
Submitted:
21 January 2026
Posted:
22 January 2026
You are already at the latest version
Abstract
Background/Objectives: Metabolic dysfunction–associated steatotic liver disease (MASLD) and its progressive inflammatory/fibrotic form, metabolic dysfunction–associated steatohepatitis (MASH), represent a growing global health burden. Theis progression driven by complex, interconnected mechanisms involving metabolic dysregulation, chronic inflammation, oxidative stress, and progressive fibrosis. Despite advances in understanding disease pathophysiology, effective pharmacological therapies remain limited. Pentacyclic triterpenes, a structurally diverse class of natural compounds widely distributed in nature, have attracted increasing attention due to their broad biological activities and ability to modulate multiple molecular pathways implicated in chronic liver disease development. This review aims to provide a mechanistic overview of the potential role of pentacyclic triterpenes in MASLD and MASH. Methods: A comprehensive review of the literature was conducted using major scientific databases (Pubmed, Scielo, Web of Science) to identify experimental studies investigating pentacyclic triterpenes in the context of metabolic liver diseases. The selected studies were analyzed according to triterpene structural classification, reported bioactivities, molecular targets, and experimental evidence from in vitro and in vivo models of MASLD/MASH or specific processes involved in the disease progression. Results: Pentacyclic triterpenes, specially ursolic acid, oleanolic acid and glycyrrhizin, exhibit pleiotropic hepatoprotective effects, including regulation of lipid metabolism, attenuation of oxidative and endoplasmic reticulum stress, suppression of pro-inflammatory signaling, inhibition of inflammasome activation, and reduction of hepatic stellate cell activation and extracellular matrix deposition. These effects are mediated through modulation of key signaling pathways such as AMPK, NF-κB, NLRP3, TGF-β, FXR, MAPK, and related metabolic and inflammatory cascades. Preclinical evidence consistently demonstrates improvements in steatosis, inflammation, and fibrosis across multiple experimental models. Conclusions: By integrating mechanistic and biological evidence, this review highlights pentacyclic triterpenes as potent multitarget modulators of MASH. However, a transition from preclinical models to well-designed clinical trials is essential to validate their safety and efficacy in humans.
Keywords:
pentacyclic triterpenes
; metabolic liver diseases
; AMPK–mTOR signaling
; NLRP3 inflammasome and pyroptosis
; hepatic fibrogenesis
; oxidative stress–driven inflammation
Introduction
Metabolic dysfunction–associated steatotic liver disease (MASLD) and its progressive inflammatory stage, metabolic dysfunction–associated steatohepatitis (MASH), represent major and growing challenges in hepatology, driven by complex interactions between metabolic imbalance, chronic inflammation, oxidative stress, and progressive fibrogenesis. Despite their increasing prevalence, therapeutic options capable of effectively targeting the multifactorial molecular mechanisms underlying disease initiation and progression remain limited. In this context, pentacyclic triterpenes have emerged as promising bioactive compounds due to their pleiotropic capacity to modulate key signaling pathways involved in lipid metabolism, inflammatory responses, redox homeostasis, and hepatic stellate cell activation. This review aims to integrate current evidence on the role of triterpenes in MASLD and MASH by first outlining the structural characteristics, biological activities and molecular targets of pentacyclic triterpenes. The discussion then contextualizes the pathophysiology of MASLD and MASH, culminating in an analysis of preclinical evidence supporting the direct application of triterpenes in experimental models of metabolic liver disease. This structured approach provides a mechanistic framework that connects triterpene chemistry to biological function and, ultimately, to disease modulation.
Methods
A comprehensive literature search was conducted in major scientific databases (e.g., PubMed, and Web of Science) to identify experimental studies evaluating pentacyclic triterpenes in MASLD/MASH or in relevant pathogenic processes (steatosis, inflammation, oxidative/ER stress, and fibrogenesis). Eligible records were screened for mechanistic endpoints, molecular targets, and in vitro and in vivo outcomes. The selected evidence was organized by triterpene class, primary bioactivities, and pathways implicated in metabolic liver disease progression.
Results
1. Classification and Structural Characteristics of Pentacyclic Triterpenes
Triterpenes constitute a vast class of bioactive compounds characterized by a thirty-carbon skeleton. Synthesized by a wide range of organisms, including bacteria, fungi, and animals, they reach their greatest structural diversity within the plant kingdom, with over 20,000 identified variants [1,2]. Characterized as complex molecules, triterpenes can be found either (i) in their free form, as non-volatile lipophilic substances soluble in organic solvents, acting as components of cellular membranes or as signaling molecules, or (ii) in oxygenated, esterified, or glycosylated derivative forms, which may confer protection to plants against pathogens and pests [3,4].
The fundamental terpene architecture arises from the sequential conjugation of isoprene units (CH₂=C(CH₃)-CH=CH₂. Terpenes are classified according to the number of isoprene units they contain, as follows: hemiterpenes (one isoprene unit), monoterpenes (two isoprene units), sesquiterpenes (three isoprene units), diterpenes (four isoprene units), sesterpenes (five isoprene units), triterpenes (six isoprene units), tetraterpenes (eight isoprene units), and polyterpenoids (more than eight isoprene units) [5]. Among these, the pentacyclic triterpenes - characterized by five fused rings - stand out for their remarkable pharmacological potential in modulating metabolic and inflammatory pathways. Representative structures of these groups are illustrated in Figure 1.
The biosynthetic precursors of terpenes are generated through two independent metabolic pathways: the mevalonic acid (MVA) pathway, which involves the condensation of acetyl-CoA units, and the 1-deoxy-D-xylulose-5-phosphate (DXP) pathway, which proceeds through the condensation of pyruvate and glyceraldehyde-3-phosphate. Regardless of the pathway, both routes yield two isomeric C5 building blocks: isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) (Figure 2) [6,7,8,9].
Although both pathways produce identical C5 units, they are compartmentalized and contribute differently to terpene biosynthesis. The MVA pathway occurs predominantly in the cytosol and is mainly responsible for the formation of sesquiterpenes and triterpenes, whereas the plastid-localized DXP pathway primarily supports the biosynthesis of mono-, di-, and tetraterpenes [10].
Terpene biosynthesis is driven by a series of sequential condensation reactions. The process initiates with the condensation of DMAPP and one IPP unit to form geranyl pyrophosphate (GPP), the fundamental precursor of monoterpenes, in a reaction catalyzed by geranyl diphosphate synthase (GDPS). GPP subsequently undergoes condensation with an additional IPP unit to generate farnesyl pyrophosphate (FPP), the C15 precursor of sesquiterpenes and triterpenes, mediated by farnesyl diphosphate synthase (FDPS). Through the action of geranylgeranyl diphosphate synthase, FPP may further condense with IPP to form geranylgeranyl diphosphate (GGPP), the C20 precursor of di- and tetraterpenes.
A critical branch point occurs when two FPP molecules undergo a reductive head-to-head condensation, catalyzed by squalene synthase (SQS), to produce squalene. This hydrocarbon is subsequently epoxidized into 2,3-oxidosqualene, the primary substrate for oxidosqualene cyclases (OSCs), which orchestrate the complex cyclization reactions that define the diverse triterpene skeletons. The stereochemical outcome is dictated by the folding conformation of the substrate: while the chair–boat–chair (CBC) conformation leads to sterol biosynthesis, the chair–chair–chair (CCC) conformation generates the dammarenyl cation. This key intermediate undergoes further ring expansion and rearrangements to yield the characteristic five-fused-ring architecture that defines the pentacyclic triterpene family (Figure 3) [1,8,10,11,12,13].
Structurally, these pentacyclic triterpenes consist of thirty carbon atoms arranged into five fused rings (A–E), a configuration that allows for immense chemical diversity through functionalization (Figure 4). Based on their core carbocyclic skeletons, these compounds are primarily categorized into three dominant groups: oleananes, ursanes, and lupanes [14]. The distinction between oleanane and ursane types lies in the specific arrangement of methyl substituents on ring E: oleananes feature a gem-dimethyl group at the C20 position, whereas ursanes possess one methyl group at C19 and another at C20. In contrast, lupane-type triterpenes are structurally unique, characterized by a five-membered E ring and a trans-fused D/E ring junction [15,16,17,18].
Due to their ubiquitous presence in the plant kingdom and their remarkably broad spectrum of biological activities, pentacyclic triterpenes have emerged as pivotal bioactive natural products. Beyond their ecological roles, these compounds are increasingly recognized as promising lead scaffolds for the development of novel therapeutic agents, particularly in the context of chronic metabolic and inflammatory diseases [19,20].
2. Bioactivity of Triterpenes
2.1. Anti-Inflammatory Action
The therapeutic application of plants rich in pentacyclic triterpenes is deeply rooted in traditional medicine, particularly for managing inflammatory conditions [21,22]. Modern pharmacological research has validated these traditional uses, identifying triterpenoids as potent modulators of the inflammatory cascade. Among these, glycyrrhizin - a triterpenoid saponin abundantly found in licorice root (Glycyrrhiza spp.) - stands out due to its broad anti-inflammatory properties. Upon oral administration, glycyrrhizin undergoes microbial transformation in the gut into glycyrrhetinic acid, an oleanane-type aglycone that serves as the primary effector molecule [23,24]. Glycyrrhizin exerts anti-inflammatory effects through a sophisticated, multi-target network It directly inhibits key pro-inflammatory enzymes, including 5-lipoxygenase and cyclooxygenase (COX)-2, thereby reducing the biosynthesis of leukotrienes and prostaglandins [25,26,27]. A hallmark of its activity is the suppression of the NF-κB and MAPK signaling axes, which orchestrate the expression of inducible nitric oxide synthase (iNOS) and critical cytokines such as TNF-α, IL-1β, and IL-6. Notably, glycyrrhizin possesses a unique ability to bind and neutralize to high-mobility group box 1 (HMGB1), a potent damage-associated molecular pattern (DAMP) that drives sterile inflammation by antagonizing HMGB1 activity and modulating Toll-like receptor (TLR) signaling, glycyrrhizin effectively limits neutrophil activation, reduces reactive oxygen species (ROS) production, and downregulates adhesion molecules. These pleiotropic actions collectively attenuate leukocyte infiltration and tissue damage, positioning glycyrrhizin as a formidable candidate for mitigating the chronic inflammatory milieu characteristic of MASH and other metabolic liver disorders [28,29,30].
Similarly, celastrol, a bioactive triterpenoid isolated from Tripterygium wilfordii, exhibits potent anti-inflammatory and immunomodulatory properties by targeting multiple regulatory nodes. Its mechanism of action is uniquely characterized by the of K63-linked deubiquitination of the NLR family pyrin domain containing 3 (NLRP3) inflammasome [31]. Furthermore, celastrol effectively suppresses the HMGB1/NF-κB axis, dampening the propagation of pro-inflammatory signals [32]. Advanced systems pharmacology and multi-omics analyses have elucidated that celastrol targets key pro-inflammatory mediators, including TNF-α, and IL-6, while regulating pathways such as phosphoinositide 3-kinase (PI3K)/ protein kinase B (Akt), MAPK, transforming growth factor (TGF)-β, and IL signaling. By attenuating immune cell activation, fibroblast invasiveness, cytokine production, and synovial inflammation, celastrol demonstrates a broad anti-inflammatory profile [33]. Beyond its anti-inflammatory profile, celastrol exerts significant anti-proliferative and pro-apoptotic effects. It potentiates TNF-induced apoptosis by downregulating a suite of survival genes, such as IAP1/2, Bcl-2, and c-FLIP, while simultaneously inhibiting mediators of invasion and angiogenesis, including MMP-9 and VEGF [34], By attenuating cytokine production, fibroblast activation, and oxidative stress, celastrol emerges as a robust therapeutic candidate for chronic inflammatory disorders, particularly those where inflammation and fibrogenesis are closely intertwined, such as MASH.
A pivotal anti-inflammatory mechanism of pentacyclic triterpenes involves the activation of the AMP-activated protein kinase (AMPK) pathway, a master regulator of energy homeostasis and immune signaling. A diverse array of compounds, including oleanolic acid (OA), ursolic acid (UA), betulinic acid (BA), glycyrrhetinic acid, and asiatic acid (AA), has been identified as potent AMPK activators, underscoring their therapeutic potential in metabolic-driven inflammation [35].
Specifically, recent investigations into Δ-OA have highlighted its efficacy as a robust AMPK activator. Comparative assays using 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) - a gold-standard AMPK agonist - demonstrated that Δ-OA achieves similar pharmacological outcomes in modulating inflammatory responses. This equivalence to established activators positions Δ-OA and related triterpenoids as promising drug candidates for conditions where AMPK dysregulation is central, such as the chronic inflammatory milieu of MASH [35].
Recent studies have highlighted the anti-inflammatory activity of ursane-type triterpenes, particularly UA with potential applications in the treatment of neuroinflammation. UA has been shown to exert immunomodulatory effects and promote neuronal repair through direct remyelination during the chronic progressive stage of multiple sclerosis [36]. Additionally, UA has demonstrated anti-inflammatory activity in models of Parkinson’s disease [37], arthritis [38], type 2 diabetes mellitus (T2DM) [39], and myasthenia gravis [40]. Administration of UA prior to disease onset also prevented encephalopathy development by inhibiting Th17 cell differentiation in a mice model of the disease [41]. Several mechanisms have been associated with its anti-inflammatory effects, including reductions in pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α levels, and malondialdehyde (MDA) level, one of the products of lipid peroxidation and a biomarker of liver oxidative damage, along with increases in superoxide dismutase (SOD) and glutathione (GSH) levels, that are key components of the cellular antioxidant system [42].
Another ursane-type triterpene under investigation is boswellic acid. Ammon (2006) demonstrated that boswellic-type triterpenes inhibit the synthesis of the pro-inflammatory enzyme 5-lipoxygenase and its products, including 5-hydroxyeicosatetraenoic acid and leukotriene B4 [43]. This compound also modulates several pathways associated with obesity and T2DM, which are discussed in later sections.
A lupane-type triterpene that has attracted attention due to its anti-inflammatory, antioxidant, and antifibrotic activities is ganoderic acid A, the major triterpenoid extracted from Ganoderma mushrooms. In a study conducted by Ma et al. (2021), this compound demonstrated significant biological activity [44].
Despite extensive investigation, it remains unclear whether the carbon skeleton of triterpenes significantly influences their anti-inflammatory efficacy. While some studies suggest that oleanane-type structures may exhibit higher activity than ursane-type compounds [45], other reports indicate comparable effects among oleanane-, lupane-, and ursane-type skeletons [46,47].
2.2. Antioxidant Activity
Oxidative stress is a central driver in the progression of MASLD to MASH, contributing significantly to hepatocyte injury and fibrogenesis. Several studies have demonstrated that pentacyclic triterpenes possess significant antioxidant potential, which directly contributes to their hepatoprotective profile.
Early investigations, such as thoseAllouche et al. (2010) evaluated the antioxidant and antithrombotic activities of uvaol, erythrodiol, OA, and maslinic acid added to isolated human low-density lipoprotein (LDL), aiming to assess their cardiovascular protective effects. The results indicated that maslinic acid, uvaol, and erythrodiol by assessing their ability to protect isolated human LDL from oxidation [48].
The antioxidant effect of maslinic acid had been previously reported [49,50]. Wang et al. (2006) demonstrated that maslinic acid strongly inhibited copper-induced LDL oxidation in vitro, reducing the formation of conjugated dienes by approximately 21% compared with the control group [49]. According to Allouche et al. (2010), this effect was significant at a concentration of approximately 100 μM, while no effect was initially detected at 10 μM. However, the same study later reported that even 10 μM maslinic acid exhibited antioxidant properties [48].
The relevance of this mechanism to hepatic disease was highlighted by Lim et al. (2024) who investigated the protective effects of four triterpenes — AA, asiaticoside, madecassic acid, and madecassoside — in different cell culture models, including Hs68 (human skin fibroblasts), HepG2 (human hepatocytes), RAW 264.7 (murine macrophages), and EA.hy926 (human umbilical vein endothelial cells). Hs68 cells were exposed to UVB radiation, while HepG2 and EA.hy926 cells were challenged with tert-butyl hydroperoxide (TBHP) and hydrogen peroxide, respectively. RAW 264.7 macrophages were stimulated with lipopolysaccharide (LPS). Pretreatment with triterpenes attenuated UVB-induced ROS and MDA generation and preserved GSH levels in skin fibroblasts, while increasing intracellular collagen content. In HepG2 cells, pretreatment significantly improved cell viability and reduced TBHP-induced ROS, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) levels. In EA.hy926 cells, triterpenes reduced ROS levels and decreased MDA and GSH expression. In RAW 264.7 macrophages, treatment significantly reduced nitric oxide, TNF-α, and IL-6 production [50].
In another study, Mamadalieva et al. (2021) evaluated the antioxidant activity of fifteen cycloartane-type triterpenes using cell-free assays, including phosphomolybdenum, 1,1-diphenyl-2-picrylhydrazyl (DPPH•), 2,2′-azinobis(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS•⁺), cupric ion-reducing antioxidant capacity (CUPRAC), ferric-reducing antioxidant power (FRAP), and metal-chelating activity assays. Most of the compounds exhibited antioxidant activity, despite lacking typical phenolic or catecholic moieties commonly associated with antioxidant effects in natural products such as flavonoids [51].
Ostapiuk et al. (2021) investigated the antioxidant mechanisms of betulin and BA, two lupane-type triterpenes, using triterpene-rich extracts. The results demonstrated a significant reduction in hydrogen peroxide and superoxide anion formation, as well as decreased lipid peroxidation [52].
As observed for anti-inflammatory effects, no clear structure–activity relationship has been established for the antioxidant activity of triterpenes. Nevertheless, most compounds within this class exhibit antioxidant effects to varying extents, underscoring their therapeutic potential in diseases and conditions associated with oxidative stress.
2.3. Antiadipogenic Activity and T2DM Control
Triterpenes have demonstrated considerable potential as antiadipogenic agents. Excessive adipose tissue accumulation is associated with multiple metabolic disorders, including insulin resistance, dyslipidemia, hepatic steatosis, coagulopathies, and hypertension [53].
Peng et al. (2022) evaluated the antiadipogenic effects of lanostane-type triterpenoids by testing forty compounds for their ability to inhibit the differentiation of 3T3-L1 preadipocytes into adipocytes. Several compounds significantly reduced adipocyte differentiation, particularly those containing an A-seco-23→26 lactone skeleton [54].
Mohsen et al. (2019) demonstrated that BA effectively reduced lipid accumulation in 3T3-L1 cells by downregulating the expression of peroxisome proliferator–activated receptor gamma (PPARγ) and liver X receptor alpha (LXRα). The study also showed enhanced interaction of steroid receptor coactivator-1 (SRC-1) with PPARγ and reduced SRC-3 levels [55]. These findings were supported by Melo et al. (2009), who investigated the effects of BA extracted from Clusia nemorosa L. (Clusiaceae) in a mouse model of obesity. The treatment resulted in significant reductions in body weight, abdominal fat accumulation, blood glucose levels, plasma triglycerides, and total cholesterol, primarily through inhibition of pancreatic lipase [56].
Betancourt et al. (2021) demonstrated that an extract composed of a mixture of UA, OA, and UA lactone reduced lipid accumulation in 3T3-L1 cells, accompanied by decreased leptin expression [57]. These findings were corroborated by Acín et al. (2021), who showed that C57BL/6J mice fed a high-fat diet and treated with this extract exhibited improved glucose tolerance, reduced body weight, decreased hepatic and adipose fat content, lower plasma lipid levels, and reduced hepatic mRNA expression of PPARα, CPT1A, and sterol regulatory element-binding protein (SREBP)-1 [58].
In in vitro studies using HepG2 cells treated with free fatty acids, UA reduced lipid accumulation and modulated the expression of genes involved in lipid synthesis and degradation, concomitant with increased AMPK phosphorylation. These effects were reversed by the addition of an AMPK inhibitor, confirming that AMPK activation is essential for UA activity. Molecular docking studies further demonstrated binding of UA to AMPK at the 4CFH site, similarly to the agonist Pound-Thirsk compound 1 (PT1) [59].
Bermúdez-Contreras et al. (2025) demonstrated that extracts of Agave durangensis, containing approximately 30% triterpenes, reduced triglyceride levels by 59.28% and very-low-density lipoprotein (VLDL) cholesterol by 60.27% in a mouse model of T2DM [60]. Liou et al. (2019) investigated maslinic acid extracted from olive peels and reported reductions in obesity in mice treated with doses between 10 and 20 mg/kg, along with improvements in hepatic steatosis. These effects were associated with suppression of TNF-α gene expression in the liver and epididymal adipose tissue, reduced serum TNF-α levels, downregulation of SREBP-1c, PPARγ, CCAAT/Enhancer-binding protein (C/EBP)β, and fatty acid synthase (FAS), and increased expression of adipose triglyceride lipase (ATGL), Sirtuin (Sirt)1, and AMPK phosphorylation [61].
