Submitted:
21 January 2026
Posted:
22 January 2026
You are already at the latest version
Abstract
This review highlights the pathophysiology and pathogenesis of diabetic retinopathy, the main complication of diabetes. The Neurovascular Unit (NVU) is brought to the surface for its importance to retinal physiological function. Diabetes impairs the NVU leading to the production of causative factors, such as ischemia, oxidative stress and excitotoxicity. The interplay between members of the above triad leads to the main pathological factors of diabetic retinopathy, namely neurodegeneration, neuroinflammation and vasculopathy. Emphasis is given to the pathology of the early stage of diabetic retinopathy (ESDR) and the putative new therapeutic treatments that will prevent/delay the development of the advanced stage of DR in which vision is compromised. NADPH Oxidases (NOX1-NOX5), whose main function is to produce reactive oxygen species (ROS) and induce oxidative/nitrative stress will be presented as novel therapeutic targets for the impaired neurovascular unit. The knowledge of the molecular mechanisms involved in the neuroprotection induced by novel specific inhibitors of NOX2 and NOX4 against the diabetic insults will confer the hope that therapeutic treatments for ESDR will evolve in the near future and be beneficial to the millions of patients who are in the early stage of diabetic retinopathy, as well as patients with other complications of diabetes.
Keywords:
diabetic retinopathy
; neurovascular unit
; neurodegeneration
; vasculopathy
; neuroinflammation
; NADPH oxidases
; NOX inhibitors
; neuroprotection
1. Introduction
Diabetes mellitus is a chronic, metabolic disorder which is rapidly affecting individuals around the world. Data from the International Diabetes Federation (IDF) indicate that in 2024, 588.7 million people globally had been diagnosed with diabetes, while this number is expected to rise to 852.5 million by 2050 [1]. The detrimental impact of chronic hyperglycemia to blood vessel physiology and integrity leads to the development of macrovascular and microvascular complications in multiple organs [2].
Proliferative Diabetic Retinopathy (PDR) is the most common microvascular complication of diabetes and the leading cause of preventable blindness in the working age population in developed countries [3,4,5].It has been estimated that in 2020, 103,12 million people were living with DR, 28,54 million people suffered from vision threatening diabetic retinopathy (VTDR) and 18,83 million were diagnosed with diabetic macular edema (DME). These numbers are expected to rise by 2045 to 160,50, 44,82 and 28,61 million for DR, VTDR and DME, respectively [6].
Chronic exposure to hyperglycemia and other causal factors, such as hypertension and lipid abnormalities, is believed to initiate a cascade of biochemical and physiological alterations that ultimately lead to microvascular damage and retinal dysfunction. The clinical hallmarks of PDR include increased vascular permeability and macular edema due to the breakdown of the blood-retinal barrier (BRB), which causes vascular microaneurysms and endothelial cell proliferation, all of which lead to neovascularization. Due to these pathological conditions, DR was originally considered a microangiopathy, an exclusively microvascular disease [4].
Increasing evidence has suggested that neurodegeneration is also involved in the pathophysiology of DR. Investigations in patients [7,8]and animal models of DR [9,10,11]reported that neurodegeneration is an early event in the timeline of DR and it may also precede microvascular complications. Neurodegeneration underlies the mechanisms leading to retinal cell death, suboptimal visual acuity and blindness [4,12,13].Clinical observations have revealed that hyperglycemia affects the hemodynamic response of retinal vasculature, under the form of reduced blood flow, which constitutes one of the earliest pathophysiological signs of the disease [14,15]. This early impaired hemodynamic response, in conjunction with the hyperglycemia-induced alterations in various biochemical pathways lead to oxidative damage of micro-vessels in the retina. The pathways involve the formation of advanced glycation end products (AGE) and the activation of its receptor, RAGE, activation of hexosamine, polyol and protein kinase C (PKC) pathways [16].
Based on fundus detection of these microvascular lesions, DR was categorized as non proliferative DR (NPDR), which is characterized by microaneurysms, hemorrhages, hard exudates [17] and PDR, in which excessive ischemia triggers an abnormal neovascularization response. The International Council of Ophthalmology also published a classification of DR, employed by clinicians, dependent on the recognition of specific anatomical disturbances. This classification further separates NPDR in mild, moderate and severe [18].More recent clinical observations pertaining to the symptoms observed in the different stages of DR, along with preclinical studies in animal models of the disease, contributed to the development of a novel system for DR classification, placed in the early and advanced stage of diabetic retinopathy, ESDR and ASDR, respectively.
The advanced stage of DR includes severe NPDR, PDR [13], also known as vision threatening diabetic retinopathy (VTDR) [6,19]and diabetic macular edema (DME). No visible lesions were noted under the traditional fundoscopic examination of patients in the ESDR, but there were evidence consistent with early neuro-retinal deterioration [20,21]and small scale microvascular complications [22].Thus, ESDR includes a phase without symptoms, mild and moderate NPDR. The timely identification (screening) of preclinical DR is essential in order for the clinician to select a) diabetic patients who are at a greater risk for developing ASDR, and b) the most effective management of the disease.
The therapeutic treatment for ESDR is based solely in the control of risk factors, such as blood glucose, dyslipidemia and hypertension. Insulin, fibrates (fenofibrate) and antihypertensive treatment have a significant effect on type 2 diabetic patients [23].However, some diabetic patients with tight glycemic and blood pressure control still develop DR [24].The FIELD [25] and ACCORD STUDY [26] concluded that fenofibrate, a peroxisome proliferator activated–receptor alpha (PPAR) agonist, a treatment for dyslipidemia, reduced the risk of development and progression of DR in patients with type2diabetes.However, other investigations reported that fenofibrate mediates anti-apoptotic activity, improvement of endothelial function, prevention of BRB breakdown, anti-angiogenic activity, antioxidant and anti-inflammatory events via other mechanisms [27,28,29,30].
Current pharmacological treatments focus on the ASDR [31],at the stage that vision has already been compromised. Anti-vascular endothelial growth factor (anti-VEGF) agents, administered intravitreally, treat only the microangiopathy/vasculopathy, whereas corticosteroids reduce inflammation. However, these agents have adverse side effects, such as cataract formation, retinal detachment, vitreous hemorrhage and potential loss of neural retinal cells [32]. Most importantly, they do not cause total regression of neovascularization, and do not reverse visual loss. Other commonly used treatments include laser photocoagulation, and invasive vitreoretinal surgical procedures [33,34].
The asymptomatic nature of the initial phase of ESDR, in combination with the lack of effective treatments, may be a major cause for increasing the prevalence of ASDR. These put an enormous burden on the healthcare systems and impair the quality of life of diabetic patients [35]. It is therefore necessary to develop novel therapeutic treatments for ESDR in order to postpone the development of the disease to the vision threatening advanced stage.
2. Neurovascular Unit and Diabetes
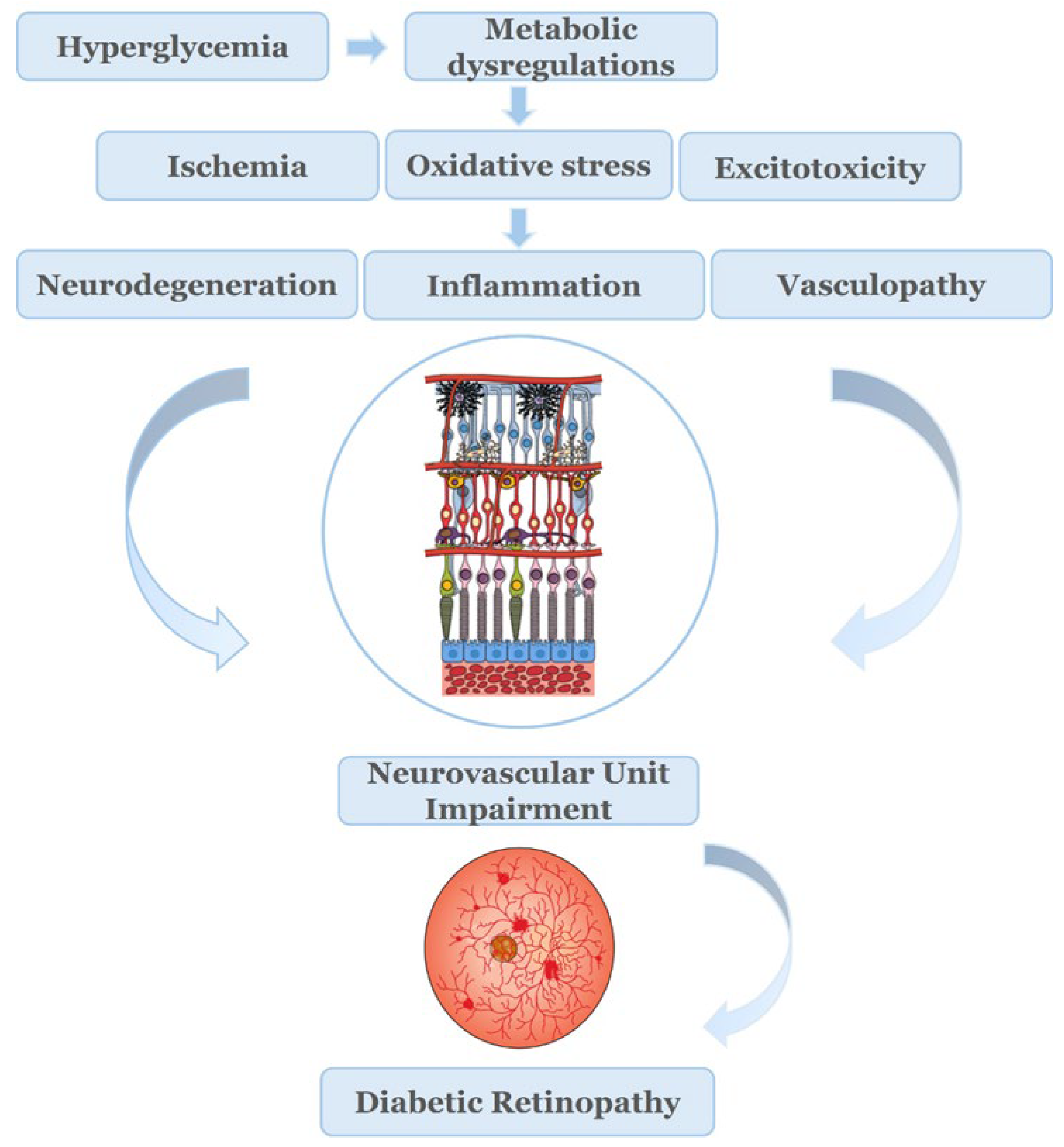
Hyperglycemia induces the dysregulation of different metabolic pathways [36].Chronic exposure to hyperglycemia, along with other factors, such as hypertension and lipid abnormalities, mentioned earlier, initiates a cascade of events that lead to the dysregulation of metabolic pathways, and production of causative factors, such as ischemia, oxidative stress and excitotoxicity [37,38].The interplay between members of this triad leads to neurodegeneration, inflammation and vasculopathy.
