initial results confirmed its ability to induce a cancer-selective pharmacodynamic response without adverse side effects.
5. Conclusion and Future Perspectives
As elucidated in this review, the strategic positioning of MKK4 and MKK7 as the “gatekeepers” of the JNK signaling architecture offers a unique therapeutic opportunity to modulate maladaptive stress responses with greater precision than direct JNK inhibition. While both kinases share high sequence identity and a canonical bilobal fold, their distinct physiological roles—MKK4 as a regenerative brake and MKK7 as an inflammatory driver—have necessitated divergent medicinal chemistry strategies.
For MKK4, the field has witnessed a paradigm shift from oncology-focused exploration to regenerative medicine. The successful advancement of HRX215 into clinical trials marks a pivotal milestone, providing the first human proof-of-concept that pharmacological inhibition of MKK4 is safe and effective for unlocking liver regeneration. From a structural perspective, the evolution of scaffolds highlights the triumph of structure-based drug design in overcoming the “selectivity challenge” against p38 MAPK and BRAF. Future efforts in this domain are required to refine physicochemical properties to balance liver accumulation with systemic safety.
In contrast, the development of MKK7 inhibitors has been defined by the exploitation of the rare Cys218 residue within the hinge region. This unique structural feature has enabled the design of highly potent covalent inhibitors that offer prolonged target residence time, a property particularly advantageous for treating chronic fibrotic and inflammatory conditions. Moreover, the application of advanced techniques like LSF via click chemistry showcases the power of modern chemical biology in accelerating inhibitor development.
Looking forward, several frontiers remain to be explored. First, while ATP-competitive inhibitors dominate the current landscape, the development of allosteric inhibitors—as hinted by the binding mode of ibrutinib—could offer superior specificity by targeting non-conserved pockets. Second, the application of proteolysis-targeting chimeras (PROTACs) remains an untapped potential for MKK4/7. Given the solvent-exposed exit vectors identified in recent SAR studies (e.g., the para-position of phenyl rings in MKK4 inhibitors), designing bifunctional degraders could eliminate the scaffolding functions of these kinases, potentially yielding distinct biological outcomes compared to kinase inhibition alone. Finally, the emerging class of dual MKK4/7 covalent inhibitors offers a rational combination strategy for aggressive cancers where pathway redundancy limits the efficacy of single-target agents.
In summary, the medicinal chemistry of MKK4 and MKK7 has matured significantly over the past decade. As research transitions from the discovery of chemical probes to the validation of clinical candidates, these upstream kinases stand poised to transform the treatment landscape for acute liver failure, tissue fibrosis, and specific malignancies.
Acknowledgments
This work was supported by the Doctoral Science Foundation of Xinjiang Second Medical College (XGYK2025-102).
Abbreviation
Aβ, amyloid-beta; AD, Alzheimer’s disease; ALF, severe acute liver failure; ATF2,activating transcription factor 2; ASH, alcoholic steatohepatitis; ASO, antisense oligonucleotides; BBB, blood-brain barrier; CETSA, cellular thermal shift assay; CLD, chronic liver diseases; CuAAC, copper(I)-catalyzed alkyne–azide cycloaddition; D domain, docking domain; DVD, domain for versatile docking; FLT3, FMS-like tyrosine kinase 3; FP, fluorescence polarization; GSC, glioma stem cell; HCC, hepatocellular carcinoma; HTS, high-throughput screening; ICW, In-Cell Western assay; ITC, Isothermal titration calorimetry; JIP1, JNK-interacting protein 1; JNK, c-Jun N-terminal kinase; KLF4, Kruppel-like factor 4; LLE, ligand lipophilic efficiency; LPS, lipopolysaccharides; LSF, late-stage functionalization; MAPK, mitogen-activated protein kinase; MAP2Ks, mitogen-activated protein kinase kinases; MLM, mouse liver microsomes; MMP, matrix metalloproteinase; NSCLC, non-small cell lung cancer; PD, Parkinson’s diseases; PK, pharmacokinetic; PLK4, Polo-like kinase 4; POC, percentage of control; POSH, plenty of SH3; PQA, prenylated quinolinecarboxylic acid; PROTACs, proteolysis-targeting chimeras; RA, rheumatoid arthritis; RASPELD, Robotics-Assisted Screening Platform for Efficient Ligand Discovery; RSK, ribosomal S6 kinase; RNAi, RNA interference; SAR, structure-activity relationship; T-ALL, T-cell acute lymphoblastic leukemia; TCKIs, targeted covalent kinase inhibitors; TH, tyrosine hydroxylase; TSA, thermal shift assay; VS, virtual screening.
References
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Atay, O.; Skotheim, J.M. Spatial and temporal signal processing and decision making by MAPK pathways. J Cell Biol 2017, 216, 317–330. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front Pharmacol 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Lu, Y.; Yao, Z.; Wang, P.; Chen, Z.; Zhou, L.; Yang, Q.; Sang, X.; Wang, K.; Cao, G. The role of JNK signaling pathway in organ fibrosis. J Adv Res 2025, 74, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Kumariya, S.; Katekar, R.; Verma, S.; Goand, U.K.; Gayen, J.R. JNK signaling pathway in metabolic disorders: An emerging therapeutic target. Eur J Pharmacol 2021, 901, 174079. [Google Scholar] [CrossRef] [PubMed]
- de Los Reyes Corrales, T.; Losada-Perez, M.; Casas-Tinto, S. JNK Pathway in CNS Pathologies. Int J Mol Sci 2021, 22, 3883. [Google Scholar] [CrossRef]
- Yan, H.; He, L.; Lv, D.; Yang, J.; Yuan, Z. The Role of the Dysregulated JNK Signaling Pathway in the Pathogenesis of Human Diseases and Its Potential Therapeutic Strategies: A Comprehensive Review. Biomolecules 2024, 14, 243. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhou, Y.; Xiong, Z.; Zou, L.; Jiang, M.; Luo, Z.; Wen, S.; Liu, W.; Liu, S.; Li, W. Involvement of JNK in the embryonic development and organogenesis in zebrafish. Mar Biotechnol (NY) 2013, 15, 716–725. [Google Scholar] [CrossRef]
- Zhu, Y.; Shuai, W.; Zhao, M.; Pan, X.; Pei, J.; Wu, Y.; Bu, F.; Wang, A.; Ouyang, L.; Wang, G. Unraveling the Design and Discovery of c-Jun N-Terminal Kinase Inhibitors and Their Therapeutic Potential in Human Diseases. J Med Chem 2022, 65, 3758–3775. [Google Scholar] [CrossRef]
- Bhujbal, S.P.; Hah, J.M. Advances in JNK inhibitor development: therapeutic prospects in neurodegenerative diseases and fibrosis. Arch Pharm Res 2025, 48, 858–886. [Google Scholar] [CrossRef]
- Katzengruber, L.; Sander, P.; Laufer, S. MKK4 Inhibitors-Recent Development Status and Therapeutic Potential. Int J Mol Sci 2023, 24, 7495. [Google Scholar] [CrossRef]
- Park, J.G.; Aziz, N.; Cho, J.Y. MKK7, the essential regulator of JNK signaling involved in cancer cell survival: a newly emerging anticancer therapeutic target. Ther Adv Med Oncol 2019, 11, 1758835919875574. [Google Scholar] [CrossRef]
- Kishimoto, H.; Nakagawa, K.; Watanabe, T.; Kitagawa, D.; Momose, H.; Seo, J.; Nishitai, G.; Shimizu, N.; Ohata, S.; Tanemura, S.; Asaka, S.; Goto, T.; Fukushi, H.; Yoshida, H.; Suzuki, A.; Sasaki, T.; Wada, T.; Penninger, J.M.; Nishina, H.; Katada, T. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. J Biol Chem 2003, 278, 16595–16601. [Google Scholar] [CrossRef]
- Lisnock, J.; Griffin, P.; Calaycay, J.; Frantz, B.; Parsons, J.; O’Keefe, S.J.; LoGrasso, P. Activation of JNK3 alpha 1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry 2000, 39, 3141–3148. [Google Scholar] [CrossRef]
- Lawler, S.; Fleming, Y.; Goedert, M.; Cohen, P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr Biol 1998, 8, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev 2001, 15, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Caliz, A.D.; Vertii, A.; Fisch, V.; Yoon, S.; Yoo, H.J.; Keaney, J.F., Jr.; Kant, S. Mitogen-activated protein kinase kinase 7 in inflammatory, cancer, and neurological diseases. Front Cell Dev Biol 2022, 10, 979673. [Google Scholar] [CrossRef]
- Sasaki, T.; Wada, T.; Kishimoto, H.; Irie-Sasaki, J.; Matsumoto, G.; Goto, T.; Yao, Z.; Wakeham, A.; Mak, T.W.; Suzuki, A.; Cho, S.K.; Zuniga-Pflucker, J.C.; Oliveira-dos-Santos, A.J.; Katada, T.; Nishina, H.; Penninger, J.M. The stress kinase mitogen-activated protein kinase kinase (MKK)7 is a negative regulator of antigen receptor and growth factor receptor-induced proliferation in hematopoietic cells. J Exp Med 2001, 194, 757–768. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Ko, C.I.; Zhang, L.; Puga, A.; Xia, Y. Distinct signaling properties of mitogen-activated protein kinase kinases 4 (MKK4) and 7 (MKK7) in embryonic stem cell (ESC) differentiation. J Biol Chem 2012, 287, 2787–2797. [Google Scholar] [CrossRef]
- Wang, X.; Destrument, A.; Tournier, C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta 2007, 1773, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Zwirner, S.; Abu Rmilah, A.A.; Klotz, S.; Pfaffenroth, B.; Kloevekorn, P.; Moschopoulou, A.A.; Schuette, S.; Haag, M.; Selig, R.; Li, K.; Zhou, W.; Nelson, E.; Poso, A.; Chen, H.; Amiot, B.; Jia, Y.; Minshew, A.; Michalak, G.; Cui, W.; Rist, E.; Longerich, T.; Jung, B.; Felgendreff, P.; Trompak, O.; Premsrirut, P.K.; Gries, K.; Muerdter, T.E.; Heinkele, G.; Wuestefeld, T.; Shapiro, D.; Weissbach, M.; Koenigsrainer, A.; Sipos, B.; Ab, E.; Zacarias, M.O.; Theisgen, S.; Gruenheit, N.; Biskup, S.; Schwab, M.; Albrecht, W.; Laufer, S.; Nyberg, S.; Zender, L. First-in-class MKK4 inhibitors enhance liver regeneration and prevent liver failure. Cell 2024, 187, 1666–1684 e26. [Google Scholar] [CrossRef]