Zaitone et al. (2015) demonstrated that oral administration of boswellic acids reduced food intake, body weight gain, and improved energy expenditure. These effects were associated with reduced pro-inflammatory mediators, decreased systemic insulin resistance, and attenuation of hepatic lipid accumulation, leading to improved liver function [62].
Han et al. (2025) treated human skin fibroblasts with β-boswellic acid, an ursane-type triterpene, and demonstrated safety at concentrations up to 40 μM. Treatment reduced high-glucose–induced ferroptosis and lipid peroxidation while improving cell migration. Western blot analysis revealed increased levels of phosphorylated signal transducer and activator of transcription (STAT)3. In vivo, β-boswellic acid (10 mg/kg, subcutaneous) accelerated wound closure, reduced inflammatory cell infiltration, improved granulation tissue organization, and increased collagen deposition in diabetic rat wounds [63].
Given the strong association between obesity and T2DM, these findings collectively highlight the promising potential of triterpenes for the treatment of metabolic disorders.
2.4. Antifibrotic Activity
Fibrosis is a pathological process resulting from dysregulated tissue repair following persistent or severe injury, most commonly associated with chronic inflammatory conditions. It is characterized by excessive accumulation of extracellular matrix (ECM) components, including collagen and fibronectin [64]. In cases of repetitive injury, continuous ECM deposition leads to progressive disruption of tissue architecture, organ dysfunction, and ultimately organ failure [65,66].
Among the most extensively studied antifibrotic triterpenes is AA, an ursane-type compound isolated from Centella asiatica, widely used in traditional African, Ayurvedic, and Chinese medicine. AA exhibits anti-inflammatory, antioxidant, antitumor, and wound-healing properties [67]. In vitro studies have shown that AA protects hepatocytes and Kupffer cells from injury by modulating mitochondrial stress and enhancing cellular antioxidant defenses [68].
Tang et al. (2012) demonstrated in vitro that AA effectively blocks TGF-β1-induced myofibroblast differentiation and collagen I expression in hepatic stellate cells (HSCs), suggesting direct antagonism of profibrotic signaling [69]. These findings align with earlier evidence indicating the involvement of TGF-β pathways in hepatic fibrosis [70]. The antifibrotic potential of AA was further confirmed in vivo in a rat model of carbon tetrachloride–induced hepatic fibrosis, where treatment significantly attenuated liver fibrosis and preserved hepatic function [71].
UA has also been extensively investigated for its antifibrotic properties. Wang et al. (2011) demonstrated in vitro that UA selectively induces apoptosis in activated HSCs, as evidenced by increased caspase-3 cleavage, while sparing hepatocytes [72]. Additionally, UA undergoes in vivo biotransformation into an epoxy derivative capable of covalently interacting with Cys-163 in caspase-3, leading to enzyme inactivation [73]. UA treatment was also associated with increased mitochondrial membrane permeability and inhibition of NF-κB activation, reinforcing its dual pro-apoptotic and anti-inflammatory mechanisms [72].
Synthetic triterpenoids have also demonstrated strong antifibrotic activity. Wei et al. (2013) evaluated the synthetic oleanane-type triterpenoid CDDO (2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid) using in vivo fibrosis models [74]. In a bleomycin-induced skin fibrosis model mimicking the inflammatory phase of systemic sclerosis, CDDO treatment significantly attenuated dermal fibrosis and inflammatory cell infiltration [75,76].
Given the anti-inflammatory properties of CDDO, these effects may result from immunosuppression, inhibition of TGF-β signaling, or both. To isolate its direct antifibrotic action, a complementary inflammation-independent, TGF-β–driven scleroderma model was employed. In this model, CDDO abolished dermal thickening and myofibroblast accumulation. Consistently, in vitro experiments using explanted skin fibroblasts showed that CDDO directly antagonized TGF-β–induced fibrotic responses [77].
Additional evidence for the antifibrotic activity of lupane-type triterpenes was provided by Chang et al. (2021), who evaluated an enriched extract from Momordica charantia leaves containing kuguacin R and 3β,7β,25-trihydroxycucurbita-5,23-dien-19-al. Oral administration of the extract to ICR mice, concomitantly treated with carbon tetrachloride, significantly suppressed hepatic necroinflammation and collagen deposition. Treatment also reduced α-smooth muscle actin (α-SMA) expression, TGF-β1 levels, pro-inflammatory cytokines (IL-6 and TNF-α), TLR4 expression, and intrahepatic infiltration of Ly6C⁺ monocytes [78].
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease of unknown etiology, influenced by genetic susceptibility, epigenetic modifications, aging, and environmental factors [79]. Multiple signaling pathways, including TGF-β, PPARγ, mTOR, and Wnt, are implicated in IPF progression.
BA, a lupane-type triterpene, has demonstrated antifibrotic activity in pulmonary fibrosis models. BA inhibits Wnt3a-induced fibroblast activation by suppressing Wnt/β-catenin signaling. In vivo, BA treatment was beneficial in a bleomycin-induced pulmonary fibrosis mouse model and exhibited superior pharmacodynamic effects compared with pirfenidone. These findings suggest that BA and its derivatives represent promising candidates for antifibrotic drug development targeting pulmonary fibrosis [80].
2.5. Antitumor Activity
Pentacyclic triterpenes have been extensively investigated for their antitumor properties, largely due to their ability to modulate key signaling pathways involved in cell proliferation, apoptosis, angiogenesis, and metastasis. In vitro studies have consistently demonstrated that triterpenes such as OA, UA, and BA exert cytotoxic effects against a broad spectrum of cancer cell lines, including hepatocellular carcinoma, breast, prostate, colon, and lung cancer cells [81,82,83]. These effects are frequently associated with cell cycle arrest and induction of apoptosis through both intrinsic and extrinsic pathways.
Mechanistically, in vitro evidence indicates that UA and BA induce mitochondrial dysfunction, characterized by loss of mitochondrial membrane potential, cytochrome c release, and activation of caspase-9 and caspase-3 [84,85]. These compounds also downregulate anti-apoptotic proteins such as Bcl-2 and Bcl-xL while upregulating pro-apoptotic mediators including Bax and Bad. Additionally, triterpenes have been shown to suppress pro-survival signaling cascades, notably NF-κB, PI3K/Akt, and STAT3 pathways, thereby sensitizing tumor cells to apoptotic stimuli [86].
Beyond apoptosis, triterpenes interfere with tumor cell proliferation by modulating cell cycle regulators. In vitro studies have demonstrated that BA induces G₀/G₁ or G₂/M cell cycle arrest, depending on the tumor cell type, through downregulation of cyclins (cyclin D1, cyclin B1) and cyclin-dependent kinases, accompanied by increased expression of cyclin dependent kinase (CDK) inhibitors such as p21 and p27 [87]. Moreover, triterpenes inhibit angiogenesis-related processes by suppressing VEGF expression and endothelial cell migration, further limiting tumor growth and metastatic potential [88].
The antitumor efficacy of triterpenes has been corroborated in multiple in vivo animal models. BA treatment significantly reduced tumor growth in xenograft mouse models of melanoma, breast cancer, and hepatocellular carcinoma, with concomitant increases in tumor cell apoptosis and reductions in microvessel density [89,90,91]. Similarly, UA administration inhibited tumor progression in chemically induced and xenograft cancer models, effects that were associated with decreased inflammatory signaling and oxidative stress within the tumor microenvironment [92].
Synthetic triterpenoids have also shown enhanced antitumor potency in vivo. Compounds such as CDDO and its derivatives (CDDO-Me and CDDO-Im) markedly suppressed tumor growth and metastasis in preclinical models of breast, lung, and pancreatic cancer [93]. These effects were attributed to simultaneous inhibition of NF-κB signaling, activation of Nrf2-mediated cytoprotective responses, and induction of apoptosis in malignant cells, highlighting the therapeutic potential of structural modification of naturally occurring triterpenes.
Although robust in vitro and in vivo data support the antitumor activity of pentacyclic triterpenes, clinical evidence remains limited. Early-phase clinical studies evaluating synthetic triterpenoids, particularly CDDO derivatives, have demonstrated acceptable safety profiles but modest efficacy, underscoring the need for improved bioavailability, optimized dosing strategies, and combination approaches to enhance translational success [94].
Collectively, the bioactivity of triterpenes highlights the substantial therapeutic potential of this class of compounds for the treatment of a wide range of diseases. Among these, metabolic dysfunction-associated fatty liver disease (MASLD) and metabolic dysfunction–associated steatohepatitis (MASH) are particularly relevant, as their pathogenesis encompasses several of the biological processes modulated by triterpenes. These processes operate at different stages of disease development, acting as predisposing factors - such as obesity and T2DM - as integral components of disease progression, including hepatic steatosis, chronic inflammation, oxidative stress, and fibrogenesis, or as long-term complications, such as hepatocellular carcinoma. In the following section, the pathophysiology of MASH is discussed, with emphasis on the key cellular events and signaling pathways that drive disease initiation and progression.
3. Mechanisms of Metabolic Dysfunction–Associated Steatotic Liver Disease (MASLD) and Metabolic Dysfunction–Associated Steatohepatitis (MASH)
Chronic liver diseases (CLDs) and cirrhosis represent major global health challenges, accounting for substantial morbidity and mortality worldwide [93]. According to the Global Burden of Disease Study 2017, viral hepatitis B and C remain strongly associated with the rising prevalence of cirrhosis and other CLDs. In parallel, metabolic dysfunction–associated steatotic liver disease (MASLD) and its progressive form, metabolic dysfunction–associated steatohepatitis (MASH), have emerged as leading contributors to the global burden of liver disease, driving an increasing incidence of cirrhosis and hepatocellular carcinoma (HCC). Autoimmune and genetic disorders further contribute to the complex etiology of CLDs [94].
MASLD is a multisystemic disorder in which insulin resistance and metabolic dysfunction play central roles in disease initiation and progression [95]. The diagnosis is established when more than 5% of hepatocytes exhibit steatosis in the presence of defined metabolic abnormalities, including obesity, T2DM , or dyslipidemia [96]. Clinically, MASLD encompasses a broad spectrum ranging from isolated triglyceride accumulation in hepatocytes — termed metabolic dysfunction–associated steatotic liver (MASL) — to MASH, which is characterized by lobular or portal inflammation and hepatocellular ballooning [97,98]. In advanced stages, persistent injury may culminate in fibrosis, cirrhosis, and ultimately HCC [97].
The early pathophysiology of MASLD is complex and multifactorial [95]. To account for this complexity, the “multiple-hit” hypothesis was proposed (Figure 5), postulating that disease progression results from the simultaneous action of several pathogenic stimuli. These include genetic susceptibility, dietary factors, obesity, insulin resistance, altered lipid metabolism, lipotoxicity, oxidative stress, mitochondrial and endoplasmic reticulum dysfunction, intestinal dysbiosis, and dysregulated cytokine and adipokine production [99]. Collectively, these factors promote activation of pro-inflammatory cascades and inflammasomes, particularly NLRP3, leading to hepatocellular injury, cell death, and fibrogenesis — hallmarks of MASH. Concomitantly, activation of hepatic stellate cells (HSCs) and immune cells enhances extracellular matrix (ECM) deposition, accelerating fibrosis progression [94,95,100].
Within this context, dysregulated fatty acid (FA) metabolism has been closely linked to cellular stress and lipotoxicity, triggering inflammation, immune cell recruitment, and hepatocyte death — key drivers of the transition from MASLD to MASH and more advanced liver disease stages [101]. Elevated levels of free FA and cholesterol exacerbate insulin resistance, promote adipocyte dysfunction, and alter gut microbiota composition, leading to increased intestinal permeability and enhanced translocation of lipopolysaccharides (LPS). These events further amplify hepatic inflammation via the gut–liver axis [102].
In the liver, excessive lipid influx and de novo lipogenesis induce oxidative stress, mitochondrial dysfunction, and inflammasome activation, culminating in hepatocyte injury and apoptosis [103]. Lipotoxicity activates pro-inflammatory signaling through Toll-like receptors (TLRs), resulting in increased production of cytokines such as TNF-α, IL-6, and IL-1β. These mediators amplify inflammatory responses and drive the transdifferentiation of HSCs into myofibroblasts, thereby promoting fibrosis [101]. TLR4, abundantly expressed in Kupffer cells (KCs) and HSCs, is activated by bacterial LPS and FA, triggering the MyD88/NF-κB pathway and sustaining chronic inflammation.
In parallel, oxidative stress arising from mitochondrial and endoplasmic reticulum dysfunction activates the NLRP3 inflammasome, further intensifying inflammatory and profibrotic signaling [94]. Persistent activation of the IKK/NF-κB and JNK pathways in HSCs, along with TLR5-mediated NF-κB and MAPK signaling, contributes to chronic inflammation, tissue remodeling, and fibrosis progression [101].
More recently, pyroptosis has been recognized as an additional driver of hepatic inflammation and fibrogenesis. Pyroptosis is a pro-inflammatory form of programmed cell death mediated by caspase activation and gasdermin D (GSDMD)–dependent pore formation in the plasma membrane, resulting in the release of inflammatory cytokines, lactate dehydrogenase (LDH), osmotic swelling, and cell lysis. This process occurs through canonical (caspase-1–dependent) and non-canonical (caspase-4/5/11–dependent) pathways and has been shown to promote macrophage activation and fibrosis progression [104,105,106].
Activation of these interconnected pathways compromises hepatic regenerative capacity and promotes excessive ECM deposition, ultimately leading to progressive fibrosis [98]. Regardless of etiology, liver fibrosis shares common molecular mechanisms, including hepatocyte death, chronic inflammation, HSC activation, and disruption of epithelial and endothelial barriers, all of which are closely associated with liver-related mortality [107].
Under physiological conditions, HSCs remain quiescent, storing vitamin A and producing type IV collagen. However, chronic liver injury induces a phenotypic switch characterized by increased proliferation, contractility, and synthesis of type I and III collagen, typical of cirrhotic tissue [103]. This transition is driven by sustained inflammation, whereby hepatocyte death releases damage-associated molecular patterns (DAMPs) and apoptotic bodies that further activate HSCs [94].
TGF-β signaling plays a pivotal role in hepatic fibrogenesis by activating the SMAD pathway and inducing the expression of genes encoding type I collagen, fibronectin, and proteoglycans — key components of the ECM [96]. Additional pathways, including platelet-derived growth factor (PDGF) and VEGF, contribute to HSC proliferation, migration, survival, and ECM production, while also enhancing TGF-β signaling [108]. NF-κB activation by inflammatory stimuli and inflammasome signaling further reinforces the profibrotic response, perpetuating excessive ECM accumulation [94,95].
The hepatic ECM undergoes dynamic remodeling during fibrogenesis and plays a critical role in CLD progression [109]. Its proteome, comprising structural proteins, proteases, and protease inhibitors, evolves with disease severity, influencing cell–cell communication and inflammatory signaling [108]. Maintenance of ECM homeostasis depends on the balance between MMPs and their tissue inhibitors (TIMPs). Disruption of this balance - through increased TIMP activity or reduced MMP function - results in excessive collagen deposition, increased liver stiffness, and fibrosis progression [110].
As fibrosis advances, ECM deposition predominates due to elevated TIMP-1 expression, which also protects activated HSCs from apoptosis, thereby sustaining fibrogenesis. Activated macrophages and persistent MAPK and NF-κB signaling further exacerbate ECM dysregulation, aggravating liver injury [108,110]. Ultimately, prolonged fibrotic remodeling leads to cirrhosis, an advanced and often irreversible stage of CLDs characterized by extensive ECM accumulation and regenerative nodule formation, resulting in architectural distortion and functional impairment of the liver [111,112].
Cirrhosis arises from continuous cycles of hepatocyte necrosis and regeneration, leading to sinusoidal capillarization, reduced functional parenchyma, altered hepatic blood flow, and portosystemic shunting. These changes underpin the development of portal hypertension and its complications, including ascites, hepatic encephalopathy, and pulmonary, renal, and cardiac dysfunction, as well as hyponatremia [113].
MASLD is also closely linked to alterations in gut microbiota composition and intestinal barrier integrity, positioning the gut–liver axis as a central component of disease pathophysiology. Metabolic and dietary factors promote intestinal dysbiosis, characterized by depletion of commensal bacteria and expansion of pro-inflammatory Gram-negative species. Inflammatory and hypoxic conditions disrupt tight junction integrity, increasing intestinal permeability and facilitating bacterial translocation and passage of microbial products such as LPS into the portal circulation. These molecules activate KCs via pattern recognition receptors, particularly TLRs, triggering inflammatory cascades mediated by cytokines and ROS [114].
NADPH oxidase 2 (NOX2), a major source of ROS in phagocytes, and the NLRP3 inflammasome are activated by signals derived from the dysbiotic microbiota and impaired epithelial barrier, modulating hepatic inflammation and fibrosis progression [115]. Clinically, cirrhosis presents as a heterogeneous condition with two major prognostic stages: a compensated phase, characterized by preserved liver function, and a decompensated phase marked by complications such as ascites, variceal bleeding, infections, and hepatic encephalopathy [116]. Disease progression may culminate in severe outcomes, including hepatorenal syndrome, acute-on-chronic liver failure (ACLF), and multiple organ failure [117].
4. Current Applications of Triterpenes for MASLD and MASH
Accumulating preclinical evidence supports pentacyclic triterpenes as promising multitarget modulators of metabolic dysfunction–associated steatotic liver disease (MASLD) and metabolic dysfunction–associated steatohepatitis (MASH). Although these compounds differ in chemical structure and primary molecular targets, they consistently converge on key pathogenic processes, including dysregulated lipid metabolism, oxidative and endoplasmic reticulum stress, chronic inflammation, inflammasome activation, and hepatic fibrogenesis. The multitarget mechanisms by which pentacyclic triterpenes modulate MASLD/MASH pathophysiology are summarized in Figure 6.
Pentacyclic triterpenes, including UA, BA, AA, OA, celastrol, and glycyrrhizin/glycyrrhetinic acid, modulate key pathogenic pathways involved in MASLD/MASH progression. These compounds converge on metabolic regulation (AMPK, FXR, lipogenesis), inflammatory signaling (TLR–NF-κB, NLRP3 inflammasome), oxidative and endoplasmic reticulum stress, and hepatic fibrogenesis (TGF-β/SMAD, HSC activation), resulting in reduced steatosis, inflammation, and fibrosis and improved hepatic function.
A comparative overview of the main triterpenes investigated to date, including their molecular mechanisms, experimental models, therapeutic outcomes, and key supporting references, is provided in Table 1. The following subsections focus on compound-specific mechanistic features and translational insights that extend beyond the comparative framework summarized in the table.
4.1. Ursolic Acid (UA)
Beyond its well-established hepatoprotective effects summarized in Table 1, UA exhibits a distinctive capacity to simultaneously modulate metabolic reprogramming and fibrogenic signaling.
UA has long been recognized for its hepatoprotective and antifibrotic properties, with early evidence of its therapeutic potential in acute liver injury models dating back to the 1980s [118]. In a seminal study, Martín-Aragón et al. (2001) demonstrated that pretreatment with UA in a rat model of CCl₄-induced acute liver injury significantly reduced lipid peroxidation and serum transaminase levels, while enhancing hepatic antioxidant defenses, including superoxide dismutase, catalase, GSH reductase, GSH peroxidase, and total GSH content [119].
Subsequent studies extended these findings to chronic injury and metabolic contexts. Saravanan et al. (2006) reported that UA administration attenuated ethanol-induced liver injury in rats, reducing serum AST, ALT, and bilirubin levels, lowering lipid peroxidation markers, and restoring circulating antioxidants, with efficacy comparable to silymarin [120]. In diet-induced steatosis models, oral UA administration mitigated hepatic inflammation and fibrosis, in part by increasing decorin expression, a key regulator of extracellular matrix homeostasis, thereby promoting matrix repair and remodeling [121]. Consistently, UA was shown to reduce hepatic lipid accumulation, adipocyte hypertrophy, and circulating lipid levels through activation of AMPK signaling, highlighting its metabolic regulatory capacity [59].
The prophylactic potential of UA against MASLD has also been explored in the context of excessive fructose intake. Using a developmental “multiple-hit” model, Mukonowenzou et al. (2021) demonstrated that early-life UA supplementation significantly attenuated fructose-induced hepatic lipid accumulation and long-term metabolic dysfunction, suggesting a role for UA in metabolic programming and disease prevention [122].