The American Diabetes Association (ADA) has defined DR as a highly tissue-specific neurovascular complication [39]. The term “Neurovascular Unit” (NVU) was first described in brain, but was also introduced in retina. NVU describes the complex interactions, interdependency and functional coupling of neurons, glia cells (macro/microglia) and the components of vasculature (endothelial cells, pericytes) [4].
The different neural cell types (retinal ganglion cells (RGCs), bipolar, horizontal and amacrine cells), together with glia cells (Müller cells, astrocytes and microglia) constitute the “neural part” of the NVU, while endothelial cells and pericytes compose the “vascular part”. Each specific cell type of the NVU has a distinct role. Their coordinated function and close communication ensures the maintenance of the inner Blood Retinal Barrier (iBRB), as well as the regulation of blood flow locally, depending on the metabolic status of neural retina. Communication between the different cell types is achieved either directly, through physical interactions (e.g., endothelial cells and pericytes) in paracrine and/or autocrine manner [40,41,42,43,44].
Under pathological conditions, such as diabetes, a number of serious factors, such as excitotoxicity, imbalance in neurotrophic factors, glial dysfunction, and apoptotic cell death, can occur that lead to severe dysregulation and disruption of the NVU homeostasis. This dysregulation subsequently causes BRB breakdown, structural and functional impairments on neurons and the vasculature [33,40,41,45].The activation of macro and microglia and the release of inflammatory cytokines introduce a hyperglycemia induced inflammatory component [46],which provides the link between the neural and vascular impairment of the NVU [47].
Thus, neurodegeneration, inflammation and vasculopathy appear to be responsible for the impairment of the NVU and BRB disruption, both critical for the pathogenesis of the early pathologies of DR. It has also been reported that retinal dysfunction is present in patients with diabetes lacking microvascular abnormalities. These latter findings led to the suggestion of renaming the microangiopathy disease, diabetic retinopathy (DR) to Diabetic Retinal Disease (DRD), a name in which both neurodegenerative and microvascular pathologies are involved [39,48].
New knowledge regarding the role of neurodegeneration and the impairment of NVU in the development of DR has led to a better understanding of the pathogenesis of ESDR [42]. In addition, the development of animal models of ESDR to assess the pharmacological profile of new therapeutic strategies targeting both neurodegeneration and microvascular abnormalities in diabetic animals has been established. The streptozotocin (STZ) model of diabetic retinopathy in rodents (rat/mice) has been employed in a large number of investigations, in which the therapeutic agents under study, peptides [49,50,51],microneurotrophins [52,53], endocannabinoids [54],NADPH Oxidase inhibitors [55,56] and others, are administered topically, as eye drops or intravitreally in different chronological (length of treatment) protocols. Db/db mice were also employed as a useful model for the study of neuroprotection against retinal neurodegeneration, by different pharmacological agents [57,58].The findings of the above mentioned investigations suggested that ESDR models and routes of administration of the putative therapeutics under study protected the diabetic retina against neurodegeneration and vasculopathy. Most importantly, it has been determined that topical administration, as eye-drops, of diverse therapeutic targets, is an effective route of administration with many advantages, including the limitation of systemic side effects, the avoidance of invasive methods and patient friendly treatment [59].
This review focuses on the role of NADPH Oxidases (NOX1, NOX2, NOX4) in the early stage of DR and as targets for the development of novel therapeutics for the pathologies instigated by the impairment of the neurovascular unit.
3. NADPH Oxidases (NOX): Physiology and Function
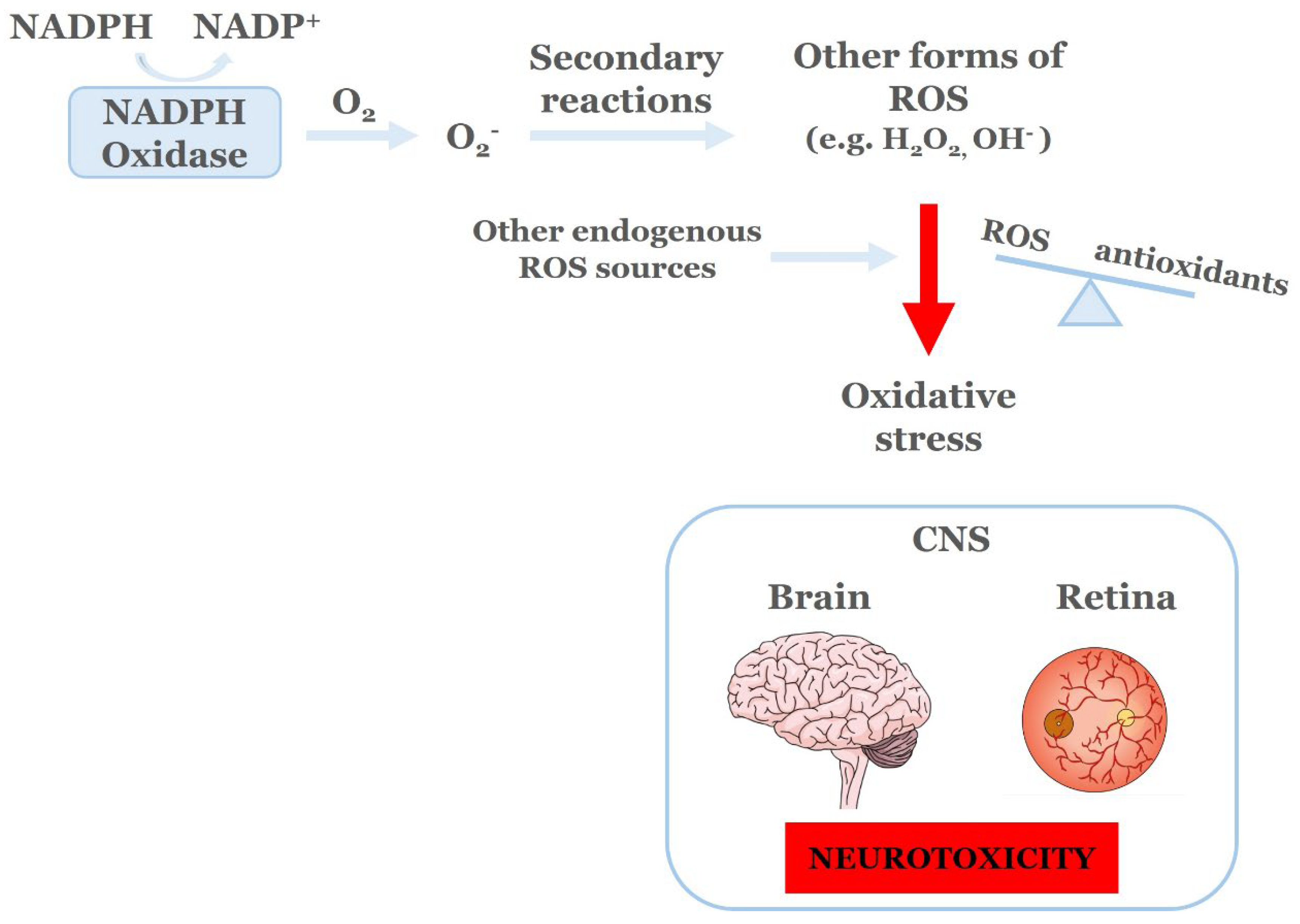
NADPH Oxidases (NOX)is afamily of enzymes that includes seven known isoforms, namely NOX1-NOX5, DUOX1/2 [60]. The main role of NOX is to produce reactive oxygen species (ROS) by catalyzing the transfer of one electron from NADPH to molecular oxygen, leading to the production of superoxide (O2.). The newly formed superoxide, through secondary reactions, will lead to the subsequent production of other forms of ROS, including hydrogen peroxide (H2O2) and hydroxyl radicals (OH.). Normally, the NOX derived ROS, as well as those produced from other endogenous sources of ROS (mitochondria, xanthine oxidase, cytochrome P450 oxygenases, lipoxygenase and cyclooxygenase), are essential for a plethora of molecular and cellular processes, including host defense and the regulation of gene expression and cell signaling [61].
Most cell types, in brain and retina, express multiple NOX isoforms that are differentially regulated, have distinct sub-cellular localizations and serve unique roles. Apart from their roles in normal physiology of brain and retina, NOX isoforms are correlated with diverse CNS pathologies. In brain, NOX4, located in endothelial cells and neurons, is linked to the breakdown of the blood-brain barrier and neuronal cell death that leads to ischemic stroke [62]. In retina, ΝOΧ1, NOX2, and NOX4 are expressed in rat retinal ganglion cells, amacrine and endothelial cells, pericytes, and macro- and microglia. Their activation under pathological conditions leads to vascular dysfunction and neurodegeneration. The role of NOX in the development of inflammation was also reported by many investigators [57,63,64,65,66].
NOX 1 isoform: It represents the first identified homolog of NOX2 [67].The NOX1 enzymatic complex includes the trans membrane subunits NOX1 and p22phox, along with the cytosolic subunits NOX organizer 1 (NOXO1)[61]. It also requires the recruitment of Rac1 regulatory protein for the activation of the complex and the subsequent generation of superoxide [68,69].
