
Submitted:
22 January 2026
Posted:
23 January 2026
You are already at the latest version
Abstract
Huntington’s disease (HD) is characterized by progressive striatal atrophy and complex proteomic changes in the central nervous system. Using the ultrasensitive NULISA proteomic platform, we analyzed cerebrospinal fluid (CSF) from 88 persons with HD to dissect the biological correlates of gray matter loss. We identified a two-signal pattern of pathology. The first track, marked by the axonal damage protein Neurofilament Light (NEFL), showed a strong negative correlation with putamen volume (Pearson r = -0.53, p < 0.001), consistent with prior work supporting NEFL as a proxy for neurodegeneration and brain atrophy in HD. The second track was defined by a positive association between the immune regulator TNFRSF8 (CD30) and putamen volume (Pearson r = 0.36, p < 0.001), indicating a loss of putative immune-regulatory signal as atrophy progresses. TNFRSF8 was pre-specified for follow-up given its immune-regulatory role and interpretability in the context of immune dysregulation consistent with an immune-exhaustion-like profile. TNFRSF8 showed an independent association with striatal volume (Beta = 0.24, p = 0.008) after controlling for NEFL, CAG-Age-Product (CAP) score, and sex. These findings suggest that reduced immune-regulatory signaling is a distinct pathological correlate in HD, separable from general cytoskeletal damage, and warrants evaluation in longitudinal and interventional studies.
Keywords:
Huntington’s disease
; proteomics
; NULISA
; neurodegeneration
; striatum
; neuroinflammation
; biomarkers
1. Introduction
Huntington’s disease (HD) is a fatal neurodegenerative disorder driven by a Cytosine-Adenine-Guanine (CAG) repeat expansion in the huntingtin (HTT) gene [1]. A hallmark of HD pathology is the selective vulnerability and progressive degeneration of striatal medium spiny neurons in the caudate and putamen [2]. Striatal atrophy begins well before the onset of diagnostic motor manifestations, providing a long prodromal window during which neurodegeneration can be tracked using structural magnetic resonance imaging (MRI) [3,4,5,6]. While MRI offers a macroscopic readout of tissue loss, fluid biomarkers are needed to interrogate the molecular processes associated with this degeneration and its modulation over time [7].
Current biomarker research has largely emphasized cytoskeletal markers such as neurofilament light chain (NEFL), which increase in cerebrospinal fluid (CSF) and blood in association with neurodegeneration and track disease intensity and progression in HD [8]. Although NEFL provides a robust gauge of aggregate axonal damage, it is less informative regarding upstream biological processes—such as inflammation and immune dysregulation—that may contribute to or modulate degeneration in HD [9,10]. Advances in high-sensitivity, high-multiplex proteomic technologies now enable improved characterization of low-abundance inflammatory and immune mediators in biofluids. In particular, the NULISA (Next-Gen Ultra-Sensitive Immunoassay) platform supports attomolar-sensitive, multiplexed quantification of large protein panels—including cytokines and other immune-related targets—offering a new window into the immune microenvironment associated with neurodegeneration [11].
HD progression shows substantial inter-individual variability, and mounting evidence implicates immune dysregulation as one contributor to phenotypic heterogeneity and progression rates. High-sensitivity CSF proteomics provides an opportunity to quantify low-abundance immune-regulatory proteins that may index distinct biological “tracks” of neurodegeneration beyond aggregate cytoskeletal injury. In this study, we test whether immune-regulatory signaling measured in CSF explains variance in striatal atrophy that is statistically independent of neuroaxonal damage markers.
2. Results
2.1. Validation of Striatal Atrophy
Volumetric screening confirmed significant, disease-specific atrophy in the striatum. The strongest correlation between gray matter volume and disease burden (CAG-Age-Product [CAP] score) was observed in the putamen (Spearman rho = -0.64, p < 0.001), followed by the caudate (Spearman rho = -0.44, p < 0.001). Measures of global brain volume showed weaker associations, validating the striatum as the primary locus of degeneration in this cohort (Table 1). These patterns are consistent with established longitudinal imaging findings in HD cohorts [4,5,12,13,14].
2.2. Divergent Proteomic Signatures
We identified two distinct biological signals tracking with striatal atrophy. Screening results are provided in Table S1 (Spearman rho) whereas the region-of-interest associations below are reported as Pearson r.
The Damage Signal (NEFL): Levels of NEFL were strongly negatively correlated with putamen volume (Pearson r = -0.53, p < 0.001). As striatal tissue is lost, CSF NEFL concentrations rise, reflecting neuroaxonal injury and release of cytoskeletal debris, consistent with extensive prior work supporting NEFL as a biomarker of neurodegeneration in HD [8,15,16].
The Immune Signal (TNFRSF8): In a divergent pattern, the immune regulator TNFRSF8 (CD30) showed a positive correlation with putamen volume (Pearson r = 0.36, p < 0.001). As the striatum degenerates, levels of this putative immune-regulatory signal decline. TNFRSF8/CD30 is a member of the tumor necrosis factor (TNF) receptor superfamily with immune-regulatory functions, supporting its interpretation as an immune-associated signal in this context [17,18].
The correlations differed in direction (Figure 1). Additionally, the difference between dependent correlations was statistically significant (Steiger’s Z = -5.94, p < 0.001). Taken together, these patterns support opposing biological behaviors rather than a single underlying factor [19].