Strong antifibrotic effects of UA have been consistently demonstrated in chemically induced fibrosis models. In thioacetamide (TAA)-induced liver fibrosis, UA treatment reduced fibrotic area, hepatic hydroxyproline content, and expression of α-SMA, Col1a2, and TIMP1, while inducing apoptosis of activated hepatic stellate cells, as evidenced by reduced TUNEL and α-SMA staining [123]. These findings were corroborated by Li et al. (2023), who showed that UA attenuated TAA-induced fibrosis by inhibiting TGF-β₁ signaling, resulting in reduced HSC activation and collagen deposition [20].
Similarly, in CCl₄-induced fibrosis models, UA administration consistently reduced fibrosis scores, collagen accumulation, and serum transaminases, while modulating gut microbiota composition toward a profile resembling that of healthy controls, particularly increasing Firmicutes and Bacteroidetes populations [124]. Comparable antifibrotic effects were observed in a methionine- and choline-deficient (MCD) diet model [115]. Additional studies demonstrated that UA suppresses fibrogenesis by restoring the balance between MMP and their inhibitors, reducing pyroptosis, inhibiting caspase-1 and gasdermin D activation, and attenuating inflammatory cytokine release [104].
In experimental MASH models, UA exhibited both preventive and therapeutic efficacy, reducing oxidative stress, inflammation, fibrosis, and hepatic dysfunction. These effects were accompanied by downregulation of NADPH oxidase (NOX) isoforms and suppression of NLRP3 inflammasome activation, reinforcing the role of UA in targeting key oxidative and inflammatory pathways involved in disease progression [125]. Collectively, these findings support UA as a potent modulator of oxidative stress, inflammation, and fibrogenesis, with significant therapeutic potential for MASLD and MASH.
4.2. Betulinic Acid (BA)
While the comparative metabolic and antifibrotic outcomes of betulinic acid (BA) are outlined in Table 1, mechanistic studies highlight BA as a potent regulator of lipid synthesis and bile acid signaling.
The first evidence supporting the therapeutic potential of BA in hepatic steatosis was provided in the beginning of the 2010s. Quan et al. (2013) demonstrated that BA reduced lipid accumulation in HepG2 cells by inhibiting SREBP1 maturation and nuclear translocation, leading to downregulation of lipogenic genes and upregulation of lipid oxidation–related pathways. These effects were mediated by activation of the CAMKK–AMPK axis and subsequent suppression of the mTOR/S6K pathway and were both dose- and time-dependent [126]. These molecular mechanisms were later confirmed in vivo in high-fat diet (HFD)-fed mice, validating the relevance of AMPK signaling in BA-mediated metabolic regulation.
In another study, Zhao et al. (2021) demonstrated that BA treatment of TGF-β1–induced HSCs reduced cell activation, as evidenced by decreased Col1a1 and α-SMA expression, and impaired cell proliferation and migration, as shown by reduced wound closure in a scratch assay. These effects were associated with suppression of Lck expression and activity, a Src-family tyrosine kinase that promotes hepatic stellate cell activation and profibrotic JAK/STAT signaling, along with increased expression of SOCS1, a key negative regulator of cytokine signaling that limits JAK/STAT-driven inflammation and fibrogenesis [127].
Kim et al. (2019) showed that BA administration attenuated HFD-induced obesity, improved insulin sensitivity, and enhanced energy expenditure in mice. These systemic metabolic benefits were associated with AMPK activation across multiple metabolic tissues, including liver, adipose tissue, and skeletal muscle, resulting in suppression of lipogenic pathways and improved energy balance [128].
The relevance of BA in MASLD has been further substantiated by Gu et al. (2019), who demonstrated that BA significantly attenuated hepatic steatosis in murine models induced by either a HFD or an MCD diet. These effects were mechanistically linked to activation of the farnesoid X receptor (FXR) and subsequent suppression of endoplasmic reticulum stress pathways, including protein kinase RNA-like ER kinase (PERK)/eukaryotic initiation factor (eIF)2α/activating transcription factor (ATF)4/ C/EBP homologous Protein (CHOP) signaling. Importantly, the protective effects of BA were largely abolished in FXR-deficient mice, underscoring the central role of FXR in mediating BA activity [129].
BA has also shown robust antifibrotic activity in experimental models of liver fibrosis. In TAA-induced fibrosis models, BA treatment markedly improved liver histology and function, reducing collagen deposition and suppressing profibrotic mediators such as TGF-β1, Col1α1, and α-SMA, while restoring antioxidant enzyme activity, thereby highlighting its hepatoprotective and antifibrotic profile [130]. These results were corroborated in an experimental model of CCl4-induced fibrosis in mice, in which BA treatment reduced ALT and AST levels and fibrotic index, associated with decrease in α-SMA and Col1α1 levels [127]. Mu et al. (2020) also demonstrated that BA markedly protected against hepatic steatosis, inflammation, and fibrosis in both HFD- and MCD-induced MASLD/MASH models. The hepatoprotective effects were attributed to inhibition of de novo lipogenesis via suppression of the YY1/FAS signaling axis, thereby limiting triglyceride synthesis and lipid accumulation in hepatocytes [131].
More recently, Zhang et al. (2022) performed the rational design of BA derivatives as intestine-selective antagonists of the FXR for the treatment of MASH. Through structure–activity relationship optimization, a new compound was identified as a potent FXR antagonist with preferential intestinal accumulation and minimal systemic exposure. This compound selectively inhibits intestinal FXR signaling, leading to compensatory activation of hepatic FXR, increased bile acid synthesis, reduced hepatic cholesterol levels, and suppression of NLRP3 inflammasome activation. In two mice models of diet-induced MASH (Gubra Amylin NASH - GAN diet, or high fat methionine choline deficient - HFMCD diet), this compound reduced hepatic steatosis, inflammation, fibrosis, and improved glucose homeostasis, and insulin sensitivity, outperforming the FXR antagonist glycoursodeoxycholic acid (GUDCA). Collectively, these findings establish intestinal FXR antagonism as a viable therapeutic strategy for MASH and highlight BA-derivatives as promising gut-restricted FXR modulators [132].
Taken together, these studies consistently demonstrate that BA exerts hepatoprotective, antifibrotic, and metabolic regulatory effects through modulation of AMPK, FXR, lipid metabolism, oxidative stress, and inflammatory signaling pathways, supporting its potential as a therapeutic candidate for MASLD and MASH.
4.3. Asiatic Acid (AA)
In contrast to other pentacyclic triterpenes, AA displays moderate but consistent efficacy in experimental MASH models, with mechanistic actions primarily linked to oxidative stress and ER stress modulation.
Similarly to BA, the potential use of AA to attenuate MASH development has been reported since the early 2010s. Yan et al. demonstrated that AA treatment (10 or 20 mg/kg/day, oral gavage, for 10 weeks) in HFD-fed mice reduced body weight, water and feed intake, epididymal fat, and plasma and hepatic triglyceride levels, which was associated with downregulation of key lipogenic enzymes (coenzyme A carboxylase - ACC1, FAS, and stearoyl CoA desaturase - SCD-1) and the transcription factor SREBP-1c. In parallel, AA attenuated hepatic steatosis, inflammation, and oxidative stress by suppressing pro-inflammatory cytokine production (IL-1β, IL-6, TNF-α) and NF-κB signaling while increasing catalase and GSH-peroxidase activities and GSH level, and improving insulin sensitivity [133]. Concordant findings were reported by Wang et al., in which HFD-fed mice were treated with AA (4 or 8 mg/Kg/day, oral gavage for 10 weeks). Besides attenuation of hepatic steatosis and liver injury, the authors demonstrated reduction on myeloperoxidase (MPO) and MDA levels, while increasing SOD, GSH-peroxidase and GSH-reductase activities. It was also demonstrated that the treatment with AA lead to the reduction of glucose-regulated protein 78 (GRP78), PERK, eIF2α and CHOP expression in liver tissue, all of these proteins are correlated to the endoplasmic reticulum stress [134].
In a rat model of CCl₄-induced MASH, Wei et al. (2018) demonstrated that AA treatment (5 or 15 mg/kg/day, oral gavage, for six weeks) resulted in modest but consistent hepatoprotective effects, including reductions in serum AST, ALT, and total bilirubin levels, as well as decreases in fibrosis index and circulating fibrosis markers - hyaluronic acid (HA), laminin (LN), collagen type IV (IV-C), and procollagen III N-terminal peptide (PIIINP) - and downregulation of Col1a1 and Col1a3 mRNA expression. In line with findings from HFD-fed mouse models, AA treatment dose-dependently reduced hepatic malondialdehyde (MDA) levels and pro-inflammatory mediators, including TGF-β, COX-2, TNF-α, IL-6, and IL-1β, while enhancing antioxidant defenses through increased SOD and GSH-peroxidase activities. AA also attenuated CCl₄-induced hepatocyte apoptosis, as evidenced by reduced caspase-3 levels, and inhibited hepatic stellate cell activation, reflected by decreased α-SMA expression and lower levels of TIMP-1 and TIMP-2. Mechanistically, these protective effects were associated with inhibition of the PI3K/AKT/mTOR signaling pathway [135].
Although studies with AA consistently demonstrate beneficial effects in experimental MASH, the magnitude of improvement does not appear to have been sufficient to justify more in-depth investigations of this compound for the treatment of MASLD and MASH. One plausible explanation is the poor aqueous solubility of AA, reported to be approximately 0.1 mg/mL [136], which may limit its oral bioavailability and, consequently, its therapeutic efficacy. While several studies have explored structural modifications of asiatic acid to enhance its water solubility [137,138,139,140], to the best of our knowledge, no investigations have yet evaluated these derivatives in the context of MASLD or MASH, but these are potential molecules to this use.
4.4. Oleanolic Acid (OA)
OA is an oleanane-type pentacyclic triterpene widely investigated for its effects on lipid metabolism, insulin sensitivity, oxidative stress, and inflammatory responses, particularly in experimental models of metabolic-associated steatotic liver disease, obesity, and liver injury.
In vitro evidence demonstrates that OA directly modulates hepatocellular lipid accumulation under metabolic stress conditions. OA significantly reduced intracellular triglyceride and total cholesterol levels and attenuated lipid droplet formation in hepatocytes exposed to free fatty acids, indicating a cell-autonomous anti-steatotic effect [141]. Lin et al. (2018) further showed that, in hepatic and intestinal cell models treated with OA for 24 h or up to 14 days, OA suppressed ligand-induced lipogenesis by inhibiting LXRα- and PXR-mediated transcriptional activity. Specifically, OA reduced the expression of lipogenic markers including SREBP-1c, FAS, SCD-1, ATP-citrate lyase (ACLY), ACC1, and Fatty acid elongase (FAE) in hepatic cells, while preserving or enhancing ATP-binding cassette transporter (ABC)A1 and ABCG1 expression in intestinal cells. These effects were accompanied by increased AMPK phosphorylation in hepatic cells and differential recruitment of nuclear receptor coregulators, characterized by decreased SRC-1 and increased small heterodimer partner–interacting leucine zipper protein (SMILE) binding to the SREBP-1c promoter, as assessed by reporter assays, qRT-PCR, Western blotting, and DNA affinity precipitation analyses [142].
Consistent with these cellular findings, Liu et al. (2013) reported that oral administration of OA (25 mg/kg/day, oral gavage, once daily for 10 weeks) markedly attenuated liquid fructose–induced hepatic triglyceride accumulation in rats. This was accompanied by reductions in hepatocyte ballooning, hepatic collagen deposition, and serum insulin levels, and was mechanistically associated with downregulation of hepatic SREBP-1/1c mRNA and nuclear protein levels, as well as decreased expression of FAS, SCD1, and ACC1. In contrast, Carbohydrate response element–binding protein (ChREBP) expression and PPARα and PPARγ signaling were not altered, indicating selective suppression of de novo lipogenesis [143].
Djeziri et al. (2018) further demonstrated that OA administration (15–60 mg/kg/day, oral gavage, once daily for four weeks) significantly attenuated hepatic steatosis in HFD–fed rodents, reducing liver weight and hepatic lipid content and improving histopathological features. These effects were associated with improvements in serum lipid parameters, including reductions in total cholesterol, triglycerides, and LDL, and activation of hepatic AMPK signaling [144].
Nyakudya et al. (2018) provided evidence that early-life exposure to OA exerts long-term metabolic effects. In this developmental programming model, neonatal oral administration of OA (60 mg/kg/day, once daily for 7 days) significantly attenuated the later development of high-fructose diet–induced MASLD in adult rats. Early OA treatment prevented hepatic lipid accumulation, steatosis, low-grade inflammation, fibrosis, increased MASLD activity scores, visceral and epididymal adiposity, impaired glucose metabolism, and excessive body weight gain, demonstrating sustained metabolic modulation originating from neonatal exposure [145].
Wang et al. (2013) reported that OA improves hepatic insulin resistance in leptin receptor–deficient Lep^db/db mice. In this model, OA administration (20 mg/kg/day, intraperitoneal injection, once daily for two weeks) restored body, liver, and fat weights and normalized liver morphology and function. OA lowered fasting blood glucose, improved glucose and insulin tolerance, enhanced hepatic insulin signaling, and suppressed gluconeogenesis. These effects were accompanied by reductions in serum triglycerides, total cholesterol, LDL, free fatty acids, and pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), increased HDL levels, attenuation of hepatic lipid accumulation, and decreased mitochondrial ROS production in the liver [146].
Gamede et al. (2019) similarly demonstrated that OA administration in a diet-induced pre-diabetes rat model (80 mg/kg, oral administration, every third day for 12 weeks following 20 weeks of high-fat high-carbohydrate feeding) significantly reduced the liver-to-body weight ratio, plasma triglycerides, and VLDL cholesterol. OA treatment lowered plasma SREBP levels, intrahepatic triglyceride content, ALT and AST activities, and malondialdehyde levels, while increasing superoxide dismutase and glutathione peroxidase activities. Histological analyses confirmed attenuation of hepatic lipid accumulation and structural abnormalities [147].
Yao et al. (2023) investigated OA in fructose-induced models of hepatic steatosis. OA administration in rats (25 mg/kg/day, oral gavage, once daily for 5 weeks) and in mice (75 mg/kg/day, once daily for 7–11 days) significantly reduced hepatic triglyceride accumulation and lipid droplet deposition across multiple genotypes, including SREBP1c⁺/⁺, SREBP1c⁻/⁻, SCD1⁺/⁺, and SCD1⁻/⁻ mice. OA consistently suppressed SCD1 mRNA and protein expression, while SREBP1c and ChREBP expression remained unchanged under fasting conditions. In fructose plus oleic acid–supplemented models, OA additionally reduced SREBP1c, FAS, and ACC1 expression and altered hepatic fatty acid composition, decreasing oleic acid levels and the C18:1/C18:0 ratio. OA also increased AMPK phosphorylation and CPT1α expression in SCD1⁻/⁻ mice, indicating modulation of fatty acid oxidation pathways under specific metabolic contexts [148].
Xue et al. (2021) demonstrated that OA targets the gut–liver axis in high-fat diet–induced obesity and NAFLD. OA administration (25–100 mg/kg/day, oral gavage, once daily for 8 weeks following 12 weeks of HFD feeding) reduced hepatic lipid accumulation, improved serum lipid profiles, and decreased ALT and AST levels. OA suppressed hepatic expression of LXRα, SREBP-1c, FAS, and ACCα, while increasing PPARα, CPT1, and AMPK phosphorylation. Additionally, OA reduced hepatic and systemic levels of TNF-α, IL-1β, and IL-6 and inhibited TLR4/NF-κB signaling. In the intestine, OA enhanced barrier integrity by increasing occludin, claudin-1, and ZO-1 expression, reduced circulating LPS levels, and modulated gut microbiota composition, including reductions in the Firmicutes/Bacteroidetes ratio [149].
Pi et al. (2024) reported that OA alleviates ischemia–reperfusion injury in a rat model of severe steatotic liver IRI. OA was administered orally by gavage at 21 mg/kg/day for 7 consecutive days prior to surgery, followed by 80 min of ischemia and 24 h of reperfusion. OA reduced Suzuki histological scores, serum ALT and AST levels, and circulating IL-6, TNF-α, and IL-1β. OA also decreased hepatic Fe²⁺, MDA, 4-hydroxynonenal, and TLR4 levels, while increasing glutathione content and GPX4 expression. These effects were associated with activation of the KEAP1/NRF2/ARE signaling pathway, including increased nuclear NRF2 and elevated NQO1 and GCLC expression [150].
In a very recent study published in 2026, Pi et al. demonstrated that OA attenuates intestinal injury secondary to hepatic ischemia–reperfusion under steatotic conditions. Male Sprague–Dawley rats fed a high-fat diet for 20 weeks received OA (21 mg/kg/day, oral gavage, once daily for 7 days) prior to ischemia–reperfusion. OA reduced liver and intestinal injury scores, serum ALT and AST levels, and circulating IL-6, TNF-α, and IL-1β. OA preserved intestinal barrier integrity by increasing ZO-1 and occludin expression and reducing D-lactate, diamine oxidase, and LPS levels. Mechanistically, OA activated PPARG signaling and promoted macrophage polarization toward an M2 phenotype, evidenced by increased CD206 and ARG1 and reduced iNOS and CD86 expression. Pharmacological inhibition of PPARG with GW9662 reversed these effects, confirming PPARG-dependent signaling in this recently described model [151].
Collectively, these findings demonstrate the substantial potential of oleanolic acid for the prevention, control, and attenuation of disease progression in MASLD and MASH.
4.5. Celastrol
Early in vitro studies demonstrate that celastrol directly modulates hepatocellular pathways involved in lipid accumulation, bile acid metabolism, inflammation, and insulin resistance, corresponding to initial stages of MASLD development.
Hu et al. (2025) employed HepG2 hepatocytes to validate targets identified by multiomic and network pharmacology analyses, showing that celastrol-induced regulation of bile acid metabolism was dependent on CYP7B1, as siRNA-mediated knockdown of CYP7B1 abolished these effects. Celastrol increased CYP7B1 expression and modulated FXR and LXR signaling, indicating activation of the alternative bile acid synthetic pathway; markers analyzed included CYP7B1, CYP27A1, FXR, LXR, and bile acid–related metabolic signatures assessed by RT-qPCR and Western blotting [152]. Complementarily, Li et al. (2025) demonstrated that celastrol reduced lipid accumulation in palmitic-acid–treated AML-12 cells and primary murine hepatocytes while downregulating Notch2 and osteopontin (OPN) expression, thereby suppressing hepatocyte-driven activation of hepatic stellate cells in co-culture systems [153]. In oleic-acid–induced steatosis models, celastrol reduced lipid droplet accumulation and triglyceride content in HepG2 cells , with effects reversed by the PPARγ agonist rosiglitazone, implicating PPARγ-dependent signaling [154]. Additional HepG2 experiments showed that celastrol (0.1–0.4 µg/mL, 24 h) decreased intracellular triglycerides and cholesterol while increasing FGF21, phosphorylated AMPK, PGC-1α, PPARγ, and SIRT3, linking celastrol to mitochondrial and energy-sensing pathways relevant to steatosis control [155].
Inflammatory signaling and insulin resistance - key drivers of MASLD progression - were also addressed in vitro. Han et al. (2018) established a steatotic HepG2 model using free fatty acids (palmitate:oleate) and demonstrated that celastrol reduced triglyceride accumulation and suppressed TLR4/MyD88/NF-κB signaling, with decreased IL-1β and TNF-α protein levels; TLR4 siRNA further potentiated these effects [156]. Zhang et al. (2018) modeled lipid-induced insulin resistance by treating HepG2 cells with palmitic acid and adding celastrol, restoring insulin-stimulated glucose uptake and implicating the TLR4/MD2 axis in the regulation of insulin signaling [157]. Although performed in renal cells, Kim et al. (2013) further demonstrated that celastrol suppressed fatty acid– and high-glucose–induced inflammatory responses via inhibition of TLR4/MyD88/NF-κB, pathways that are central to inflammation-driven insulin resistance in MASLD [158]. Additionally, Ma et al. (2015) showed that celastrol activated HSF1, leading to increased PGC-1α transcription and enhanced mitochondrial and oxidative metabolism gene expression, establishing a mechanistic link to lipid oxidation and systemic insulin sensitivity [159].