NOX2 isoform: It was the prototypic enzyme of the NADPH oxidase family. NOX2 consists of two membrane subunits, gp91phox and p22phox, that form the NOX2 core, known as cytochrome b558, and three cytosolic subunits, p47phox, p67phox and p40phox.Upon activation, phosphorylation of p47phox leads to the interaction with the p22phox membrane subunit and the subsequent recruitment of the cytosolic subunitsp67phox and p40phox to the NOX2 core. Rac1 or Rac2 regulatory protein binds directly to p67phox, resulting in the formation of the NOX2 activated enzymatic complex that catalyzes the production of superoxide [61,70,71].
NOX4 isoform: Unlike the other isoforms, it is constitutively active. It exhibits a unique ROS generation pattern, with its rapid conversion of superoxide to hydrogen peroxide [72]. The NOX4 dependent production of ROS also requires interaction with the membrane subunit p22phox, but the function of the NOX4 isoform seems to be independent of other cytosolic regulatory proteins, including the rac1 GTPase [73]. Interaction between p22phox and NOX4 in the endoplasmic reticulum suggested a unique mechanism of NADPH oxidase complex formation [74]. Interaction of the regulatory protein poldip2 with p22phox has been correlated with increased NOX4 enzymatic activity and its constitutive character [75], something that has to be further investigated.
Most recently, NOX3 expression and function in retinal ganglion cells and amacrine cells was reported [76].
3.1. NADPH Oxidasesas Critical Regulators ofOxidative Stress and Retinal Ischemia
Excessive accumulation of ROS, as a result of increased formation from the ROS producing systems and/or limited or deregulated function of the enzymatic and non enzymatic antioxidants systems is described as oxidative stress (OS), a condition responsible for tissue injury and organ abnormalities. In the CNS, OS leads to neurotoxicity and has been identified as one of the major mechanisms underlying the pathogenesis of vasculopathy, neurodegeneration and neuroinflammation in both brain and retina.The two major sources of ROS are the mitochondria (oxidative phosphorylation, electron transport chain, cytochrome P450, xanthine oxidase, nitric oxide synthase [77] and NADPH Oxidases [61,78].
Figure 2.
NADPH Oxidases (NOX) function and oxidative stress. ROS; Reactive Oxygen Species, CNS; Central Nervous System [61].
Figure 2.
NADPH Oxidases (NOX) function and oxidative stress. ROS; Reactive Oxygen Species, CNS; Central Nervous System [61].
Ischemia is the underlying cause of many ocular diseases, including diabetic retinopathy that leads to retinal cell loss, neovascularization and blindness. Retinal ischemia induces the release of excess glutamate, from retinal photoreceptors, bipolar, and ganglion cells. Glutamate is the leading neurotransmitter in retina. It initiates a cascade of events that lead to excitotoxicity via the activation of different receptor subtypes belonging to ionotropic, N-methyl-D-aspartate (NMDA), (RS)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid hydrobromide (AMPA), kainate and metabotropic receptors [79,80]. Glutamate activation of NMDA receptors leads to an increase in calcium ions (Ca2+), activation of nitric oxide synthetase and the release of reactive oxygen and nitrogen species (ROS/RNS) leading to neuronal cell death. NMDA excitotoxicity has been used extensively as a model for retinal neurodegenerative disease, since NMDA receptors are located on retinal ganglion cells (RGCs), whose axons are responsible for forming the optic nerve. NMDA excitotoxicity leads of RGC and visual loss [81], thus it may be considered a model of the ASDR.
Besides the Ca2+permeable NMDA receptors, activation of other glutamatergic receptors, mentioned above, the non-NMDA receptors, AMPA and kainate, are also involved in retinal excitotoxicity. Their use as experimental models has played an important role in the evaluation of ischemia related cell death and neuroprotection in retinal disease. The toxic effects of AMPA induced the attenuation of the number of viable retinal cells, activated macroglia (Müller cells) and microglia [82]. AMPA receptors are composed of tetrameric subunits, GluA1–4 that form homomeric or heteromeric complexes that are differentially expressed in retinal neurons [83]. AMPA receptors lacking the GluA2 subunit are permeable to calcium ions (Ca2+) [84,85], and their activation does lead to excitotoxicity.
Administration of AMPA intravitreally in rats affected retinal cell viability of cholinergic [choline acetyl transferase; (ChAT-IR), nitric oxide synthetase; (bNOS-IR)] expressing amacrine cells and horizontal cells (calbindin-IR), but had no effect on photoreceptors, bipolar or ganglion cells [86]. These findings led to the conclusion that the AMPA model of excitotoxicity is an effective model for the study of the early events of retinal ischemia, and in the assessment of the neuroprotective properties of new pharmacological agents. The results of additional studies [87,88,89] suggested that AMPA excitotoxicity not only leads to neurodegeneration, but also to neuroinflammation, in agreement to the concept that glutamate excitotoxicity is primarily associated with neurodegeneration, but also with neuroinflammation in the CNS.
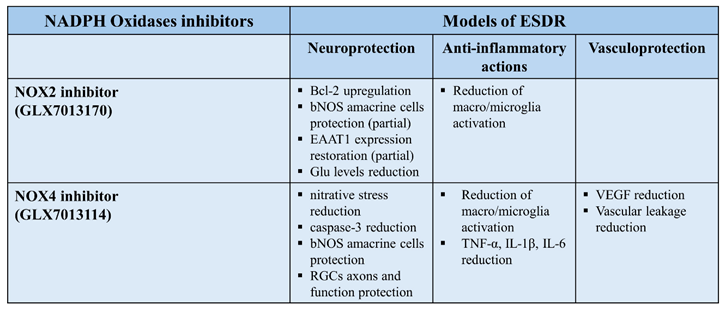
NADPH Oxidases were reported to mediate an increase in ROS levels in rat retina in the presence of AMPA. Neuronal loss of nitric oxide synthetase (NOS) expressing amacrine cells and a significant increase in the activation of both micro- and macroglia, respectively, was observed. NOX1,2, and 4 isoforms increased AMPA induced excitotoxicity. NOX inhibitors afforded differential neuroprotective patterns. Blockade of a) NOX1(ML171) afforded neuroprotective and anti-inflammatory actions, b) NOX2 (GLX7013170) partially protected NOS+ retinal amacrine cells and attenuated the AMPA induced increase in macro and microglia, and c) NOX4 (GLX7013114), attenuated only the AMPA induced increase in macro- and microglia, providing anti-inflammatory actions [90].These findings proposed that excitotoxicity affects the activation of micro/macroglia, and its subsequent inflammatory phenotype, through a mechanism that implicates NOX derived ROS and oxidative stress.
4. NADPH Oxidase Involvement in the Development of Diabetic Retinopathy
Studies using different in vitro and in vivo models of DR have correlated hyperglycemia with increased expression and enzymatic activity of NADPH oxidases, including NOX1/4 [63,91], NOX2 [92] and NOX4 [56,66]isoforms present in retinal cells. Moreover, in humans, a genome-wide association study has been linked the NOX4 gene with increased risk of severe diabetic retinopathy in diabetes type 2 patients, suggesting the involvement of NADPH oxidases in the diabetes induced retinal abnormalities [93]. An important mechanism that is associated with the upregulation of NOX isoforms in DR is the hyperglycemia induced activation of PKC, a significant upstream regulator of NADPH oxidases that leads to their over activation and subsequent increase ROS production in different retinal cells [77]. In brain, glutamate excitotoxic insults also increased the expression of the NOX2 isoform in reactive microglia cells [94].
Glutamate excitotoxicity and OS are two major playersin DR. The relationship between diabetes and excitotoxicity was first addressed by Ambati et al.[94].It was reported that the levels of glutamate and other agents, gamma-aminobutyric acid (GABA) and vascular endothelial growth factor (VEGF), are increased in the vitreous of patients with proliferative diabetic retinopathy (PDR). Diabetes increases glutamate levels in humans and rats and alters glutamate uptake, thus interfering with retinal glutamate homeostasis, leading to retinal cell death [95,96,97]. Investigations that examined the effect of diabetes on glutamate metabolism, the glutamate transporter and glutamate receptors reported that diabetes produced enzymatic abnormalities in glutamate metabolism in retina [98], as well as dysfunction of the glutamate transporter [99] and alterations of ionotropic glutamate receptors [100,101]. All these dysfunctions have been observed in ESDR [102].
The relationship between OS and diabetes has also been addressed. It was reported that an increase retinal superoxide production was observed in diabetic mice (2 and 8-months) via various mechanisms that included NOX signaling. Apomycin (NOX inhibitor) suppressed the diabetes-induced increase of superoxide levels, specifically in the 8-month diabetic model, and reduced capillary degeneration [103]. Other investigations aimed in deciphering the inter-relationship amongst the increase of retinal glutamate levels, oxidative stress and nitric oxide in diabetes and the effect and therapeutic potential of anti-oxidants in diabetic retinopathy [97,104,105].
The above mentioned results recommend for the first time that the above triad, diabetes, excitotoxicity and oxidative stress is interrelated. OS represents a putative link between diabetes and glutamate metabolism abnormalities in retina, which are further induced by excitotoxic insults.
5. Diabetic Retinopathy: NOX2 and NOX4
As mentioned above, the expression of NOX1, NOX2, and NOX4 isoforms of the NADPH oxidase family has been identified in different types of retinal cells, including macro- and microglia, pericytes, endothelial cells [63,106,107,108].The loss of amacrine cells was also observed in ischemic retinal disease, including diabetic retinopathy [109] and is believed to represent the early events of retinal ischemia.
A real-time PCR analysis, employing the experimental STZ ESDR (2wk) paradigm, revealed a statistically significant increase in NOX2 and NOX4 mRNA expression in retinas of diabetic rats. These findings support the alterations on the expression of at least two NOX isoforms in retina, as early as two weeks after STZ administration, and highlight the important role they play in diabetic retina. In contrast, the levels of NOX1 mRNA were reduced in diabetic retina, something that needs to be investigated further [55].In the same study, NOX2 and NOX4 increased the nitration of protein tyrosine residues [nitrotyrosine (NT-IR)], suggesting an increase in ROS levels in ESDR. Diabetes induced oxidative stress in rat retina, causing mitochondrial damage, one of the primary mechanisms implicated in the pathology of neurodegeneration, and a major player in the development of DR [110,111,112].Increased oxidative stress and protein nitration has also been reported in other paradigms of ESDR, with different time scales (e.g., 15d, 20d or 4 week models) and in retinas of human diabetic patients [55,113,114]. NT-positive cells are localized in different retinal layers (INL and GCL), including the photoreceptor layer and retinal pigment epithelium. Based on these results, and the reports of others [115],it was concluded that NOX2 and NOX4 isoforms are implicated in the induction of nitrative stress and cell damage in diabetic retina [116].