2.3. Independent Contributions to Atrophy
To determine whether the immune signal provides unique information, we performed an independence test using multivariable regression (Figure 2). A baseline model with CAP score, sex, and NEFL explained 35.6% of the variance in putamen volume (R2 = 0.356). Adding TNFRSF8 increased explained variance to 40.9% (R2 = 0.409; ΔR2 = 0.053), and it significantly improved model fit (p = 0.008). TNFRSF8 was statistically significant as an independent predictor (Beta = 0.24, p = 0.008) even after controlling for the robust effect of NEFL.
Representative individual-level examples are shown in Figure 3, illustrating two distinct biomarker signatures (high NEFL vs. low TNFRSF8) within the putamen atrophy quartile.
In prespecified disease-burden model comparisons on the same complete-case sample (N=88), the CAP-adjusted model showed Adj R2 = 0.380 (CV RMSE = 0.108), compared with the age-adjusted model (Adj R2 = 0.350; CV RMSE = 0.111) and the no-burden model (Adj R2 = 0.355; CV RMSE = 0.110). CAP-adjusted models showed the best performance and modestly outperformed the age-adjusted and no-burden specifications (Table 2).
To mitigate optimism from using the same cohort for screening and modeling, we estimated confidence intervals for Beta via bootstrap resampling (B = 1,000). Internal validation confirmed the stability of the TNFRSF8 effect (Beta = 0.24, 95% Bootstrap CI: [0.06, 0.46]) [20].
2.4. Mediation Analysis
Cross-sectional mediation of CAP effects via TNFRSF8 was not strongly supported (Sobel p = 0.106; bootstrap indirect effect small and borderline; see Table S2).
3. Discussion
Our findings propose a “Two-Track” model of HD pathology. Track 1, represented by NEFL, is the familiar signal of structural disintegration—a readout of neuroaxonal injury that is increasingly used in HD biomarker research. [4,5,30] Track 2, represented by TNFRSF8 (CD30), suggests a distinct immune-associated signal that declines with advancing striatal degeneration. This distinction is a hallmark of immune dysregulation consistent with an immune-exhaustion-like profile. TNFRSF8/CD30 is a TNF receptor superfamily member with known roles in immune signaling and regulation, consistent with an interpretation involving reduced immune-regulatory signaling as atrophy progresses [17,18].
This immune interpretation is biologically plausible given a broader literature supporting immune alterations in HD, including immune activation detectable prior to clinical onset, neuroinflammatory pathways implicated in disease progression, and evidence of microglial activation in persons with HD before clinical motor diagnosis [10,21,22,23,24,25]. The clinical implication is that targeting the immune system in HD may require a different approach than nonspecific suppression of inflammation. Rather, future research should evaluate whether preserving or restoring immune-regulatory signaling pathways (e.g., TNF/TNFR family superfamily) yields benefit, ideally using designs that can test temporal ordering and within-person coupling [26].
Consistent with CAP as a disease-burden construct [27], CAP-adjusted models provided better overall performance than age-adjusted models in this cohort (Adj R2 0.380 vs 0.350; CV RMSE 0.108 vs 0.111).
Consistent with this separability, cross-sectional mediation of CAP effects via TNFRSF8 was not strongly supported in this sample (Sobel p = 0.106; bootstrap indirect effect small and borderline; see Table S2).
Furthermore, the independence of the TNFRSF8 signal suggests that a composite biomarker (NEFL + TNFRSF8) could provide a more comprehensive picture of disease state than either marker alone, potentially aiding stratification for future clinical trials and biomarker-guided enrichment strategies [8,15,16].
Despite these striking findings, this study presents several limitations that may inform future research. Cross-sectional mediation analysis may be biased in cases where the underlying causal process is dynamic or complex. Additionally, the comparatively small sample size – 88 persons with HD – may limit the statistical power and generalizability of our findings. Future work should therefore extend this cross-sectional analysis using larger longitudinal datasets with repeated CSF collection paired to structural MRI. Such designs would enable (i) mixed-effects trajectory models to quantify within-person change in TNFRSF8 and striatal volume, (ii) tests of temporal precedence using time-lagged associations (e.g., whether TNFRSF8 decline precedes accelerated putamen atrophy), and (iii) longitudinal mediation or dynamic path models that are not identifiable from cross-sectional data and can reduce bias inherent in single-timepoint mediation inference [28,29,30]. With larger longitudinal samples, multi-marker approaches (e.g., TNFRSF8 + NEFL) could be evaluated for prognostic enrichment, stratification of progression subtypes, and sensitivity to therapeutic target engagement in interventional studies.
4. Materials and Methods
4.1. Participants
We analyzed data from 88 people with HD (CAG repeats ≥36) spanning the stages before clinical motor diagnosis and the early stages after clinical motor diagnosis [31,32]. Demographics and clinical characteristics are summarized in Table 1. The cohort had a mean age of 39.0 years (SD=11.8) and a mean CAP score of 336.5 (SD=92.3) [27,33].

Table 3.