Consistent with these cellular findings, extensive in vivo evidence demonstrates that celastrol attenuates disease progression from hepatic steatosis to steatohepatitis and fibrosis. Hu et al. (2025) induced MASH in mice using Western diet or HFD feeding and administered celastrol (100–300 µg/kg/day, i.p. for 4 weeks or 0.75–1.5 mg/kg/day, oral gavage for 28 days), resulting in reduced hepatic steatosis, liver fibrosis index, serum ALT, AST, triglycerides, cholesterol, and total bile acids, alongside correction of the 12α-hydroxylated/non-12-hydroxylated bile acid ratio; these effects were associated with restoration of CYP7B1 expression and modulation of FXR/LXR signaling [152]. Canová et al. (2025) further showed that celastrol (200 µg/kg i.p., every second day for 4 weeks) administered during either early or late stages of WD/fructose-induced disease attenuated MASLD-to-MASH progression, reducing hepatic triglycerides, lipid peroxidation, liver weight, and improving glucose tolerance, with regulation of SREBP-dependent lipogenic markers and hepatic GalR2 expression [160]. Benmouna et al. (2025) reported that celastrol (100 µg/kg/day i.p. for 4 weeks) reduced hepatic SREBP-1c, FAS, and ACC1 expression and lowered liver triglyceride and cholesterol content, alongside decreased IL-6 and TNF-α, supporting its role in early MASLD [161].
Progression to steatohepatitis and fibrosis was addressed in more severe models. Li et al. (2025) demonstrated that in a Western diet plus low-dose CCl₄–induced NASH model, celastrol (100 or 250 µg/kg/day i.p. for 2 weeks) reduced hepatic triglycerides, collagen deposition, α-SMA expression, and Sirius Red–positive fibrosis through suppression of Notch2/OPN signaling, with hepatocyte-specific Notch2 overexpression abolishing these effects [153]. Xue et al. (2024) showed that celastrol (1–5 mg/kg/day by gavage for 12 weeks) attenuated HFD-induced MASLD, reducing hepatic steatosis, ALT, AST, triglycerides, and inflammatory cytokines while improving mitochondrial ultrastructure, ATP content, and antioxidant status via activation of the FGF21/AMPK/PGC-1α pathway [155]. In a combined T2DM–MASLD model, Sun et al. (2020) demonstrated that celastrol (10–100 mg/kg/day orally for 6 weeks) reduced hepatic lipid accumulation, oxidative stress (reduced MDA and increased SOD), hepatocyte apoptosis, and improved liver histology through activation of the Nrf2/HO-1 pathway [162]. Finally, systemic insulin resistance and its hepatic consequences were corroborated in vivo by Zhang et al. (2018), who showed that celastrol reduced hepatic steatosis, suppressed SREBP-1c, activated AMPK/LKB1, and inhibited NF-κB signaling in HFD-fed mice [157,163], and by Kim et al. (2013), who demonstrated that celastrol (1 mg/kg/day i.p. for 2 months) improved HOMA-IR, fasting glucose, HbA1c, and reduced hepatic lipid accumulation and inflammatory cytokine expression in db/db mice [158].
Collectively, these in vitro and in vivo findings demonstrate that celastrol consistently targets key pathogenic mechanisms across the MASLD–MASH spectrum - attenuating hepatocellular lipid accumulation, bile acid dysregulation, inflammatory and insulin-resistance signaling, mitochondrial dysfunction, and fibrogenic activation - thereby limiting disease initiation and progression under diverse metabolic stress conditions.
4.6. Glycyrrhizin and Glycyrrhetinic Acid
Glycyrrhizin and its active metabolite glycyrrhetinic acid have been investigated in experimental settings relevant to MASLD and MASH, with early evidence indicating direct effects on hepatic lipid handling and bile acid–related pathways.
Initial in vitro observations by Yamamoto et al. (1970) demonstrated that direct exposure of rat liver slices to glycyrrhizin increased the incorporation of acetate and mevalonate into cholesterol, suggesting stimulation of hepatic cholesterol synthesis. In parallel in vivo experiments, intramuscular pretreatment with glycyrrhizin (10–100 mg/kg/day for 5 days) accelerated biliary and fecal excretion of cholesterol and bile acids and reduced plasma cholesterol and triglyceride levels in cholesterol-fed rats, indicating a coordinated effect on hepatic cholesterol turnover and bile acid elimination rather than lipid accumulation per se [164]. Complementarily, Negishi et al. (1991) identified a specific, high-affinity binding site for glycyrrhetinic acid in rat liver membrane fractions, supporting a direct liver-targeted mechanism of action for glycyrrhizin, although this study did not evaluate steatosis, inflammation, or fibrotic endpoints [165].
Subsequent in vivo studies addressed early metabolic alterations that precede MASLD development, focusing on systemic and hepatic lipid homeostasis. Lim et al. (2009) reported that short-term oral administration of glycyrrhizin (50 mg/kg/day, oral gavage, for 7 days) increased lipoprotein lipase (LPL) expression in multiple tissues, including the liver, and improved circulating lipid profiles, as evidenced by reductions in serum triglycerides, total cholesterol, and LDL, alongside increased HDL levels. These changes were accompanied by reduced lipid deposition in peripheral tissues assessed by Oil Red O staining, although hepatic lipid reduction did not reach statistical significance within the short treatment window [166]. These findings were corroborated by studies of Eu et al. (2010) that demonstrated that longer glycyrrhizin treatment (100 mg/kg/day, oral gavage, for 28 days) in HFD–induced obese rats significantly improved insulin sensitivity, reflected by reduced fasting glucose and HOMA-IR, while also lowering serum triglycerides, total cholesterol, LDL, and free fatty acids. These metabolic improvements were accompanied by reduced lipid deposition in several tissues, including the liver, and tissue-specific regulation of LPL expression, interpreted in the context of altered PPAR-associated lipid metabolism [167].
Additional in vivo evidence under combined dietary and stress-related metabolic challenge was provided by Yaw et al. (2015). In this study, glycyrrhizin (100 mg/kg/day, oral gavage for 14 or 28 days) administered to rats fed a high-calorie diet and exposed to short- or long-term stress selectively modulated the expression of lipid metabolism–related genes, including PPARα, PPARγ, and LPL, as well as fatty acid elongases and desaturases (ELOVL5 and ELOVL6), in hepatic and extrahepatic tissues. While stress exposure markedly influenced lipid metabolic pathways, glycyrrhizin did not significantly alter hepatic elongase or desaturase expression, suggesting that its hepatic effects under these conditions were primarily related to lipid uptake and handling rather than de novo fatty acid synthesis or modification. These molecular findings were supported by analyses of serum lipid profiles and tissue fatty acid composition [168].
Shi et al. (2020) employed in vitro and in vivo approaches centered on retinoid metabolism. In HepG2 cells, glycyrrhizin directly inhibited the activity of aldo–keto reductase family 1 member B10 (AKR1B10), a key enzyme involved in vitamin A metabolism, as demonstrated by enzymatic kinetics, molecular docking, and loss-of-function experiments using AKR1B10-knockout hepatocytes. glycyrrhizin treatment restored intracellular retinoic acid levels, whereas glycyrrhetinic acid exerted comparable effects only after conversion to glycyrrhizinic acid. Markers analyzed included AKR1B10 expression and activity, retinol and retinoic acid species, and retinoid-related metabolic genes assessed by RNA sequencing, RT-qPCR, and HPLC. In vivo, oral administration of GL (15, 30, or 60 mg/kg/day, once daily for 4 weeks) significantly reduced hepatic lipid accumulation and inflammatory lesions in both HFD–induced MASLD and MCD diet–induced MASH models. These effects were accompanied by normalization of hepatic retinoid metabolism, reduced serum ALT and AST activities, and decreased hepatic triglyceride and cholesterol levels, without evidence of classical PPARα or PPARγ agonist activation [169].
Finally, recent studies have linked glycyrrhizin to regulation of inflammatory steatosis through modulation of the hepatic ACE2 axis. Zhou et al. (2025) demonstrated in vitro that glycyrrhizin attenuated lipid accumulation and inflammatory injury in HepG2 hepatocytes exposed to free fatty acids (palmitic acid/oleic acid) combined with LPS. Glycyrrhizin reduced intracellular triglyceride content, lipid droplet accumulation, and ALT and AST release into the culture medium. Molecular analyses showed restoration of ACE2 and Mas receptor expression, suppression of Ang II and SREBP-1c overexpression, increased phosphorylation of AMPKα, and reduced total and phosphorylated MDM2 levels. Silencing of ACE2 abolished glycyrrhizin-induced reductions in lipid accumulation and enzyme release, indicating ACE2-dependent signaling [171]. Consistently, in vivo experiments by Zhou et al. (2024) showed that oral glycyrrhizin administration (5 or 30 mg/kg/day, once daily for 3 days) significantly attenuated hepatic injury in an LPS/D-galactosamine–induced steatohepatitis mouse model. glycyrrhizin reduced serum ALT and AST activities, hepatic triglyceride accumulation, and histopathological liver damage, while restoring hepatic ACE2/Ang-(1–7)/Mas signaling and modulating AMPK and MDM2 expression in liver tissue [172].
These studies indicate that glycyrrhizin and glycyrrhetinic acid exert consistent hepatoprotective effects across early and intermediate stages of MASLD and MASH. The available evidence supports a mechanistic framework in which these compounds modulate hepatic lipid and bile acid metabolism, improve insulin sensitivity, and suppress metabolic inflammation through defined molecular pathways, including regulation of LPL and PPAR-associated lipid handling, AKR1B10-mediated retinoid metabolism, and ACE2/Ang-(1–7)/Mas signaling coupled to AMPK activation and MDM2 inhibition.
Taken together, the sections above highlight that pentacyclic triterpenes can reproducibly attenuate key pathogenic steps along the MASLD–MASH development - ranging from early hepatocellular lipid accumulation and insulin resistance to inflammatory amplification, oxidative/ER stress, and fibrogenic activation - through pleiotropic but convergent mechanisms across multiple experimental models. Nevertheless, despite the breadth of preclinical efficacy and the mechanistic depth now available for several scaffolds, translation remains challenged by compound-dependent pharmaceutical limitations that can substantially constrain systemic exposure and, ultimately, therapeutic effect. Accordingly, the next section summarizes contemporary strategies being pursued to overcome the main barriers that currently limit clinical development - particularly poor aqueous solubility, low and variable oral bioavailability, and inconsistencies in in vivo potency across models and dosing regimens.
5. Strategies for Enhancing Triterpenes Bioavailability and Efficacy
Across pentacyclic triterpenes, one major route to improve drug-like exposure has been chemical derivatization, typically aimed at increasing polarity, tuning ionization, and/or strengthening target engagement so that efficacious dosing becomes feasible despite intrinsically low aqueous solubility. For OA, examples include simple scaffold modification such as 3-acetylation (3-acetyl-oleanolic acid) and target-oriented derivatives (e.g., HA-20) designed to enhance functional activity and thereby support practical administration in vivo, as well as more systematic structure-optimization programs that introduce amino acid–type conjugation (e.g., 12β-O-γ-glutamyl OA derivatives) to obtain FXR antagonist/modulator profiles for MASH-oriented applications. In parallel, broader surveys of OA derivatives emphasize the strategy of tailoring the OA scaffold to modulate metabolic nuclear receptors, reflecting a medicinal-chemistry approach in which optimized potency and physicochemical properties can jointly improve the likelihood of sufficient exposure at tolerable doses [173,174,175,176].
A second, widely used strategy focuses on formulation-driven solubility and dissolution enhancement, which can markedly improve oral performance for triterpenes that are otherwise poorly absorbed. Multiple studies illustrate “drug–carrier” approaches that keep the parent triterpene unchanged while improving its apparent solubility and release: solid dispersions (e.g., UA in Gelucire 50/13) and self-micelle solid-dispersion systems (e.g., based on inositol hexanicotinate) are used to increase drug dispersion at the intestinal interface and facilitate uptake; cyclodextrin complexation (e.g., hydroxypropyl-γ-cyclodextrin complexes with OA/UA) provides host–guest inclusion to improve aqueous compatibility; and more recently, OA has been incorporated into cyclodextrin metal–organic frameworks (MOFs) via co-crystallization, representing an emerging encapsulation platform intended to improve handling and delivery of highly lipophilic triterpenes. Collectively, these approaches primarily address the “front-end” barrier of poor dissolution/solubilization that limits oral absorption [177,178,179,180].
Beyond solubilization, a substantial body of work has applied nanotechnology-based carriers to enhance triterpene delivery by improving dispersion stability, protecting the payload, and enabling more controllable biodistribution—features that can translate into higher effective exposure and more reliable administration. For UA, reported platforms include phospholipid nanoparticles (including freeze-dried preparations to support handling and dosing), polymeric nanoparticles (including block-copolymer systems), liposomes engineered for extended circulation and pH sensitivity, chitosan-based nanoparticles (including ligand-directed variants such as folate-functionalized systems), PLGA nanoparticles, and nanocrystals where particle-size control is used to increase dissolution rate and thereby improve bioavailability. Related engineering efforts extend to prodrug-like dendrimeric constructs for targeted delivery and to nanoemulsions or other nanoplatforms designed for controlled release in specialized settings. Although several of these examples were developed for non-hepatic indications, they collectively exemplify generalizable delivery principles for triterpenes: increasing apparent solubility, improving stability, and enabling administration via formulations that overcome absorption and distribution constraints [181,182,183,184,185,186,187,188,189,190].
Newer studies illustrate how targeted or biomimetic delivery systems can be leveraged for particularly challenging triterpenes (notably celastrol) where oral dosing and tissue selectivity are limiting, and how route-of-administration engineering can help when absorption is inherently poor (as in glycyrrhizin). Celastrol has been formulated into albumin-based nanoparticles (including biodegradable and ligand-decorated versions), polysaccharide-derived hybrid micelles (e.g., chondroitin sulfate–based), and co-assembly systems where hyaluronic acid functions as a self-assembly “chaperone,” all aiming to improve dispersibility, reduce nonspecific exposure, and support effective dosing. A particularly translationally oriented approach is the design of oral celastrol micelles that form a HDL corona to promote hepatocyte targeting, directly addressing both oral delivery and liver selectivity. Separately, for glycyrrhizin—classically recognized as poorly absorbed—systematic evaluation of non-vascular administration routes has been explored to enhance systemic availability when oral absorption is insufficient, providing a complementary “delivery-by-route” solution distinct from chemical derivatization or nanocarriers [191,192,193,194,195,196].
Conclusions
Pentacyclic triterpenes emerge from the available evidence as a coherent class of multitarget modulators capable of intercepting key pathogenic processes across the MASLD-MASH continuum. By converging on central nodes of disease progression - namely dysregulated lipid metabolism, oxidative and endoplasmic reticulum stress, chronic inflammation, inflammasome activation, and hepatic stellate cell–driven fibrogenesis - compounds such as ursolic acid, oleanolic acid, betulinic acid, asiatic acid, celastrol, and glycyrrhizin consistently attenuate steatosis, inflammatory signaling, and fibrotic remodeling in diverse experimental models. Despite structural diversity, these triterpenes share common mechanistic themes, including activation of energy-sensing pathways (e.g., AMPK), suppression of lipogenic transcriptional programs (e.g., SREBP-1c), inhibition of pro-inflammatory cascades (e.g., TLR–NF-κB and NLRP3 inflammasome signaling), and modulation of profibrotic axes (e.g., TGF-β/SMAD), supporting the concept that multitarget intervention is particularly suited to the multifactorial nature of MASH.
However, translation of these promising preclinical findings into effective clinical therapies remains constrained by compound-specific pharmaceutical and biological limitations. Poor aqueous solubility, low and variable oral bioavailability, and heterogeneity of experimental models limit direct extrapolation to human disease. Future progress will require rational prioritization of triterpenes based on translational readiness, coupled with optimized medicinal chemistry, formulation strategies, and delivery platforms that improve systemic exposure and tissue selectivity. Equally important is the need for well-designed, mechanism-informed clinical trials that incorporate appropriate patient stratification, pharmacokinetic–pharmacodynamic assessment, and validated histological and noninvasive endpoints. Within this framework, pentacyclic triterpenes should be viewed not as single-target agents, but as adaptable scaffolds for the development of next-generation therapies aligned with the complex biology of MASLD and MASH.
Funding Information
This work was supported by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG; grant no. APQ-01209-22) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant no. 436526/2018-2). I.C.Z. and J.A.L. received fellowships from FAPEMIG (PROBIC) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, DS), respectively.
Author Contributions
Conceptualization: D.S.F; Methodology: D.S.F and A.E.M.F.M.O; Writing – Original Draft Preparation: D.S.F., A.E.M.F.M.O., I.M.S., J.A.L, and I.C.S.; Writing - Figures preparation: J.A.L, and I.C.S; Writing - Reviewing and Editing: D.S.F., A.E.M.F.M.O., J.A.L, and I.C.S; Supervision: D.S.F.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene Biosynthesis in Plants. Annual Review of Plant Biology 2014, 65, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Goddard, Z.R.; Searcey, M.; Osbourn, A. Advances in Triterpene Drug Discovery. Trends in Pharmacological Sciences 2024, 45, 964–968. [Google Scholar] [CrossRef]
- Bishop, G.J.; Koncz, C. Brassinosteroids and Plant Steroid Hormone Signaling. The Plant Cell 2002, 14 (Suppl), S97–S110. [Google Scholar] [CrossRef]
- Tarasova, E. V.; Luchnikova, N. A.; Grishko, V. V.; Ivshina, I. B. Actinomycetes as Producers of Biologically Active Terpenoids: Current Trends and Patents. Pharmaceuticals (Basel) 2023, 16(6), 872. [Google Scholar] [CrossRef]
- Krivoruchko, A.; Nielsen, J. Production of Natural Products through Metabolic Engineering of Saccharomyces cerevisiae. Current Opinion in Biotechnology 2015, 35, 7–15. [Google Scholar] [CrossRef]
- Silva, F.C.; Duarte, L.P.; Vieira Filho, S.A. Celastraceae Family: Source of Pentacyclic Triterpenes with Potential Biological Activity. Revista Virtual de Química 2014, 6(5), 1205–1220. [Google Scholar] [CrossRef]
- Sato, T. Unique Biosynthesis of Sesquarterpenes (C35 Terpenes). Bioscience, Biotechnology, and Biochemistry 2013, 77(6), 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Ninkuu, V.; Zhang, L.; Yan, J.; Fu, Z.; Yang, T.; Zeng, H. Biochemistry of Terpenes and Recent Advances in Plant Protection. International Journal of Molecular Sciences 2021, 22(11), 5710. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.C.O.; Ferreira, M.K.A.; Silva, A.W.d.; Matos, M.G.C.; Magalhães, F.E.A.; Silva, P.T.d.; Bandeira, P.N.; Menezes, J.E.S.A.d.; Santos, H.S. Bioactivities of Plant-Isolated Triterpenes: A Brief Review. Revista Virtual de Química 2020, 12(1), 234–247. [Google Scholar] [CrossRef]
- Kirby, J.; Keasling, J.D. Biosynthesis of Plant Isoprenoids: Perspectives for Microbial Engineering. Annual Review of Plant Biology 2009, 60, 335–355. [Google Scholar] [CrossRef]
- Thibodeaux, C.J.; Liu, H.-W. The Type II Isopentenyl Diphosphate:Dimethylallyl Diphosphate Isomerase (IDI-2): A Model for Acid/Base Chemistry in Flavoenzyme Catalysis. Archives of Biochemistry and Biophysics 2017, 632, 47–58. [Google Scholar] [CrossRef]
- Phillips, D.R.; Rasbery, J.M.; Bartel, B.; Matsuda, S.P.T. Biosynthetic Diversity in Plant Triterpene Cyclization. Current Opinion in Plant Biology 2006, 9(3), 305–314. [Google Scholar] [CrossRef] [PubMed]
- Parveen, I.; Wang, M.; Lee, J.; Zhao, J.; Zhu, Y.; Chittiboyina, A.G.; Khan, I.A.; Pan, Z. Identification and Functional Characterization of Oxidosqualene Cyclases from Medicinal Plant Hoodia gordonii. Plants (Basel) 2024, 13(2), 231. [Google Scholar] [CrossRef]
- Dzhemileva, L.U.; Tuktarova, R.A.; Dzhemilev, U.M.; D’yakonov, V.A. Pentacyclic Triterpenoids-Based Ionic Compounds: Synthesis, Study of Structure–Antitumor Activity Relationship, Effects on Mitochondria and Activation of Signaling Pathways of Proliferation, Genome Reparation and Early Apoptosis. Cancers 2023, 15(3), 756. [Google Scholar] [CrossRef] [PubMed]
- Ames, T.R.; Halsall, T.G.; Jones, E.R.H. The Chemistry of the Triterpenes. Part VII. An Interrelationship between the Lupeol and the β-Amyrin Series. Elucidation of the Structure of Lupeol. Journal of the Chemical Society 1951, 450–457. [Google Scholar] [CrossRef]
- Halsall, T.G.; Aplin, R.T. A Pattern of Development in the Chemistry of Pentacyclic Triterpenes. Fortschritte der Chemie Organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products 1964, 22, 153–202. [Google Scholar]
- Arpino, P.; Albrecht, P.; Ourisson, G. Studies on the Organic Constituents of Lacustrine Eocene Sediments. Possible Mechanisms for the Formation of Some Geolipids Related to Biologically Occurring Terpenoids. In Advances in Organic Geochemistry; von Gaertner, H.R., Wehner, H. (Eds.); Pergamon Press: Oxford, 1972; pp. 173–187.