5.1. NOX2 Inhibitors and Mechanisms of Action in the Neuroprotection of Early Diabetic Insults
Diabetes affects retinal neurons leading to neurodegeneration and retinal cell death. The selective NOX2 inhibitor GLX7013170 (Glucox A.E., Sweden),reversed the diabetes induced low levels of the anti-apoptotic protein Bcl-2 in the ESDR paradigm. This finding suggested that NOX2 plays a major role in the regulation of apoptosis in ESDR [56], in agreement with other studies [117]. NOX2 is located in amacrine, ganglion cells and axons in retina, and its activation leads to cell death of nitric oxide synthetase (NOS) expressing amacrine cells and RGC axons [neurofilament (NFL)], respectively. GLX7013170 (NOX2 blockade) partially protected NOS-expressing amacrine cells, but it had no effect on RGC axons, in the ESDR model [56]. The first reports on the involvement of the role played by NOX2 activation on oxidative stress were obtained in studies examining the induction of neuronal abnormalities by NOX due to astroglial Fo-κB mediating oxidative stress [118]and neuroprotection by the deletion of NOX2 isoform [119] in a model of ischemia/reperfusion injury.
NOX2 was found to be involved in AMPA-induced glutamate excitotoxicity and diabetic retinopathy [56,90]. A statistically significant increase in glutamate levels in retinas of diabetic non-treated animals was observed as an early diabetic insult. This effect was due to the diabetes induced impairment in the expression of glutamate transporters. A significant reduction in the expression of the excitatory amino acid transporter 1 (EAAT1) in the diabetic non-treated retinas was observed [56], in agreement with other studies who reported similar observations in diabetic retina and brain, two weeks after the onset of diabetes [120]. EAAT1 represents the predominant glutamate transporter expressed in Müller cells, being primarily responsible for the removal of excess glutamate from the extracellular space [121]. Changes in its expression and function are correlated with retinal neuronal loss due to excitotoxicity [122]. EAAT1 is found to be vulnerable to oxidative damage. This leads to the impairment of its physiological function in ESDR [99]. GLX7013170 blockade of NOX2 partially restored the expression of EAAT1 and, subsequently, reduced glutamate levels in retinas of diabetic non-treated animals.The above mentioned findings recommend that there is an interplay amongst the triad, NOX2, oxidative stress, and glutamate excitotoxicity, which promotes neurodegeneration and neuroinflammation in diabetic retinopathy. Selective blockade of the NOX2 isoform protects the retina and NOX2 inhibitors, such as GLX7013170, are putative neuroprotectants, anti-inflammatory agents and potential therapeutics of retinal diseases, including ESDR [56]. Thus, oxidative stress and NOX2 affect glutamate metabolism in the diabetic retina. Studies in brain have also identified NOX2 as a mediator for the excessive release of glutamate and the induction of excitotoxicity in pathological conditions such as stroke [123]
5.2. NOX4 Inhibitors as Antioxidants in ESDR
It has been reported that NOX4 is over expressed in hypoxia and in high-glucose treated primary bovine retinal capillary endothelial cells and in db/db mouse retina [57]. A genome-wide association study supported the involvement of the NOX4 gene in severe DR of type 2 diabetes [93].GLX7013114, the NOX4 selective inhibitor, was reported to improve β-islet mitochondrial activity and survival in a model of short-term stress, suggesting that it may be a putative therapeutic for Type 2 diabetes [124]. The results of these studies recommend that NOX4 inhibition is an important strategy to pursue for the development of new therapeutics for diabetes and diabetic retinopathy.
NOX4 blockade reversed the diabetes effects on OS, ROS levels and caspase-3 expression in hypoxic human retinal Müller cells [66].GKT13783 (dual NOX1/NOX4 inhibitor) was reported to attenuate the diabetes-induced increase in NOX4 mRNA and OS [91] in diabetic (STZ-treated) retinas. The colocalization of anti nitrotyrosine (NT), a marker of oxidative stress, with the nuclear marker DAPI, in the ESDR model suggested that part of the OS damage is neuronal. GLX7013114 (NOX4 inhibitor), administered as eye drops, blocked the early events of DR with respect to nitrative stress [55], in agreement with the antioxidant and neuroprotective effect of GLX7013114 in the retina AMPA excitotoxicity model [90]. NT-IR stain could also be found in vessels due to the location of NOX4 in retinal endothelial cells, in addition to neurons, glia, and pericytes [125].
5.3. NOX4-Neuronal Cell Death and Neuroprotection
An experimental intervention with the novel NOX4 selective inhibitor GLX7013114, employing two paradigms of ESDR, paradigm A: preventive and paradigm B: therapeutic, led to neuroprotective, anti-inflammatory, and antivascular actions. GLX7013114 reversal of the diabetes-induced attenuation of the number of NOS+-expressing retinal amacrine cells (INL), supports the presence of the neuronal staining of NT in the INL [55].The depletion of NOS-expressing amacrine cells in the diabetic retina has previously being reported in the ESDR STZ-DR models [52,53,126].
In a recent review, Barber A.J. [127] examined the form and function of retinal ganglion cells in diabetes. A key pathological feature of DR pertains to the marked decrease of NFL immunoreactivity (IR), a marker of retinal ganglion cell axons. NOX4 blockade was efficacious in blocking or reversing the diabetes-induced attenuation of NFL-IR, as early as two weeks, as well as the thickness or shrinkage of the area corresponding to (GCL and IPL) [55].Despite the reduction in NFL thickness and intensity, as assessed by NFL- IR, no statistically significant reduction in the thickness of the GCL was observed nor in the number of retinal ganglion cells (RGCs) between control and diabetic animals [54]. Martyn et al.[73] also reported that the number of RGCs, as well as the relative thickness of GCL remained unaffected two weeks post-STZ-induced diabetes in a murine (mouse) model. However, a statistically significant reduction was observed 10-12 weeks post-diabetes induction. Changes in NFL thickness represent one of the earliest structural changes at the neuronal level of the diabetic retina, thus this will play an important role on the further development of DR. NFL-IR may be a useful biomarker for predicting the fate of RGCs in optical neuropathies, including DR. Progressive loss of ganglion cells was reported after axotomy of the optic nerve [128]. The existence of a large number of RGCs, with no detectable retrograde Fluoro-Gold staining, was reported in a mouse model of glaucoma, suggesting that part of their axons was damaged, but RGCs remained functional and expressed various RGC genes. The authors suggested that despite the degeneration of their distal part, the proximal portion of the axons remained unaffected one-month post-insult, thus contributing to RGCs’ survival [129]. Pattern electroretinography (pERG) analysis, a sensitive measure of RGC function, was performed in all groups of the “therapeutic” paradigm B (see above). NOX4 blockade with GLX7013114 reversed the diabetic insult and protected the function of RGCs [55].
All the aforementioned findings suggested that GLX7013114 provides neuroprotection to retinal neurons (amacrine and RGCs) and RGC axons against the diabetic insults of ESDR and recommend that treatment of the diabetes-induced NFL deficits in the ESDR will be beneficial towards the viability of ganglion cells and the postponement of the advanced stage of DR. Similar alterations in the morphology of NFL were also reported in a five-week [52], a two and five-week [54] ESDR models. Nerve fiber layer thinning in patients with preclinical retinopathy [130] and reduction of RNFL- and increased INL/OPL thickness, in human diabetic patients without DR or with initial DR suggested early alterations in the inner retina [11,131].
5.4. NOX4 Blockade and Vascular Leakage
VEGF is responsible for vascular permeability and neovascularization in the diabetic retina. Oxidative stress activates a VEGF autocrine loop in Müller cells via a mechanism involving transcription factors responsible for the transcription, translation, and release of VEGF and VEGFR2. Therefore, oxidative stress not only triggers the VEGF autocrine loop but also sustains it [131]. VEGF is involved in both the early and late stage of DR, being regulated via autocrine and paracrine mechanisms. However, there are other important mechanisms besides oxidative stress that cause upregulation of VEGF during the development of DR, including hypoxia, advanced glycation end products, and proinflammatory cytokines [13,132].
VEGF is believed to have a dual role in DR, an initial neuroprotectant of retinal neurons against the hyperglycemia-induced OS and as a subsequent facilitator or enhancer of the progression of DR [133,134]. Diabetes induced an increase in VEGF levels in both paradigms of ESDR (2/5wk). However, blockade of NOX4 with GLX7013114 attenuated the diabetes-induced increase in VEGF levels in a statistically significant manner only in the 5-wk paradigm [55]. Li J. et al. [57] reported a NOX4-dependent upregulation of VEGF in retinas of db/db mice 6 weeks after the initiation of their study. A significant increase of vascular leakage [Evans blue assay] in the diabetic non treated retinas compared to control was reported in the 5-wk paradigm [55]. Ocular topical administration of GLX7013114 reduced vascular leakage in a statistically significant manner suggesting a protective role of this NOX4 inhibitor against diabetes-induced disruption of BRB integrity. These findings imply that GLX7013114 plays a role in the attenuation of VEGF levels and vascular leakage produced in ESDR [55]. Increased VEGF levels and vascular leakage were reported in other ESDR models at one week [135], two weeks [136] and six months [137].
A recent review by Wang et al. [138] focused on the role of NOX4 and its regulatory mechanisms in diabetic microangiopathies. In an epigenetic regulation study, regarding DR, it was reported that miR-99a-5p improves DR via NOX4 by inhibiting abnormal proliferation and migration of human retinal microvascular endothelial cells. Regarding NOX4 inhibitors as therapeutics for treating diabetic microvascular complications, GLX7013114 was noted as a “specific inhibitor of NOX4 that has demonstrated promising effects in the treatment of DR”.