- Hindson, J. Development of MKK4 inhibitors for liver regeneration. Nat Rev Gastroenterol Hepatol 2024, 21, 376. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Shirakabe, K.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Matsuda, S.; Moriguchi, T.; Gotoh, Y.; Matsumoto, K.; Nishida, E. TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem 1997, 272, 8141–8144. [Google Scholar] [CrossRef]
- Liu, Y.; Gorospe, M.; Yang, C.; Holbrook, N.J. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun N-terminal kinase activity and AP-1-dependent gene activation. J Biol Chem 1995, 270, 8377–8380. [Google Scholar] [CrossRef] [PubMed]
- Takekawa, M.; Tatebayashi, K.; Saito, H. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell 2005, 18, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Dai, T.; Deak, J.C.; Kyriakis, J.M.; Zon, L.I.; Woodgett, J.R.; Templeton, D.J. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature 1994, 372, 798–800. [Google Scholar] [CrossRef]
- Huang, J.; Tu, Z.; Lee, F.S. Mutations in protein kinase subdomain X differentially affect MEKK2 and MEKK1 activity. Biochem Biophys Res Commun 2003, 303, 532–540. [Google Scholar] [CrossRef]
- Deacon, K.; Blank, J.L. Characterization of the mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 pathways regulated by MEK kinases 2 and 3. MEK kinase 3 activates MKK3 but does not cause activation of p38 kinase in vivo. J Biol Chem 1997, 272, 14489–14496. [Google Scholar] [CrossRef] [PubMed]
- Fleming, Y.; Armstrong, C.G.; Morrice, N.; Paterson, A.; Goedert, M.; Cohen, P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem J 2000, 352, 145–154. [Google Scholar] [CrossRef]
- Lombard, C.K.; Davis, A.L.; Inukai, T.; Maly, D.J. Allosteric Modulation of JNK Docking Site Interactions with ATP-Competitive Inhibitors. Biochemistry 2018, 57, 5897–5909. [Google Scholar] [CrossRef]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 2004, 15, 661–675. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Hibi, M.; Lin, A.; Smeal, T.; Minden, A.; Karin, M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993, 7, 2135–2148. [Google Scholar] [CrossRef]
- Blake, M.J.; Steer, C.J. Liver Regeneration in Acute on Chronic Liver Failure. Clin Liver Dis 2023, 27, 595–616. [Google Scholar] [CrossRef]
- Wirth, K.M.; Kizy, S.; Steer, C.J. Liver Regeneration in the Acute Liver Failure Patient. Clin Liver Dis 2018, 22, 269–287. [Google Scholar] [CrossRef]
- Wuestefeld, T.; Pesic, M.; Rudalska, R.; Dauch, D.; Longerich, T.; Kang, T.W.; Yevsa, T.; Heinzmann, F.; Hoenicke, L.; Hohmeyer, A.; Potapova, A.; Rittelmeier, I.; Jarek, M.; Geffers, R.; Scharfe, M.; Klawonn, F.; Schirmacher, P.; Malek, N.P.; Ott, M.; Nordheim, A.; Vogel, A.; Manns, M.P.; Zender, L. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell 2013, 153, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Stress kinase MKK7: savior of cell cycle arrest and cellular senescence. Cell Cycle 2004, 3, 577–579. [Google Scholar] [CrossRef]
- Ura, S.; Nishina, H.; Gotoh, Y.; Katada, T. Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol Cell Biol 2007, 27, 5514–5522. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, X.; Cao, J.; Zhu, T.; Liang, S.; Du, L.; Cao, M.; Wang, H.; Zhang, Y.; Zhou, Y.; Shen, B.; Feng, J.; Zhang, J.; Wang, J.; Jin, J. Components of the JNK-MAPK pathway play distinct roles in hepatocellular carcinoma. J Cancer Res Clin Oncol 2023, 149, 17495–17509. [Google Scholar] [CrossRef] [PubMed]
- Schramek, D.; Kotsinas, A.; Meixner, A.; Wada, T.; Elling, U.; Pospisilik, J.A.; Neely, G.G.; Zwick, R.H.; Sigl, V.; Forni, G.; Serrano, M.; Gorgoulis, V.G.; Penninger, J.M. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet 2011, 43, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Saito, H.; Takekawa, M. SAPK pathways and p53 cooperatively regulate PLK4 activity and centrosome integrity under stress. Nat Commun 2013, 4, 1775. [Google Scholar] [CrossRef]
- Hubner, A.; Mulholland, D.J.; Standen, C.L.; Karasarides, M.; Cavanagh-Kyros, J.; Barrett, T.; Chi, H.; Greiner, D.L.; Tournier, C.; Sawyers, C.L.; Flavell, R.A.; Wu, H.; Davis, R.J. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc Natl Acad Sci U S A 2012, 109, 12046–12051. [Google Scholar] [CrossRef]
- Atala, A. Re: JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. J Urol 2013, 189. [Google Scholar] [CrossRef]
- Shen, Y.; Park, C.S.; Suppipat, K.; Mistretta, T.A.; Puppi, M.; Horton, T.M.; Rabin, K.; Gray, N.S.; Meijerink, J.P.P.; Lacorazza, H.D. Inactivation of KLF4 promotes T-cell acute lymphoblastic leukemia and activates the MAP2K7 pathway. Leukemia 2017, 31, 1314–1324. [Google Scholar] [CrossRef]
- Qiu, F.; Yang, L.; Lu, X.; Chen, J.; Wu, D.; Wei, Y.; Nong, Q.; Zhang, L.; Fang, W.; Chen, X.; Ling, X.; Yang, B.; Zhang, X.; Zhou, Y.; Lu, J. The MKK7 p.Glu116Lys Rare Variant Serves as a Predictor for Lung Cancer Risk and Prognosis in Chinese. PLoS Genet 2016, 12, e1005955. [Google Scholar] [CrossRef]
- Sakai, H.; Sato, A.; Aihara, Y.; Ikarashi, Y.; Midorikawa, Y.; Kracht, M.; Nakagama, H.; Okamoto, K. MKK7 mediates miR-493-dependent suppression of liver metastasis of colon cancer cells. Cancer Sci 2014, 105, 425–430. [Google Scholar] [CrossRef]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; van Duin, M.; Zoppoli, P.; Jaxa-Chamiec, A.; Thotakura, A.K.; Dyson, J.; Walker, B.A.; Leonardi, A.; Chambery, A.; Driessen, C.; Sonneveld, P.; Morgan, G.; Palumbo, A.; Tramontano, A.; Rahemtulla, A.; Ruvo, M.; Franzoso, G. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef]
- Rega, C.; Russo, R.; Foca, A.; Sandomenico, A.; Iaccarino, E.; Raimondo, D.; Milanetti, E.; Tornatore, L.; Franzoso, G.; Pedone, P.V.; Ruvo, M.; Chambery, A. Probing the interaction interface of the GADD45beta/MKK7 and MKK7/DTP3 complexes by chemical cross-linking mass spectrometry. Int J Biol Macromol 2018, 114, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Jun, S.Y.; Na, H.J.; Kim, H.T.; Jung, S.Y.; Ha, G.H.; Park, Y.H.; Long, L.Z.; Yu, D.Y.; Kim, J.M.; Kim, J.H.; Ko, J.H.; Kim, C.H.; Kim, N.S. Inhibition of MKK7-JNK by the TOR signaling pathway regulator-like protein contributes to resistance of HCC cells to TRAIL-induced apoptosis. Gastroenterology 2012, 143, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, J.; Pei, Y.; Wang, Q.; Wu, Y.; Qiu, G.; Zhang, D.; Lv, M.; Li, W.; Zhang, J. Neddylation controls basal MKK7 kinase activity in breast cancer cells. Oncogene 2016, 35, 2624–2633. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, W.; Wang, J.; Feng, J.; Wang, Q.; Jin, J.; Lv, M.; Li, X.; Li, Y.; Ma, Y.; Shen, B.; Zhang, J. Receptor for activated C kinase 1 promotes hepatocellular carcinoma growth by enhancing mitogen-activated protein kinase kinase 7 activity. Hepatology 2013, 57, 140–151. [Google Scholar] [CrossRef]
- Takahashi, S.; Ebihara, A.; Kajiho, H.; Kontani, K.; Nishina, H.; Katada, T. RASSF7 negatively regulates pro-apoptotic JNK signaling by inhibiting the activity of phosphorylated-MKK7. Cell Death Differ 2011, 18, 645–655. [Google Scholar] [CrossRef]
- Caliz, A.D.; Yoo, H.J.; Vertii, A.; Dolan, A.C.; Tournier, C.; Davis, R.J.; Keaney, J.F., Jr.; Kant, S. Mitogen Kinase Kinase (MKK7) Controls Cytokine Production In Vitro and In Vivo in Mice. Int J Mol Sci 2021, 22, 9364. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Boyle, D.L.; Berdeja, A.; Firestein, G.S. Regulation of inflammatory arthritis by the upstream kinase mitogen activated protein kinase kinase 7 in the c-Jun N-terminal kinase pathway. Arthritis Res Ther 2012, 14, R38. [Google Scholar] [CrossRef]
- Tournier, C.; Whitmarsh, A.J.; Cavanagh, J.; Barrett, T.; Davis, R.J. The MKK7 gene encodes a group of c-Jun NH2-terminal kinase kinases. Mol Cell Biol 1999, 19, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.T.; Bardwell, A.J.; Grewal, S.; Iverson, C.; Bardwell, L. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem 2006, 281, 13169–13179. [Google Scholar] [CrossRef]
- Asaoka, Y.; Nishina, H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J Biochem 2010, 148, 393–401. [Google Scholar] [CrossRef]
- Li, X.; MacLeod, R.; Dunlop, A.J.; Edwards, H.V.; Advant, N.; Gibson, L.C.; Devine, N.M.; Brown, K.M.; Adams, D.R.; Houslay, M.D.; Baillie, G.S. A scanning peptide array approach uncovers association sites within the JNK/beta arrestin signalling complex. FEBS Lett 2009, 583, 3310–3316. [Google Scholar] [CrossRef]
- Ho, D.T.; Bardwell, A.J.; Abdollahi, M.; Bardwell, L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem 2003, 278, 32662–32672. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jiang, B.; He, Z.; Ficarro, S.B.; Che, J.; Marto, J.A.; Gao, Y.; Zhang, T.; Gray, N.S. Discovery of Covalent MKK4/7 Dual Inhibitor. Cell Chem Biol 2020, 27, 1553–1560 e8. [Google Scholar] [CrossRef]
- Deibler, K.K.; Mishra, R.K.; Clutter, M.R.; Antanasijevic, A.; Bergan, R.; Caffrey, M.; Scheidt, K.A. A Chemical Probe Strategy for Interrogating Inhibitor Selectivity Across the MEK Kinase Family. ACS Chem Biol 2017, 12, 1245–1256. [Google Scholar] [CrossRef]