Participant Demographics.
| Characteristic | Cohort (N=88) |
|---|---|
| Age, years (SD) | 39.01 (11.83) |
| Education, years (SD) | 15.29 (2.16) |
| CAP1 score (SD) | 336.48 (92.25) |
| CAG2 Repeats (SD) | 42.74 (2.93) |
| Sex, n (%) | |
| Female | 58 (65.9%) |
| Male | 30 (34.1%) |
1 CAG-Age-Product, 2 Cytosine-Adenine-Guanine.
4.2. Neuroimaging
T1-weighted MRI scans were acquired on 3T scanners using harmonized protocols across sites and processed with FastSurfer to generate automated segmentations of subcortical structures [34]. Caudate and putamen volumes were normalized to intracranial volume (ICV) using the ratio method (Volume/ICV). For the present analysis, which required paired CSF and MRI, we restricted the sample to participants with both structural MRI and CSF available from the University of Iowa and the University of Wisconsin–Madison.
4.3. CSF Collection and Handling
CSF samples included in this analysis were collected at the University of Iowa and the University of Wisconsin–Madison under standardized protocols, processed according to study procedures, and stored until assay. Samples were shipped to the BioSpecimen Exchange for Neurological Disorders (BioSEND) repository at Indiana University for centralized handling, where they were blinded and aliquoted before shipment to the University of Wisconsin Alzheimer’s Disease Research Center (ADRC) Biomarker Lab for analysis [35]. Following completion of laboratory assays, results were unblinded, enabling linkage of CSF measures to imaging-derived outcomes for the present conjoined analyses.
4.4. Proteomics
Proteomic profiling was performed using the NULISAseq Central Nervous System (CNS) Disease Panel 120 and Inflammation Panel 250 (Alamar Biosciences), targeting a combined total of approximately 370 proteins [11,36,37]. Data were normalized using internal and inter-plate controls and log2-transformed to the NULISA Protein Quantification (NPQ) scale [37]. Values below the limit of detection (LOD) were retained (as NPQ values) consistent with manufacturer recommendations, and low-detectability analytes were filtered prior to screening, yielding 268 proteins for downstream analysis [37].
4.5. Proteomics
Sex was included a priori as a biological covariate in volumetric models when available. Other potential covariates (education, study site/scanner, race/ethnicity, and clinical severity measures such as total motor score) were not included in the primary model because they can be sparse/collinear in this sample and, for some measures, may be downstream of neurodegeneration. These were reserved for sensitivity analyses.
Screening procedure: To identify candidate biomarkers of striatal neurodegeneration, we screened the 268 retained proteins by computing Spearman’s rank correlations between protein abundance and normalized putamen volume. P-values were adjusted using the Benjamini–Hochberg false discovery rate (FDR) procedure [38]. Significance was defined as q<0.05, and analytes meeting a suggestive threshold (q<0.10) were advanced for multivariable characterization. TNFRSF8 was pre-specified for follow-up given its immune-regulatory role and interpretability in the context of immune dysregulation consistent with an immune-exhaustion-like profile; in this screen, it met the FDR significance threshold (q = 0.040).
Multivariable modeling: Analytes meeting the suggestive FDR threshold (q<0.10) were entered into multivariable linear regression models. In addition, TNFRSF8 was included a priori as a pre-specified immune-regulatory target given its interpretability in the context of immune dysregulation. All continuous predictors (CAP score, NEFL, and candidate immune markers) were Z-score standardized prior to modeling to enable direct comparison of standardized regression coefficients (Beta). The primary model was specified as: Putamen_Volume ~ CAP + Sex + NEFL + TNFRSF8. While the screening step used Spearman correlations to reduce sensitivity to outliers, region-of-interest matrices and multivariable models used Pearson correlations and ordinary least squares (OLS) regression on standardized data to satisfy linear modeling assumptions.
Disease-burden model comparison: We compared three prespecified models on a common complete-case sample (N = 88): (i) no disease-burden covariate, (ii) age-adjusted, and (iii) CAP-adjusted. Models were compared using adjusted R2, AIC/BIC, and k-fold cross-validated RMSE/MAE (k = 10 when available). CAP was retained as the primary disease-burden covariate because it provided superior fit and/or lower cross-validated error relative to age.
Model diagnostics: Multicollinearity was assessed using the variance inflation factor (VIF) [39]; all predictors had VIF < 3 (max VIF = 1.80). Residuals were evaluated for normality (Shapiro–Wilk) and homoscedasticity (Breusch–Pagan) [40,41]. Influential observations were assessed using Cook’s distance (threshold 4/N) [42].
Validation and limitations: To mitigate optimism from using the same cohort for screening and modeling, confidence intervals for regression coefficients were estimated via bootstrap resampling (B=1,000) [20]. Mediation analysis was conducted to assess statistical mediation, evaluating whether TNFRSF8 mediated the association between CAP score and putamen volume [43,44]. All results are presented as exploratory pending external replication.
4.6. Generative Artificial Intelligence
The study employed a paper-as-code workflow, in which pre-specified analysis scripts automatically generate tables, figures, and corresponding manuscript text directly from the finalized statistical outputs. This approach ensures internal consistency, reproducibility, and traceability between the analysis code and the reported results.
Any automated text generation was deterministic and rule-based, reflecting fixed templates populated by computed results, rather than probabilistic or generative modeling. No large language model or generative AI system was used to infer, summarize, or interpret findings beyond formatting and structured insertion of results into manuscript templates. All analytical decisions, statistical modeling choices, and scientific interpretations were defined a priori by the authors and verified manually prior to submission.