5.5. NOX2 / NOX4 and Neuroinflammation
ROS derived from NOX are shown to induce the activation of microglia and macroglia and their transition to a pro-inflammatory phenotypes [73,74]. This leads to the release of pro-inflammatory cytokines, chemokines, and glutamate, as well as additional ROS, largely derived by NOX isoforms, further affecting the retina.
Microglia are the resident inflammatory cells in retina. Several reports addressed the morphology and activation of microglia (amoeboid state) after the onset of hyperglycemia. Zeng et al. [139] reported that the number of activated microglia was increased in the IPL of the diabetic retina as early as the first month of diabetes and in the IPL and outer plexiform layer (OPL) at 4 months. A paracrine mechanism was suggested to be involved in the regulation of microglia in the inner retina, whereas ganglion cell axons, photoreceptors, and other retinal cell damage was responsible for the increase in the number of activated microglia in the IPL and ONL [140]. Pro-inflammatory cytokine levels, as well as caspase-3 expression are increased by activated microglia [141].
Retinal Müller cells (macroglia) also play an important role in the inflammation process induced by diabetes and are an important source of pro-inflammatory cytokines, TNF-α, IL-1β, IL-6, and VEGF. These cytokines induce impairments of glutamate metabolism, directly affecting retinal neurons, vascular cells, and the integrity of the blood–retina barrier. VEGF mRNA expression has been reported in retinal neurons, glia, endothelial cells, pericytes, and RPE [142]. In patients with nonproliferative DR an increase in VEGF mRNA expression was reported in glial cells and the optic nerve [143]. VEGF was reported to be constitutively expressed in Müller cells and to enhance the survival of Müller cells (an autocrine role), photoreceptors and RGCs [134].
Μany reports have suggested that neighboring cells, such as neurons and vessels, may also release VEGF and increase its levels, aiding the progression of the disease to microangiopathy. The involvement of NOX2 and NOX4 isoforms in the mechanisms associated with micro/macroglia activation in retina has been supported by other researchers employing different models of DR [57,91] and other retinopathies [oxygen-induced retinopathy, (OIR)][65]. In light of these data, it appears that NADPH oxidases have a dual role regarding the function of micro/macroglia: NOX derived ROS from other retinal cells (e.g., neurons, endothelial cells) induce the activation of micro/macroglia, which leads to further production of ROS through NOX-dependent mechanisms, and affect retinal neurons and vascular cells.
The NOX2 isoform has been mainly associated with vascular impairments that characterize diabetic retinopathy. GLX7013170, the novel NOX2 inhibitor significantly attenuated the over activation of both micro- and macroglia in the ESDR model. NOX2 had a key and partial role in neuroinflammatory and neurodegenerative impairments, respectively. Administered topically, as eye drops, GLX7013170 depicted a pharmacological profile suggesting its therapeutic potential for ESDR [56].
NOX4 inhibition of the activation of microglia and macroglia also afforded neuroprotection to the diabetic retina. GLX7013114 induced blockade of NOX4 with the subsequent reduction of the number of activated microglia in layers, GCL, IPL, and INL of diabetic retinas in both, 2 and 5wk, paradigms of ESDR [55].These findings suggested that GLX7013114 reversed the activation of microglia and subsequently provided anti-inflammatory properties to the diabetic retina.
The diabetes induced increase in GFAP expression, in the 2-week- and 5-week ESDR paradigms was reduced by NOX4 blockade (GLX7013114). Increased activation of both micro- and macroglia indicates a significant production and release pro-inflammatory cytokines. GLX7013114 reversed the diabetes-induced increase of TNF-α protein levels and mRNA, and IL-1β and IL-6 levels, in the 2/5 week ESDR paradigms, respectively [55]. In agreement with the above, GKT13783 (dual NOX1/NOX4 inhibitor) attenuated ROS and VEGF levels released by neuron and glial cell populations in a rat model of OIR [75]. In addition, NOX4 inhibition by apomycin attenuated the expression of retinal inflammatory proteins in 2-month-old diabetic mice [76].

6. Conclusions
In summary, this review highlights the mechanisms involved in diabetes induced impairment of the Neurovascular Unit and the subsequent production of ischemia, oxidative stress and excitotoxicity. The interplay between members of this triad leads to the three pathologies of the early stage of diabetic retinopathy, namely neurodegeneration, vasculopathy and neuroinflammation. This review also highlights the classification of the different phases of the disease, according to fundus detection, anatomic disturbances and symptomatology that are included in the Early and Late Stage of DR. Presently, pharmaceuticals for the treatment of the pathologies of ESDR are limited and the need for new treatments is urgent.
This review highlights the importance of NOX2 and NOX4 inhibitors in the treatment of neurodegeneration, vasculopathy and neuroinflammation in ESDR.NADPH Oxidases have emerged as novel targets for the development of new therapeutics for the treatment of neurodegenerative disease (i.e., in brain and retina). Novel specific inhibitors of NOX4 (GLX7013114) and NOX2 (GLX7013170), administered topically as eye drops in diabetic models of ESDR were beneficial in the protection of the diabetic retina against the above mentioned pathologies.
GLX7013114, the novel NOX4 selective inhibitor (IC50, 0.3 mmol/L), has the pharmacological profile of a putative therapeutic for the treatment of the early events of DR. It is characterized as a small molecule, with no affinity for other NOX isoforms present in retina, and no scavenging or assay interference properties [54,59,144]. GLX7013114 fulfills all the criteria that dictate the specificity of NOX inhibitors. NOX4 blockade protected the neural and vascular part of the neurovascular unit in rat diabetic retina. Administered as eye drops, GLX7013114 blocked all of the early pathological events of diabetic retinopathy. A pharmacokinetic study (PK) substantiated that GLX7013114 reaches the retina when administered as eye drops. The PK data justified its efficacy and suggest that a proper dose regimen of GLX7013114may be beneficial for the treatment of ESDR and maybe therapeutic in delaying the development of the advanced stage of the disease and vision loss [55].
The new knowledge evolved regarding the interplay between NOX2, oxidative stress and glutamate excitotoxicity, and the molecular mechanisms responsible for the actions of NOX2 and NOX4 inhibitors in ESDR provide a better understanding for the development of new therapeutics. These will be beneficial not only for diabetic retinopathy patients but also patients with other diabetic microvascular (diabetic cardiopathy, neuropathy, and kidney disease) and macrovascular (stroke, cardiovascular morbidity, and others) complications, since DR is considered an independent predictor of all diabetic complications. A frequent screening of DR is therefore essential and beneficial in order to elucidate the presence and severity of the disease, but also to identify patients at increased risk for micro and macrovascular complications [145].
DR is also associated with other neurodegenerative diseases due to its neurodegenerative pathology which is evident in the early stage of the disease. Individuals with type 2 diabetes were reported to have a higher risk of developing neurodegenerative disease (e.g., Alzheimer’s disease). It is considered that the two diseases share pathogeni pathways [146]. A Danish Registry-Based Nationwide Cohort Study investigated whether diabetes and DR is a risk marker of AD. It was reported that individuals with DR had a 34% higher risk of incident AD, whereas individuals with diabetes without DR were less likely to develop AD compared to persons without diabetes [147].
In closing, the information presented in this review supports even more than ever that diabetic retinopathy is a neurodegenerative disease. Novel NOX4 and putative dual NOX2/NOX4 inhibitors have a therapeutic potential for retinal and brain neurodegenerative diseases. They will alleviate the burden of vision loss and brain diseases, where no treatment is yet available. In addition, these will be efficacious for treating other micro- and macrovascular complications of diabetes.
Author Contributions
S.D. wrote parts of this review that is ac tually based on her Master’s and Ph.D. theses and also did the editing of different drafts .She also prepared Figure 1,2 and Table 1. K.T. wrote the manuscript and did the final editing.All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the University of Crete (ELKE KA 10752 to K.T.) and a donation from Glucox Biotech AB to University of Crete (ELKE KA 10879 to K.T.) and the Christina Spyraki award to S.D. The funders had no role in the design of the project and its experiments.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We would like to thank Dimitris Spyridakos for reading the manuscript and for his helpful comments. We would also like to thank our collaborator Per Wikström, CEO & Co-Founder of Glucox Biotech for the NOX2 and NOX 4 inhibitors and for his support of this work.
Conflicts of Interest
The authors have no conflict of interest.
References
- International Diabetes Foundation. IDF Diabetes Atlas, 11th Edition. 2025. Available online: http://www.diabatesatlas.org.