- Sogabe, Y.; Matsumoto, T.; Hashimoto, T.; Kirii, Y.; Sawa, M.; Kinoshita, T. 5Z-7-Oxozeaenol covalently binds to MAP2K7 at Cys218 in an unprecedented manner. Bioorg Med Chem Lett 2015, 25, 593–596. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Pei, D.S.; Guan, Q.H.; Sun, Y.F.; Liu, X.M.; Zhang, G.Y. Blockade of the translocation and activation of mitogen-activated protein kinase kinase 4 (MKK4) signaling attenuates neuronal damage during later ischemia-reperfusion. J Neurochem 2006, 98, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 2003, 22, 252–261. [Google Scholar] [CrossRef]
- Shevchenko, E.; Poso, A.; Pantsar, T. The autoinhibited state of MKK4: Phosphorylation, putative dimerization and R134W mutant studied by molecular dynamics simulations. Comput Struct Biotechnol J 2020, 18, 2687–2698. [Google Scholar] [CrossRef]
- Wolle, P.; Hardick, J.; Cronin, S.J.F.; Engel, J.; Baumann, M.; Lategahn, J.; Penninger, J.M.; Rauh, D. Targeting the MKK7-JNK (Mitogen-Activated Protein Kinase Kinase 7-c-Jun N-Terminal Kinase) Pathway with Covalent Inhibitors. J Med Chem 2019, 62, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, Y.; Hashimoto, T.; Matsumoto, T.; Kirii, Y.; Sawa, M.; Kinoshita, T. A crucial role of Cys218 in configuring an unprecedented auto-inhibition form of MAP2K7. Biochem Biophys Res Commun 2016, 473, 476–481. [Google Scholar] [CrossRef]
- Schroder, M.; Tan, L.; Wang, J.; Liang, Y.; Gray, N.S.; Knapp, S.; Chaikuad, A. Catalytic Domain Plasticity of MKK7 Reveals Structural Mechanisms of Allosteric Activation and Diverse Targeting Opportunities. Cell Chem Biol 2020, 27, 1285–1295 e4. [Google Scholar] [CrossRef]
- Ehrt, C.; Schulze, T.; Graef, J.; Diedrich, K.; Pletzer-Zelgert, J.; Rarey, M. ProteinsPlus: a publicly available resource for protein structure mining. Nucleic Acids Res 2025, 53, W478–W484. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Kikuchi, H.; Shakespear, N.; Suzuki, T.; Yamaki, J.; Homma, M.K.; Oshima, Y.; Homma, Y. Prenylated quinolinecarboxylic acid derivative prevents neuronal cell death through inhibition of MKK4. Biochem Pharmacol 2019, 162, 109–122. [Google Scholar] [CrossRef]
- Deibler, K.K.; Schiltz, G.E.; Clutter, M.R.; Mishra, R.K.; Vagadia, P.P.; O’Connor, M.; George, M.D.; Gordon, R.; Fowler, G.; Bergan, R.; Scheidt, K.A. Synthesis and Biological Evaluation of 3-Arylindazoles as Selective MEK4 Inhibitors. ChemMedChem 2019, 14, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.J.; Pham, T.N.D.; Oelschlager, H.E.; Munshi, H.G.; Scheidt, K.A. Rational Design, Optimization, and Biological Evaluation of Novel MEK4 Inhibitors against Pancreatic Adenocarcinoma. ACS Med Chem Lett 2021, 12, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Klovekorn, P.; Pfaffenrot, B.; Juchum, M.; Selig, R.; Albrecht, W.; Zender, L.; Laufer, S.A. From off-to on-target: New BRAF-inhibitor-template-derived compounds selectively targeting mitogen activated protein kinase kinase 4 (MKK4). Eur J Med Chem 2021, 210, 112963. [Google Scholar] [CrossRef]
- Pfaffenrot, B.; Klovekorn, P.; Juchum, M.; Selig, R.; Albrecht, W.; Zender, L.; Laufer, S.A. Design and synthesis of 1H-pyrazolo[3,4-b]pyridines targeting mitogen-activated protein kinase kinase 4 (MKK4)—A promising target for liver regeneration. Eur J Med Chem 2021, 218, 113371. [Google Scholar] [CrossRef]
- Juchum, M.; Pfaffenrot, B.; Klovekorn, P.; Selig, R.; Albrecht, W.; Zender, L.; Laufer, S.A. Scaffold modified Vemurafenib analogues as highly selective mitogen activated protein kinase kinase 4 (MKK4) inhibitors. Eur J Med Chem 2022, 240, 114584. [Google Scholar] [CrossRef]
- Kircher, T.; Pantsar, T.; Oder, A.; Peter von Kries, J.; Juchum, M.; Pfaffenrot, B.; Kloevekorn, P.; Albrecht, W.; Selig, R.; Laufer, S. Design and synthesis of novel fluorescently labeled analogs of vemurafenib targeting MKK4. Eur J Med Chem 2021, 209, 112901. [Google Scholar] [CrossRef]
- Katzengruber, L.; Sander, P.; Zwirner, S.; Rasch, A.; Eberlein, E.; Selig, R.; Albrecht, W.; Zender, L.; Laufer, S.A. Discovery of the First Highly Selective 1,4-dihydropyrido[3,4-b]pyrazin-3(2H)-one MKK4 Inhibitor. J Med Chem 2025, 68, 14782–14805. [Google Scholar] [CrossRef]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol 2013, 20, 146–159. [Google Scholar] [CrossRef]
- Chen, T.J.; Du, W.; Junco, J.J.; Bridges, C.S.; Shen, Y.; Puppi, M.; Rabin, K.R.; Lacorazza, H.D. Inhibition of the MAP2K7-JNK pathway with 5Z-7-oxozeaenol induces apoptosis in T-cell acute lymphoblastic leukemia. Oncotarget 2021, 12, 1787–1801. [Google Scholar] [CrossRef]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.F.; Roth, M.; Toader, D.; Saeh, J.C.; Chen, H. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem Biol 2013, 8, 643–650. [Google Scholar] [CrossRef]
- Shraga, A.; Olshvang, E.; Davidzohn, N.; Khoshkenar, P.; Germain, N.; Shurrush, K.; Carvalho, S.; Avram, L.; Albeck, S.; Unger, T.; Lefker, B.; Subramanyam, C.; Hudkins, R.L.; Mitchell, A.; Shulman, Z.; Kinoshita, T.; London, N. Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chem Biol 2019, 26, 98–108 e5. [Google Scholar] [CrossRef]
- Wolle, P.; Engel, J.; Smith, S.; Goebel, L.; Hennes, E.; Lategahn, J.; Rauh, D. Characterization of Covalent Pyrazolopyrimidine-MKK7 Complexes and a Report on a Unique DFG-in/Leu-in Conformation of Mitogen-Activated Protein Kinase Kinase 7 (MKK7). J Med Chem 2019, 62, 5541–5546. [Google Scholar] [CrossRef]
- Gehrtz, P.; Marom, S.; Buhrmann, M.; Hardick, J.; Kleinbolting, S.; Shraga, A.; Dubiella, C.; Gabizon, R.; Wiese, J.N.; Muller, M.P.; Cohen, G.; Babaev, I.; Shurrush, K.; Avram, L.; Resnick, E.; Barr, H.; Rauh, D.; London, N. Optimization of Covalent MKK7 Inhibitors via Crude Nanomole-Scale Libraries. J Med Chem 2022, 65, 10341–10356. [Google Scholar] [CrossRef] [PubMed]
- Tivon, B.; Wiese, J.; Muller, M.P.; Gabizon, R.; Rauh, D.; London, N. Computational Design of Lysine Targeting Covalent Binders Using Rosetta. J Chem Inf Model 2025, 65, 5612–5622. [Google Scholar] [CrossRef]
- Rao, S.; Gurbani, D.; Du, G.; Everley, R.A.; Browne, C.M.; Chaikuad, A.; Tan, L.; Schroder, M.; Gondi, S.; Ficarro, S.B.; Sim, T.; Kim, N.D.; Berberich, M.J.; Knapp, S.; Marto, J.A.; Westover, K.D.; Sorger, P.K.; Gray, N.S. Leveraging Compound Promiscuity to Identify Targetable Cysteines within the Kinome. Cell Chem Biol 2019, 26, 818–829 e9. [Google Scholar] [CrossRef]
- Nakamura, K.; Ito, Y.; Takenouchi, S.; Nakagawa-Saito, Y.; Suzuki, S.; Togashi, K.; Sugai, A.; Mitobe, Y.; Sonoda, Y.; Kitanaka, C.; Okada, M. Effective Targeting of Glioma Stem Cells by BSJ-04-122, a Novel Covalent MKK4/7 Dual Inhibitor. Anticancer Res 2025, 45, 2917–2924. [Google Scholar] [CrossRef]
- Kim, D.R.; Orr, M.J.; Kwong, A.J.; Deibler, K.K.; Munshi, H.H.; Bridges, C.S.; Chen, T.J.; Zhang, X.; Lacorazza, H.D.; Scheidt, K.A. Rational Design of Highly Potent and Selective Covalent MAP2K7 Inhibitors. ACS Med Chem Lett 2023, 14, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, W.S.; Yu, T.; Yi, Y.S.; Park, J.G.; Jeong, D.; Kim, J.H.; Oh, J.S.; Yoon, K.; Kim, J.H.; Cho, J.Y. Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem Pharmacol 2014, 88, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Karpenko, A.S.; Khlebnikov, A.I.; Shibinska, M.O.; Levandovskiy, I.A.; Kirpotina, L.N.; Danilenko, N.V.; Quinn, M.T. Synthesis, anticancer activity, and molecular modeling of 1,4-naphthoquinones that inhibit MKK7 and Cdc25. Eur J Med Chem 2019, 183, 111719. [Google Scholar] [CrossRef] [PubMed]
- Sandomenico, A.; Di Rienzo, L.; Calvanese, L.; Iaccarino, E.; D’Auria, G.; Falcigno, L.; Chambery, A.; Russo, R.; Franzoso, G.; Tornatore, L.; D’Abramo, M.; Ruvo, M.; Milanetti, E.; Raimondo, D. Insights into the Interaction Mechanism of DTP3 with MKK7 by Using STD-NMR and Computational Approaches. Biomedicines 2020, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Capece, D.; D’Andrea, D.; Begalli, F.; Verzella, D.; Bennett, J.; Acton, G.; Campbell, E.A.; Kelly, J.; Tarbit, M.; Adams, N.; Bannoo, S.; Leonardi, A.; Sandomenico, A.; Raimondo, D.; Ruvo, M.; Chambery, A.; Oblak, M.; Al-Obaidi, M.J.; Kaczmarski, R.S.; Gabriel, I.; Oakervee, H.E.; Kaiser, M.F.; Wechalekar, A.; Benjamin, R.; Apperley, J.F.; Auner, H.W.; Franzoso, G. Preclinical toxicology and safety pharmacology of the first-in-class GADD45beta/MKK7 inhibitor and clinical candidate, DTP3. Toxicol Rep 2019, 6, 369–379. [Google Scholar] [CrossRef] [PubMed]
Short Biography of Authors
Min Zhao received her a Medical Doctor degree in Traditional Chinese Medicine from Gansu University of Chinese Medicine in 2025. She is currently a Lecturer at the School of Pharmacy, Xinjiang Second Medical College. Her main research interests focus on the processing of Chinese Materia Medica and pharmaceutical technology.
Baojian Li received his master’s degree in traditional Chinese medicine from Shaanxi University of traditional Chinese medicine in 2024. He is currently working in the scientific and technological innovation and transformation center of Xinjiang Second Medical College. His main research interests include the extraction and bioactivity evaluation of ethnic medicine ingredients including traditional Chinese medicine.
Ying Gao obtained her master’s degree in traditional Chinese medicine from Shaanxi University of Chinese Medicine in 2025. She currently works at the Science and Technology Innovation and Transformation Center of Xinjiang Second Medical College. Her primary research focuses on the development of Chinese medicine resources and quality analysis of Chinese medicinal materials.
Liang Yan holds a bachelor’s degree from Xinjiang Normal University. She is currently an Experimentalist at Xinjiang Second Medical College, with research interests focusing on traditional Chinese medicine.