- Kimble, B.J.; Maxwell, J.R.; Philp, R.F.; Eglinton, G.; Albrecht, P.; Ensminger, A.; Arpino, A.; Ourisson, G. Tri- and Tetra-Terpenoid Hydrocarbons in the Messes Oil Shale. Geochimica et Cosmochimica Acta 1974, 38, 1165–1181. [Google Scholar] [CrossRef]
- Hodon, J.; Borkova, L.; Pokorny, J.; Kazakova, A.; Urban, M. Design and Synthesis of Pentacyclic Triterpene Conjugates and Their Use in Medicinal Research. European Journal of Medicinal Chemistry 2019, 182, 111653. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.; Ma, Y.; Zou, Y.; Cai, Y.; Gao, X.; Zhang, Y.; Zhao, Y.-J. Natural Products of Pentacyclic Triterpenoids: From Discovery to Heterologous Biosynthesis. Natural Product Reports 2023, 40(8), 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Safayhi, H.; Sailer, E.-R. Anti-Inflammatory Actions of Pentacyclic Triterpenes. Planta Medica 1997, 63(6), 487–493. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Hu, K.; Xu, Q.; Wen, X.; Cheng, K.; Chen, C.; Yuan, H.; Dai, L.; Sun, H. Synthesis and Anti-Inflammatory Activity of Saponin Derivatives of δ-Oleanolic Acid. European Journal of Medicinal Chemistry 2021, 209, 112932. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-Y.; Ha, J.Y.; Kim, K.-M.; Jung, Y.-S.; Jung, J.-C.; Oh, S. Anti-Inflammatory Activities of Licorice Extract and Its Active Compounds, Glycyrrhizic Acid, Liquiritin and Liquiritigenin, in BV2 Cells and Mice Liver. Molecules 2015, 20, 13041–13054. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A Phytochemical and Pharmacological Review. Phytotherapy Research 2018, 32(12), 2323–2339. [Google Scholar] [CrossRef]
- Bisht, D.; Rashid, M.; Arya, R.K.K.; Kumar, D.; Chaudhary, S.K.; Rana, V.S.; Sethiya, N.K. Revisiting Liquorice (Glycyrrhiza glabra L.) as Anti-Inflammatory, Antivirals and Immunomodulators: Potential Pharmacological Applications with Mechanistic Insight. Phytomedicine Plus 2022, 2(1), 100206. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Bhattacharjee, A.; Majumder, S.; Bhattacharya Majumdar, S.; Majumdar, S. Glycyrrhizic Acid Suppresses Cox-2-Mediated Anti-Inflammatory Responses during Leishmania donovani Infection. Journal of Antimicrobial Chemotherapy 2012, 67(8), 1905–1914. [Google Scholar] [CrossRef]
- Houssen, M.E.; Ragab, A.; Mesbah, A.; El-Samanoudy, A.Z.; Othman, G.; Moustafa, A.F.; Badria, F.A. Natural Anti-Inflammatory Products and Leukotriene Inhibitors as Complementary Therapy for Bronchial Asthma. Clinical Biochemistry 2010, 43(10–11), 887–890. [Google Scholar] [CrossRef]
- Račková, L.; Jančinová, V.; Petríková, M.; Drábiková, K.; Nosáľ, R.; Štefek, M.; Kováčová, M. Mechanism of anti-inflammatory action of liquorice extract and glycyrrhizin. Natural Product Research 2007, 21(14), 1234–1241. [Google Scholar] [CrossRef]
- Fatima, I.; Sahar, A.; Tariq, A.; Naz, T.; Usman, M. Exploring the Role of Licorice and Its Derivatives in Cell Signaling Pathway NF-κB and MAPK. Journal of Nutrition and Metabolism 2024, 2024, 9988167. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Choi, A.; Seo, N.; et al. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on post-contrast acute kidney injury. Scientific Reports 2021, 11, 15625. [Google Scholar] [CrossRef]
- Yan, C. Y.; Ouyang, S. H.; Wang, X.; et al. Celastrol ameliorates Propionibacterium acnes/LPS-induced liver damage and MSU-induced gouty arthritis via inhibiting K63 deubiquitination of NLRP3. Phytomedicine 2021, 80, 153398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, W.; Liu, X.; Huang, Z.; Shan, R.; Huang, C. Celastrol ameliorates inflammatory pain and modulates HMGB1/NF-κB signaling pathway in dorsal root ganglion. Neuroscience Letters 2019, 692, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yu, G.; Yang, K.; et al. Exploring the mechanism of celastrol in the treatment of rheumatoid arthritis based on systems pharmacology and multi-omics. Scientific Reports 2024, 14, 1604. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Pandey, M.K.; Aggarwal, B.B. Celastrol, a Novel Triterpene, Potentiates TNF-Induced Apoptosis and Suppresses Invasion of Tumor Cells by Inhibiting NF-κB-Regulated Gene Products and TAK1-Mediated NF-κB Activation. [published correction appears in Blood. 2013 Aug 15;122(7):1327]. Blood 2007, 109(7), 2727–2735. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Hu, K.; Xu, Q.; Wen, X.; Cheng, K.; Chen, C.; Yuan, H.; Dai, L.; Sun, H. Synthesis and anti-inflammatory activity of saponin derivatives of δ-oleanolic acid. European Journal of Medicinal Chemistry 2021, 209, 112932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Ciric, B.; Curtis, M.T.; Chen, W.J.; Rostami, A.; Zhang, G.X. A Dual Effect of Ursolic Acid in the Treatment of Multiple Sclerosis through Both Immunomodulation and Direct Remyelination. Proceedings of the National Academy of Sciences of the United States of America 2020, 117(16), 9082–9093. [Google Scholar] [CrossRef]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic Acid Rescues Mitochondrial Function in Common Forms of Familial Parkinson’s Disease. Brain 2013, 136(10), 3038–3050. [Google Scholar] [CrossRef]
- Kim, E.Y.; Sudini, K.; Singh, A.K.; Haque, M.; Leaman, D.; Khuder, S.; Ahmed, S. Ursolic Acid Facilitates Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts by Inducing SP1-Mediated Noxa Expression and Proteasomal Degradation of Mcl-1. FASEB Journal 2018, 32(11), fj201800425R. [Google Scholar] [CrossRef]
- Alqahtani, A; Hamid, K.; Kam, A.; Wong, K.H.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The Pentacyclic Triterpenoids in Herbal Medicines and Their Pharmacological Activities in Diabetes and Diabetic Complications. Current Medicinal Chemistry 2013, 20(7), 908–931. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, M.; Li, X.-L.; Li, H.; Yue, L.-T.; Zhang, X.-X.; Wang, C.-C.; Wang, S.; Duan, R.-S. Low and High Doses of Ursolic Acid Ameliorate Experimental Autoimmune Myasthenia Gravis through Different Pathways. Journal of Neuroimmunology 2015, 281, 61–67. [Google Scholar] [CrossRef]
- Xu, T.; Wang, X.; Zhong, B.; Nurieva, R.I.; Ding, S.; Dong, C. Ursolic Acid Suppresses Interleukin-17 (IL-17) Production by Selectively Antagonizing the Function of RORγt Protein. Journal of Biological Chemistry 2011, 286(26), 22707–22710. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, F.; Tang, Z.; Yang, X.; Liu, Y.; Wang, F.; Chen, B. Anti-Inflammatory and Antioxidant Activity of Ursolic Acid: A Systematic Review and Meta-Analysis. Frontiers in Pharmacology 2023, 14, 1256946. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.P.T. Boswellic acids in chronic inflammatory diseases. Planta Medica 2006, 72(12), 1100–1116. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-Q.; Zhang, Y.-J.; Tian, Z.-K. Anti-oxidant, Anti-inflammatory and Anti-fibrosis Effects of Ganoderic Acid A on Carbon Tetrachloride Induced Nephrotoxicity by Regulating the Trx/TrxR and JAK/ROCK Pathway. Chemico-Biological Interactions 2021, 344, 109529. [Google Scholar] [CrossRef]
- Figueroa-Suárez, M.Z.; Christen, J.G.; Cardoso-Taketa, A.T.; Villafuerte, M.d.C.G..; Rodríguez-López, V. Anti-Inflammatory and Antihistaminic Activity of Triterpenoids Isolated from Bursera cuneata (Schldl.) Engl. Journal of Ethnopharmacology 2019, 238, 111786. [Google Scholar] [CrossRef]
- Li, M.-M.; Su, X.-Q.; Sun, J.; Gu, Y.-F.; Huang, Z.; Zeng, K.-W.; Zhang, Q.; Zhao, Y.-F.; Ferreira, D.; Zjawiony, J.K.; Li, J.; Tu, P.-F. Anti-Inflammatory Ursane- and Oleanane-Type Triterpenoids from Vitex negundo var. cannabifolia. Journal of Natural Products 2014, 77(10), 2248–2254. [Google Scholar] [CrossRef]
- Recio, M.C.; Giner, R.M.; Máñez, S.; Ríos, J.L. Structural Requirements for the Anti-Inflammatory Activity of Natural Triterpenoids. Planta Medica 1995, 61(2), 182–185. [Google Scholar] [CrossRef] [PubMed]
- Allouche, Y.; Beltrán, G.; Gaforio, J.J.; Uceda, M.; Mesa, M.D. Antioxidant and Antiatherogenic Activities of Pentacyclic Triterpenic Diols and Acids. Food and Chemical Toxicology 2010, 48(10), 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, W.; Wang, L.; Liu, R.; Ding, Y.; Du, L. Constituents of the Flowers of Punica granatum. Fitoterapia 2006, 77(7–8), 534–537. [Google Scholar] [CrossRef]
- Lim, J.; Lee, H.; Hong, S.; Lee, J.; Kim, Y. Comparison of the Antioxidant Potency of Four Triterpenes of Centella asiatica against Oxidative Stress. Antioxidants 2024, 13, 483. [Google Scholar] [CrossRef]
- Mamadalieva, N.Z.; Youssef, F.S.; Hussain, H.; Zengin, G.; Mollica, A.; Al Musayeib, N.M.; Ashour, M.L.; Westermann, B.; Wessjohann, L.A. Validation of the Antioxidant and Enzyme Inhibitory Potential of Selected Triterpenes Using In Vitro and In Silico Studies, and the Evaluation of Their ADMET Properties. Molecules 2021, 26, 6331. [Google Scholar] [CrossRef]
- Ostapiuk, A.; Kurach, Ł.; Strzemski, M.; Kurzepa, J.; Hordyjewska, A. Evaluation of Antioxidative Mechanisms In Vitro and Triterpenes Composition of Extracts from Silver Birch (Betula pendula Roth) and Black Birch (Betula obscura Kotula) Barks by FT-IR and HPLC-PDA. Molecules 2021, 26, 4633. [Google Scholar] [CrossRef] [PubMed]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harbor Perspectives in Biology 2012, 4(9), a008417. [Google Scholar] [CrossRef]
- Peng, X.; Wang, Q.; Su, H.-G.; Zhou, L.; Xiong, W.-Y.; Qiu, M.-H. Anti-Adipogenic Lanostane-Type Triterpenoids from the Edible and Medicinal Mushroom Ganoderma applanatum. Journal of Fungi 2022, 8(4), 331. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, A.-M; Abu-Taweel, G.M.; Rajagopal, R.; Sun-Ju, K.; Kim, H.-J.; Kim, Y.O.; Mothana, R.A.; Kadaikunnan, S.; Khaled, J.M.; Siddiqui, N.A.; Al-Rehaily, A.J. Betulinic acid lowers lipid accumulation in adipocytes through enhanced NCoA1-PPARγ interaction. Journal of Infection and Public Health 2019, 12(5), 726–732. [Google Scholar] [CrossRef]
- Melo, C.L.D.; Queiroz, M.G.R.; Arruda Filho, A.C.V.; Rodrigues, A.M.; Sousa, D.F.D.; Almeida, J.G.L.; Pessoa, O.D.L.; Silveira, E.R.; Menezes, D.B.; Melo, T.S.; Santos, F.A.; Rao, V.S. Betulinic acid, a natural pentacyclic triterpenoid, prevents abdominal fat accumulation in mice fed a high-fat diet. Journal of Agricultural and Food Chemistry 2009, 57(19), 8776–8781. [Google Scholar] [CrossRef]
- Betancur, L.I.; Muñoz, D.L.; Guillen, A.; Echeverri, L.F.; Balcazar, N.; Acín, S. Major Triterpenoids from Eucalyptus tereticornis Have Enhanced Beneficial Effects in Cellular Models When Mixed with Minor Compounds Present in Raw Extract. Anais da Academia Brasileira de Ciências 2021, 93 (Suppl. 3), e20201351. [Google Scholar] [CrossRef]
- Acín, S.; Balcázar, N.; Guillen, A.; Echeverri, L.F.; Betancur, L.I.; Muñoz, D.L. Triterpene-Enriched Fractions from Eucalyptus tereticornis Leaves with Different Triterpene Composition Improve Insulin Sensitivity and Reduce Hepatic Lipid Accumulation in a Nutritional Animal Model of Prediabetes. Chemico-Biological Interactions 2021. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, Y.; Liu, Y.; Liu, D.; Liu, Y.; Guo, Y.; Wu, Z.; Li, H.; Wang, H. Ursolic acid alleviates lipid accumulation by activating the AMPK signaling pathway in vivo and in vitro. Journal of Food Science 2020, 85(11), 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Bermudes-Contreras, J.D.; Gutiérrez-Velázquez, M.V.; Delgado-Alvarado, E.A.; Torres-Ricario, R.; Cornejo-Garrido, J. Hypoglycemic and Hypolipidemic Effects of Triterpenoid Standardized Extract of Agave durangensis Gentry. Plants 2025, 14(6), 894. [Google Scholar] [CrossRef]
- Liou, C.-J.; Dai, Y.-W.; Wang, C.-L.; Fang, L.-W.; Huang, W.-C. Maslinic acid protects against obesity-induced nonalcoholic fatty liver disease in mice through regulation of the Sirt1/AMPK signaling pathway. FASEB Journal 2019, 33(11), 11791–11803. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Barakat, B.M.; Bilasy, S.E.; Fawzy, M.S.; Abdelaziz, E.Z.; Farag, N.E. Protective Effect of Boswellic Acids versus Pioglitazone in a Rat Model of Diet-Induced Non-Alcoholic Fatty Liver Disease: Influence on Insulin Resistance and Energy Expenditure. Naunyn-Schmiedeberg's Archives of Pharmacology 2015, 388(6), 587–600. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wu, W.; Bai, Z.; Xiu, Y.; Zhou, D. Beta-boswellic Acid Facilitates Diabetic Wound Healing by Targeting STAT3 and Inhibiting Ferroptosis in Fibroblasts. Frontiers in Pharmacology 2025, 16, 1578625. [Google Scholar] [CrossRef]
- Reish, R.G.; Eriksson, E.; Galiano, R.D.; Dobryansky, M.; Levine, J.P.; Gurtner, G.C. Scars: A Review of Emerging and Currently Available Therapies. Plastic and Reconstructive Surgery 2008, 122(4), 1068–1078. [Google Scholar] [CrossRef]
- Richmond, N.A.; Maderal, A.D.; Vivas, A.C. US-National Institutes of Health-Funded Research for Cutaneous Wounds in 2012. Wound Repair and Regeneration 2013, 21(6), 789–792. [Google Scholar] [CrossRef]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.-F.; Okamoto, T.; John, A.E.; et al. Genetic Variants Associated with Susceptibility to Idiopathic Pulmonary Fibrosis in People of European Ancestry: A Genome-Wide Association Study. The Lancet Respiratory Medicine 2017, 5(11), 869–880. [Google Scholar] [CrossRef]
- Friedman, S.L.; Bansal, M.B. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134(6), 1655–1669. [Google Scholar] [CrossRef]
- Inagaki, Y.; Okazaki, I.; Sanjo, A.; Aoyama, T.; Kato, H.; Fujii, H.; Sata, M.; Tsuneyama, K.; Nishimura, T. Emerging Insights into Transforming Growth Factor β/Smad Signal in Hepatic Fibrogenesis. Gut 2007, 56(2), 284–292. [Google Scholar] [CrossRef]
- Tang, L.-X.; Wu, W.-J.; Chen, X.-H.; Li, Q.; Chen, Y.; Lin, L.; Yang, F.; Fang, M.; Gao, J.; Zhang, D.-M. Asiatic Acid Inhibits Liver Fibrosis by Blocking TGF-β/Smad Signaling In Vivo and In Vitro. PLoS One 2012, 7(2), 1–13. [Google Scholar] [CrossRef]
- Dong, M.-S.; Lee, H.-S.; Park, J.-H.; Lee, S.-J.; Kim, H.-J.; Kim, D.-H. Structure-Related Cytotoxicity and Anti-Hepatofibric Effect of Asiatic Acid Derivatives in Rat Hepatic Stellate Cell-Line, HSC-T6. Archives of Pharmacal Research 2004, 27(5), 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, E.; Löser, E.; Klotz, L.-O.; Weise, C.; Gebhardt, R. Conditional Tetracycline-Regulated Expression of TGF-β1 in Liver of Transgenic Mice Leads to Reversible Intermediary Fibrosis. Hepatology 2003, 37(5), 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ikejima, K.; Kon, K.; Arai, K.; Aoyama, T.; Okumura, K.; Abe, W.; Sato, N.; Watanabe, S. Ursolic Acid Ameliorates Hepatic Fibrosis in the Rat by Specific Induction of Apoptosis in Hepatic Stellate Cells. Journal of Hepatology 2011, 55(2), 379–387. [Google Scholar] [CrossRef]
- Ma, X.-Y.; Zhang, M.; Fang, G.; Cheng, C.-J.; Wang, M.-K.; Han, Y.-M.; Hou, X.-T.; Hao, E.-W.; Hou, Y.-Y.; Bai, G. Ursolic Acid Reduces Hepatocellular Apoptosis and Alleviates Alcohol-Induced Liver Injury via Irreversible Inhibition of CASP3 in Vivo. Acta Pharmacologica Sinica 2021, 42, 1101–1110. [Google Scholar] [CrossRef]
- Wei, J.; Fang, F.; Ma, J.; Li, C.; Zhang, J.; Sun, L.; Lu, C.; Wang, X.; Wu, Y.; Xu, Q. A Synthetic PPAR-γ Agonist Triterpenoid Ameliorates Experimental Fibrosis: PPAR-γ-Independent Suppression of Fibrotic Responses. Annals of the Rheumatic Diseases 2013, 73(2), 446–454. [Google Scholar] [CrossRef]
- Batteux, F.; LeRoy, E.C.; Assassi, S.; Denton, C.P.; Distler, O.; Varga, J. New Insights on Chemically Induced Animal Models of Systemic Sclerosis. Current Opinion in Rheumatology 2011, 23(6), 511–518. [Google Scholar] [CrossRef]