- Yamagishi, S.I.; Imaizumi, T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr. Pharmaceut. Design 2005, 11, 2279–2299. [Google Scholar] [CrossRef]
- Yau, J.W.; Rogers, S.I.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Meta-Analysis for Eye Disease (META-EYE) Study Group, Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S; Simó, R. Diabetic retinopathy. Nat. Rev. Dis.Primers 2016, 2, 16012. [Google Scholar] [CrossRef]
- Teo, Z. L.; Tham, Y. C.; Yu, M.; Chee, M. L.; Rim, T. H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global prevalence of diabetic retinopathy and projection of burden through 2045: systematic review and meta analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Tavares Ferreira, J.; Alves, M.; Dias-Santos, A.; Costa, L.; Oliveira Santos, B.; Cunha, J.P.; Papoila, A.L.; Abegão Pinto, L. Retinal neurodegeneration in diabetic patients without diabetic retinopathy. Investig. Ophthalmol. 2016, 57, 6455–6460. [Google Scholar] [CrossRef]
- Jonsson, K.B.; Frydkjaer-Olsen, U.; Grauslund, J. Vascular Changes and Neuro-degeneration in the Early Stages of Diabetic Retinopathy: Which Comes First? Ophthalmic Res. 2016, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Invest. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Barber, A.J.; Gardner, T.W.; Abcouwer, S.F. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1156–116. [Google Scholar] [CrossRef]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.J.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. (USA) 2016, 113, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; Mckay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammens, H.P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 1, 156–186. [Google Scholar] [CrossRef]
- Simó, R.; Hernández, C. New insights into treating early and advanced stage diabetic retinopathy. Int. J. Mol. Sci. 2022, 23, 8513. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, T.; Matsuyoshi, M.; Emoto, M.; Taniwaki, H.; Kanda, H.; Okuno, Y.; Inaba, M.; Ishimura, E.; Nishizawa, Y.; Morii, H. Impaired endothelium-dependent vascular responses of retinal and intrarenal arteries in patients with type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2509–2516. [Google Scholar] [CrossRef]
- Gilmore, E.D; Hudson, C.; Nrusimhadevara, R.K.; Ridout, R.K.; Harvey, P.T.; Mandelcorn, M.; Lam, W-C.; Devenyi, R.G. Retinal arteriolar hemodynamic response to a combined isocapnic hyperoxia and glucose provocation in early sight-threatening diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Nawaz, M.I.; Siddiquei, M.M.; Al-Amro, S.; Abu El-Asrar, A.M. Recent advances in understanding the biochemical and molecular mechanism of diabetic retinopathy. J Diabetes Complications 2012, 26, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Nayak, J.; Bhat, P.S.; Acharya, R.P.; Lim, C.M.; Kagathi, M. Automated identification of diabetic retinopathy stages using digital fundus images. J. Med. Syst. 2008, 32, 107–115. [Google Scholar] [CrossRef]
- Yang, Z.; Tan, T-E.; Shao, Y.; Wong, T.Y.; Li, X.R. Classification of diabetic retinopathy: Past, present and future. Front. Endocrinol. 2022, 13, 1079217. [Google Scholar] [CrossRef]
- Zhang, X.; Saaddine, J.B.; Chou, C-F; Cotch, M.F.; Cheng, Y.J.; Geiss, L.S.; Gregg, E.W.; Albright, A.L.; Klein, B.E.K.; Klein, R. Prevalence of Diabetic Retinopathy in the United States, 2005-2008. JAMA 2010, 304, 649–656. [Google Scholar] [CrossRef]
- Toprak, I.; Fenkci, S.M.; Fidan Yaylali, G.; Martin, C.; Yaylali, V. Early retinal neurodegeneration in preclinical diabetic retinopathy: a multifactorial investigation. Eye 2020, 34, 1100–1107. [Google Scholar] [CrossRef]
- Park, J.C.; Chen, Y. F.; Liu, M.; Liu, K.; McAnany, J.J. Structural and functional abnormalities in early-stage diabetic retinopathy. Current eye research 2020, 45, 975–985. [Google Scholar] [CrossRef]
- Cao, D.; Yang, D.; Huang, Z.; Zeng, Y.; Wang, J.; Hu, Y.; Zhang, L. Optical coherence tomography angiography discerns preclinical diabetic retinopathy in eyes of patients with type 2 diabetes without clinical diabetic retinopathy. Acta diabetologica 2018, 55, 469–477. [Google Scholar] [CrossRef]
- Dissanayake, H.A.; Kiire, C.A.; Preiss, D.; Tan, G.D.J. The use of fenofibrate in diabetic retinopathy: Narrativereview. Diabet. Complications 2025, 39, 109135. [Google Scholar] [CrossRef]
- Zhang, L.; Krzentowski, G.; Albert, A.; Lefebvre, P.J. Risk of developing retinopathy in Diabetes Control and Complications Trial type 1 diabetic patients with good or poor metabolic control. Diabetes care 2001, 24, 1275–1279. [Google Scholar] [CrossRef]
- Keech, A.C.; Mitschell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.E.; Moffitt, M.S.; Taskinen; Simes, M.R.; Tse, R.J.; Williamson, D.E. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial study): a randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A; Lovato, J.F.; Perdue, L.H. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233e244. [Google Scholar]
- Wong, T.Y.; Simó, R.; Mitchell, P. Fenofibrate - A Potential Systemic Treatment for Diabetic Retinopathy? Am. J. Ophthalmol. 2012, 154, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahn, J.H.; Kim, J.H.; Yu, Y.S.; Kim, H.S.; Ja, J.; Shinn, S.H.; Oh, Y.S. Fenofibrate regulates retinal endothelial cell survival through the AMPK signal transduction pathway. Exp. Eye. Res. 2007, 84, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Roy, S.; Behar-Cohen, F.; Keech, A.; Mitchell, P.; Wong, T.Y. Fenofibrate: a new treatment for diabeticretinopathy. Molecular mechanisms and future perspectives. Curr Med Chem. 2013, 20, 3258–3266. [Google Scholar] [CrossRef]
- Roy, S.; Kim, D.; Hernández, C.; Simó, R.; Roy, S. Beneficial effects of fenofibric acid on overexpression of extracellular matrix components, COX-2, and impairment of endothelial permeability associated with diabeticretinopathy. Exp. Eye Res. 2015, 140, 124–129. [Google Scholar] [CrossRef]
- Mounirou, B.A.; Adam, N.D.; Yakoura, A.K.; Aminou, M.S.; Liu, Y. T.; Tan, L. Y. Diabetic retinopathy: an overview of treatments. Indian J. Endocrinol. Metabol. 2022, 26, 111–118. [Google Scholar] [CrossRef]
- Simó, R.; Hernández, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Ret. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic Retinopathy: Current Understanding, Mechanisms, and Treatment Strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef]
- Tomita, Y.; Lee, D.; Tsubota, K.; Negishi, K.; Kurihara, T. Updates on the Current Treatments for Diabetic Retinopathy and Possibility of Future Oral Therapy. J. Clin. Med. 2021, 10, 4666. [Google Scholar] [CrossRef] [PubMed]
- Lundeen, E. A.; Kim, M.; Rein, D. B.; Wittenborn, J. S.; Saaddine, J.; Ehrlich, J. R.; Holliday, C. S. Trends in the prevalence and treatment of diabetic macular edema and vision-threatening diabetic retinopathy among commercially insured adults aged<65 years. Diabetes Care 2023, 46, 687–696. [Google Scholar]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vis. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.E.; Pulido, J.S; Erie, J.C.; Arroyo, J.; Bertram, K.; Lu, M-J.; Shippy, S.S. A role for excitatory amino acids in diabetic eye disease. Exp Diabetes Res. 2007, 2007, 36150. [Google Scholar] [CrossRef]
- Hammes, H.P. Diabetic retionopathy: hyperglycemia, oxidate stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef]
- Solomon, S.D.; Chew, E.; Duh, E.J; Sobrin, L.; Sun, J.K.; VanderBeek, B.L.; Wykoff, C.; Gardner, T.W. Diabetic Retinopahy: a position statement by the American Diabetes Association. Diabetes Care 2017, 40, 412–418. [Google Scholar] [CrossRef]
- Gardner, T.W.; Davila, J.R. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 2017, 255, 1–6. [Google Scholar] [CrossRef]
- Zafar, S.; Sachdeva, M.; Frankfort, B.J.; Channa, R. Current Diabetes Reports. 2019, 19, 2–13. [Google Scholar]
- Simo, R.; Simo-Servat, O.; Bogdanov, P.; Hernandez, C. Neurovascular Servat, Patricia Bogdanov, “NeurovascularUnit: A New Target for Treating Early Stages of Diabetic Retinopathy. Pharmaceutics 2021, 13, 1320. [Google Scholar] [CrossRef]
- Yang, I.; Zexin, X.; Xu, H.M.; Chen, Y.; Cao, M.; Yi, M.; Fu, Min M. Autophagy in the Retinal Neurovascular Unit: New Perspectives into Diabetic Retinopathy. J.Diabetes 2023, 15, 382–396. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Augustine, J.; Evan, P.; Troendle, E.P.; Barabas, P.; McAleese, C.A.; Friedel, T.; Stitt, A.; Curtis, T.M. The Role of Lipoxidation in the Pathogenesis of Diabetic Retinopathy. Front. Endocrinol. 2021, 11, 621938. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef]
- Ramos, H.; Hernández, C.; Simó, R.; Simó-Servat, O. Inflammation: The Link between Neural and Vascular Impairment in the Diabetic Retina and Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 8796. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Hernández, C. Topical ocular administration of DPP-IV inhibitors: a new approach for treating diabetes induced retinal neurodegeneration. Neural Regen. Res. 2024, 19, 713–714. [Google Scholar] [CrossRef]
- Hernández, C.; García-Ramírez, M.; Corraliza, L.; Fernández-Carneado, J.; Farrera-Sinfreu, J.; Ponsati, B.; González-Rodríguez, A.; Valverde, A.M.; Simó, R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes 2013, 62, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, P.; Simó-Servat, O.; Sampedro, J.; Solà-Adell, C.; Garcia-Ramírez, M.; Ramos, H.; Guerrero, M.; Suñé-Negre, J.M.; Ticó, J.R.; Montoro, B.; et al. Topical administration of bosentan prevents retinal neurodegeneration in experimental diabetes. Int. J. Mol. Sci. 2018, 19, 3578. [Google Scholar] [CrossRef] [PubMed]