Nanqi Shao is an undergraduate student majoring in Clinical Medicine at Xinjiang Second Medical College, enrolled in 2023. His research interest focuses on clinical pharmacology and therapeutics of fibrotic diseases.
Xinbo Shi obtained a Master’s degree in traditional Chinese medicine from Shaanxi University of Chinese Medicine. He currently works at the Shaanxi Collaborative Innovation Center of Chinese Medicine Resources Industrialization. His primary research focuses on the development of Chinese medicine resources and quality analysis of Chinese medicinal materials.
Jie Li obtained a Medical Doctor degree in Traditional Chinese Medicine from Gansu University of Chinese Medicine in 2025. She is currently employed as a lecturer at the School of Pharmacy, Xinjiang Second Medical College. Her main research direction is the prevention and treatment of cardiovascular and drug-induced liver injuries.
Figure 1.
Schematic representation of the JNK signaling architecture. The pathway exhibits a funnel-like topology where diverse upstream stimuli converge upon the central gatekeepers, MKK4 and MKK7.
Figure 1.
Schematic representation of the JNK signaling architecture. The pathway exhibits a funnel-like topology where diverse upstream stimuli converge upon the central gatekeepers, MKK4 and MKK7.
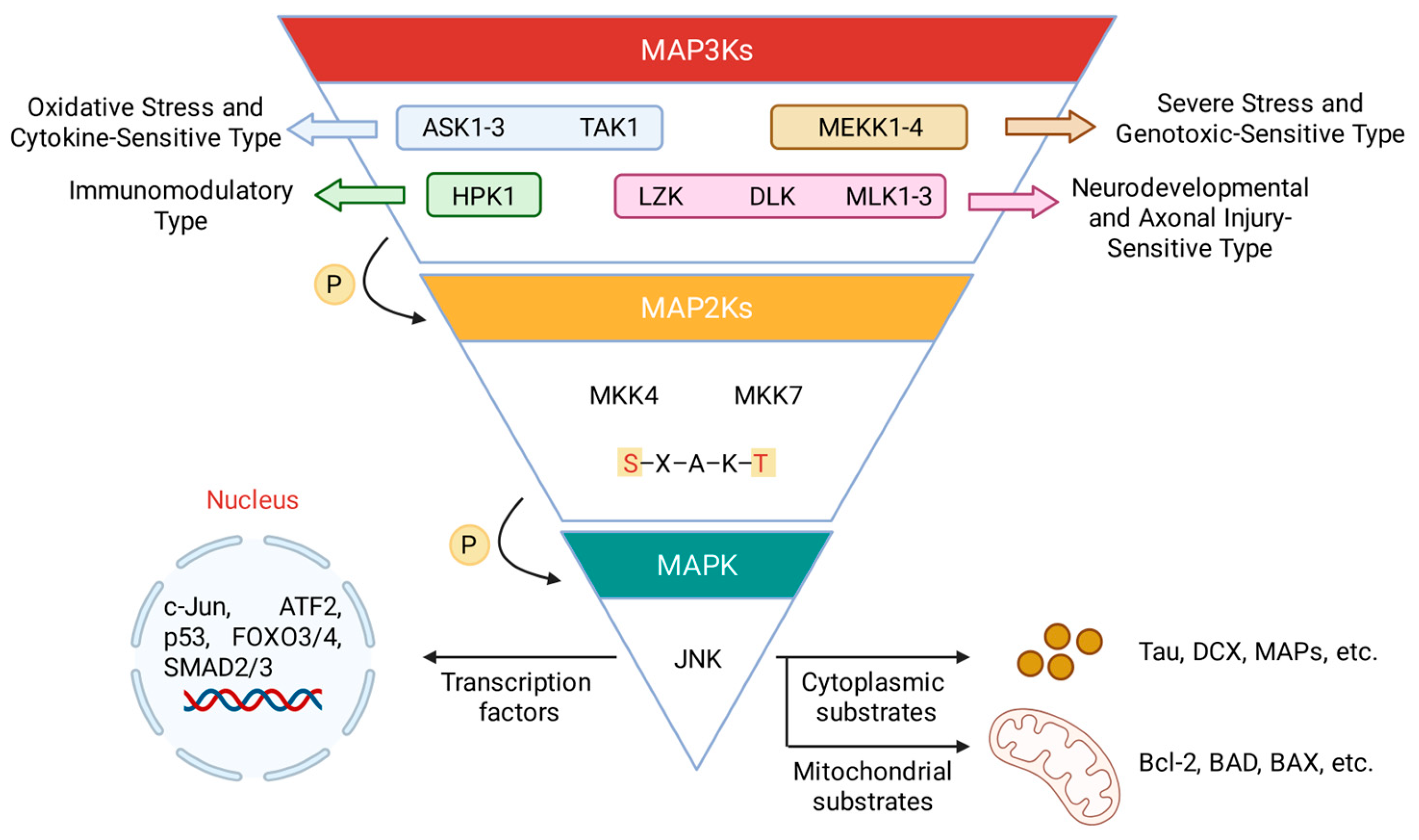
Figure 2.
The role of MKK4 in liver protection and regeneration.
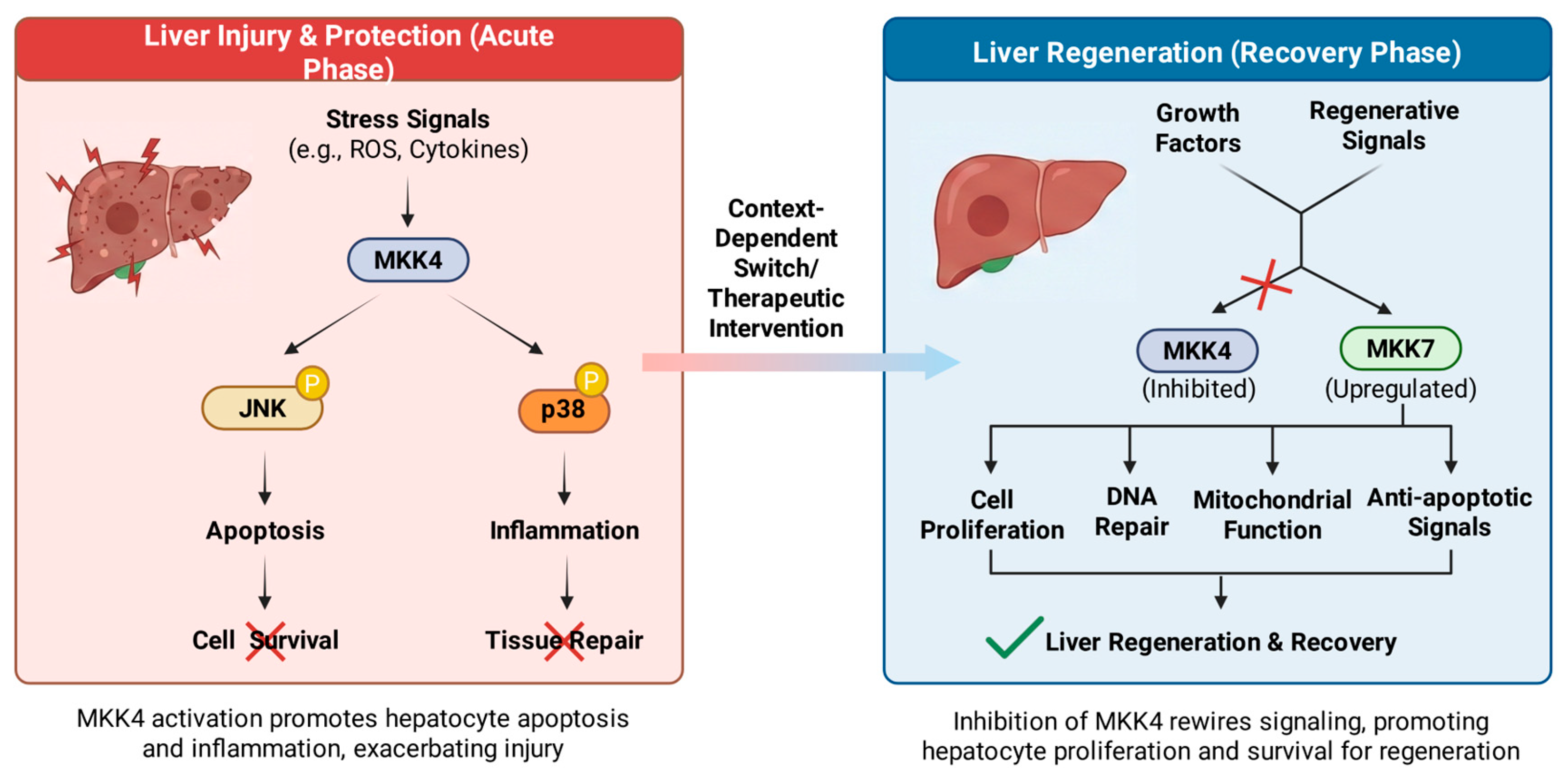
Figure 3.
(A) The dual role of MKK7 in cancer progression. (B) Schematic representation of the MKK7 signaling pathway in the inflammatory response.
Figure 3.
(A) The dual role of MKK7 in cancer progression. (B) Schematic representation of the MKK7 signaling pathway in the inflammatory response.