5. Conclusions
In summary, our study finds that reduced immune-regulatory signaling is a distinct pathological correlate in HD, separable from general cytoskeletal damage. Our findings support a “Two-Track” model of HD pathology: Track 1, represented by NEFL, signals structural disintegration; Track 2, represented by TNFRSF8 (CD30), suggests a distinct immune-associated signal that declines with advancing striatal degeneration. This distinction is a sign of immune dysregulation consistent with an immune-exhaustion-like profile. This study enhances our understanding of the upstream biological processes that may contribute to or modulate degeneration in HD, and its findings warrant evaluation in longitudinal and interventional studies. Future work should extend the present analysis using larger longitudinal samples, preparing the way for multi-marker approaches that may be appropriate for prognostic enrichment, subtype stratification, and sensitivity to therapeutic target engagement in interventional studies for persons with HD.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Proteomic Screening Results; Table S2: Cross-sectional mediation analysis (CAP → TNFRSF8 → putamen).
Author Contributions
Conceptualization, H.J.B., J.D.C., and J.S.P.; methodology, H.J.B. and V.D.C.; formal analysis, H.J.B. and J.D.C.; writing—original draft preparation, H.J.B.; writing—review and editing, J.S.P., V.D.C., and J.D.C.; supervision, J.S.P. and V.D.C. All authors have read and agreed to the published version of the manuscript.
Funding
The Predict-HD study was supported by NINDS/NIH and CHDI Foundation funding: "Neurobiological Predictors of Huntington's Disease" (R01NS040068); CHDI Grants A3917 and A6266. The PREVENT-HD study is supported by NINDS/NIH funding: "Statistical disease modeling and clinimetrics to prepare for preventative trials in Huntington's disease" (U01NS103475); "Preparing for preventative clinical trials in Huntington's disease" (U01NS105509).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and it was approved by the Institutional Review Board (IRB) of the University of Iowa and participating sites (IRB# 2020-1254). The studies involving human participants were reviewed and approved by the IRB or ethics committee at each participating institution for the Predict-HD and PREVENT-HD protocols.
Informed Consent Statement
All participants provided written informed consent prior to participation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request, in accordance with the data sharing policies of the Predict-HD and PREVENT-HD studies.
Acknowledgments
We thank the study participants, the site investigators, and coordinators. We appreciate administrative assistance from Deven K. Burks.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADRC | Alzheimer’s Disease Research Center |
| BioSEND | BioSpecimen Exchange for Neurological Disorders |
| CAG | Cytosine-Adenine-Guanine |
| CAP | CAG-Age-Product |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| FDR | False discovery rate |
| HD | Huntington’s Disease |
| HTT | Huntingtin gene |
| ICV | Intracranial volume |
| LOD | Limit of detection |
| MRI | Magnetic resonance imaging |
| NEFL | Neurofilament light |
| NPQ | NULISA Protein Quantification |
| NULISA | Next-Gen Ultra-Sensitive Immunoassay |
| OLS | Ordinary least squares |
| TNF | Tumor necrosis factor |
| VIF | Variance inflation factor |
References
- Tong, H.; Yang, T.; Xu, S.; Li, X.; Liu, L.; Zhou, G.; Yang, S.; Yin, S.; Li, X.J.; Li, S. Huntington's Disease: Complex Pathogenesis and Therapeutic Strategies. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Ehrlich, M.E. Huntington's disease and the striatal medium spiny neuron: cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 2012, 9, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Biglan, K.M.; Ross, C.A.; Langbehn, D.R.; Aylward, E.H.; Stout, J.C.; Queller, S.; Carlozzi, N.E.; Duff, K.; Beglinger, L.J.; Paulsen, J.S.; et al. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT-HD study. Mov Disord 2009, 24, 1763–1772. [Google Scholar] [CrossRef]
- Li, K.; Furr-Stimming, E.; Paulsen, J.S.; Luo, S.; Group, P.-H.I.o.t.H.S. Dynamic Prediction of Motor Diagnosis in Huntington's Disease Using a Joint Modeling Approach. J Huntingtons Dis 2017, 6, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef]
- Roze, E.; Cahill, E.; Martin, E.; Bonnet, C.; Vanhoutte, P.; Betuing, S.; Caboche, J. Huntington's Disease and Striatal Signaling. Front Neuroanat 2011, 5, 55. [Google Scholar] [CrossRef]
- Fang, Q.; Strand, A.; Law, W.; Faca, V.M.; Fitzgibbon, M.P.; Hamel, N.; Houle, B.; Liu, X.; May, D.H.; Poschmann, G.; et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics 2009, 8, 451–466. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. The Lancet Neurology 2017, 16, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Zetterberg, H.; Wild, E.J. Cerebrospinal Fluid Inflammatory Biomarkers Reflect Clinical Severity in Huntington's Disease. PLoS One 2016, 11, e0163479. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington's disease. Trends Immunol 2015, 36, 364–373. [Google Scholar] [CrossRef]
- Feng, W.; Beer, J.C.; Hao, Q.; Ariyapala, I.S.; Sahajan, A.; Komarov, A.; Cha, K.; Moua, M.; Qiu, X.; Xu, X.; et al. NULISA: a proteomic liquid biopsy platform with attomolar sensitivity and high multiplexing. Nat Commun 2023, 14, 7238. [Google Scholar] [CrossRef] [PubMed]