- Liao, D.; Yu, X.; Chen, M.; Sun, H.; Wang, Y.; Zhang, J.; Li, H.; Zhang, L.; Xu, J.; Li, Q.; et al. Targeted Therapeutic Remodeling of the Tumor Microenvironment Improves an HER-2 DNA Vaccine and Prevents Recurrence in a Murine Breast Cancer Model. Cancer Research 2011, 71(17), 5688–5696. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of Canonical Wnt Signalling Is Required for TGF-β-Mediated Fibrosis. Nature Communications 2012, 3(1), 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-L.; Lin, Y.-T.; Kung, H.-N.; Hou, Y.-C.; Liu, J.-J.; Pan, M.-H.; Chen, H.-L.; Yu, C.-H.; Tsai, P.-J. A Triterpenoid-Enriched Extract of Bitter Melon Leaves Alleviates Hepatic Fibrosis by Inhibiting Inflammatory Responses in Carbon Tetrachloride-Treated Mice. Food and Function 2021, 12(17), 7805–7815. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Zhou, Y.; Gaggar, A.; Duncan, S.R.; Osei, E.T.; Jenkins, R.G. Fibrosis: Ultimate and Proximate Causes. Journal of Clinical Investigation 2014, 124(11), 4673–4677. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Zhang, Y.; Chen, J.; Li, H.; Sun, Q.; Zhao, Y.; Wang, X.; Zhang, L. Betulinic Acid Attenuated Bleomycin-Induced Pulmonary Fibrosis by Effectively Intervening Wnt/β-Catenin Signaling. Phytomedicine 2021, 81, 153428. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Razboršek, M.; Voncina, D.; Doleček, V.; Voncina, E. Determination of Oleanolic, Betulinic and Ursolic Acid in Lamiaceae and Mass Spectral Fragmentation of Their Trimethylsilylated Derivatives. Chromatographia 2008, 67, 433–440. [Google Scholar] [CrossRef]
- Cantley, L.C.; Baselga, J. The Era of Cancer Discovery. Cancer Discovery 2011, 1, 1–4. [Google Scholar] [CrossRef]
- Yasui, W. Future Perspectives of Gastric Cancer Treatment – From Bench to Bedside. Pathobiology 2011, 78, 293–294. [Google Scholar] [CrossRef]
- Muffler, K.; Leipold, D.; Scheller, M.C.; Haans, C.; Steingroewer, J.; Bley, T.; Neuhaus, H.E.; Mirata, M.A.; Schrader, J.; Ulber, R. Biotransformation of Triterpenes. Process Biochemistry 2011, 46, 1–15. [Google Scholar] [CrossRef]
- Huang, L.; Chen, T.; Ye, Z.; Chen, G. Use of Liquid Chromatography–Atmospheric Pressure Chemical Ionization–Ion Trap Mass Spectrometry for Identification of Oleanolic Acid and Ursolic Acid in Anoectochilus roxburghii (Wall.) Lindl. Journal of Mass Spectrometry 2007, 42, 910–917. [Google Scholar] [CrossRef]
- Shang, J.-H.; Sun, W.-J.; Zhu, H.-T.; Wang, D.; Yang, C.-R.; Zhang, Y.-J. New Hydroperoxylated and 20,24-Epoxylated Dammarane Triterpenes from the Rot Roots of Panax notoginseng. Journal of Ginseng Research 2020, 44(3), 405–412. [Google Scholar] [CrossRef]
- Xie, J. Anti-inflammatory and Analgesic Effects and the Mechanism of Actions of Ginsenoside Rg1. Chinese Journal of Hospital Pharmacy 2013, 33, 1592. [Google Scholar]
- Ahn, S.; Siddiqi, M. H.; Noh, H. Y.; Kim, Y. J.; Jin, C. G.; Yang, D. C. Anti-inflammatory Activity of Ginsenosides in LPS-Stimulated RAW264.7 Cells. Science Bulletin 2015, 60(8), 773–784. [Google Scholar] [CrossRef]
- He, W.; Li, X.; Xia, S. Lupeol triterpene exhibits potent antitumor effects in A427 human lung carcinoma cells via mitochondrial mediated apoptosis, ROS generation, loss of mitochondrial membrane potential and downregulation of m-TOR/PI3K/AKT signalling pathway. Journal of BUON 2018, 23(3), 635–640. [Google Scholar]
- Ali, S.; Khan, M. A.; Khan, M. R.; Anwar, F.; Khan, A.; Shah, N.; Ullah, R.; Rahman, A. Evaluation of the Cytotoxic Potential of a New Pentacyclic Triterpene from Rhododendron arboreum Stem Bark. Pharmaceutical Biology 2017, 55(1), 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Kuo, P.-L.; Lin, L.-T.; Lin, C.-C. Proliferative Inhibition, Cell-Cycle Dysregulation, and Induction of Apoptosis by Ursolic Acid in Human Non-Small Cell Lung Cancer A549 Cells. Life Sciences 2004, 75(19), 2303–2316. [Google Scholar] [CrossRef]
- Xu, J.-H.; Yu, Y.-Y.; Xu, X.-Y. Management of chronic liver diseases and cirrhosis: current status and future directions. Chinese Medical Journal (Engl.) 2020, 133(22), 2647–2649. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T. F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9(4), 875. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C. D.; Tilg, H. MASLD: a systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Mladenić, K.; Lenartić, M.; Marinović, S.; Polić, B.; Wensveen, F. M. The “Domino effect” in MASLD: The inflammatory cascade of steatohepatitis. European Journal of Immunology 2024, 54, e2149641. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, V.; Papatheodoridis, G. V. Natural history of metabolic dysfunction-associated steatotic liver disease. European Journal of Internal Medicine 2024, 122, 3–10. [Google Scholar] [CrossRef]
- Israelsen, M.; Johansen, S.; Torp, N.; et al. Steatotic liver disease. Lancet 2024, 404, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. International Journal of Molecular Sciences 2016, 17, 1575. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M. V.; Roma, M. G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cellular and Molecular Life Sciences 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Tourkochristou, E.; Assimakopoulos, S. F.; Thomopoulos, K.; Marangos, M.; Triantos, C. NAFLD and HBV interplay—related mechanisms underlying liver disease progression. Frontiers in Immunology 2022, 13, 965548. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E. A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65(8), 1038–1048. [Google Scholar] [CrossRef]
- Bansal, S. K.; Bansal, M. B. Pathogenesis of MASLD and MASH—role of insulin resistance and lipotoxicity. Alimentary Pharmacology & Therapeutics 2024, 59(1), S10–S22. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, W.; Huang, C.; Jian, J.; Zhang, Y.; Liu, Q.; Chen, P.; Zhu, X. Ursolic acid alleviates Kupffer cells pyroptosis in liver fibrosis by the NOX2/NLRP3 inflammasome signaling pathway. International Immunopharmacology 2022, 113, 109321. [Google Scholar] [CrossRef]
- Shu, B.; Zhou, Y.-X.; Li, H.; Zhang, R.-Z.; He, C.; Yang, X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discovery 2021, 7(1), 368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.; Zhang, M.; Dong, X.; Zheng, L.; Li, G.; Han, X.; Yao, Z.; Han, T.; Hong, W. Silencing lncRNA Lfar1 alleviates the classical activation and pyroptosis of macrophage in hepatic fibrosis. Cell Death & Disease 2020, 11(2), 132. [Google Scholar] [CrossRef]
- Berumen, J.; Baglieri, J.; Kisseleva, T.; Mekeel, K. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mechanisms of Disease 2021, 13, e1499. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Hepatic inflammatory responses in liver fibrosis. Nature Reviews Gastroenterology & Hepatology 2023, 20, 633–646. [Google Scholar] [CrossRef]
- Kurzepa, J.; Mądro, A.; Czechowska, G.; Kurzepa, J.; Celiński, K.; Kazmierak, W.; Słomka, M. Role of MMP-2 and MMP-9 and their natural inhibitors in liver fibrosis, chronic pancreatitis and non-specific inflammatory bowel diseases. Hepatobiliary & Pancreatic Diseases International 2014, 13(6), 570–579. [Google Scholar] [CrossRef]
- Sabir, U.; Gu, H.-M.; Zhang, D.-W. Extracellular matrix turnover: phytochemicals target and modulate the dual role of matrix metalloproteinases (MMPs) in liver fibrosis. Phytotherapy Research 2023, 37, 4932–4962. [Google Scholar] [CrossRef]
- Jalan, R.; D'Amico, G.; Trebicka, J.; Moreau, R.; Angeli, P.; Arroyo, V. New clinical and pathophysiological perspectives defining the trajectory of cirrhosis. Journal of Hepatology 2021, 75(1), S14–S26. [Google Scholar] [CrossRef]
- Sharma, B.; John, S. Hepatic Cirrhosis. StatPearls Publishing 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482419/.
- Yoshiji, H.; Nagoshi, S.; Akahane, T.; Asaoka, Y.; Ueno, Y.; Ogawa, K.; Kawaguchi, T.; Kurosaki, M.; Sakaida, I.; Shimizu, M.; Taniai, M.; Terai, S.; Nishikawa, H.; Hiasa, Y.; Hidaka, H.; Miwa, H.; Chayama, K.; Enomoto, N.; Shimosegawa, T.; Takehara, T.; Koike, K. Evidence-Based Clinical Practice Guidelines for Liver Cirrhosis 2020. Journal of Gastroenterology 2021, 56, 593–619. [Google Scholar] [CrossRef]
- Benedé-Ubieto, R.; Cubero, F. J.; Nevzorova, Y. A. Breaking the barriers: the role of gut homeostasis in Metabolic-Associated Steatotic Liver Disease (MASLD). Gut Microbes 2024, 16(1), 2331460. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, L.-X.; Li, B.-M.; Zhang, W.; Zhang, Y.; Chen, P.; Huang, C.-K.; Nie, Y.; Zhu, X. Exploring the mechanism of ursolic acid in preventing liver fibrosis and improving intestinal microbiota based on NOX2/NLRP3 inflammasome signaling pathway. Chemico-Biological Interactions 2025, 405, 111305. [Google Scholar] [CrossRef]
- Fortea, J. I.; Crespo, J.; Puente, Á. Cirrhosis, a Global and Challenging Disease. Journal of Clinical Medicine 2022, 11(21), 6512. [Google Scholar] [CrossRef]
- Engelmann, C.; Clària, J.; Szabo, G.; Bosch, J.; Bernardi, M. Pathophysiology of Decompensated Cirrhosis: Portal Hypertension, Circulatory Dysfunction, Inflammation, Metabolism and Mitochondrial Dysfunction. Journal of Hepatology 2021, 75(1), S49–S66. [Google Scholar] [CrossRef] [PubMed]
- Ma, X. H.; Zhao, Y. C.; Yin, L.; Xu, R. L.; Han, D. W.; Wang, M. S. Studies on the preventive and therapeutic effects of ursolic acid (UA) on acute hepatic injury in rats. Yao Xue Xue Bao 1986, 21(5), 332-335. https://pubmed.ncbi.nlm.nih.gov/3776541/. [Google Scholar]
- Martin-Aragón, S.; de las Heras, B.; Sanchez-Reus, M. I.; Benedi, J. Pharmacological Modification of Endogenous Antioxidant Enzymes by Ursolic Acid on Tetrachloride-Induced Liver Damage in Rats and Primary Cultures of Rat Hepatocytes. Experimental and Toxicologic Pathology 2001, 53(2–3), 199–206. [Google Scholar] [CrossRef]
- Saravanan, R.; Viswanathan, P.; Pugalendi, K. V. Protective Effect of Ursolic Acid on Ethanol-Mediated Experimental Liver Damage in Rats. Life Sciences 2006, 78(7), 713–718. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Huang, C.; Zhao, L.; Chen, Y.; Liu, F. Regulation of Decorin by Ursolic Acid Protects against Non-Alcoholic Steatohepatitis. Biomedicine & Pharmacotherapy 2021, 143, 112166. [Google Scholar] [CrossRef]
- Mukonowenzou, N. C.; Dangarembizi, R.; Chivandi, E.; Nkomozepi, P.; Erlwanger, K. H. Administration of ursolic acid to new-born pups prevents dietary fructose-induced non-alcoholic fatty liver disease in Sprague Dawley rats. Journal of Developmental Origins of Health and Disease 2021, 12(1), 101–112. [Google Scholar] [CrossRef]
- Wang, X.; Ikejima, K.; Kon, K.; Arai, K.; Aoyama, T.; Okumura, K.; Abe, W.; Sato, N.; Watanabe, S. Ursolic Acid Ameliorates Hepatic Fibrosis in the Rat by Specific Induction of Apoptosis in Hepatic Stellate Cells. Journal of Hepatology 2011, 55(2), 379–387. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Huang, C.; Wang, A.; Zhu, X. Ursolic Acid Improves the Bacterial Community Mapping of the Intestinal Tract in Liver Fibrosis Mice. PeerJ 2020, 8, e9050. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.; Zhang, W.; Huang, C.; Chen, J.; He, W.; Wang, A.; Li, B.; Zhu, X. Ursolic Acid Ameliorates CCl₄-Induced Liver Fibrosis through the NOXs/ROS Pathway. Journal of Cellular Physiology 2018, 233(10), 6799–6813. [Google Scholar] [CrossRef] [PubMed]
- Quan, H. Y.; Kim, D. Y.; Kim, S. J.; Jo, H. K.; Kim, G. W.; Chung, S. H. Betulinic acid alleviates non-alcoholic fatty liver by inhibiting SREBP1 activity via the AMPK–mTOR–SREBP signaling pathway. Biochemical Pharmacology 2013, 85, 1330–1340. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Zhang, Y.; Feng, S.; Ding, Y.; Deng, X.; Feng, R.; Li, J.; Ma, T.; Huang, C. Betulinic acid prevents liver fibrosis by binding Lck and suppressing Lck in HSC activation and proliferation. Journal of Ethnopharmacology 2022, 296, 115459. [Google Scholar] [CrossRef]
- Kim, K. D.; Jung, H. Y.; Ryu, H. G.; Kim, B.; Jeon, J.; Yoo, H. Y.; Park, C. H.; Choi, B. H.; Hyun, C. K.; Kim, K. T.; Fang, S.; Yang, S. H.; Kim, J. B. Betulinic acid inhibits high-fat diet-induced obesity and improves energy balance by activating AMPK. Nutrition, Metabolism & Cardiovascular Diseases 2019, 29, 409–420. [Google Scholar] [CrossRef]
- Gu, M.; Zhao, P.; Zhang, S.; Fan, S.; Yang, L.; Tong, Q.; Ji, G.; Huang, C. Betulinic acid alleviates endoplasmic reticulum stress-mediated nonalcoholic fatty liver disease through activation of farnesoid X receptors in mice. British Journal of Pharmacology 2019, 176, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, Y.-L.; Lian, L.-H.; Xie, W.-X.; Li, X.; OuYang, B.-Q.; Bai, T.; Li, Q.; Yang, N.; Nan, J.-X. The anti-fibrotic effect of betulinic acid is mediated through the inhibition of NF-κB nuclear protein translocation. Chemico-Biological Interactions 2012, 195, 215–223. [Google Scholar] [CrossRef]
- Mu, Q.; Wang, H.; Tong, L.; Fang, Q.; Xiang, M.; Han, L.; Jin, L.; Yang, J.; Qian, Z.; Ning, G.; Zhang, Y.; Zhang, Z. Betulinic acid improves nonalcoholic fatty liver disease through YY1/FAS signaling pathway. The FASEB Journal 2020, 34, 13033–13048. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Wang, Y.; Ge, X.; Jiao, T.; Yin, J.; Wang, K.; Li, C.; Guo, S.; Xie, X.; Xie, C.; Nan, F. Discovery of betulinic acid derivatives as potent intestinal farnesoid X receptor antagonists to ameliorate nonalcoholic steatohepatitis. Journal of Medicinal Chemistry 2022, 65, 13452–13472. [Google Scholar] [CrossRef]
- Yan, S. L.; Yang, H. T.; Lee, Y. J.; Lin, C. C.; Chang, M. H.; Yin, M. C. Asiatic acid ameliorates hepatic lipid accumulation and insulin resistance in mice consuming a high-fat diet. Journal of Agricultural and Food Chemistry 2014, 62, 4625–4631. [Google Scholar] [CrossRef]
- Wang, D.; Lao, L.; Pang, X.; Qiao, Q.; Pang, L.; Feng, Z.; Bai, F.; Sun, X.; Lin, X.; Wei, J. Asiatic acid from Potentilla chinensis alleviates non-alcoholic fatty liver by regulating endoplasmic reticulum stress and lipid metabolism. International Immunopharmacology 2018, 65, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chen, Q.; Guo, A.; Fan, J.; Wang, R.; Zhang, H. Asiatic acid attenuates CCl₄-induced liver fibrosis in rats by regulating the PI3K/AKT/mTOR and Bcl-2/Bax signaling pathways. International Immunopharmacology 2018, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-K.; Kim, J.-H.; Park, K.-M.; Oh, K.-H.; Oh, U.; Hwang, S.-J. Preparation and evaluation of a titrated extract of Centella asiatica injection in the form of an extemporaneous micellar solution. International Journal of Pharmaceutics 1997, 146, 63–70. [Google Scholar] [CrossRef]
- Lu, Y. H.; Chen, M. C.; Liu, F.; Xu, Z.; Tian, X. T.; Xie, Y.; Huang, C. G. Synthesis and cytotoxic activity of novel C-23-modified asiatic acid derivatives. Molecules 2020, 25, 3709. [Google Scholar] [CrossRef]
- Li, J. F.; Huang, R. Z.; Yao, G. Y.; Ye, M. Y.; Wang, H. S.; Pan, Y. M.; Xiao, J. T. Synthesis and biological evaluation of novel aniline-derived asiatic acid derivatives as potential anticancer agents. European Journal of Medicinal Chemistry 2014, 86, 175–188. [Google Scholar] [CrossRef]
- Jing, Y.; Wang, G.; Ge, Y.; Xu, M.; Gong, Z. Synthesis, anti-tumor and anti-angiogenic activity evaluations of asiatic acid amino acid derivatives. Molecules 2015, 20, 7309–7324. [Google Scholar] [CrossRef]
- Jew, S. S.; Yoo, C. H.; Lim, D. Y.; Kim, H.; Mook-Jung, I.; Jung, M. W.; Choi, H.; Jung, Y. H.; Kim, H.; Park, H. G. Structure-activity relationship study of asiatic acid derivatives against beta amyloid (Aβ)-induced neurotoxicity. Bioorganic & Medicinal Chemistry Letters 2000, 10, 119–121. [Google Scholar] [CrossRef]
- Nyakudya, T,T,; Mukwevho, E.; Nkomozepi, P.; Erlwanger, K.H. Neonatal intake of oleanolic acid attenuates the subsequent development of high fructose diet-induced non-alcoholic fatty liver disease in rats. Journal of Developmental Origins of Health and Disease 2018, 9, 500–510. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Chang, H.-Y.; Wang, C. C. N.; Chu, F.-Y.; Shen, H.-Y.; Chen, C.-J.; Lim, Y.-P. Oleanolic acid inhibits liver X receptor alpha and pregnane X receptor to attenuate ligand-induced lipogenesis. Journal of Agricultural and Food Chemistry 2018, 66, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Zuo, G.; Xu, W.; Gao, H.; Yang, Y.; Yamahara, J.; Wang, J.; Li, Y. Oleanolic acid diminishes liquid fructose-induced fatty liver in rats: role of modulation of hepatic sterol regulatory element-binding protein-1c–mediated expression of genes responsible for de novo fatty acid synthesis. Evidence-Based Complementary and Alternative Medicine 2013, 2013, 534084. [Google Scholar] [CrossRef] [PubMed]
- Djeziri, F. Z.; Belarbi, M.; Murtaza, B.; Hichami, A.; Benammar, C.; Khan, N. A. Oleanolic acid improves diet-induced obesity by modulating fat preference and inflammation in mice. Biochimie 2018, 152, 110–120. [Google Scholar] [CrossRef]