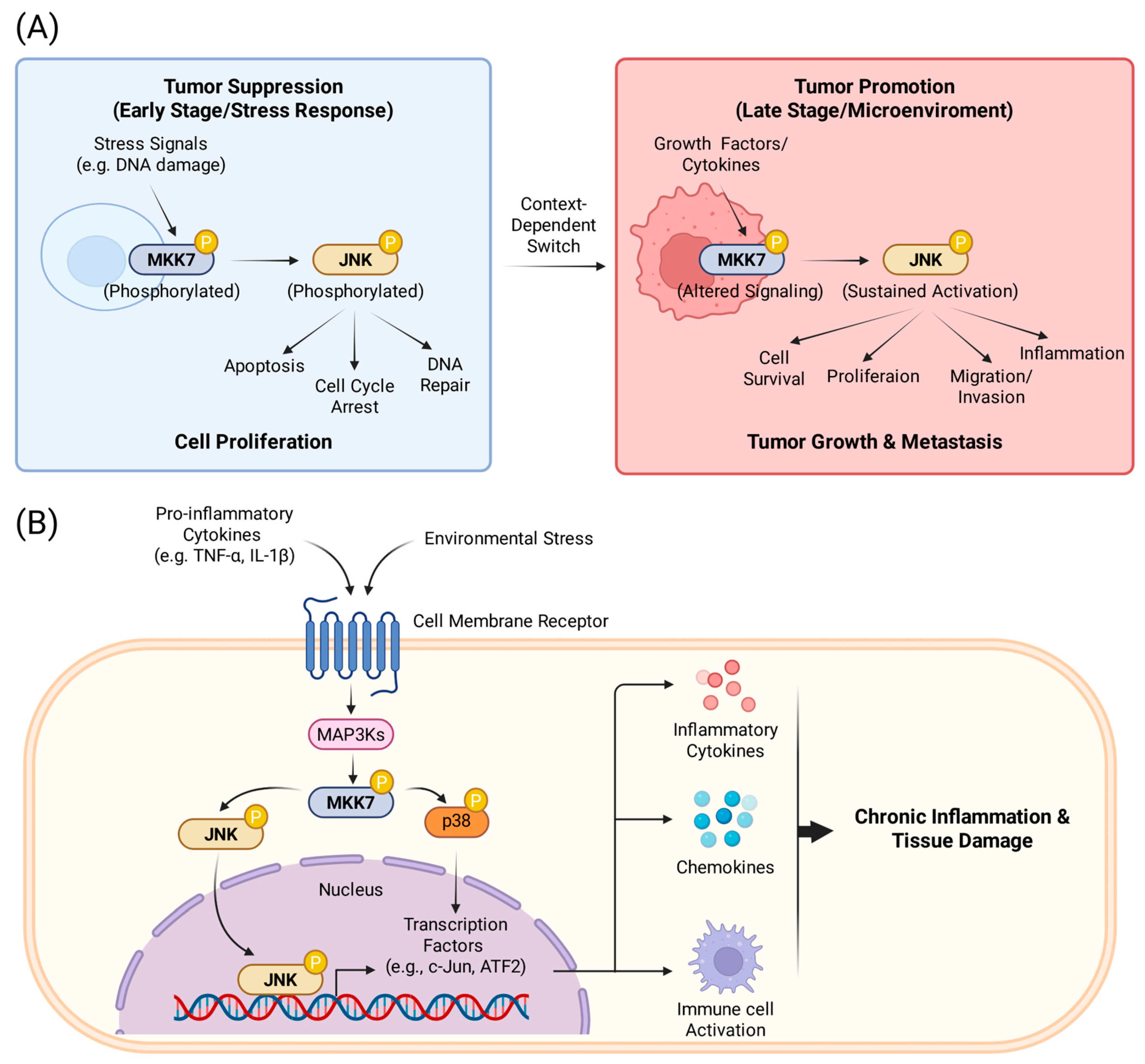
Figure 4.
(A) Schematic illustration of MKK4 and MKK7. The two kinases contain several functional domains for activation and binding of other proteins (D, docking domain; DVD, domain of versatile docking; P, phosphorylation site). They are activated by phosphorylation of two residues in their kinase domain (MKK4: Ser257 and Thr261; MKK7: Ser271 and Thr275). Cysteine residues for covalent binding are also highlighted. (B) Crystal structure of human non-phosphorylated MKK4 kinase domain complexed with AMP-PNP (PDB: 3ALN). The kinase domain is colored in magenta, and the DVD domain is colored in light blue. However, the currently known crystal structures of MKK4 and MKK7 are missing the D domain in the N-lobe. (C) Three-dimensional structural models of the ATP-binding pockets of MKK2 (purple), MKK4 (wheat), and MKK7 (green) are displayed. Quantitative characterization of the ATP-binding pockets, including surface area (Å2), volume (Å3), depth (Å), and hydrophobicity, is computed using ProteinPlus [71]. The corresponding data are summarized in the table below. (D) The ATP-binding pockets of MKK4 (cyan, PDB: 3ALN) and MKK7 (magenta, PDB: 6QFT) are superimposed, with key divergent residues highlighted. Corresponding amino acid pairs are labeled: S115/Q149, I108/M142, S185/C218, and D186/E219. Sequence motifs in the entrance of ATP-binding pocket are also indicated: GTCG (positions 111′–114′) in MKK4 and GAYG (positions 145′–148′) in MKK7. The motif of MKK7 points inward toward the binding pocket, while that of MKK4 extends outward, which may explain the small volume and shallow depth of the ATP-binding pocket in MKK7.
Figure 4.
(A) Schematic illustration of MKK4 and MKK7. The two kinases contain several functional domains for activation and binding of other proteins (D, docking domain; DVD, domain of versatile docking; P, phosphorylation site). They are activated by phosphorylation of two residues in their kinase domain (MKK4: Ser257 and Thr261; MKK7: Ser271 and Thr275). Cysteine residues for covalent binding are also highlighted. (B) Crystal structure of human non-phosphorylated MKK4 kinase domain complexed with AMP-PNP (PDB: 3ALN). The kinase domain is colored in magenta, and the DVD domain is colored in light blue. However, the currently known crystal structures of MKK4 and MKK7 are missing the D domain in the N-lobe. (C) Three-dimensional structural models of the ATP-binding pockets of MKK2 (purple), MKK4 (wheat), and MKK7 (green) are displayed. Quantitative characterization of the ATP-binding pockets, including surface area (Å2), volume (Å3), depth (Å), and hydrophobicity, is computed using ProteinPlus [71]. The corresponding data are summarized in the table below. (D) The ATP-binding pockets of MKK4 (cyan, PDB: 3ALN) and MKK7 (magenta, PDB: 6QFT) are superimposed, with key divergent residues highlighted. Corresponding amino acid pairs are labeled: S115/Q149, I108/M142, S185/C218, and D186/E219. Sequence motifs in the entrance of ATP-binding pocket are also indicated: GTCG (positions 111′–114′) in MKK4 and GAYG (positions 145′–148′) in MKK7. The motif of MKK7 points inward toward the binding pocket, while that of MKK4 extends outward, which may explain the small volume and shallow depth of the ATP-binding pocket in MKK7.
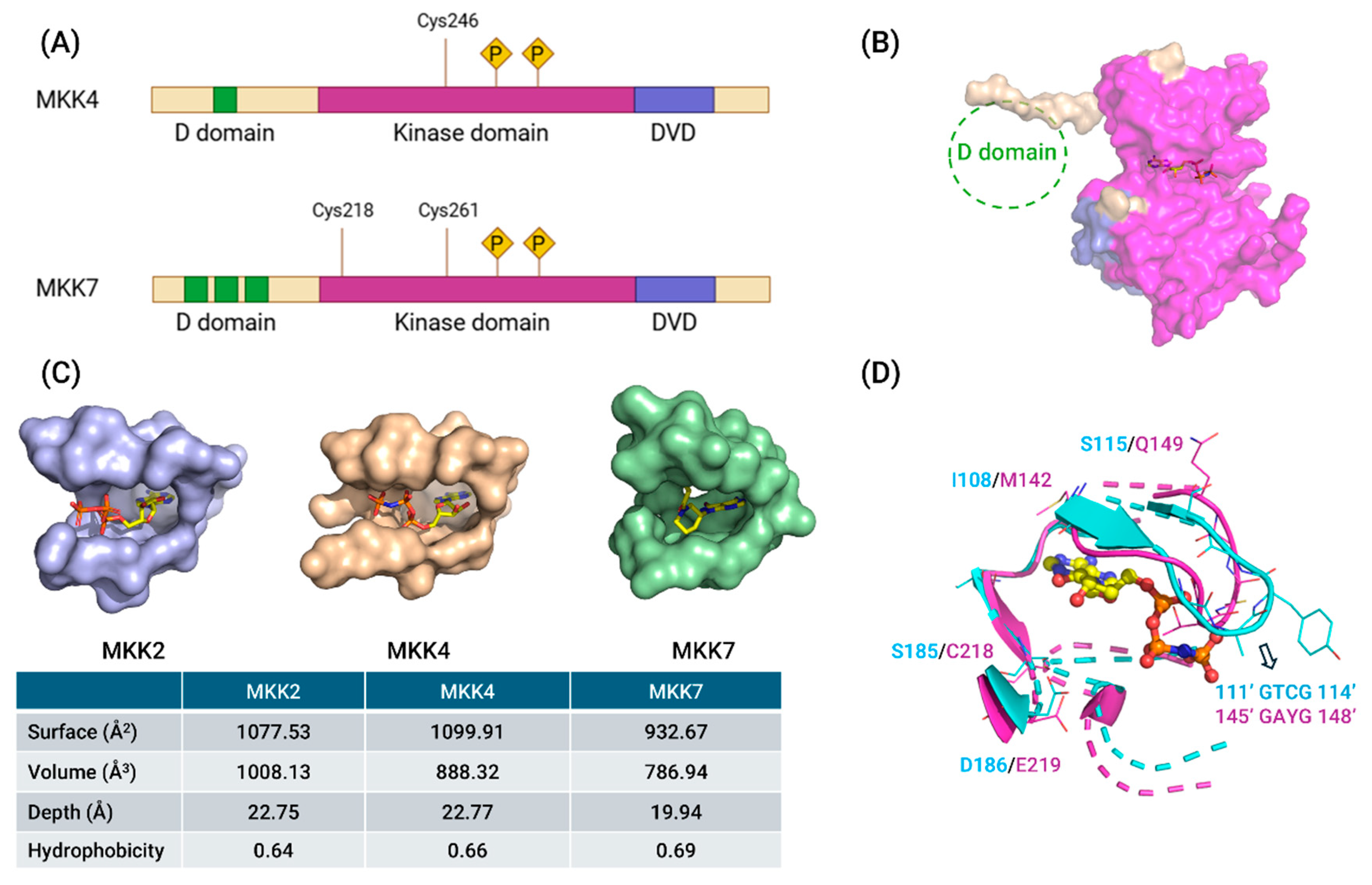
Figure 5.
(A) Structures, affinities, and cellular activities of prenylated quinolinecarboxylic acid-derived MKK4 Inhibitors 1 and 2. (B) Structures, inhibitory activities, and selectivity of 3-arylindole-derived MKK4 inhibitors 3-6.
Figure 5.
(A) Structures, affinities, and cellular activities of prenylated quinolinecarboxylic acid-derived MKK4 Inhibitors 1 and 2. (B) Structures, inhibitory activities, and selectivity of 3-arylindole-derived MKK4 inhibitors 3-6.