- Aylward, E.H. Magnetic resonance imaging striatal volumes: a biomarker for clinical trials in Huntington's disease. Mov Disord 2014, 29, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Ghazaleh, N.; Houghton, R.; Palermo, G.; Schobel, S.A.; Wijeratne, P.A.; Long, J.D. Ranking the Predictive Power of Clinical and Biological Features Associated With Disease Progression in Huntington's Disease. Front Neurol 2021, 12, 678484. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Long, J.D.; Johnson, H.J.; Aylward, E.H.; Ross, C.A.; Williams, J.K.; Nance, M.A.; Erwin, C.J.; Westervelt, H.J.; Harrington, D.L.; et al. Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci 2014, 6, 78. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Tortelli, R.; Johnson, E.B.; Wijeratne, P.A.; Arridge, M.; De Vita, E.; Ghazaleh, N.; Houghton, R.; Furby, H.; et al. Mutant huntingtin and neurofilament light have distinct longitudinal dynamics in Huntington's disease. Sci Transl Med 2020, 12. [Google Scholar] [CrossRef]
- Caron, N.S.; Haqqani, A.S.; Sandhu, A.; Aly, A.E.; Findlay Black, H.; Bone, J.N.; McBride, J.L.; Abulrob, A.; Stanimirovic, D.; Leavitt, B.R.; et al. Cerebrospinal fluid biomarkers for assessing Huntington disease onset and severity. Brain Commun 2022, 4, fcac309. [Google Scholar] [CrossRef]
- Muta, H.; Podack, E.R. CD30: from basic research to cancer therapy. Immunol Res 2013, 57, 151–158. [Google Scholar] [CrossRef]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: a historical perspective providing insight into future directions. Blood Cancer J 2017, 7, e603. [Google Scholar] [CrossRef]
- Steiger, J.H. Testing Pattern Hypotheses On Correlation Matrices: Alternative Statistics And Some Empirical Results. Multivariate Behav Res 1980, 15, 335–352. [Google Scholar] [CrossRef]
- Efron, B. Estimation and Accuracy after Model Selection. J Am Stat Assoc 2014, 109, 991–1007. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The role of the immune system in Huntington's disease. Clin Dev Immunol 2013, 2013, 541259. [Google Scholar] [CrossRef]
- Li, X.; Tong, H.; Xu, S.; Zhou, G.; Yang, T.; Yin, S.; Yang, S.; Li, X.; Li, S. Neuroinflammatory Proteins in Huntington's Disease: Insights into Mechanisms, Diagnosis, and Therapeutic Implications. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial Activation in the Pathogenesis of Huntington's Disease. Front Aging Neurosci 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov 2013, 12, 147–168. [Google Scholar] [CrossRef]
- Warner, J.H.; Long, J.D.; Mills, J.A.; Langbehn, D.R.; Ware, J.; Mohan, A.; Sampaio, C. Standardizing the CAP Score in Huntington's Disease by Predicting Age-at-Onset. J Huntingtons Dis 2022, 11, 153–171. [Google Scholar] [CrossRef]
- Cain, M.K.; Zhang, Z.; Bergeman, C.S. Time and Other Considerations in Mediation Design. Educ Psychol Meas 2018, 78, 952–972. [Google Scholar] [CrossRef]
- Mackinnon, D.P.; Lockwood, C.M.; Williams, J. Confidence Limits for the Indirect Effect: Distribution of the Product and Resampling Methods. Multivariate Behav Res 2004, 39, 99. [Google Scholar] [CrossRef]
- Maxwell, S.E.; Cole, D.A. Bias in cross-sectional analyses of longitudinal mediation. Psychol Methods 2007, 12, 23–44. [Google Scholar] [CrossRef]
- Kinnunen, K.M.; Mullin, A.P.; Pustina, D.; Turner, E.C.; Burton, J.; Gordon, M.F.; Scahill, R.I.; Gantman, E.C.; Noble, S.; Romero, K.; et al. Recommendations to Optimize the Use of Volumetric MRI in Huntington's Disease Clinical Trials. Front Neurol 2021, 12, 712565. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Leavitt, B.R.; Macdonald, D.; Thompson, L.M.; Huntington's Disease Nomenclature Working, G. Towards Standardizing Nomenclature in Huntington's Disease Research. J Huntingtons Dis 2024, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.H.; Sampaio, C. Modeling Variability in the Progression of Huntington's Disease A Novel Modeling Approach Applied to Structural Imaging Markers from TRACK-HD. CPT Pharmacometrics Syst Pharmacol 2016, 5, 437–445. [Google Scholar] [CrossRef]
- Henschel, L.; Conjeti, S.; Estrada, S.; Diers, K.; Fischl, B.; Reuter, M. FastSurfer - A fast and accurate deep learning based neuroimaging pipeline. Neuroimage 2020, 219, 117012. [Google Scholar] [CrossRef]
- Price, R. NINDS-supported biospecimen repositories: BioSEND and NHCDR. Alzheimers Dement 2024, 20. [Google Scholar] [CrossRef]
- Alamar Biosciences. NULISAseq CNS Disease Panel 120 (product page). 2023. [Google Scholar]
- Alamar Biosciences. Technical note: NPQ reporting and normalization. 2023. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kim, J.H. Multicollinearity and misleading statistical results. Korean J Anesthesiol 2019, 72, 558–569. [Google Scholar] [CrossRef]
- Barker, L.E.; Shaw, K.M. Best (but oft-forgotten) practices: checking assumptions concerning regression residuals. Am J Clin Nutr 2015, 102, 533–539. [Google Scholar] [CrossRef]
- Ghasemi, A.; Zahediasl, S. Normality tests for statistical analysis: a guide for non-statisticians. Int J Endocrinol Metab 2012, 10, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Ibrahim, J.G.; Cho, H. Perturbation and Scaled Cook's Distance. Ann Stat 2012, 40, 785–811. [Google Scholar] [CrossRef]
- Imai, K.; Keele, L.; Tingley, D. A general approach to causal mediation analysis. Psychol Methods 2010, 15, 309–334. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, D.P.; Pirlott, A.G. Statistical approaches for enhancing causal interpretation of the M to Y relation in mediation analysis. Pers Soc Psychol Rev 2015, 19, 30–43. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Divergent 'Two-Track' signatures of striatal neurodegeneration. Scatterplots show normalized putamen volume (y-axis) versus CSF biofluid markers (x-axis). (Left) Track 1: Neuroaxonal damage marker NEFL increases as putamen volume declines. (Right) Track 2: Immune regulator TNFRSF8 (CD30) decreases as putamen volume declines. Across panels, up indicates larger putamen volume and right indicates higher biomarker level. This divergence is consistent with immune dysregulation in an immune-exhaustion-like profile.