- Nyakudya, T. T.; Mukwevho, E.; Nkomozepi, P.; Erlwanger, K. H. Neonatal intake of oleanolic acid attenuates the subsequent development of high fructose diet-induced non-alcoholic fatty liver disease in rats. Journal of Developmental Origins of Health and Disease 2018, 9, 500–510. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Zhang, W.; Zhang, X.; Liao, N.; Wang, Z.; Li, W.; Qin, X.; Hai, C. Oleanolic acid improves hepatic insulin resistance via antioxidant, hypolipidemic and anti-inflammatory effects. Molecular and Cellular Endocrinology 2013, 376, 70–80. [Google Scholar] [CrossRef]
- Gamede, M.; Mabuza, L.; Ngubane, P.; Khathi, A. Plant-derived oleanolic acid ameliorates markers associated with non-alcoholic fatty liver disease in a diet-induced pre-diabetes rat model. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 2019, 12, 1953–1962. [Google Scholar] [CrossRef]
- Yao, L.; Wang, M.; Zhang, J.; Luo, X.; Yuan, C.; Bai, R.; Wang, T.; Xi, Y.; Li, C.; Ke, D.; Yamahara, J.; Li, Y.; Yi, Y.; Wang, S.; Wang, J. Oleanolic acid inhibits SCD1 gene expression to ameliorate fructose-induced hepatosteatosis through SREBP1c-dependent and -independent mechanisms. Molecular Nutrition & Food Research 2023, 67, e2200533. [Google Scholar] [CrossRef]
- Xue, C.; Li, Y.; Lv, H.; Zhang, L.; Bi, C.; Dong, N.; Shan, A.; Wang, J. Oleanolic acid targets the gut–liver axis to alleviate metabolic disorders and hepatic steatosis. Journal of Agricultural and Food Chemistry 2021, 69, 7884–7897. [Google Scholar] [CrossRef]
- Pi, X.; Zuo, H.; Wang, Y.; Zheng, W.; Zhou, H.; Deng, L.; Song, H. Oleanolic acid alleviates ischemia–reperfusion injury in rat severe steatotic liver via KEAP1/NRF2/ARE signaling. International Immunopharmacology 2024, 138, 112617. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Wang, Y.; Guo, Q.; Zheng, W.; Zhou, H.; Deng, L.; Xu, N.; Song, H. Oleanolic acid alleviates intestinal injury after hepatic ischemia–reperfusion under steatosis via PPARG-dependent M2 macrophage polarization. International Immunopharmacology 2026, 171, 116162. [Google Scholar] [CrossRef]
- Hu, M.; Yang, J.; Chen, Y.; Zhang, Y.; Liu, C. H.; Feng, Y.; Ye, B.; Li, D. Celastrol ameliorates metabolic dysfunction–associated steatohepatitis by regulating the CYP7B1-mediated alternative bile acid synthetic pathway. Phytomedicine 2025, 147, 157172. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Ye, C.; Lin, B.; Zhang, T.; Chen, Q.; Yu, C.; Wan, X. Celastrol ameliorates fibrosis in Western diet/tetrachloromethane-induced nonalcoholic steatohepatitis by suppressing Notch/osteopontin signaling. Phytomedicine 2025, 137, 156369. [Google Scholar] [CrossRef]
- Luo, M.; Wang, Y.; Ma, Y.; Li, J.; Wang, J.; Liu, C. Celastrol stabilizes glycolipid metabolism in hepatic steatosis by binding and regulating the peroxisome proliferator-activated receptor γ signaling pathway. Metabolites 2024, 14, 64. [Google Scholar] [CrossRef]
- Xue, J.; Liu, Y.; Liu, B.; Jia, X.; Fang, X.; Qin, S.; Zhang, Y. Celastrus orbiculatus Thunb. extracts and celastrol alleviate NAFLD by preserving mitochondrial function through activating the FGF21/AMPK/PGC-1α pathway. Frontiers in Pharmacology 2024, 15, 1444117. [Google Scholar] [CrossRef]
- Han, L. P.; Sun, B.; Li, C. J.; Xie, Y.; Chen, L. M. Effect of celastrol on toll-like receptor 4–mediated inflammatory response in free fatty acid–induced HepG2 cells. International Journal of Molecular Medicine 2018, 42, 2053–2061. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Ge, H. Y.; Gu, Y. J.; Cao, F. F.; Yang, C. X.; Uzan, G.; Peng, B.; Zhang, D. H. Celastrol reverses palmitic acid–caused TLR4–MD2 activation-dependent insulin resistance via disrupting MD2-related cellular binding to palmitic acid. Journal of Cellular Physiology 2018, 233, 6814–6824. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. E.; Lee, M. H.; Nam, D. H.; Song, H. K.; Kang, Y. S.; Lee, J. E.; Kim, H. W.; Cha, J. J.; Hyun, Y. Y.; Han, S. Y.; Han, K. H.; Han, J. Y.; Cha, D. R. Celastrol, an NF-κB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLOS ONE 2013, 8, e62068. [Google Scholar] [CrossRef]
- Ma, X.; Xu, L.; Alberobello, A. T.; Gavrilova, O.; Bagattin, A.; Skarulis, M.; Liu, J.; Finkel, T.; Mueller, E. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1–PGC1α transcriptional axis. Cell Metabolism 2015, 22, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Canová, N.; Šípková, J.; Arora, M.; Pavlíková, Z.; Kučera, T.; Šeda, O.; Šopin, T.; Vacík, T.; Slanař, O. Effects of celastrol on the heart and liver galaninergic system expression in a mouse model of Western-type diet-induced obesity and metabolic dysfunction–associated steatotic liver disease and steatohepatitis. Frontiers in Pharmacology 2025, 16, 1476994. [Google Scholar] [CrossRef]
- Benmouna, M.; Benammar, C.; Khan, A. S.; Djeziri, F. Z.; Hichami, A.; Khan, N. A. Celastrol improves preference for a fatty acid, and taste bud and systemic inflammation in diet-induced obese mice. Nutrients 2025, 17, 1308. [Google Scholar] [CrossRef]
- Sun, J.; Wang, H. J.; Yu, J.; Li, T.; Han, Y. Protective effect of celastrol on type 2 diabetes mellitus with nonalcoholic fatty liver disease in mice. Food Science & Nutrition 2020, 8, 6207–6216. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, C.; Liu, X.; Li, M.; Gao, M.; Liu, X.; Fang, F.; Chang, Y. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Molecular Metabolism 2017, 6, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Takeuchi, N.; Kotani, S.; Kumagai, A. Effects of glycyrrhizin and cortisone on cholesterol metabolism in the rat. Endocrinologia Japonica 1970, 17, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Negishi, M.; Irie, A.; Nagata, N.; Ichikawa, A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochimica et Biophysica Acta 1991, 1066, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lim, W. Y.; Chia, Y. Y.; Liong, S. Y.; Ton, S. H.; Kadir, K. A.; Husain, S. N. Lipoprotein lipase expression, serum lipid and tissue lipid deposition in orally administered glycyrrhizic acid-treated rats. Lipids in Health and Disease 2009, 8, 31. [Google Scholar] [CrossRef]
- Eu, C. H.; Lim, W. Y.; Ton, S. H.; Abdul Kadir, K. Glycyrrhizic acid improved lipoprotein lipase expression, insulin sensitivity, serum lipid and lipid deposition in high-fat diet-induced obese rats. Lipids in Health and Disease 2010, 9, 81. [Google Scholar] [CrossRef]
- Yaw, H. P.; Ton, S. H.; Chin, H. F.; Karim, M. K.; Fernando, H. A.; Kadir, K. A. Modulation of lipid metabolism in glycyrrhizic acid-treated rats fed on a high-calorie diet and exposed to short- or long-term stress. International Journal of Physiology, Pathophysiology and Pharmacology 2015, 7, 61–75. [Google Scholar]
- Shi, L.; Guo, S.; Zhang, S.; Gao, X.; Liu, A.; Wang, Q.; Zhang, T.; Zhang, Y.; Wen, A. Glycyrrhetinic acid attenuates disturbed vitamin A metabolism in non-alcoholic fatty liver disease through AKR1B10. European Journal of Pharmacology 2020, 883, 173167. [Google Scholar] [CrossRef]
- Wang, C.; Duan, X.; Sun, X.; Liu, Z.; Sun, P.; Yang, X.; Sun, H.; Liu, K.; Meng, Q. Protective effects of glycyrrhizic acid from edible botanical Glycyrrhiza glabra against non-alcoholic steatohepatitis in mice. Food & Function 2016, 7, 3716–3723. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, S.; Wei, Y.; Sun, Y.; Yang, Y.; Lin, B.; Li, Y.; Wang, C. Glycyrrhizic acid restores the downregulated hepatic ACE2 signaling in the attenuation of mouse steatohepatitis. European Journal of Pharmacology 2024, 967, 176365. [Google Scholar] [CrossRef]
- Zhou, L.; Yi, Y.; Lin, B.; Qiu, Z.; Wang, C.; Li, Y. Glycyrrhizic acid mitigates hepatocyte steatosis and inflammation through ACE2 stabilization via dual modulation of AMPK activation and MDM2 inhibition. European Journal of Pharmacology 2025, 1002, 177817. [Google Scholar] [CrossRef]
- Ou-Yang, Q.; Xuan, C. X.; Wang, X.; Luo, H. Q.; Liu, J. E.; Wang, L. L.; Li, T. T.; Chen, Y. P.; Liu, J. 3-Acetyl-oleanolic acid ameliorates non-alcoholic fatty liver disease in high fat diet-treated rats by activating AMPK-related pathways. Acta Pharmacologica Sinica 2018, 39, 1284–1293. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Shen, Q.; Wu, J.; Li, J. X. Oleanolic acid derivative HA-20 inhibits adipogenesis in a manner involving PPARγ-FABP4/aP2 pathway. Journal of Molecular Endocrinology 2021, 66, 245–258. [Google Scholar] [CrossRef]
- Ma, H.; Bao, Y.; Niu, S.; Wang, S.; Li, Y.; He, H.; Zhang, N.; Fang, W. Structure optimization of 12β-O-γ-glutamyl oleanolic acid derivatives resulting in potent FXR antagonist/modulator for NASH therapy. Pharmaceuticals 2023, 16, 758. [Google Scholar] [CrossRef]
- Radwan, M. O.; Kadasah, S. F.; Aljubiri, S. M.; Alrefaei, A. F.; El-Maghrabey, M. H.; El Hamd, M. A.; Tateishi, H.; Otsuka, M.; Fujita, M. Harnessing oleanolic acid and its derivatives as modulators of metabolic nuclear receptors. Biomolecules 2023, 13, 1465. [Google Scholar] [CrossRef]
- de Oliveira Eloy, J.; Saraiva, J.; de Albuquerque, S.; Marchetti, J. M. Solid dispersion of ursolic acid in Gelucire 50/13: a strategy to enhance drug release and trypanocidal activity. AAPS PharmSciTech 2012, 13, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Sun, C.; Zhu, X.; Xu, W.; Yu, J.; Zhang, Q.; Dushkin, A. V.; Su, W. Inositol hexanicotinate self-micelle solid dispersion is an efficient drug delivery system in the mouse model of non-alcoholic fatty liver disease. International Journal of Pharmaceutics 2021, 602, 120576. [Google Scholar] [CrossRef] [PubMed]
- Soica, C.; Oprean, C.; Borcan, F.; Danciu, C.; Trandafirescu, C.; Coricovac, D.; Crăiniceanu, Z.; Dehelean, C. A.; Munteanu, M. The synergistic biologic activity of oleanolic and ursolic acids in complex with hydroxypropyl-γ-cyclodextrin. Molecules 2014, 19, 4924–4940. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hu, D.; Zhang, D.; Xu, G.; Wu, D.; Gao, C.; Meng, L.; Feng, X.; Cheng, W.; Wang, Z.; Yang, Y.; Tang, X. Encapsulation of oleanolic acid into cyclodextrin metal-organic frameworks by co-crystallization: preparation, structure characterization and its effect on a zebrafish larva NAFLD model. Food Research International 2025, 204, 115936. [Google Scholar] [CrossRef]
- Zhou, X. J.; Hu, X. M.; Yi, Y. M.; Wan, J. Preparation and body distribution of freeze-dried powder of ursolic acid phospholipid nanoparticles. Drug Development and Industrial Pharmacy 2009, 35, 305–310. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Ding, J.; Xu, H.; Dai, X.; Hou, Z.; Zhang, K.; Sun, K.; Sun, W. Delivery of ursolic acid in polymeric nanoparticles effectively promotes the apoptosis of gastric cancer cells through enhanced inhibition of cyclooxygenase-2. International Journal of Pharmaceutics 2013, 441, 261–268. [Google Scholar] [CrossRef]
- Caldeira de Araújo Lopes, S.; Novais, M. V. M.; Teixeira, C. S.; Honorato-Sampaio, K.; Pereira, M. T.; Ferreira, L. A.; Braga, F. C.; Oliveira, M. C. Preparation, physicochemical characterization, and cell viability evaluation of long-circulating and pH-sensitive liposomes containing ursolic acid. BioMed Research International 2013, 2013, 467147. [Google Scholar] [CrossRef]
- Dong, H.; Yang, X.; Xie, J.; Xiang, L.; Li, Y.; Ou, M.; Chi, T.; Liu, Z.; Yu, S.; Gao, Y.; Chen, J.; Shao, J.; Jia, L. UP12, a novel ursolic acid derivative with potential for targeting multiple signaling pathways in hepatocellular carcinoma. Biochemical Pharmacology 2015, 93, 151–162. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Z.; Xie, X.; Wang, C.; You, J.; Mo, F.; Jin, B.; Chen, J.; Shao, J.; Chen, H.; Jia, L. Dendrimeric anticancer prodrugs for targeted delivery of ursolic acid to folate receptor-expressing cancer cells. European Journal of Pharmaceutical Sciences 2015, 70, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, H. L.; Abrego, G.; Garduño-Ramirez, M. L.; Clares, B.; Calpena, A. C.; García, M. L. Design and optimization of oleanolic/ursolic acid-loaded nanoplatforms for ocular anti-inflammatory applications. Nanomedicine 2015, 11, 521–530. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, D.; Ding, J.; Xu, H.; Li, X.; Sun, W. Efficient delivery of ursolic acid by poly(N-vinylpyrrolidone)-block-poly(ε-caprolactone) nanoparticles for inhibiting the growth of hepatocellular carcinoma in vitro and in vivo. International Journal of Nanomedicine 2015, 10, 1909–1920. [Google Scholar] [CrossRef]
- Jin, H.; Pi, J.; Yang, F.; Wu, C.; Cheng, X.; Bai, H.; Huang, D.; Jiang, J.; Cai, J.; Chen, Z. W. Ursolic acid-loaded chitosan nanoparticles induce potent anti-angiogenesis in tumor. Applied Microbiology and Biotechnology 2016, 100, 6643–6652. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Liu, Z.; Wang, H.; Gu, X.; Wang, S.; Zhang, B.; Luan, H.; Zhu, Z. Ursolic acid nanocrystals for dissolution rate and bioavailability enhancement: influence of different particle size. Current Drug Delivery 2016, 13, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Baishya, R.; Nayak, D. K.; Kumar, D.; Sinha, S.; Gupta, A.; Ganguly, S.; Debnath, M. C. Ursolic acid loaded PLGA nanoparticles: in vitro and in vivo evaluation to explore tumor targeting ability on B16F10 melanoma cell lines. Pharmaceutical Research 2016, 33, 2691–2703. [Google Scholar] [CrossRef]
- Fan, N.; Zhao, J.; Zhao, W.; Zhang, X.; Song, Q.; Shen, Y.; Shum, H. C.; Wang, Y.; Rong, J. Celastrol-loaded lactosylated albumin nanoparticles attenuate hepatic steatosis in non-alcoholic fatty liver disease. Journal of Controlled Release 2022, 347, 44–54. [Google Scholar] [CrossRef]
- Fan, N.; Zhao, J.; Zhao, W.; Shen, Y.; Song, Q.; Shum, H. C.; Wang, Y.; Rong, J. Biodegradable celastrol-loaded albumin nanoparticles ameliorate inflammation and lipid accumulation in diet-induced obese mice. Biomaterials Science 2022, 10, 984–996. [Google Scholar] [CrossRef]
- Zheng, T.; Gan, X.; Luo, J.; Shi, Z.; Zhao, Y.; Wang, Y.; Chen, J.; Yu, C. Hyaluronic acid act as drug self-assembly chaperone and co-assembled with celastrol for ameliorating non-alcoholic steatohepatitis. International Journal of Biological Macromolecules 2024, 282, 137289. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhang, Y.; Ouyang, H.; Gong, T.; Zhang, Z.; Cao, X.; Fu, Y. Targeted delivery of celastrol via chondroitin sulfate-derived hybrid micelles for alleviating symptoms in nonalcoholic fatty liver disease. ACS Applied Bio Materials 2023, 6, 4877–4893. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhu, H.; Wang, K.; Hu, H.; Wei, X.; Xu, D.; Zhang, J.; Liu, Y.; Chen, J.; Shen, Y.; Qiu, N.; Xu, X. Oral celastrol micelles forming high-density lipoprotein corona targeting hepatocytes for MASLD treatment. Advanced Science 2025, 12, e00854. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yonebayashi, S.; Yoshida, M.; Shimizu, K.; Aotsuka, T.; Takayama, K. Improvement in the bioavailability of poorly absorbed glycyrrhizin via various non-vascular administration routes in rats. International Journal of Pharmaceutics 2003, 265, 95–102. [Google Scholar] [CrossRef]
Figure 1.
Representative structures of each group of terpenes.
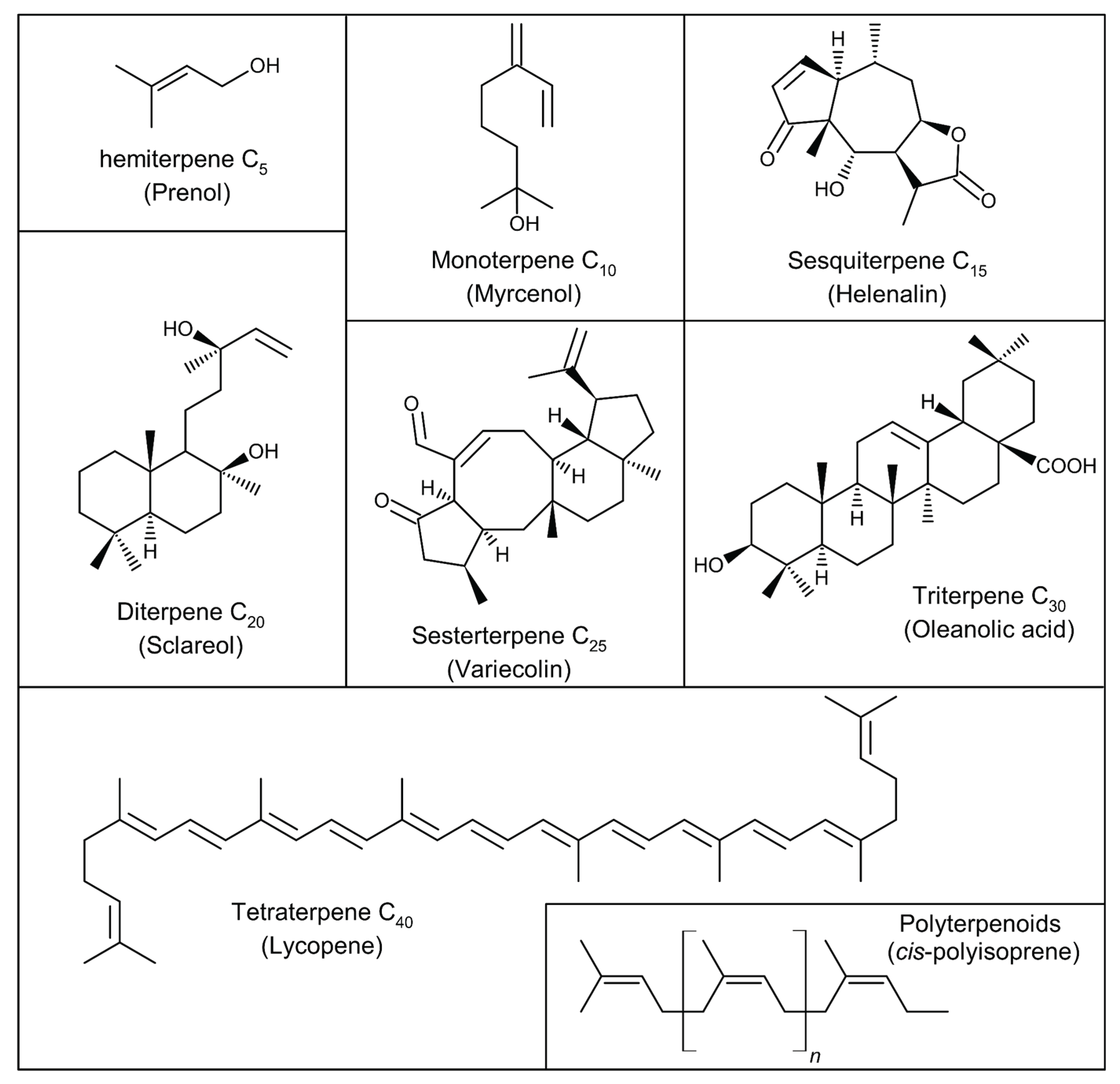
Figure 2.
Schematic representation of the synthesis of the direct precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) involved in terpenoid biosynthesis. The mevalonate (MVA) pathway occurs in the cytosol and provides precursors for the production of sesqui- and triterpenes, whereas the plastid-localized 1-deoxy-D-xylulose-5-phosphate (DXP) pathway supports the biosynthesis of mono-, di-, and tetraterpenes. Abbreviations: GDPS, geranyl diphosphate synthase; FDPS, farnesyl diphosphate synthase; GGDPS, geranylgeranyl diphosphate synthase; SQS, squalene synthase.
Figure 2.
Schematic representation of the synthesis of the direct precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) involved in terpenoid biosynthesis. The mevalonate (MVA) pathway occurs in the cytosol and provides precursors for the production of sesqui- and triterpenes, whereas the plastid-localized 1-deoxy-D-xylulose-5-phosphate (DXP) pathway supports the biosynthesis of mono-, di-, and tetraterpenes. Abbreviations: GDPS, geranyl diphosphate synthase; FDPS, farnesyl diphosphate synthase; GGDPS, geranylgeranyl diphosphate synthase; SQS, squalene synthase.