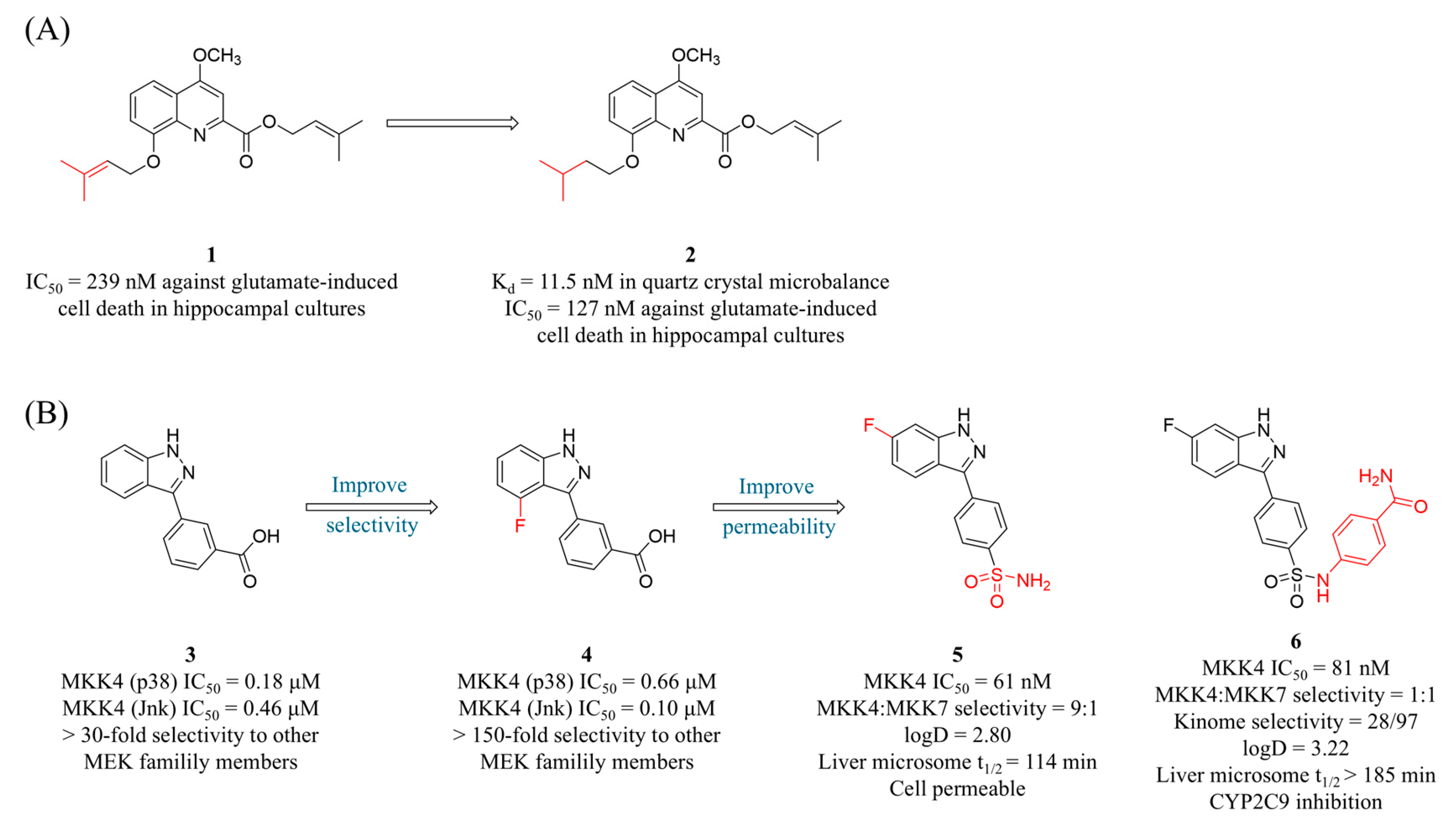
Figure 6.
(A) The structures and inhibitory activities of MKK4 inhibitors 7-11. (B) The SAR diagram of MKK4 inhibitors featuring a 7-azaindole or 1H-pyrazolo[3,4-b]pyridine scaffold.
Figure 6.
(A) The structures and inhibitory activities of MKK4 inhibitors 7-11. (B) The SAR diagram of MKK4 inhibitors featuring a 7-azaindole or 1H-pyrazolo[3,4-b]pyridine scaffold.
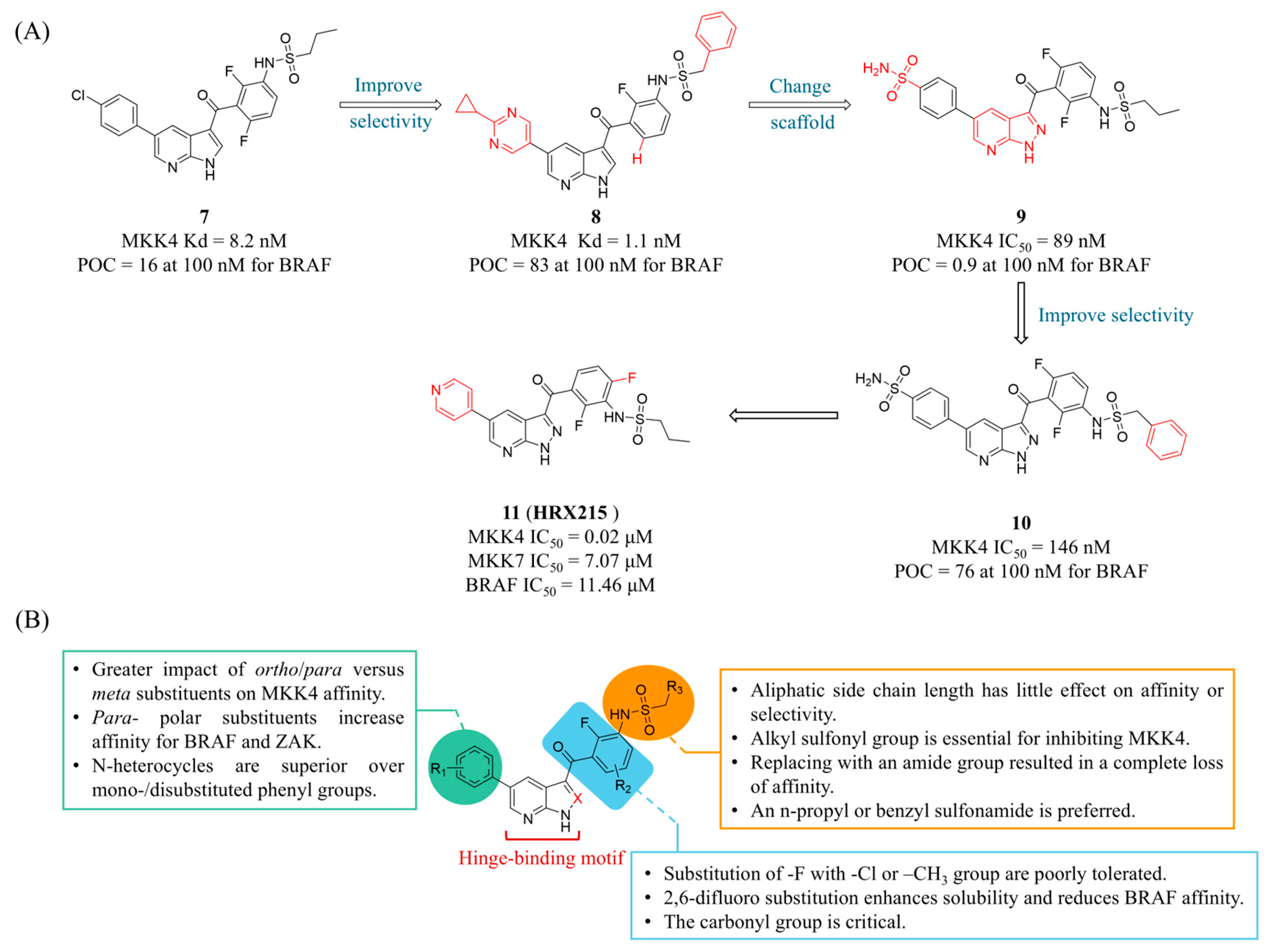
Figure 7.
(A) The structures and inhibitory activities of MKK4 inhibitors 12, 13 and 14. (B) The structures, inhibitory activities, selectivity and SAR studies of compounds 15 and 16 featuring a 1,4-dihydropyrido[3,4-b]pyrazin-3(2H)-one scaffold.
Figure 7.
(A) The structures and inhibitory activities of MKK4 inhibitors 12, 13 and 14. (B) The structures, inhibitory activities, selectivity and SAR studies of compounds 15 and 16 featuring a 1,4-dihydropyrido[3,4-b]pyrazin-3(2H)-one scaffold.
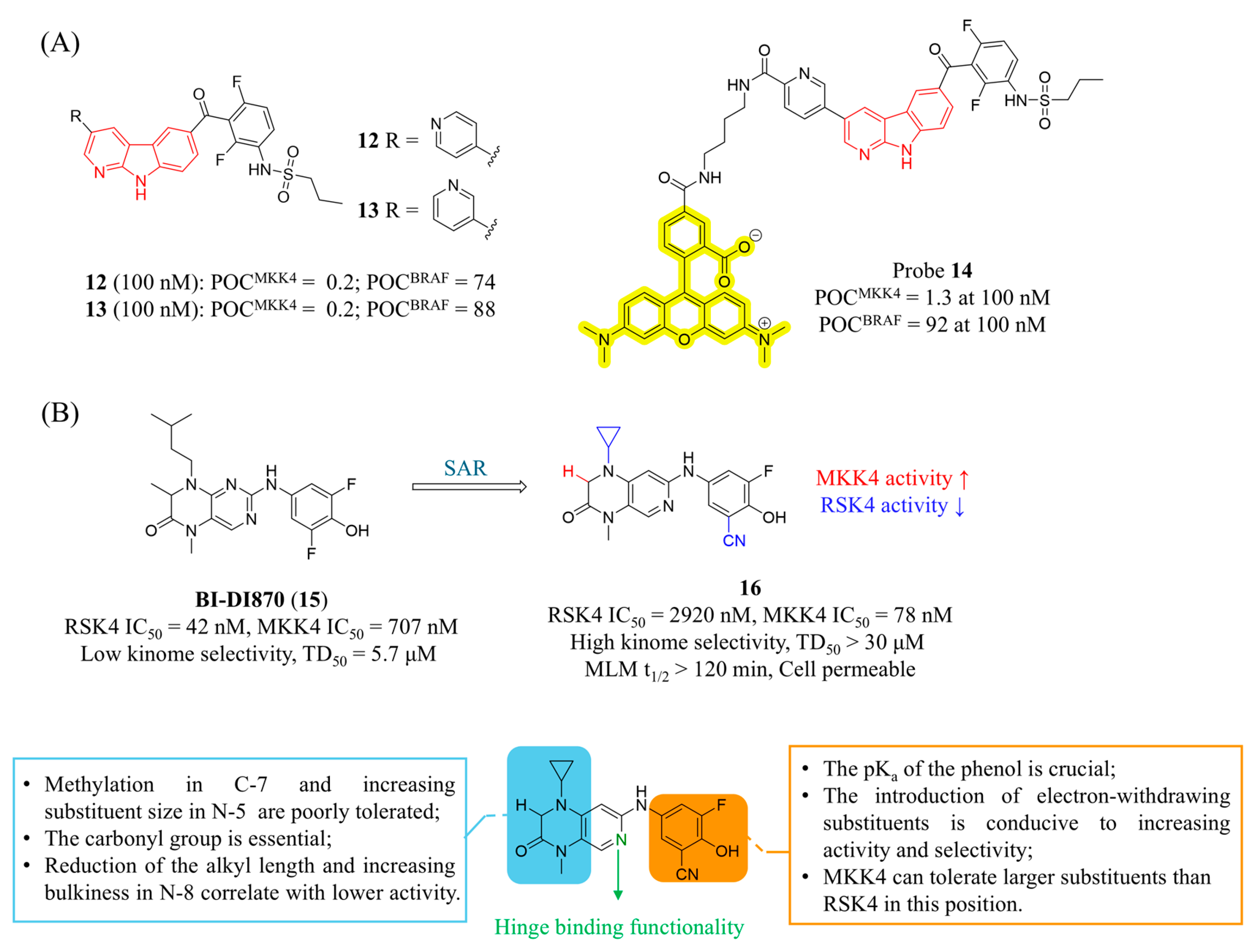
Figure 8.
(A) The structure and inhibitory activities of MKK7 covalent inhibitor 17. The dashed box indicates its co-crystal structure with MKK7 (PDB: 3wzu). (B) The structures and inhibitory activities of covalent inhibitors 18, 19 and 20. The dashed box indicates the co-crystal structure of 19 and MKK7 (PDB: 5z1d). Key amino acids are shown as sticks, with hydrogen bonds depicted as magenta dashed lines.