Figure 1.
Divergent 'Two-Track' signatures of striatal neurodegeneration. Scatterplots show normalized putamen volume (y-axis) versus CSF biofluid markers (x-axis). (Left) Track 1: Neuroaxonal damage marker NEFL increases as putamen volume declines. (Right) Track 2: Immune regulator TNFRSF8 (CD30) decreases as putamen volume declines. Across panels, up indicates larger putamen volume and right indicates higher biomarker level. This divergence is consistent with immune dysregulation in an immune-exhaustion-like profile.
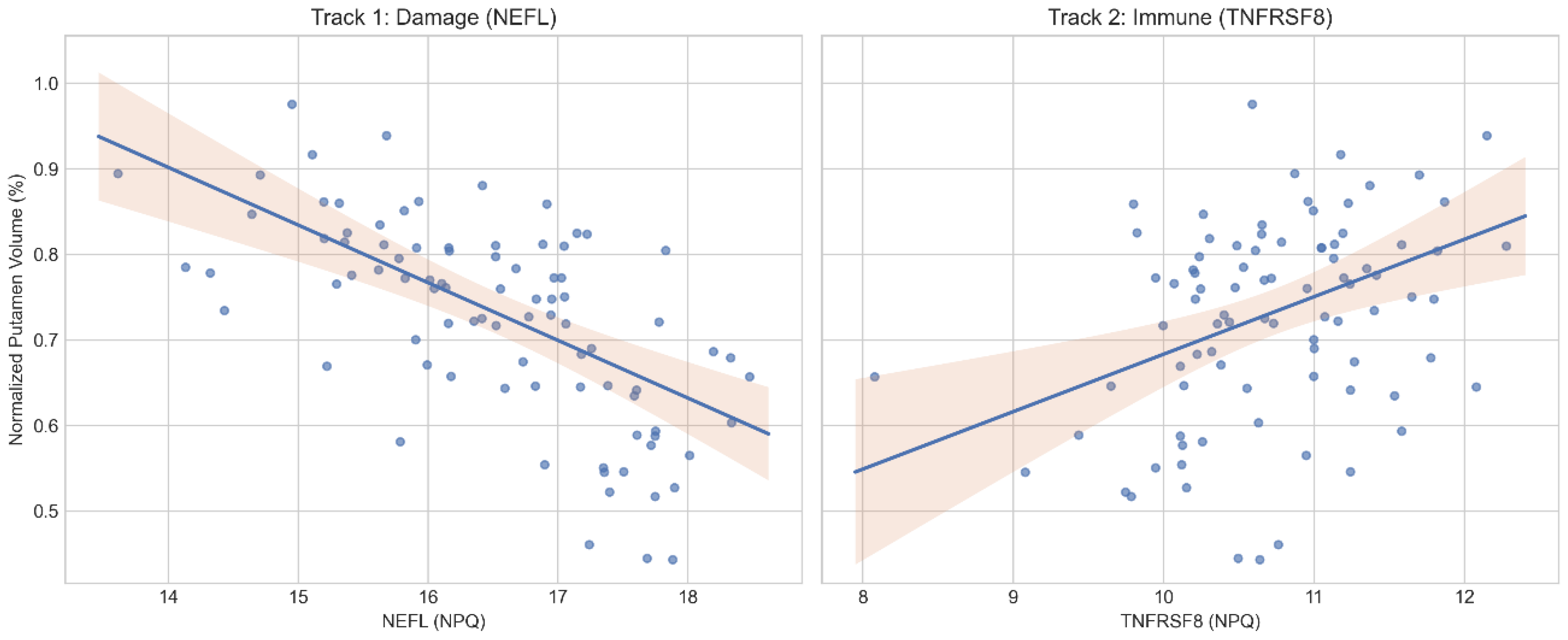
Figure 2.
Standardized regression coefficients for the multivariable model predicting normalized putamen volume. Coefficients are adjusted for CAP score (and sex when available), highlighting separable associations for NEFL and TNFRSF8.
Figure 2.
Standardized regression coefficients for the multivariable model predicting normalized putamen volume. Coefficients are adjusted for CAP score (and sex when available), highlighting separable associations for NEFL and TNFRSF8.