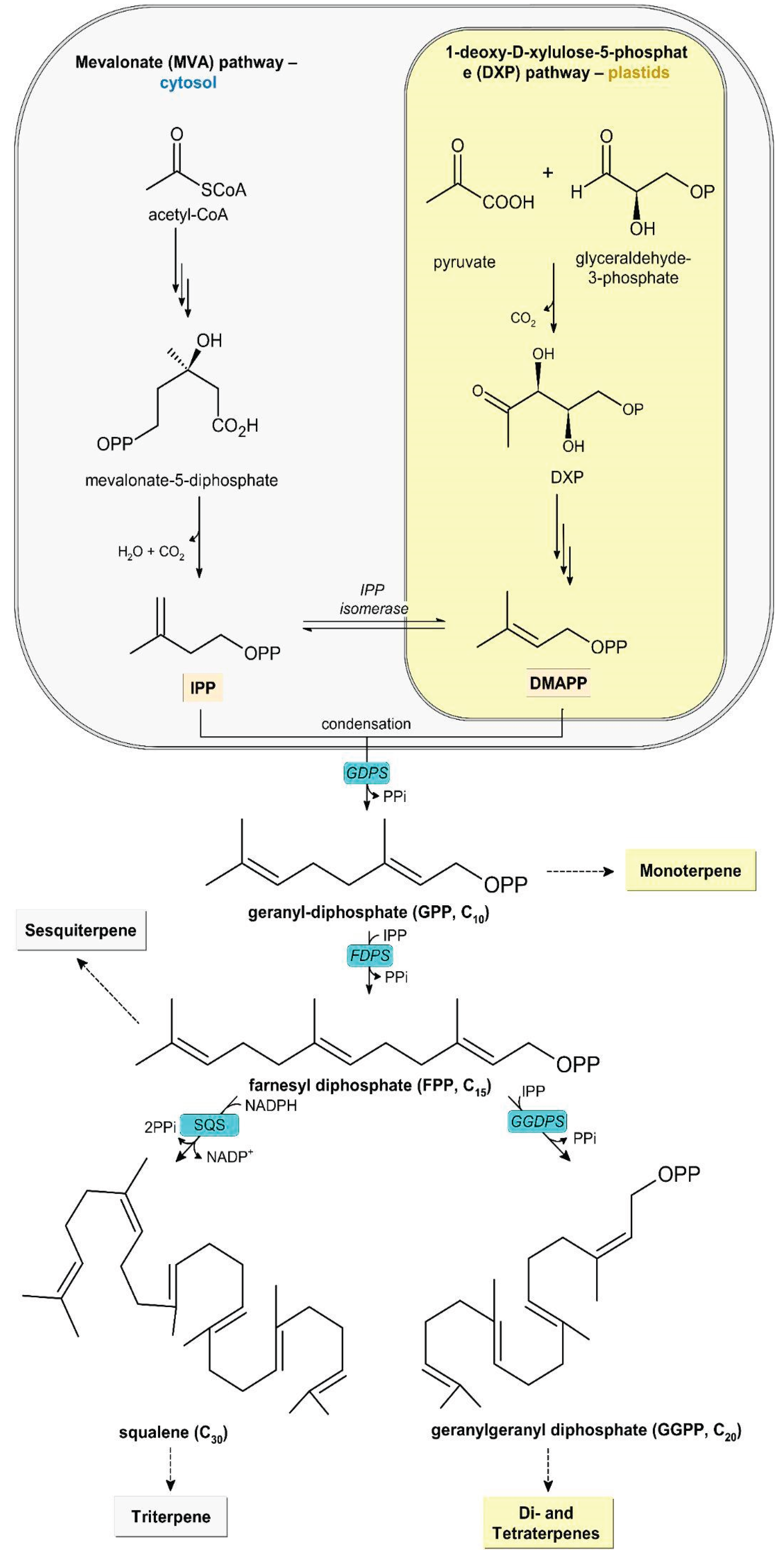
Figure 3.
Oxidosqualene cyclase (OSC)-catalyzed 2,3-oxidosqualene cyclization pathways leading to sterols and triterpene compounds through protosteryl and dammarenyl carbocation intermediates, respectively. Abbreviations: OSC, oxidosqualene cyclase; SQE, squalene epoxidase.
Figure 3.
Oxidosqualene cyclase (OSC)-catalyzed 2,3-oxidosqualene cyclization pathways leading to sterols and triterpene compounds through protosteryl and dammarenyl carbocation intermediates, respectively. Abbreviations: OSC, oxidosqualene cyclase; SQE, squalene epoxidase.
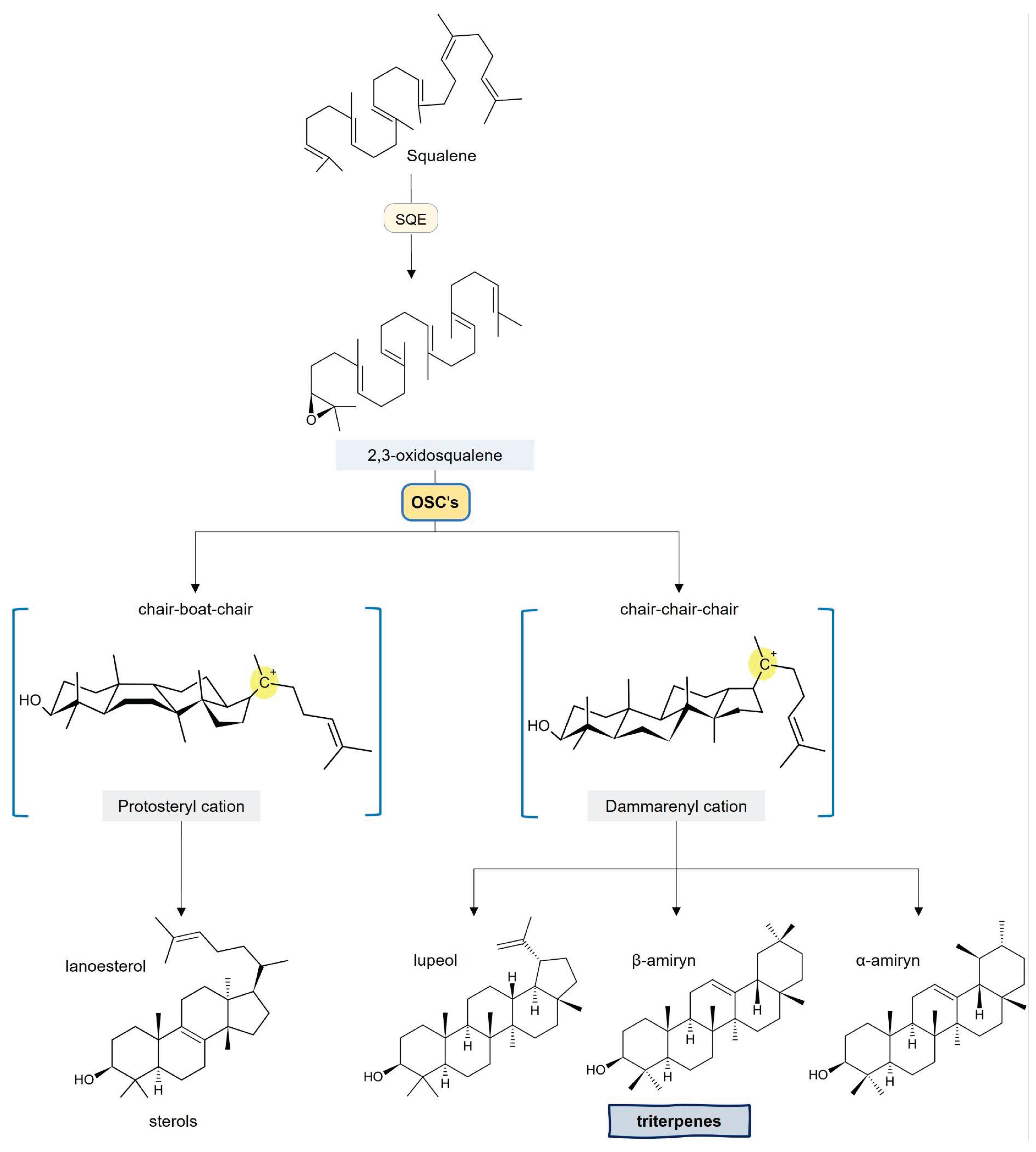
Figure 4.
General structure of pentacyclic triterpenes classified according to their carbon skeletons into oleananes, ursanes, and lupanes.
Figure 4.
General structure of pentacyclic triterpenes classified according to their carbon skeletons into oleananes, ursanes, and lupanes.
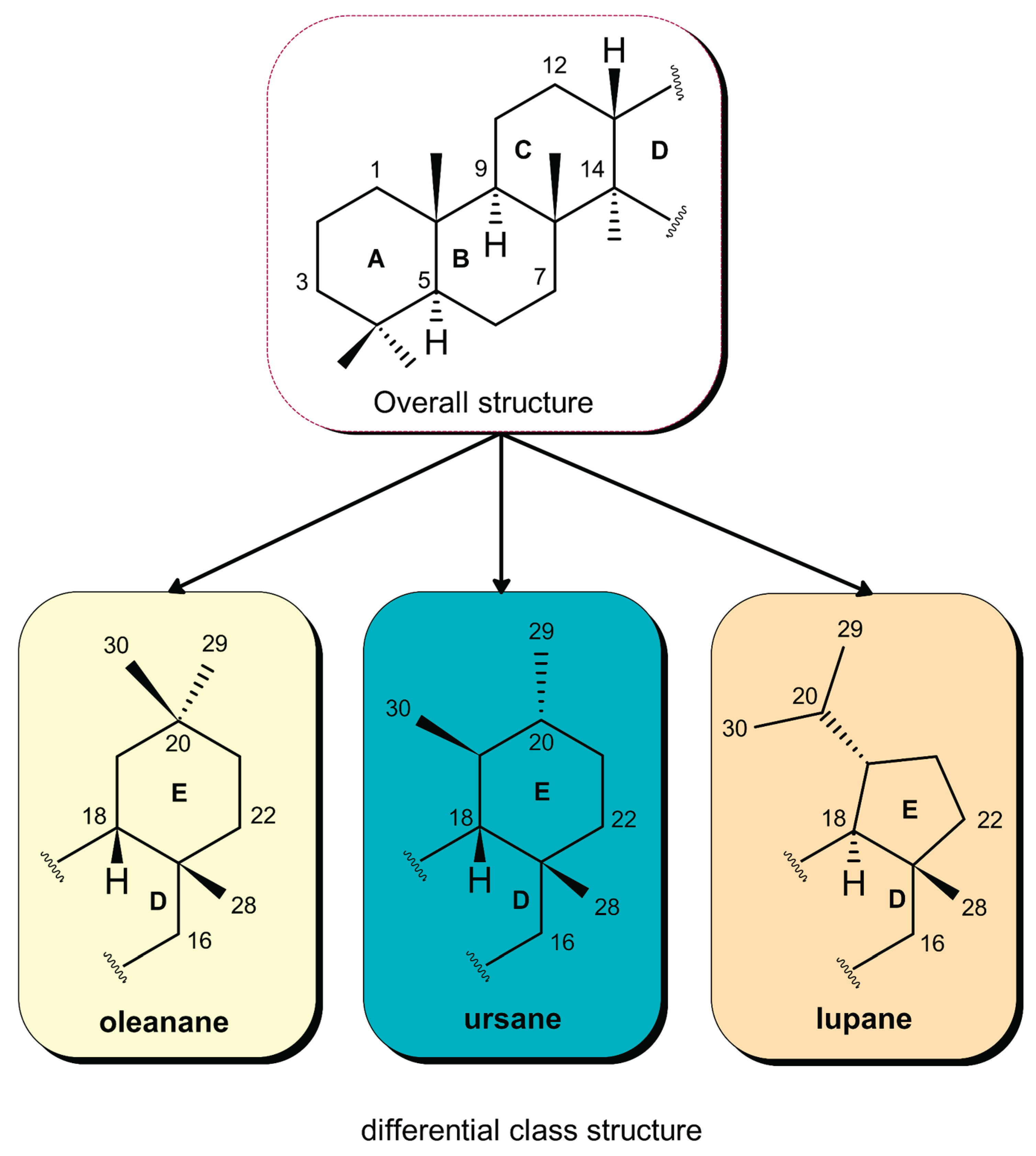
Figure 5.
Main mechanisms involved in the development of MASLD/MASH and its complications.
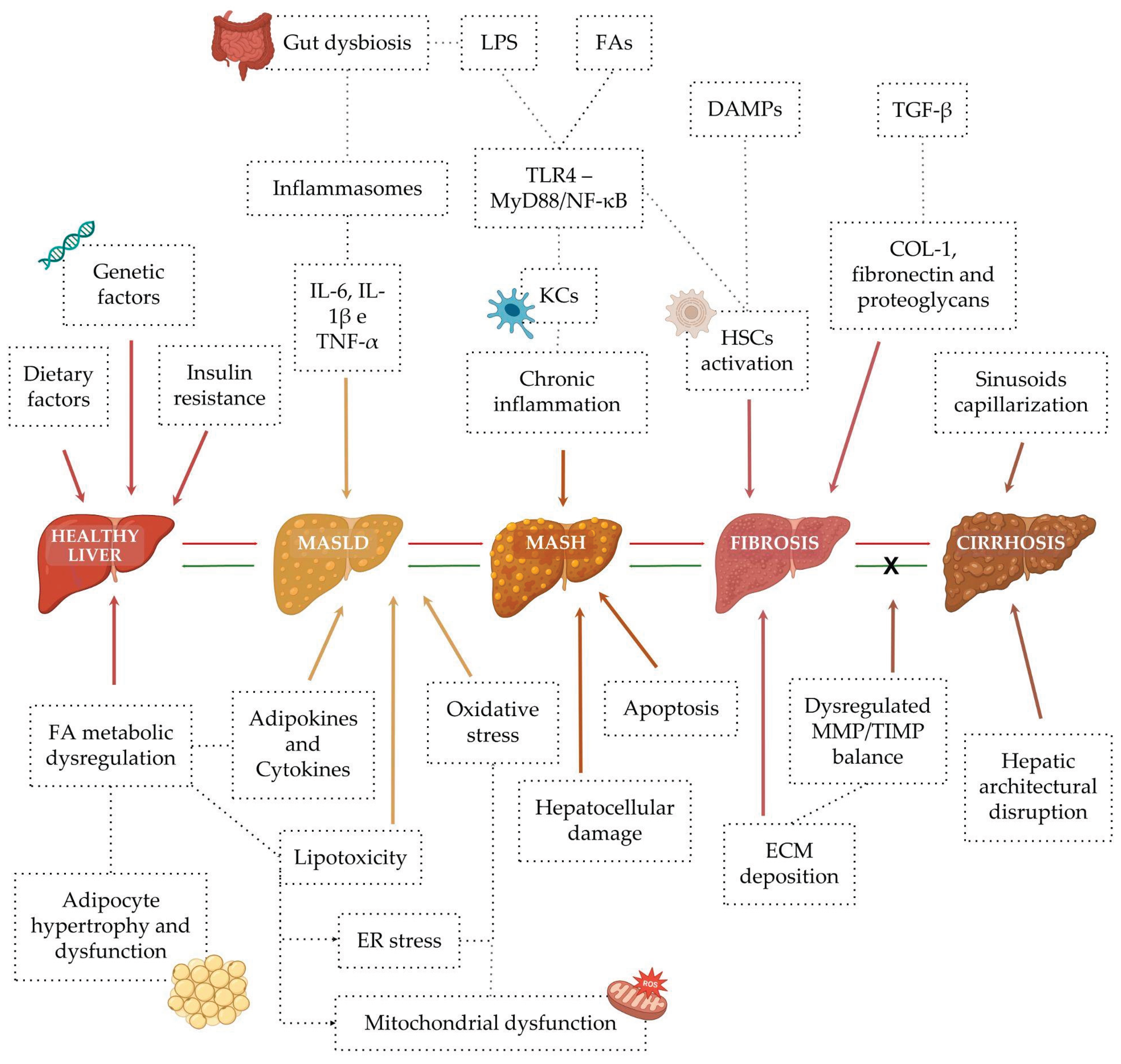
Figure 6.
Multitarget mechanisms of pentacyclic triterpenes in MASLD and MASH.
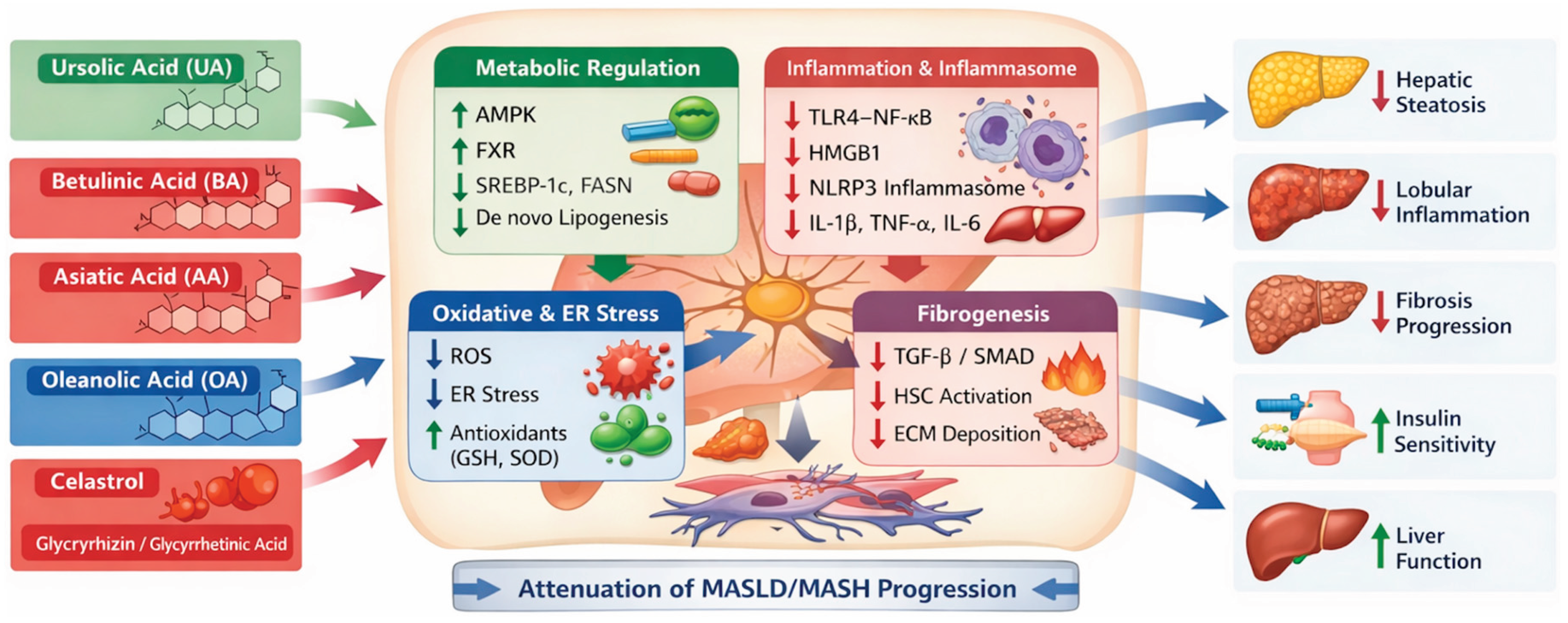

Table 1.
Summary of pentacyclic triterpenes in MASLD/MASH models.
| Compound | Triterpene class | Main molecular mechanisms | Experimental model | Main outcomes | Key references |
|---|---|---|---|---|---|
| Ursolic acid | Ursane | AMPK activation; inhibition of TGF-β/SMAD and NLRP3; antioxidant effects | HFD, MCD, CCl₄, TAA MASLD/MASH models | Reduced steatosis, inflammation, fibrosis; improved liver enzymes | [20,119,120] |
| Betulinic acid | Lupane | AMPK and FXR activation; inhibition of SREBP-1c, YY1/FAS; antifibrotic | HFD- and MCD-fed mice; TAA/CCl₄ fibrosis | Attenuation of steatosis, insulin resistance, fibrosis | [126,129,131] |
| Asiatic acid | Ursane | NF-κB and PI3K/AKT/mTOR inhibition; ER stress reduction | HFD-fed mice; CCl₄-induced MASH | Reduced lipid accumulation, inflammation, oxidative stress | [133,134,135] |
| Oleanolic acid | Oleanane | PPARα activation; NF-κB inhibition; lipogenesis suppression | High-fructose and HFD steatosis models | Improved insulin sensitivity; reduced hepatic lipid content | [142,143,149] |
| Celastrol | Quinone methide triterpenoid | AMPK activation; SREBP-1c/FASN suppression; NLRP3 and HMGB1/NF-κB inhibition | Diet-induced MASH mouse models | Reduced steatosis, inflammation and fibrosis | [152,155,157] |
| Glycyrrhizin / Glycyrrhetinic acid | Oleanane-type saponin/metabolite | NF-κB, MAPK and NLRP3 inhibition; bile acid homeostasis modulation | MCD-induced MASH; chronic hepatitis clinical use | Attenuation of inflammation and fibrosis; hepatoprotection | [164,169,171,172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