Figure 8.
(A) The structure and inhibitory activities of MKK7 covalent inhibitor 17. The dashed box indicates its co-crystal structure with MKK7 (PDB: 3wzu). (B) The structures and inhibitory activities of covalent inhibitors 18, 19 and 20. The dashed box indicates the co-crystal structure of 19 and MKK7 (PDB: 5z1d). Key amino acids are shown as sticks, with hydrogen bonds depicted as magenta dashed lines.
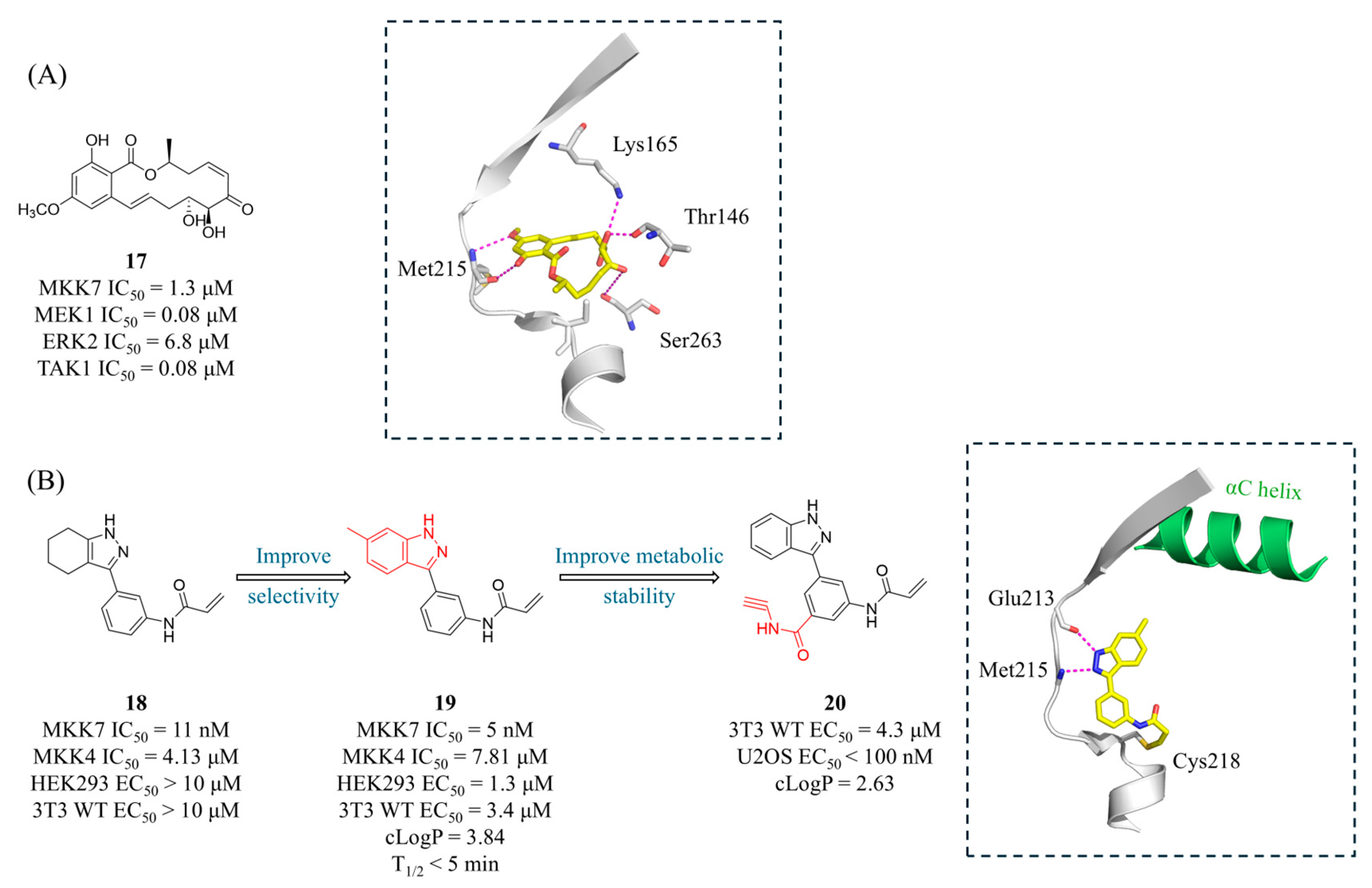
Figure 9.
The structures of compounds 21-25, along with their inhibitory activities against targets and cells. The co-crystal structures are located below each compound: 22 (PDB: 6ib0), 23 (PDB: 6ib2), 24 (PDB: 6qg4), and 25 (PDB: 6qhr). The α-C helix of MKK7 is shown in green, with interacting residues represented as sticks and hydrogen bonds depicted as magenta dashed lines.
Figure 9.
The structures of compounds 21-25, along with their inhibitory activities against targets and cells. The co-crystal structures are located below each compound: 22 (PDB: 6ib0), 23 (PDB: 6ib2), 24 (PDB: 6qg4), and 25 (PDB: 6qhr). The α-C helix of MKK7 is shown in green, with interacting residues represented as sticks and hydrogen bonds depicted as magenta dashed lines.
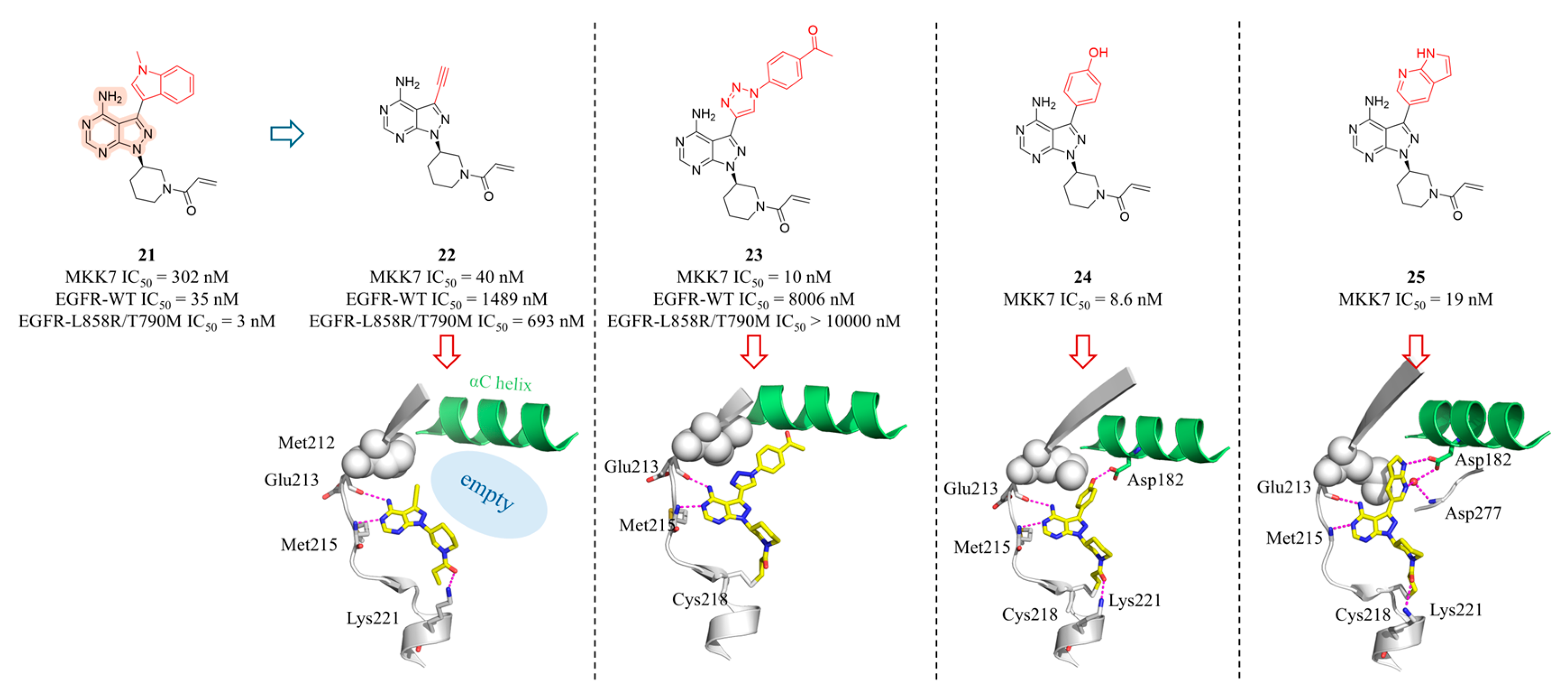
Figure 10.
(A) The CuAAC reaction for LSF of compound 22. The dashed box shows the co-crystal structure of 26 with MKK7 (PDB: 7ovj), where Asp182 is displaced due to steric hindrance. (B) The CuAAC reaction for LSF of compound 20. The dashed box shows the co-crystal structure of 27 with MKK7 (PDB: 7ovl) with interacting residues represented as sticks and hydrogen bonds depicted as magenta dashed lines. (C) The structures and inhibitory activities of Lys221-targeting covalent compounds 28 and 29. The dashed box shows the co-crystal structure of 28 with MKK7 (PDB: 9hz0).
Figure 10.
(A) The CuAAC reaction for LSF of compound 22. The dashed box shows the co-crystal structure of 26 with MKK7 (PDB: 7ovj), where Asp182 is displaced due to steric hindrance. (B) The CuAAC reaction for LSF of compound 20. The dashed box shows the co-crystal structure of 27 with MKK7 (PDB: 7ovl) with interacting residues represented as sticks and hydrogen bonds depicted as magenta dashed lines. (C) The structures and inhibitory activities of Lys221-targeting covalent compounds 28 and 29. The dashed box shows the co-crystal structure of 28 with MKK7 (PDB: 9hz0).