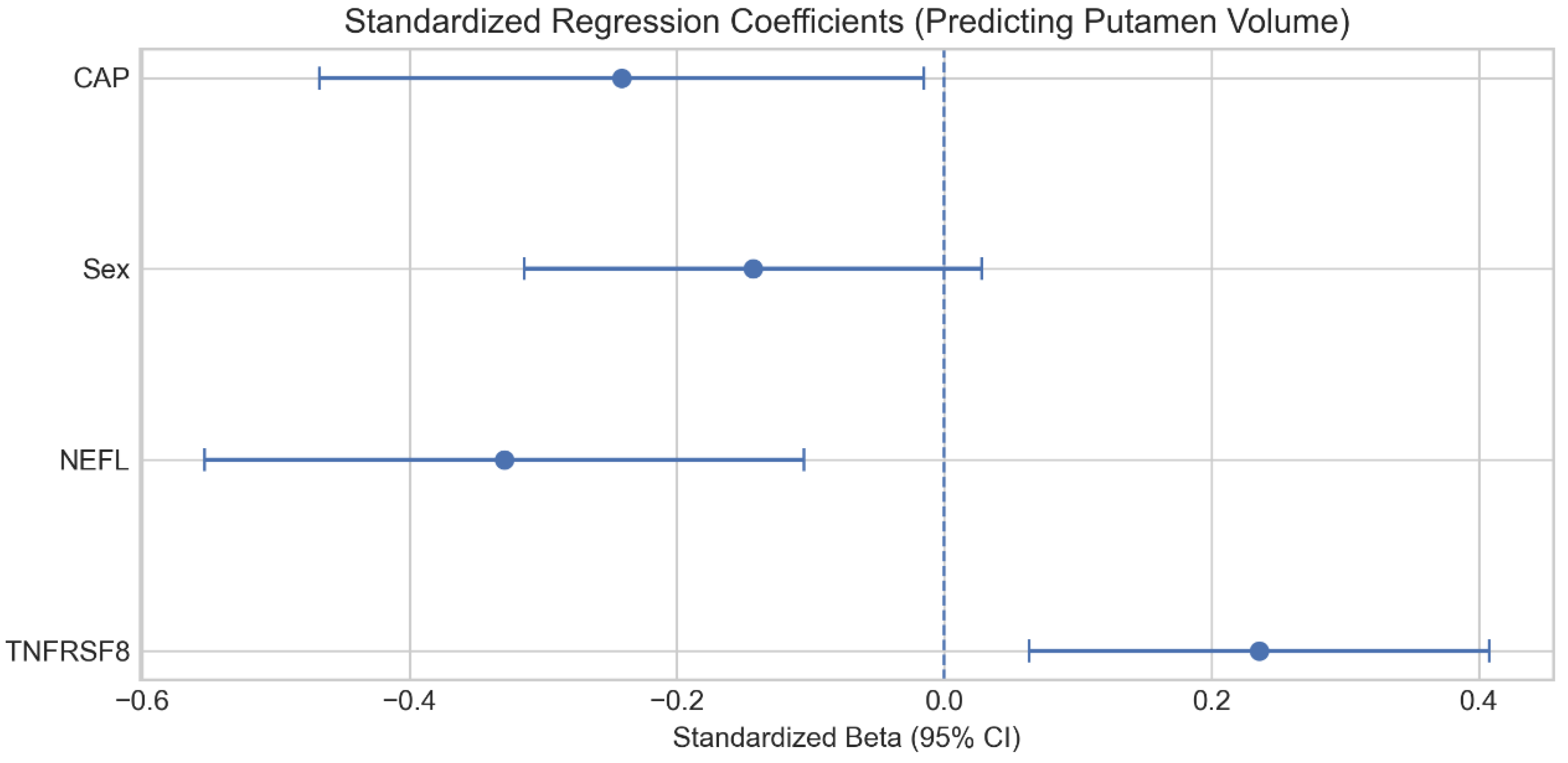
Figure 3.
Individual-level exemplars of the divergent biomarker tracks. Axial slices show putamen segmentation overlays and chip-style annotations for normalized putamen volume (left/right), NEFL, and TNFRSF8. Exemplars are selected from the putamen atrophy quartile to illustrate high-NEFL and low-TNFRSF8 profiles.
Figure 3.
Individual-level exemplars of the divergent biomarker tracks. Axial slices show putamen segmentation overlays and chip-style annotations for normalized putamen volume (left/right), NEFL, and TNFRSF8. Exemplars are selected from the putamen atrophy quartile to illustrate high-NEFL and low-TNFRSF8 profiles.