- HernándezC; BogdanovP; CorralizaL; García-Ramírez, M.; Solà-Adell, C.; Arranz, J.A.; Arroba, A.I.; Valverde, A.M.; Simó, R. Topical administration of GLP-1 receptor agonists prevents retinal neurodegeneration in experimental diabetes. Diabetes 2016, 65, 172–187. [Google Scholar] [CrossRef]
- Ibán-Arias, R.; Lisa, S.; Mastrodimou, N.; Kokona, D; Koulakis, E.; Iordanidou, P.; Kouvarakis, A.; Fothiadaki, M.; Papadogkonaki, S.; Sotiriou, A.; et al. BNN27 affects retinal function in rats with streptozotocin-induced diabetes. Diabetes 2018, 67, 321–333. [Google Scholar] [CrossRef]
- Ibán-Arias, R.; Lisa, S.; Poulaki, S.; Mastrodimou, N.; Charalampopoulos, I.; Gravanis, A.; Thermos, K. Effect of topical administration of the microneurotrophin BNN27 in the diabetic rat retina. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 429–2436. [Google Scholar] [CrossRef] [PubMed]
- Spyridakos, D.; Mastrodimou, N.; Vemuri, K.; Ho, T.C.; Nikas, S.P.; Makriyannis, A.; Thermos, K. Blockade of CB1 or Activation of CB2 Cannabinoid Receptors Is Differentially Efficacious in the Treatment of the Early Pathological Events in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2023, 24, 240. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Wikstrom, P.; Bucolo, C.; Romano, G.L.; Micale, V.; Svensson, R.; Spyridakos, D.; Mastrodimou, N.; Georgakis, S.; Verginis, P.; et al. Topically Administered NOX4 Inhibitor, GLX7013114, Is Efficacious in Treating the Early Pathological Events of Diabetic Retinopathy. Diabetes 2023, 72, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Dionysopoulou, S.; Wikstrom, P.; Walum, E.; Georgakis, S.; Thermos, K. Investigation of the Effects of a Novel NOX2 Inhibitor, GLX7013170, against Glutamate Excitotoxicity and Diabetes Insults in the Retina. Pharmaceuticals 2024, 17, 393. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.J.; Yu, Q.; Chen, K.; Mahadev, K.; Zhang, S.X. Inhibition of reactive oxygen species by lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes 2010, 59, 1528–1538. [Google Scholar] [CrossRef]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumí, J.; Ramos, D.; Ruberte, J.; Simó, R.; Hernández, C. The db/db mouse: A useful model for the study of diabetic retinal neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef]
- Thagaard; Mikkel, S.; Vergmann, A.S.; Grauslund, J. Topical Treatment of Diabetic Retinopathy: A Systematic Review. Acta Ophthalmologica 2022, 100, 136–47. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Casas, A.I.; Geuss, E.; Kleikers, P.W.M.; Stine, M.; Herrmann, A.M.; Buendia, I.; Egea, J.; Sven, G.; Meuth, M.; Lopez, G.; Kleinschnitz, C.; Schmidt, H.H.H. W. NOX4-dependent neuronal autotoxicity and BBB breakdown explain the superior sensitivity of the brain to ischemic damage. PNAS 2017, 114, 12315–12320. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.M.; Cooper, M.E.; et al. NADPH Oxidase, NOX1, Mediates Vascular Injury in Ischemic Retinopathy. Antioxid. Redox Signal. 2014, 2726–2740. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; A Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Invest. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J Neuroinflammation 2015, 12, 136. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I.; Siddiquei, M.M.; El-Asrar, AM. Apocynin ameliorates NADPH oxidase 4 (NOX4) induced oxidative damage in the hypoxic human retinal Müller cells and diabetic rat retina. Mol. Cell Biochem. 2021, 476, 2099–2109. [Google Scholar] [CrossRef]
- Suh, Y-A.; Arnold, R.S.; Lassegue, B.; Shi, J.; XuX; Sorescu, D.; Chung, A.B.; Kathy K.; Griendling, K.K.; Lambeth, J.D. Cell Transformation by the Superoxide-Generating Oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-Dependent Reactive Oxygen Generation Is Regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef]
- Ueyama, T.; Miklós, G.; Leto, T.L. Involvement of Rac1 in Activation of Multicomponent Nox1 and Nox 3 Based NADPH Oxidases. Mol. Cell. Biol. 2006, 26, 2160–2174. [Google Scholar] [CrossRef]
- Noreng, S.; Ota, N.; Sun, Y.; Ho, H.; Johnson, M.; Arthur, C.P.; Schneider, K.; Lehoux, I.; Davies, C.W.; Mortara, K.; Wong, K.; Seshasayee, D.; Masureel, M.; Payandeh, J.; Yi, T.; Koerber, J.T. Structure of the core human NADPH oxidase NOX2. Nat. Commun. 2022, 13, 6079. [Google Scholar] [CrossRef]
- Chan, E.C.; van Wijngaarden, P.; Liu, G.S.; Jiang, F.; Peshavariya, H.; Dusting, G.J. Involvement of Nox2 NADPH Oxidase in Retinal Neovascularization. Invest. Opthalmology Vis. Sci. 2013, 54, 7061–7067. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L; Cartier, L; Bedard, K; Banfi, B; Lardy, B; Plastre, O; Sienkiewicz, A; Fórró, L; Schlegel, W; Krause, KH. Biochem J 2007, 406, 105–111. [CrossRef]
- Martyn, P.M.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zana, M.; Péterfi, Z.; Kovács, H.A.; Tóth, Z.E.; Enyedi, B.; Morel, F.; Paclet, M-H.; Ágnes; Donkó, Á.; Morand, S.; Leto, T.L.; Geiszt, M. Interaction between p22phox and Nox4 in the endoplasmic reticulum suggests a unique mechanism of NADPH oxidase complex formation. Free Radic. Biol. Med. 2018, 116, 41–49. [Google Scholar] [CrossRef]
- Lyle, A.L.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassègue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–59. [Google Scholar] [CrossRef]
- Ueyama, T.; Yamaguchi, K.; Aoyama, Y.; Aoshima, K.; Onizuka, M.; Tamagawa, T.; Kitayama, S.; Ueyama, J.; Okamoto, K.; Mohri, H.; Shimazawa, M. Nox3 expression and function in retinal ganglion cells and Amacrine cells. Cell Mol Life Sci. Online ahead of print. 2025. [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- AltenhöferS; KleikersPWM; RadermacherKA; et al. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 2012, 69, 2327–2343. [Google Scholar] [CrossRef]
- Olney, J. W. Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J. Neuropathol. Exp. Neurol. 1969, 28, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.M.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef]
- Izumi, Y.; Hammerman, S.B.; Kirby, C.O.; Benz, A.M.; Olney, J.W.; Zorumski, C.F. Involvement of glutamate in ischemic neurodegeneration in isolated retina. Vis. Neurosci. 2003, 20, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Andrés, N.; Malpesa, Y.; Rodríguez, M.J.; Mahy, N. Low sensitivity of retina to AMPA-induced calcification. J. Neurosci. Res. 2003, 72, 543–548. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Zukin, R.S. Ca+2-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.M.; Yin, H.Z.; Chiang, J.; Weiss, J.H. Ca++ permeable AMPA/kainate and NMDA channels: high rate of Ca+2 influx underlies potent induction of injury. J. Neurosci. 1996, 16, 5457–5465. [Google Scholar] [CrossRef] [PubMed]
- Kiagiadaki, F.; Thermos, K. Effect of intravitreal administration of somatostatin and sst2 analogs on AMPA-induced neurotoxicity in rat retina. Invest. Ophthalmol. Vis. Sci. 2008, 49, 3080–3089. [Google Scholar] [CrossRef]
- Kokona, D.; Charalampopoulos, I.; Pediaditakis, I.; Gravanis, A.; Thermos, K. The neurosteroid dehydroepiandrosterone (DHEA) protects the retina from AMPA-induced excitotoxicity: NGF TrkA receptor involvement. Neuropharmacology 2012, 62, 2106–2117. [Google Scholar] [CrossRef]
- Kokona, D.; Thermos, K. Synthetic and endogenous cannabinoids protect retinal neurons from AMPA excitotoxicity in vivo, via activation of CB1 receptors: Involvement of PI3K/Akt and MEK/ERK signaling pathways. Exp. Eye Res. 2015, 136, 45–58. [Google Scholar] [CrossRef]
- Kokona, D.; Spyridakos, D.; Tzatzarakis, M.; Papadogkonaki, S.; Filidou, E.; Arvanitidis, K.I.; Kolios, G.; Lamani, M.; Makriyannis, A.; Malamas, M.; et al. The endocannabinoid 2-arachidonoylglycerol and dual ABHD6/MAGL enzyme inhibitors display neuroprotective and anti-inflammatory actions in the in vivo retinal model of AMPA excitotoxicity. Neuropharmacology 2021, 185, 108450. [Google Scholar] [CrossRef]
- Dionysopoulou, S.; Wikström, P.; Walum; Thermos, E.K. Effect of NADPH oxidase inhibitors in an experimental retinal model of excitotoxicity. Exp. Eye Res. 2020, 200, 108232. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH oxidase) 1, Nox4, and Nox5 promote vascular permeability and neovascularization in retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M.; Kumar, B. Diabetic Retinopathy and Transcriptional Regulation of a Small Molecular Weight G Protein, Rac1. Exp. Eye Res. 2016, 147, 72–77. [Google Scholar] [CrossRef]
- Meng, W.; Shah, K.P.; Pollack, S.; Toppila, I.; Hebert, H.L.; McCarthy, M.I.; Groop, L.; Ahlqvist, E.; Lyssenko, V.; Agardh, E.; et al. Wellcome Trust Case Control Consortium 2 (WTCCC2), Surrogate Markers for Micro- and Macro-Vascular Hard Endpoints for Innovative Diabetes Tools (SUMMIT) Study group. A genome-wide association study suggests new evidence for an association of the NADPH oxidase 4 (NOX4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol. 2018, 96, e811–e819. [Google Scholar]
- Guemez-Gamboa, A.; Estrada- Sanchez, A.M.; Montiel, T.; Paramo, B.; Massieu, L.; Moran, J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef]
- Ambati, J.; Chawla, D. K.; D’Angio, C. T.; Guillet, E.G.; Rose, S.J.; Vanderlinde, R.E.; Ambati, B.K. Elevated γ-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch. Ophthalmol. 1997, 115, 1161–1166. [Google Scholar] [CrossRef]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Engerman, R.L.; Case, G.L.; Kerns, T.S. Retinal glutamate in diabetes and effect of antioxidants. Neurochem. Int. 2001, 38, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; LaNoue, K.F.; Antonetti, D.A.; Ratz, M. Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. Exp. Eye Res. 2000, 70, 723–730. [Google Scholar] [CrossRef]
- Li, Q.; Puro, D.G. Diabetes-induced dysfunction of the glutamate transporter in retinal Müller cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3109–3116. [Google Scholar]