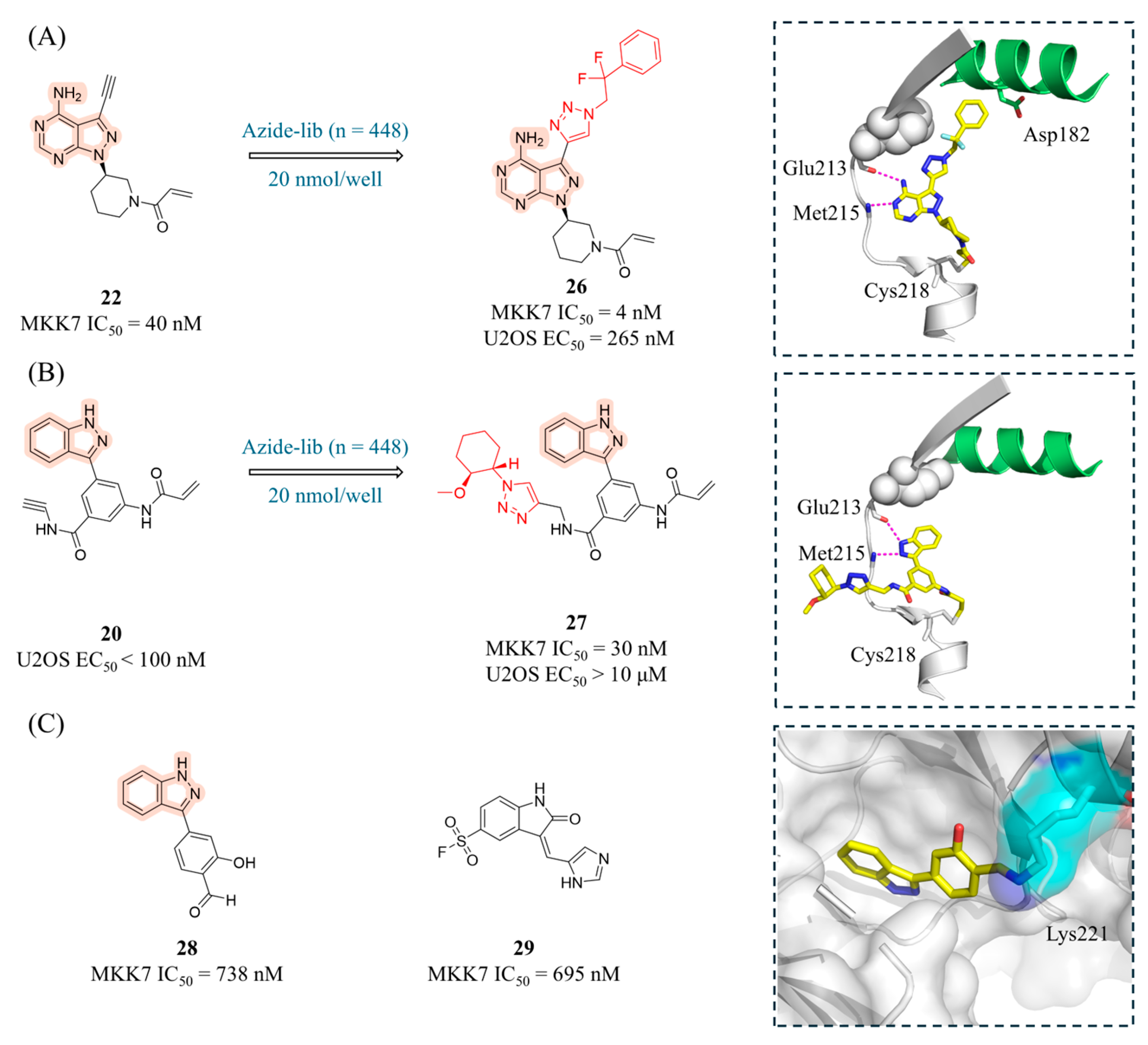
Figure 11.
(A) (B) The structures and inhibitory activities of covalent inhibitors 30-33. (C) The structures of compounds 34-36, along with their inhibitory activity against MKK7 and co-crystal structures (PDB IDs from left to right: 6yg3, 6yg6, and 6yz4). Key amino acid residues are represented as sticks, and hydrogen bonds are depicted as magenta dashed lines. The inset of ibrutinib shows structural superposition with the apo structure (pink), revealing the conformational rearrangement required for pocket formation (indicated by arrows).
Figure 11.
(A) (B) The structures and inhibitory activities of covalent inhibitors 30-33. (C) The structures of compounds 34-36, along with their inhibitory activity against MKK7 and co-crystal structures (PDB IDs from left to right: 6yg3, 6yg6, and 6yz4). Key amino acid residues are represented as sticks, and hydrogen bonds are depicted as magenta dashed lines. The inset of ibrutinib shows structural superposition with the apo structure (pink), revealing the conformational rearrangement required for pocket formation (indicated by arrows).
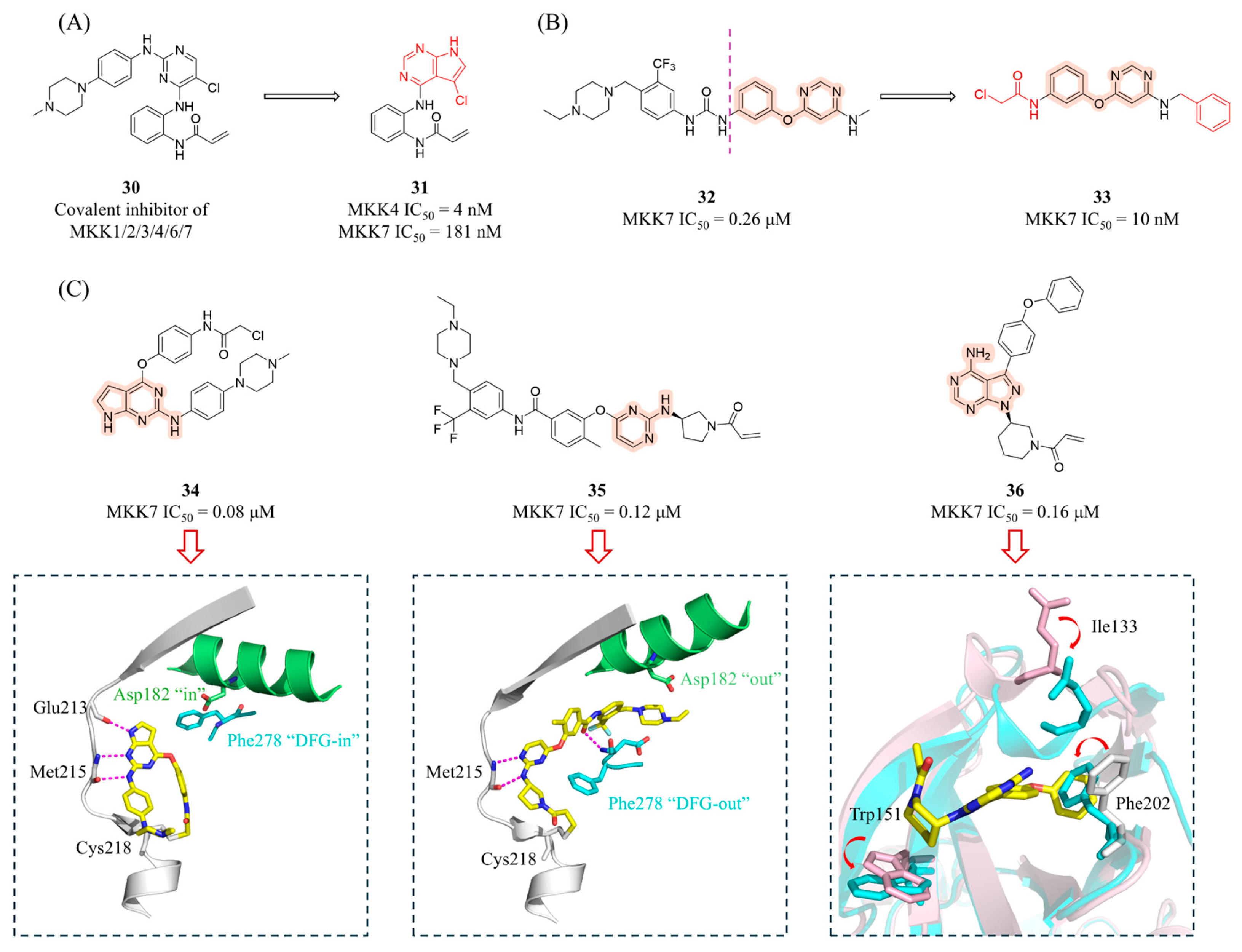
Figure 12.
Structures and target affinity of the reversible MKK7 inhibitors 37-39.
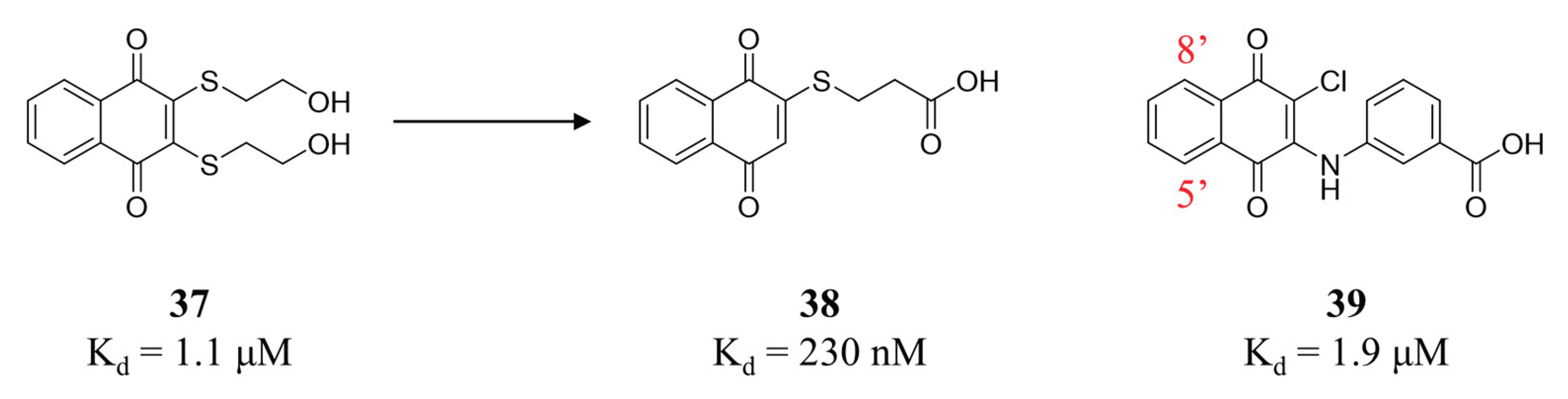

Table 1.
Summary of activation stimuli and mechanisms for major MAP3Ks.
| MAP3Ks | External/internal stimuli | Mechanism |
|---|---|---|
| TAK1 | TNF-α, IL-1, LPS, TGF-β, ROS, osmotic pressure | Sensing Lys63-linked polyubiquitinylation events at the TNF receptor. Activated through ubiquitination modifications by TRAF proteins. |
| ASK1-3 | H2O2, ROS, ER stress, TNF-α, Fasl, calcium ion | Inhibited by binding to antioxidant proteins (such as Thioredoxin) in the resting state; oxidative stress causes its dissociation and activation. |
| MLKs | TNF-α, IL-1, UV, heat shock, osmotic pressure, GPCR signals | Involved in cytoskeletal rearrangement, activated by small GTPases (such as Rac1, Cdc42). |
| MEKK1-4 | MEKK1: UV, serum deprivation, cytoskeletal perturbation MEKK2/3: TNF-α, IL-1, LPS MEKK4: osmotic pressure, cytotoxic agent |
Partially activated through recruitment by small GTPases (Rho, Rac) or receptor complexes. |
| DLK | neuronal damage (axotomy, ischemia), oxidative stress | Primarily expressed in the nervous system and involved in neurodegeneration and regeneration. |
| LZK | neuronal damage, oxidative stress | Functionally redundant with DLK, acting as a co-regulator of the JNK pathway in neurons. |
| HPK1 | lymphocyte antigen receptors, TNF-α, IL-1, LPS | Requiring own phosphorylation on tyrosine and subsequent interaction with adaptors of the SLP family. Ubiquitinated by E3 ubiquitin ligases, leading to its proteasomal degradation and limited signal duration. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