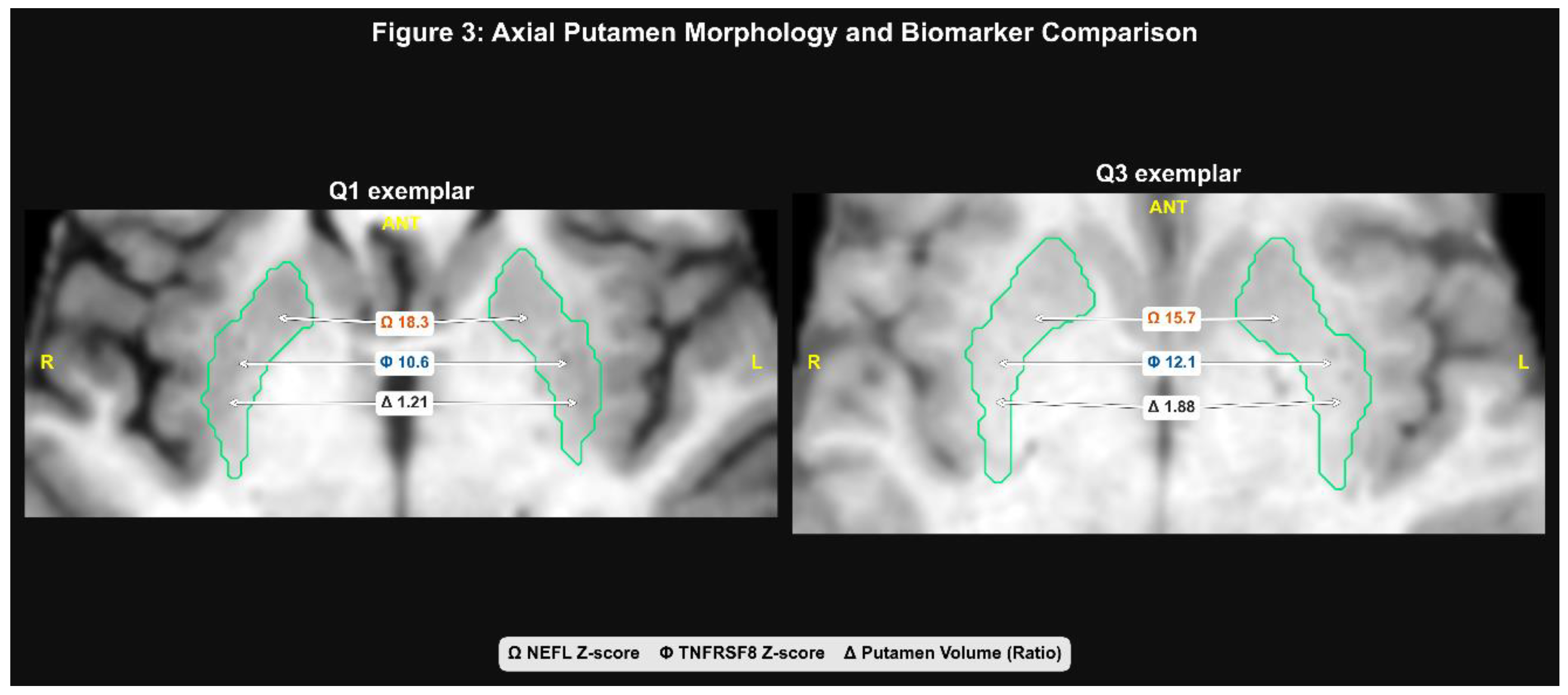
Table 1.
Volumetric Screening (Top 15 Regions).
| Region | R_CAP1 | P_CAP | Norm_Stats | -log10(p) |
| Putamen_volmm3 | -0.64 | 1.98 × 10−11 | 0.73 (0.13) | 10.70 |
| Brainsegvol | -0.54 | 5.64 × 10−8 | 197.94 (0.91) | 7.25 |
| Accumbens-area_volmm3 | -0.48 | 1.70 × 10−6 | 0.10 (0.02) | 5.77 |
| Supratentorialvol | -0.46 | 7.18 × 10−6 | 173.48 (2.59) | 5.14 |
| Caudate_volmm3 | -0.44 | 1.41 × 10−5 | 0.53 (0.11) | 4.85 |
| Pallidum_volmm3 | -0.44 | 1.88 × 10−5 | 0.29 (0.05) | 4.73 |
| SubCortGrayVol | -0.42 | 3.83 × 10−5 | 4.74 (0.47) | 4.42 |
| ctx-rh-caudalmiddlefrontal_volmm3 | -0.38 | 2.76 × 10−4 | 0.49 (0.09) | 3.56 |
| ctx-lh-postcentral_volmm3 | -0.34 | 0.001 | 0.89 (0.13) | 2.92 |
| ctx-rh-precentral_volmm3 | -0.33 | 0.002 | 1.01 (0.14) | 2.75 |
| ctx-lh-fusiform_volmm3 | -0.32 | 0.002 | 0.66 (0.09) | 2.68 |
| ctx-lh-superiorfrontal_volmm3 | -0.31 | 0.003 | 2.07 (0.27) | 2.48 |
| ctx-lh-rostralmiddlefrontal_volmm3 | -0.29 | 0.006 | 0.97 (0.14) | 2.21 |
| ctx-rh-superiorfrontal_volmm3 | -0.28 | 0.008 | 2.27 (0.26) | 2.12 |
| ctx-rh-lateraloccipital_volmm3 | -0.27 | 0.011 | 1.07 (0.13) | 1.94 |
1 CAP=CAG-Age-Product.
Table 2.
Disease-burden covariate model comparison for putamen prediction.
| Model | N_complete_case | ADJ_R2 | AIC | BIC | CV_folds | CV_RMSE | CV_MAE |
| No burden (no Age/CAP1) | 88 | 0.355 | -137.9 | -128.0 | 10 | 0.110 | 0.076 |
| Age-adjusted | 88 | 0.350 | -136.2 | -123.8 | 10 | 0.111 | 0.078 |
| CAP-adjusted | 88 | 0.380 | -140.5 | -128.1 | 10 | 0.108 | 0.075 |
1 CAP=CAG-Age-Product.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