- Santiago, A.R.; Gaspar, J.M.; Baptista, F.I.; Cristóvão, A.J.; Santos, P.F.; Kamphuis, W.; Ambrósio, A.F. Diabetes changes the levels of ionotropic glutamate receptors in the rat retina. Mol. Vis. 2009, 15, 1620–1630. [Google Scholar]
- Barber, A.J.; Ambrosio, A.F. Elevated glucose changes the expression of ionotropic glutamate receptor subunits and impairs calcium homeostasis in retinal neural cells. Invest. Ophthalmol. Vis. Sci. 2006, 47, 4130–4137. [Google Scholar]
- Carmo, A.; Lopes, C.; Santos, M.; Proença, R.; Cunha-Vaz, J.; Carvalho, A.P. Nitric oxide synthase activity and L-arginine metabolism in the retinas from streptozotocin-induced diabetic rats. Gen Pharmacol. 1998, 30, 319–324. [Google Scholar] [CrossRef]
- Du, Y.; Cramer, M.; Lee, C.A.; Tang, J.; Muthusamy, A.; Antonetti, D.A.; Jin, H.; Palczewski, K.; Kern, T.S. Adrenergic and Serotonin Receptors Affect Retinal Superoxide Generation in Diabetic Mice: Relationship to Capillary Degeneration and Permeability. FASEB J. 2015, 29, 2194–2204. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 9, 328–334. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kennedy, A. Therapeutic potential of anti-oxidants and diabetic retinopathy. Expert Opin. Investig. Drugs 2001, 10, 1665–1676. [Google Scholar] [CrossRef]
- He, M.; Pan, H.; Xiao, C.; Pu, M. Roles for redox signaling by NADPH oxidase in hyperglycemia-induced heme oxygenase-1 expression in the diabetic retina. Invest. Ophthalmol.Vis.Scie 2013, 54, 4092–4101. [Google Scholar] [CrossRef]
- Appukuttan, B.; Ma, Y.; Stempel, A.; Ashander, L.M.; Deliyanti, D.; Wilkinson-Berka, J.L.; Smith, J.R. Effect of NADPH oxidase 1 and 4 blockade in activated human retinal endothelial cells. Clin. Exp. Ophthalmol. 2018, 46, 652–660. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Grant, J.; Santos, A.R.C.; Hernandez, E.; Ivanov, D. Neuronal NAD(P)H Oxidases Contribute to ROS Production and Mediate RGC Death after Ischemia. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2823–2830. [Google Scholar] [CrossRef]
- Gastinger, M.J.; Singh, R.S.; Barber, A.J. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita diabetic mouse retina. Invest.Ophthalmol. Vis. Sci. 2006, 47, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1-RAC1 signalling axis mediated activation of NOX2 initiates mitochondrial damage and the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef]
- Simó, R.; Hernández, C. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol. Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Potilinski, M.C.; Lorenc, V.; Perisset, S.; Gallo, J.E. Mechanisms behind Retinal Ganglion Cell Loss in Diabetes and Therapeutic Approach. Int. J. Mol. Sci. 2020, 21, 2351. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Behzadian, M.A.; Abou-Mohamed, G.; Franklin, T.; Caldwell, R.W.; Caldwell, R.B. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am. J. Pathol. 2003, 162, 1995–2004. [Google Scholar] [CrossRef]
- Ali, T.K.; Matragoon, S.; Pillai, B.A.; Liou, G.I.; El-Remessy, A.B. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes 2008, 57, 889–898. [Google Scholar] [CrossRef]
- Hernández-Ramírez, E.; Sánchez-Chávez, G.; Estrella-Salazar, L.A.; Salceda, R. Nitrosative Stress in the Rat Retina at the Onset of Streptozotocin-Induced Diabetes. Cell. Physiol. Biochem. 2017, 42, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G; Duraisamy, A.J.; Kowluru, A.; Kowluru, R.A. Functional Regulation of an Oxidative Stress Mediator, Rac1, in Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 8643–8655. [Google Scholar] [CrossRef]
- George, V.T.; Brooks, G. Nox2 regulates endothelial cell cycle arrest and apoptosis via p21cip1 and p53. Free Radic. Biol. Med. 2007, 43, 976–986. [Google Scholar]
- Barakat, D.J.; Dvoriantchikova, G.; Ivanov, D.; Shestopalov, V.I. Astroglial NF-κB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. J. Neurochem. 2012, 120, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Narayanan, S.P.; Zhang, W.; Liu, H.; Rojas, M.; Xu, Z.; Lemtalsi, T.; Nagaoka, T.; Yoshida, A.; Brooks, S.E.; et al. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8123–8131. [CrossRef] [PubMed]
- Wang, Z.; Wei, X.; Liu, K.; Zhang, X.; Yang, F.; Zhang, H.; He, Y.; Zhu, T.; Li, F.; Shi, W.; et al. NOX2 deficiency ameliorates cerebral injury through reduction of complexin II-mediated glutamate excitotoxicity in experimental stroke. Free Radic. Biol. Med. 2013, 65, 942–951. [Google Scholar] [CrossRef]
- Rauen, T.; Rothstein, J.D.; Wässle, H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996, 286, 325–333. [Google Scholar] [CrossRef]
- Vorwerk, C.K.; Naskar, R.; Schuettauf, F.; Quinto, K.; Zurakowski, D.; Gochenauer, G.; Robinson, M.B.; Mackler, S.A.; Dreyer, E.B. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3615–3621. [Google Scholar]
- Brennan-Minnella, A.M.; Won, S.J.; Swanson, R.A. NADPH oxidase-2: linking glucose, acidosis, and excitotoxicity in stroke. Antioxid. Redox Signal. 2015, 22, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Elksnis, A.; Cen, J.; Wikstrom, P.; Carlsson, P.O.; Welsh, N. Pharmacological inhibition of NOX4 improves mitochondrial function and survival in human beta-cells. Biomedicines 2021, 9, 1865. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Li, B.; Gao, X.; Liou, Y.; Mao, W.; CHEN, S.-L. Resveratrol ameliorates low shear stress-induced oxidative stress by suppressing ERK/eNOS-Thr495 in endothelial cells. Mol. Med. Reports 2014, 10, 1964–1972. [Google Scholar] [CrossRef]
- Roufail, E.; Soulis, T.; Boel, E.; Cooper, M.E.; Rees, S. Depletion of nitric oxide synthase-containing neurons in the diabetic retina: reversal by aminoguanidine. Diabetologia 1998, 41, 1419–1425. [Google Scholar] [CrossRef]
- Barber, A.J. The form and Function of Retinal Ganglion Cells and Diabetes. Cell. 2025, 14, 1455. [Google Scholar] [CrossRef]
- Yi, J.; Puyang, Z.; Feng, L.; Duan, L.; Liang, P.; Backman, V.; Liu, X.; Zhang, H.F. Optical detection of early damage in retinal ganglion cells in a mouse model of partial optic nerve crush injury. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5665–5671. [Google Scholar] [CrossRef]
- Soto, I.; Oglesby, E.; Buckingham, B.P.; Son, J.L.; Roberson, E.D.; Steele, M.R.; Inman, D.M.; Vetter, M.L.; Horner, P.J.; Marsh Armstrong, N. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J. Neurosci. 2008, 28, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.H.; Lin, H.S.; Lin, S. Nerve fibre layer thinning in patients with preclinical retinopathy. Can. J. Ophthalmol. 2009, 44, 417–422. [Google Scholar] [CrossRef]
- Vujosevic, S.; Midena, E. Retinal layers changes in human preclinical and early clinical diabetic retinopathy support early retinal neuronal and Müller cells alterations. J. Diabetes Res. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Dal Monte; Casini, M.G. Oxidative stress induces a VEGF autocrine loop in the retina: relevance for diabetic retinopathy. Cells 2020, 9, 1452. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Allen, D.M.; Ferrigno, A.S.; Vellanki, S.; Pouw, C.E.; Hejny, W.A.; Tsin, A.T.C. Autocrine and paracrine secretion of vascular endothelial growth factor in the pre-hypoxic diabetic retina. Curr. Diabetes Rev. 2017, 13, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C; Young, M.J; D’Amore, P.A. Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PLoS One 2008, 3, e3554. [Google Scholar] [CrossRef]
- Bucolo, C.; Ward, K.W.; Mazzon; Cuzzocrea, S.; Drago, F. Protective effects of a coumarin derivative in diabetic rats. Invest. Ophthalmol. Vis. Sci. 2009, 50, 3846–3852. [Google Scholar] [CrossRef]
- Schrufer, T.L.; Antonetti, D.A; Sonenberg, N.; Kimball, S.R.; Gardner, T.W.; Jefferson, L.S. Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Müller cells in culture. Diabetes 2010, 59, 2107–2116. [Google Scholar]
- Qaum, T.; Xu, Q.; Joussen, A.M.; et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest. Ophthalmol. Vis. Sci. 2001, 42, 2408–2413. [Google Scholar] [PubMed]
- Wang, X.; Elksnis, A.; Wikstrom, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS One 2018, 13, e0204271. [Google Scholar] [CrossRef]
- Zeng, X.X.; Ng, Y.K.; Ling, E.A. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis.Neurosci 2000, 17, 463–471. [Google Scholar] [CrossRef]
- Rungger-Brändle, E.; Dosso, A.A.; Leuenberger, P.M. Glial reactivity, an early feature of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1971–1980. [Google Scholar]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.C.; Allen, D.M.; Cardona, A.E. The role of microglia in diabetic retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef]
- Amin, R.H.; Frank, R.N.; Kennedy, A.; Eliott, D.; Puklin, J.E.; Abrams, G.W. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 1997, 38, 36–47. [Google Scholar] [PubMed]
- Simo-Servat, O.; Hernandez, C.; Simo, R. Diabetic retinopathy in the context of patients with diabetes. Ophthalmic Res. 2019, 62, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pearce, I.; Simo, R.; Lovestan-Adrian, M.; Wong, DT. Association between diabetic eye disease and other complications. A systematic review. Diabetes Obes. Metab. 2019, 21, 467–478. [Google Scholar] [CrossRef]
- Sundstrom, J.M.; Hernandez, C.; Weber, S.; Zhao, Y.; Dunklebarger, M.; Tiberti, N.; Laremore, T.; Simó-Servat, O.; Marta Garcia-Ramirez, M.; Barber, A.J. Proteomic analysis of early diabetic retinopathy reveals mediators of neurodegerative brain disease. Invest Ophthalmol. 2018, 59, 2264–2274. [Google Scholar] [CrossRef]
- Nørregaard, F.; Pedersen, L.; Stokholm, F.; Pouwer, K.; Hass, R.; Peto, T.; Frydkjær-Olsen, U.; Suhr, A.; Thykjær, N.; Andersen, J. Diabetic Retinopathy Predicts Risk of Alzheimer’s Disease: A Danish Registry-Based Nationwide Cohort Study. J. Alzheimers Dis. 2022, 86, 451–460. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



